 Jia-Yu Xue1,2,3
Jia-Yu Xue1,2,3 Tao Zhao3Yang Liu1Yang Liu4Yong-Xia Zhang5
Tao Zhao3Yang Liu1Yang Liu4Yong-Xia Zhang5 Guo-Qiang Zhang6Hongfeng Chen7Guang-Can Zhou8*
Guo-Qiang Zhang6Hongfeng Chen7Guang-Can Zhou8* Shou-Zhou Zhang1*
Shou-Zhou Zhang1* Zhu-Qing Shao4*
Zhu-Qing Shao4*- 1Shenzhen Key Laboratory of Southern Subtropical Plant Diversity, Fairy Lake Botanical Garden, Shenzhen and Chinese Academy of Sciences, Shenzhen, China
- 2Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China
- 3VIB-UGent Center for Plant Systems Biology and Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium
- 4State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, China
- 5College of Life Sciences and Oceanography, Shenzhen University, Shenzhen, China
- 6Key Laboratory of National Forestry and Grassland Administration for Orchid Conservation and Utilization at College of Landscape Architecture, Fujian Agriculture and Forestry University, Fuzhou, China
- 7South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
- 8College of Agricultural and Biological Engineering (College of Tree Peony), Heze University, Heze, China
Orchids are one of the most diverse flowering plant families, yet possibly maintain the smallest number of the nucleotide-binding site-leucine-rich repeat (NBS-LRR) type plant resistance (R) genes among the angiosperms. In this study, a genome-wide search in four orchid taxa identified 186 NBS-LRR genes. Furthermore, 214 NBS-LRR genes were identified from seven orchid transcriptomes. A phylogenetic analysis recovered 30 ancestral lineages (29 CNL and one RNL), far fewer than other angiosperm families. From the genetics aspect, the relatively low number of ancestral R genes is unlikely to explain the low number of R genes in orchids alone, as historical gene loss and scarce gene duplication has continuously occurred, which also contributes to the low number of R genes. Due to recent sharp expansions, Phalaenopsis equestris and Dendrobium catenatum having 52 and 115 genes, respectively, and exhibited an “early shrinking to recent expanding” evolutionary pattern, while Gastrodia elata and Apostasia shenzhenica both exhibit a “consistently shrinking” evolutionary pattern and have retained only five and 14 NBS-LRR genes, respectively. RNL genes remain in extremely low numbers with only one or two copies per genome. Notably, all of the orchid RNL genes belong to the ADR1 lineage. A separate lineage, NRG1, was entirely absent and was likely lost in the common ancestor of all monocots. All of the TNL genes were absent as well, coincident with the RNL NRG1 lineage, which supports the previously proposed notion that a potential functional association between the TNL and RNL NRG1 genes.
Introduction
Plants are exposed to the threat of pathogens on a day-to-day basis in their natural habitats. In order to survive, plants have developed systems to protect themselves from invading pathogens. Specifically, plants have evolved physical barriers like the surface composed of cuticle and wax, to block pathogens or the release chemical components like phenols, terpenes and compounds containing sulfur or nitrogen, to deter or dispose of invading enemies. Moreover, plants have an innate immune system for inducing rapid defense responses. This plant-specific immune system triggers a series of hypersensitive reactions after recognizing invading pathogens, resulting in apoptosis of infected cells, which halts the replication and spread of pathogen.
The core of this defending system involves a series of specific genes, namely, disease resistance (R) genes, which detect pathogens and trigger downstream resistance reactions. Five types of R genes have been discovered, including nucleotide-binding site and leucine-reach repeats (NBS-LRR), receptor-like protein (RLP), serine/theorine kinase (STK), receptor-like kinase (RLK) genes, and other genes that do not contain regular domains. Among all types of R genes, the NBS-LRR gene family is the largest and most important, containing over 60% of characterized R genes (Meyers et al., 2005; McHale et al., 2006; Friedman and Baker, 2007; Kourelis and van der Hoorn, 2018). This type of R genes originated early in the green plant lineage (Xue et al., 2012; Shao et al., 2019), and has expanded into a large gene family in angiosperms, usually consisting of hundreds of members in an individual genome. These members actively evolved with frequent recombinations occurring between paralogs, gene duplications and losses, and high substitution rates. Since the first genome-wide analysis was conducted on NBS-LRR genes in Arabidopsis thaliana (Meyers et al., 2003a), this gene family has been comprehensively studied across tens of plant genomes, most of which belong to the rosid lineage of the eudicots and Poaceae of the monocots (Yang et al., 2008; Porter et al., 2009; Li et al., 2010a; Lozano et al., 2012; Luo et al., 2012; Andolfo et al., 2013; Wu et al., 2014; Zhong et al., 2018).
NBS-LRR genes are divided into three classes, the TIR-NBS-LRR (TNL), CC-NBS-LRR (CNL) and RPW8-NBS-LRR (RNL), which are distinguished by the presence of a Toll/Interleukin-1 Receptor-like (TIR), coiled-coil (CC) or resistance to owdery mildew8 (RPW8) domain at the N-terminus of the translated proteins (Shao et al., 2016a; Shao et al., 2019). RNL genes were long considered to be part of the CNL genes due to some similarities between the sequences of the CC and RPW8 domains (Meyers et al., 2003b), but RNLs have too few members to be easily detected. Recently, a functional characterization study found that RNLs do not to function like regular R genes (Bonardi et al., 2011). A typical R gene, such as TNL or CNL genes, usually functions as a detector of certain pathogens and trigger resistance reactions, which is the beginning of the resistance pathway (McHale et al., 2006). The recognition of pathogens by the LRR domains of TNL and CNL proteins cause conformational changes in the NBS domain, which further promotes the multimerization of TIR or CC domains that transfer defense signals, while RNL proteins appear to be more downstream and transduce signals from the TNL and CNL proteins through an undetermined pathway (Bonardi et al., 2011). Nevertheless, RNLs are clearly indispensable in the resistance pathway, otherwise resistance would be affected (Peart et al., 2005). Evolutionary studies also found strong evidence that supports RNL genes as a new class of NBS-LRR genes, equivalent to TNL and CNL genes (Wu et al., 2014; Shao et al., 2016b; Qian et al., 2017). Interestingly, although both CNL and RNL genes are always present in monocots and dicots, TNL genes are absent from monocots, which is likely due to an ancient gene loss event upon the split of this lineage (Meyers et al., 2003a; Tarr and Alexander, 2009; Andolfo et al., 2013; Shao et al., 2016b; Zhang et al., 2017b).
Diverse evolutionary patterns of NBS-LRR genes have been observed in different angiosperm lineages. For example, both Fabaceae and Rosaceae exhibit a consistently expanding pattern (Shao et al., 2014; Jia et al., 2015); whereas Brassicaceae exhibits a pattern of expansion followed by contraction (Zhang et al., 2016); Solanaceae demonstrates complicated patterns, potato shows a “consistent expansion” pattern, tomato exhibits a pattern of “first expansion and then contraction,” and pepper presents a “shrinking” pattern (Qian et al., 2017). Despite the absence of TNLs, the number of NBS-LRR genes analyzed in monocot genomes comparable to that of eudicots. For example, Asian rice Oryza sativa possesses 498 NBS-LRR genes, outnumbering most eudicots (Li et al., 2010a; Shao et al., 2016b). The discrepancy of retained gene number is drastic among species. Maize (Zea mays), a species from the same grass family as rice, possesses no more than 140 NSB-LRR genes, which shows a four-fold discrepancy between the two species and suggests an active evolutionary mode of NBS-LRR genes in Poaceae. The evolutionary history of NBS-LRR genes in Poaceae has been comprehensively studied: Li et al. used NBS-LRR genes from four sequenced genomes (Asian rice, maize, Sorghum bicolor and Brachypodium distachyon) to reconstruct the evolutionary history of NBS-LRR genes, and compared the gene tree with the systematic relationship of these four species, reconciling 496 ancestral lineages in the grass family. Varying numbers of gene gain and loss events resulted in the gene number discrepancy across these four species, indicating a shrinking pattern in this family (Li et al., 2010b).
To date, only NBS-LRR genes of the grass family have been well studied in monocots. Whether or not other monocot lineages exhibit different evolutionary patterns remains unanswered, as the sequenced genomes are not as prevalent in other monocot lineages as in Poaceae. Fortunately, in recent years the sequenced genomes in Orchidaceae (orchids) have rapidly increased and multiple genomes of this family, another monocot lineage, have been made readily available. In this study, a genome-wide analysis of NBS-LRR genes in the four sequenced orchid genomes and seven orchid transcriptomes was conducted (Figure 1). The goal of this study was to uncover the evolutionary features and modes of NBS-LRR genes in this family and further investigate the mechanisms that have shaped these evolutionary changes.
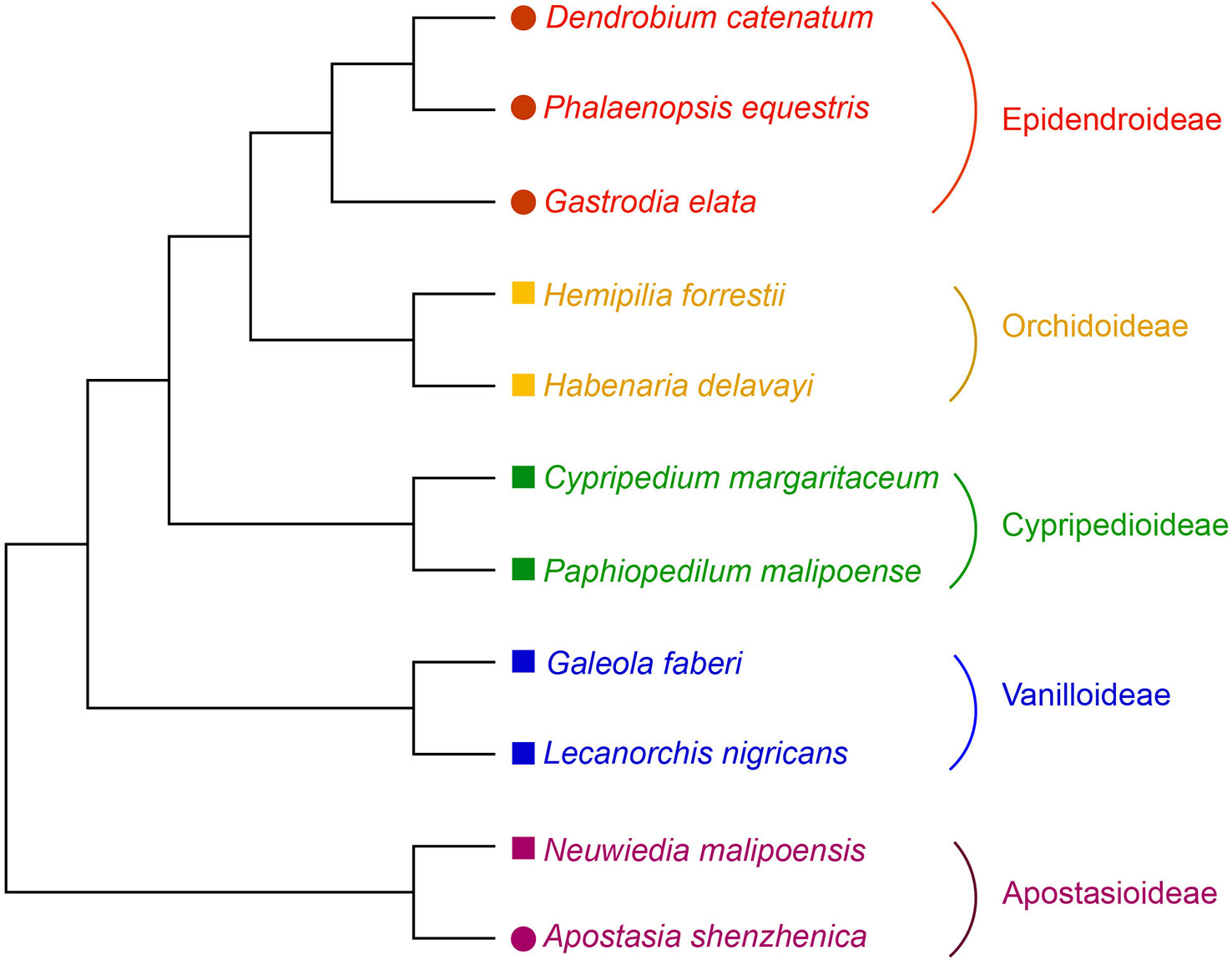
Figure 1 Phylogenetic relationship of orchid species used in this study. The phylogeny of the orchids used is from Zhang et al. (2017a). Species with genome or transcriptome information used in this study were indicated by colored solid circles or squares, respectively.
Results
Identification and Domain Combination of NBS-LRR Genes From Four Orchid Genomes
A total of 186 NBS-LRR genes (Table 1 and Table S1) were identified from four orchid genomes following previously described procedures (Xue et al., 2012; Shao et al., 2016b; Shao et al., 2019), among which, the CNL genes (182) overwhelmingly outnumbered RNL genes (4). TNL genes were absent from all four genomes, in accordance with the hypothesis that an early and thorough loss of TNLs had occurred upon the divergence of the monocot lineage (Tarr and Alexander, 2009; Shao et al., 2016b). RNL genes were found in three orchid genomes, except G. elata, but at extremely low numbers with one or two genes in each genome. Among the four orchids, the D. catenatum genome encoded the most NBS-LRR genes (115), followed by P. equestris (52) and A. shenzhenica (14), and G. elata, which encoded only five genes, the least among the four orchids and among all of the sequenced angiosperms. Since each orchid species had only one or two RNL genes, CNL genes must presumably be fully responsible for gene number variations among orchids.
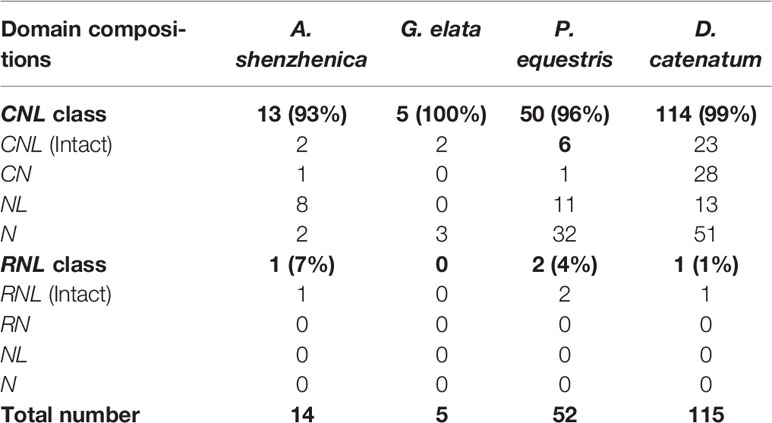
Table 1 The number of identified NBS-LRR genes in four orchid genomes.
Of the four orchids, intact NBS-LRR genes with all three domains (CC/RPW8-NBS-LRR) accounted for only 19.8% (37) of the total, whereas other genes either lacked a CC/RPW8 domain at the N-terminus, an LRR domain at the C-terminus, or lacked domains at both termini. G. elata had the highest proportion of intact genes (40.0%), while P. equestris had the lowest (15.4%). Several genomic changes, like recombination, fusion and pseudogenization, could result in real truncated genes, whereas other factors, such as sequencing, assembly errors and false annotations would elicit artificially “truncated” genes. Comparatively, the well-sequenced and annotated A. thaliana genome contains fewer (24.2%) truncated genes (Meyers et al., 2003b; Zhang et al., 2016).
Conserved Motifs of the NBS Domain in Orchids
The NBS domain contains several smaller motifs of 10 to 30 amino acids in length. including P-loop, kinase 2, kinase 3, RNBS-C, GLPL, and RNBS-D (DeYoung and Innes, 2006). Using MEME and WebLogo, these motifs in orchid CNL and RNL proteins were identified (Figure 2). Although RNL proteins are conserved along the whole NBS domain, six motifs exhibited different extents of variation in CNL proteins. Differences between the CNL and RNL proteins were observed in all six motifs, especially kinase 3 and RNBS-C, which exhibited the greatest discrepancy. These motifs can be used to distinguish orchid NBS-LRR genes without conducting phylogenetic analyses.
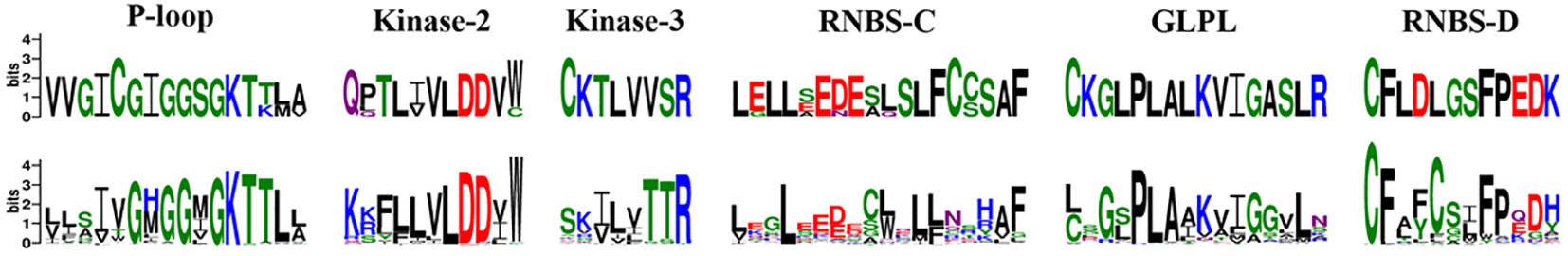
Figure 2 Conserved motifs in the NBS domain of the four orchid species. The amino acids of the six conserved motifs are extracted. Larger letters indicate higher frequency.
Phylogenetic Analysis of Orchid NBS-LRR Genes
To explore the evolutionary history of NBS-LRR genes in orchids, a phylogenetic analysis using the protein sequences of the NBS domain was conducted using three Amborella TNL proteins as outgroups. In order to obtain a more complete evolutionary pattern of NBS-LRR genes in orchid, 214 NBS-LRR genes from seven orchid transcriptome were identified and involved for phylogenetic analysis (Table S1). The phylogeny revealed a deep divergence between the RNL and CNL genes, and the evolutionary rate of RNL genes was rather low, which was reflected by the short branches among species (Figure 3). Nevertheless, the branch separating RNL genes and CNL genes was long (Figure 3), supporting the hypothesis of ancient divergence between RNL and CNL genes.
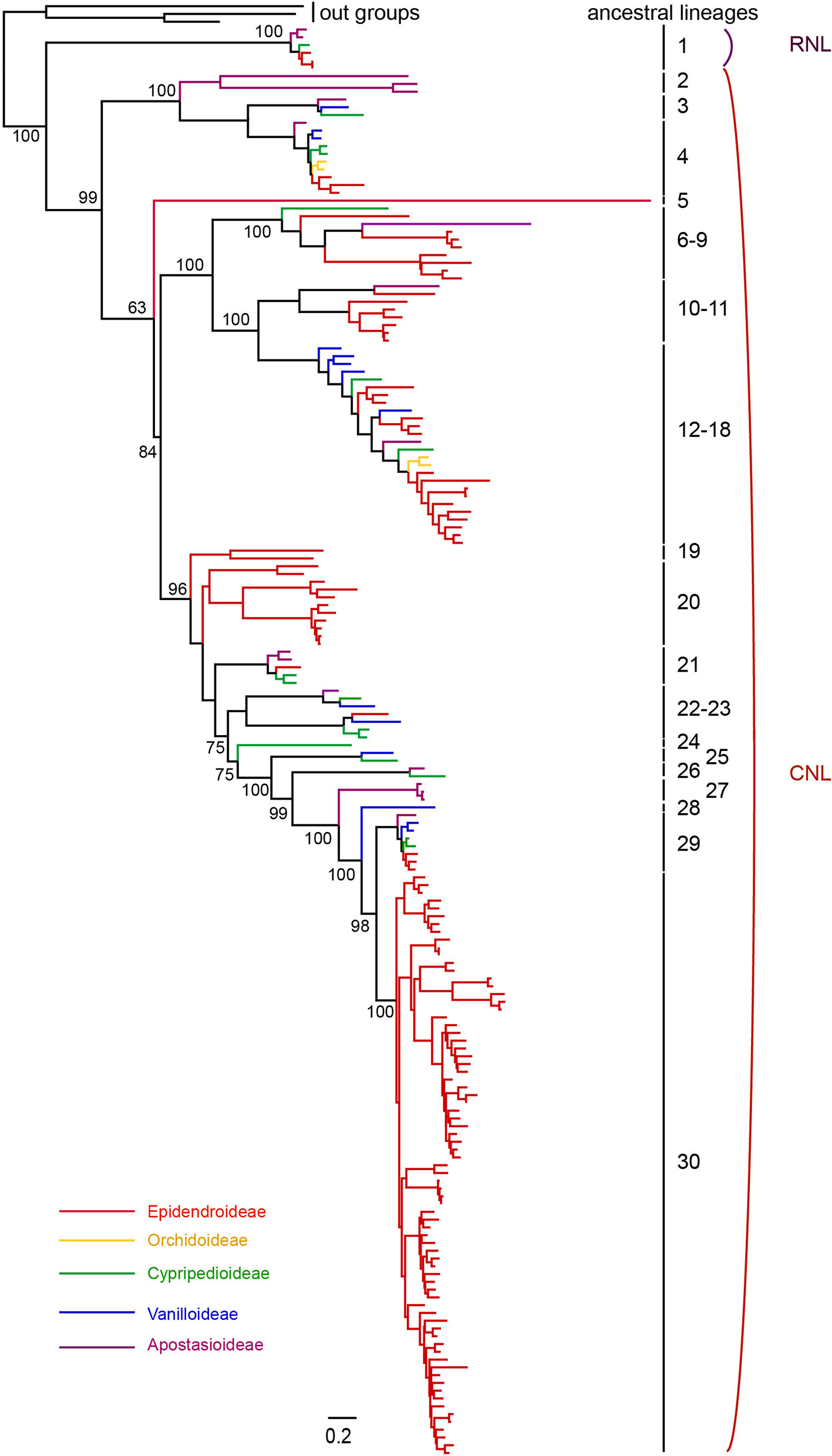
Figure 3 Phylogenetic relationships of NBS-LRR genes from orchid genomes and transcriptomes. NBS-LRR genes from different species are indicated with different colors in accordance to that of the species tree showed in Figure 1. Branch support values for two NBS-LRR classes (CNL and RNL) and each lineage are shown. The detailed phylogenetic tree is shown in Figure S2.
Reconciling the phylogeny of orchid NBS-LRR genes by the species tree recovered 30 ancestral NBS-LRR lineages, including one RNL lineage and 29 CNL lineages. This represents the minimal number of ancestral NBS-LRR genes in the common ancestor of orchids, as the full NBS-LRR repertoire from the seven orchids could not be fully recovered from their transcriptomes. The reconciled RNL gene’s phylogeny was consistent with that of the orchid species tree, suggesting that they are descendants of one ancestral gene from the common ancestor and experienced no shared gene duplication (Figures S1and S2). CNL genes exhibited a more active evolutionary pattern with far more gene duplications and losses, as well as faster evolutionary rates, which was reflected by their longer branch lengths (Figures S1 and S2). In total, 29 CNL lineages were identified from the orchid ancestor (Figure 3). Species-specific expansions were observed in different branches of the phylogenetic tree, with D. catenatum-specific expansion in Lineage 8, 9, 18, and 30, and P. equestris-specific expansion in Lineage 30 (Figures 3 and S2). Moreover, independent gene losses occurred in the evolutionary history of the orchid family, thus, none of the four species maintained all ancestral lineages. Both gene duplications and losses have contributed to the gene number variations among the different species.
The phylogenetic tree shows that 30 ancestral NBS-LRR lineages were not all retained by all four orchids, but differentially kept by different taxa (Table 2). D. catenatum maintained 17 lineages, A. shenzhenica had 10, P. equestris had seven, and G. elata had three (Table 2). Interestingly, P. equestris retained fewer ancestral lineages than A. shenzhenica, but developed more genes. For the 30 recovered ancestral lineages, 21 of them were inherited by at least one analyzed genomes. Lineage 29 is the only one lineage retained by all four orchids, 11 lineages are inherited in only one taxon, five lineages are shared by two taxa, and four lineages are reserved in three taxa.
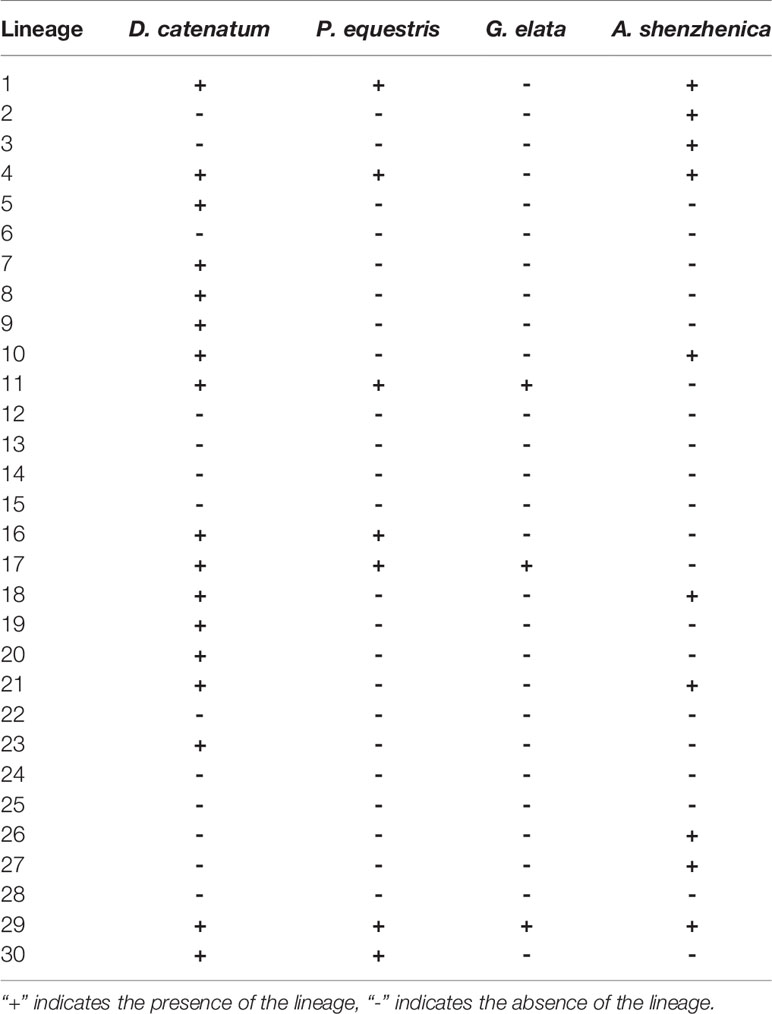
Table 2 Reservation of 30 ancestral NBS-LRR lineages in four orchids
Syntenic Analysis of NBS-LRR Genes in Orchid Genomes
The synteny analysis was performed both between and within the four orchid genomes. Results revealed that the RNLs reserved synteny among three species, except G. elata, which lost the RNL genes (Figure 4A; Table S2). These results were in accordance with the synteny analysis of the RNL genes in other angiosperms and supported the conservative evolutionary pattern of this NBS-LRR subclass (Shao et al., 2016b). Synteny of the CNL genes was also detected for some conservatively evolved CNL lineages. Lineage 29 CNL genes from the four orchid genomes were detected on syntenic blocks (Figure 4B; Table S2).
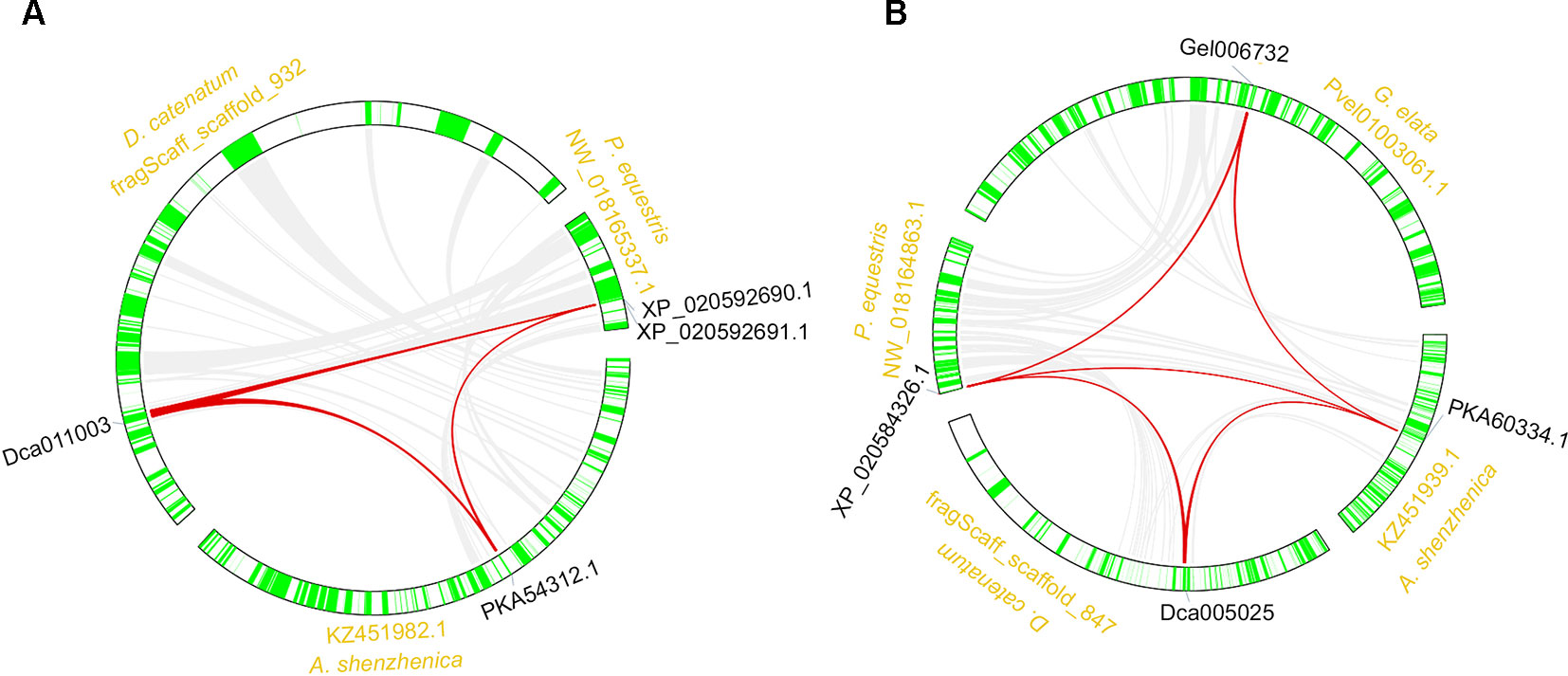
Figure 4 Synteny of orchid NBS-LRR genes. Syntenic genes of the RNL (A) and CNL (B) classes.
Within genome synteny was used to determine which lineages of the NBS-LRR genes were derived from whole genome duplications (WGDs) or segmental chromosomal duplications (Figure 4). Surprisingly, no segmentally duplicated NBS-LRR genes were identified in the four genomes, whereas 47, 22, and 7 tandemly duplicated genes were detected in D. catenatum, P. equestris, and A. shenzhenica, respectively. The remaining NBS-LRR genes from the four genomes may have been duplicated from other duplication types. Although no segmentally duplicated NBS-LRR genes were identified based on the within genome analysis, the role of this duplication mechanism in NBS-LRR gene evolution could not be ruled out. First, the syntenic relationship of NBS-LRR genes would be disrupted during long-term evolution. Second, the segmentally duplicated NBS-LRR genes may have been lost during evolution. Therefore, the contribution of segmental duplication may be underestimated in the within genome synteny analysis.
Reconciliation of Gene Losses and Gains and the Evolutionary Patterns in Orchids
Based on the phylogenetic tree, it could be inferred that many independent gene gains and losses have occurred at different stages of orchid evolution (Figure 5). Starting from 30 ancestral genes, these four species have experienced considerably different evolutionary patterns: A. shenzhenica, the first split taxon, has undergone a process of more gene losses (20) than duplications (4), resulting in 14 NBS-LRR genes in its genome today. This basal taxon overall exhibits a shrinking pattern of evolution (Figure 6). The one taxon with fewer genes than the common ancestor, G. elata, should have experienced more severe gene losses. Before its divergence, the NBS-LRR genes in the common ancestor of G. elata, D. catenatum and P. equestris was reduced to 17 and G. elata experienced additional gene loss after its split. Thus, G. elata has undergone a “consistent shrinking” pattern (Figure 6). D. catenatum and P. equestris both have more genes than the common ancestor of orchids. Along their evolutionary trajectories, these two taxa have gained more genes than they have lost and recent independent duplications have made major contribution to the gene number increase in these two species. Based on the phylogenetic tree, it is clear that that species-specific duplications have expanded the gene numbers of lineage 8, 9, 18 and 30 in D. catenatum, and Lineage 30 in P. equestris, outnumbering the other two taxa. Therefore, D. catenatum and P. equestris both exhibit an “early shrinking to recent expanding” pattern (Figure 6). Overall, the four orchids exhibit two different patterns of NBS-LRR evolution, and the discrepancy depends on whether a given taxon underwent recent expansions.
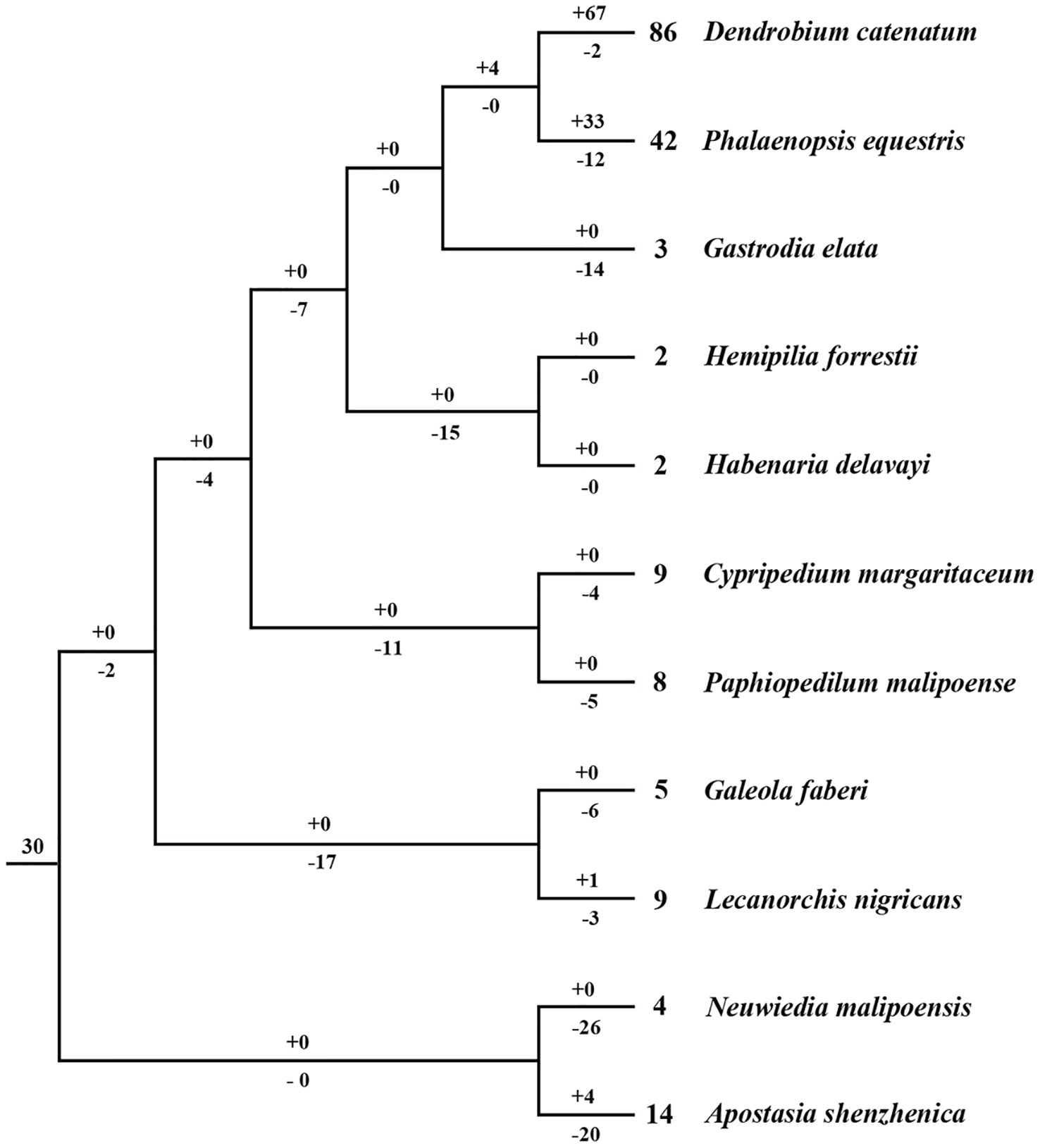
Figure 5 Loss and gain events of NBS-LRR genes across orchid evolution. Gene losses and gains are indicated by numbers with ‘–’or ‘+’ on each branch. Detailed information for gain and loss events of NBS-LRR genes is shown in Figure S3.
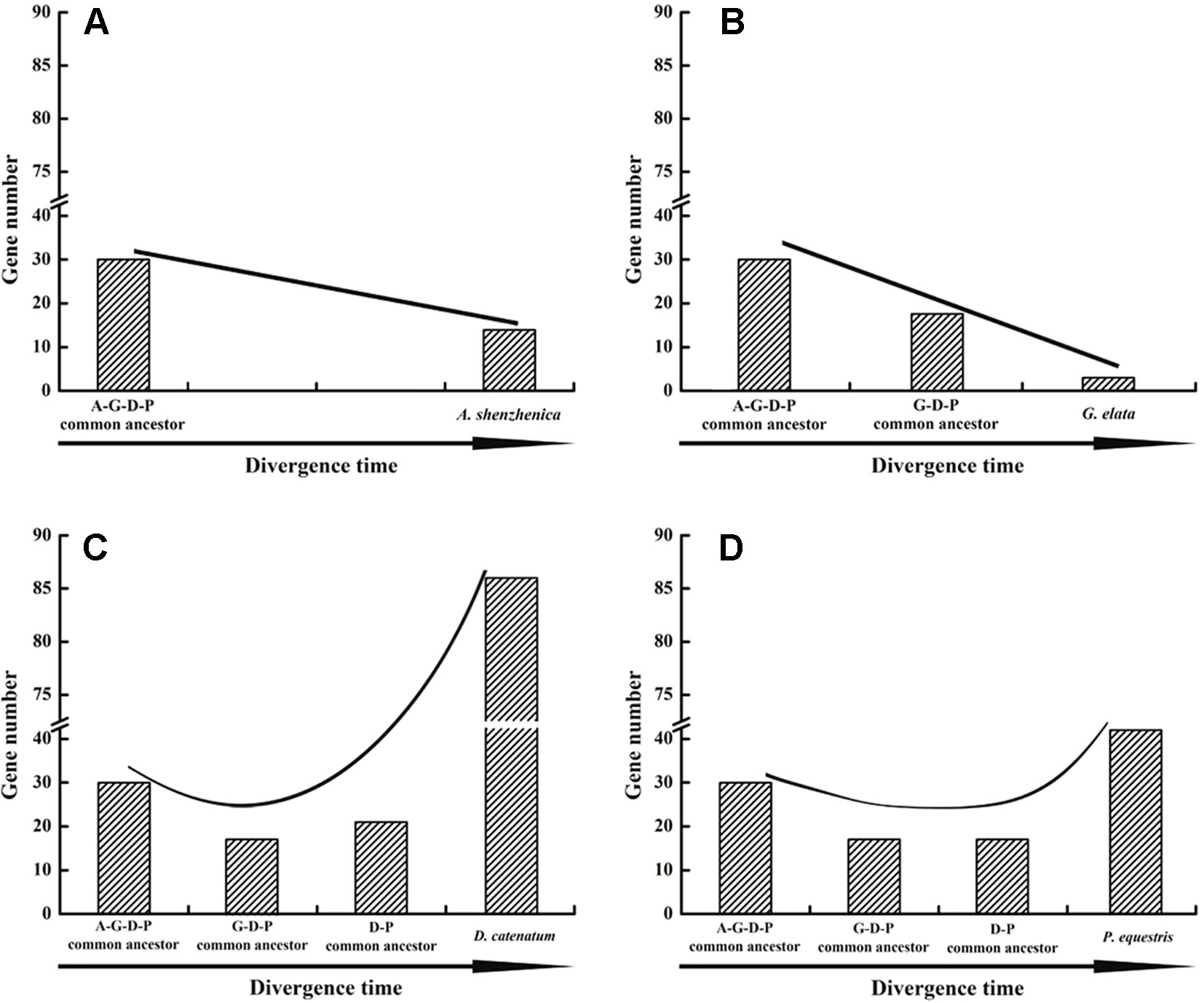
Figure 6 Evolutionary patterns of NBS-LRR genes in four orchid species. Evolutionary patterns of NBS-LRR genes in four orchids.: A. shenzhenica (A), G. elata (B), P. equestris (C), and D. catenatum (D). A-G-D-P indicates the common ancestor of all four orchids; GDP indicates the common ancestor of G. elata, P. equestris and D. catenatum, and D-P indicates the common ancestor of P. equestris and D. catenatum.
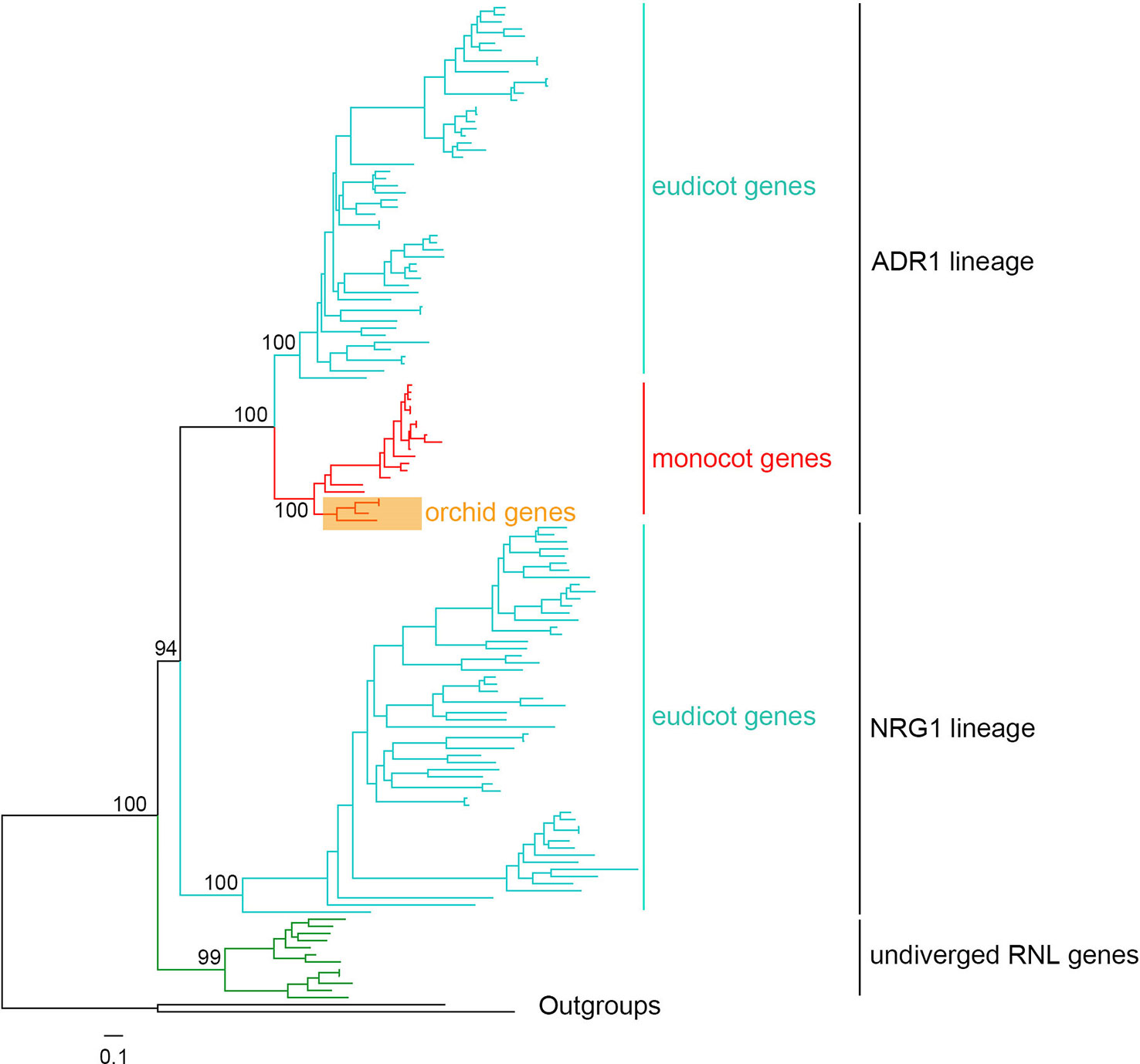
Figure 7 Phylogenetic tree of angiosperm RNL genes. A detailed phylogenetic tree of RNL genes is shown in presented Figure S3.
Discussion
The NBS-LRR Gene Number of Orchids
NBS-LRR genes belong to a large gene family in angiosperms, which includes hundreds of members. Only a small number of angiosperm taxa contain less than 100 NBS-LRR genes in their genomes. For example, Shao et al. analyzed 22 angiosperm genomes, which all had more than 100 NBS-LRR genes, except one Brassicaceae species, Thellungiella salsuginea, which had 88 genes (Shao et al., 2016b). In this study, it was discovered that three orchids also belong to the minority of plants that encode less than 100 NBS-LRR genes, and only one taxon, P. equestris, encoded over 100 genes. The number of T. salsuginea NBS-LRR genes fall below 100 because it underwent more severe gene loss events than duplications, after all, its ancestor once had d over 228 NBS-LRR genes (Zhang et al., 2016). The same situation was observed for three Cucurbitaceae species, Cucumis sativus, C. melo, and Citrullus lanatus (Lin et al., 2013). Orchids, however, are a different case. From a genetic and evolutionary perspective, as the reconciliation analysis suggests, the small number of orchid ancestral genes was mainly responsible for these results. In the orchid family, only 29 ancestral CNL genes and one RNL gene lineages were recovered as the family emerged, obviously far fewer than the 228 ancestral genes found in Brassicaceae (Zhang et al., 2016), 119 in Fabaceae (Shao et al., 2014), and 456 in Poaceae (Shao et al., 2016b). That’s why although D. catenatum and P. equestris have gained more genes than lost, they have not yet reached a large number of genes, such as rice or soybeans (Bai et al., 2002; Shao et al., 2014).
The number of NBS-LRR genes varies drastically among different taxa, even among closely-related species or subspecies. For instance, potato and tomato, both belonging to Solanaceae and have 447 and 255 NBS-LRR genes, respectively, showing a ratio of 1.75-fold difference in gene numbers (Qian et al., 2017). Intra-species variations of Oryza, Glycine and Gossypium reached a 5.4-fold discrepancy (Zhang et al., 2010). Therefore, the gene number variation observed in orchids. It is also noteworthy that the recent expansions are the main cause for this discrepancy was not surprising. Notably, recent expansions are the main cause for this discrepancy. In Fabaceae, Brasssicaceae and Solanaceae, the majority of expansions are consequence of tandem duplications (Shao et al., 2014; Zhang et al., 2016; Qian et al., 2017). In this study, D. catenatum and P. equestris appear to have undergone recent abrupt expansions, but mechanically tandem and ectopic duplications, other than WGDs are responsible for such expansions, as no syntenic genes were detected in these two species. A. shenzhenica and G. elata have not experienced sharp duplications, which explains the low number of NBS-LRR genes in these genomes. A. shenzhenica represents the earliest split of orchids, and has a rather narrow geographical distribution, as it is restricted to the Southeast Guangdong province, China (Zhang et al., 2017a). Its narrow distribution and stable habitat will likely lead fewer pathogen changes and stable pathogens diversity. Thus, A. shenzhenica has likely been battling a few of the same pathogens for a long period of time. Therefore, A. shenzhenica does not need to expand its R genes to face potential enemies. G. elata, despite its wide distribution, is an obligate mycoheterotrophic taxon, depending on a particular fungus Armillaria mellea to survive (Yuan et al., 2018), which probably does not allow G. elata to maintain many R genes. Coincidently, two other obligate mycoheterotrophic taxa, Cuscuta australis (Sun et al., 2018) and Epipogium roseum (unpublished), all seem to show a global gene loss pattern, and reduction of R gene number is only a part of the consequences.
The Evolution of RNLs in Orchids and Other Angiosperms
According to a previous WGD, angiosperm RNL genes have diverged into two lineages, ADR1 and NRG1, based on Aarbidopsis and tobacco (Collier et al., 2011; Shao et al., 2016b). In this study, the comprehensive analysis of seed plant RNL genes revealed an undiverged clade of gymnosperm genes at the basal position, followed by two diverged clades, ADR1 and NRG1, in angiosperms (Figures 7 and S3). Orchid RNL genes exclusively belong to the ADR1 lineage and have the shortest branch lengths among all of the angiosperms. Thus it is speculated that orchid RNL genes have been evolutionarily conserved since they have fewer diverse upstream signals to transduce. Orchids may be one of the plant lineages with the lowest number of R genes. The NRG1 lineage may have been lost as the origin of monocots, accompanied with the loss of an intron of the ADR1 lineage. The coincident loss of TNL genes and RNL NRG1 genes has been speculated to be due to their functional interdependence, as the resistance signals initiated by TNL genes are exclusively transduced by the NRG1 lineage (Collier et al., 2011). Several recent studies have suggested that nearly all test TNL genes are dependent on the NRG1 gene for inducing hypersensitive reactions, although potential exceptions could exist (Qi et al., 2018; Castel et al., 2019; Wu et al., 2019). As a downstream gene with a conservative function, orchid RNL genes seem unnecessary to expand. Low copies are sufficient for maintaining a functional system. This may explain why RNL genes have remained in low numbers across the evolution of angiosperms.
Materials and Methods
Identification and Classification of NBS-LRR Genes
The whole genomes of four orchid taxa, A. shenzhenica, C. elata, P. equestris and D. catenatum, were used in this study. Genomic sequences and annotation files of A. shenzhenica, P. equestris and D. catenatum were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/) (accession nos. PRJNA310678, PRJNA389183, and PRJNA262478, respectively). The genomic sequences and annotation files of C. elata were obtained from the G. elata Genome WareHouse Database (http://bigd.big.ac.cn/gwh/Assembly/129/show). The identification of NBS-LRR genes involved a two-step process. First, BLAST and hidden Markov model (HMM) searches using the NB-ARC domain (Pfam accession No.: PF00931) as a query, were performed simultaneously to identify candidate genes in each genome. For the BLAST search, the threshold expectation value was set to 1.0. For the HMM search (http://hmmer.org), default parameter settings were used. Second, all of the obtained candidate genes using BLAST or HMM searches were merged together, and the redundant hits were removed. The remaining candidate genes were submitted for an online Pfam analysis (http://pfam.sanger.ac.uk/) to further confirm the presence of the NBS domain with an E-value of 10-4. When two or more transcripts were annotated for a gene from alternative splicing, the longest form with an NBS domain was selected. All of the identified NBS-LRR genes were analyzed using the NCBI’s conserved domain database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) in order to determine the domains they possess.
Sequence Alignment and Conserved Motif Identification
The amino acid sequences of the NBS domain were extracted from the identified NBS-encoding genes and used for multiple alignments using ClustalW (Tamura et al., 2011) and Muscle (Edgar, 2004) integrated in MEGA 7.0 (Kumar et al., 2016) with default parameter settings. NBS domain sequences that were too short (i.e., shorter than two-thirds of a regular NBS domain) or too divergent (i.e. genes whose NBS domains could not be well aligned with others, and the aligned lengths are shorter than two-thirds of a regular NBS domain) were removed to prevent interference with the alignment and subsequent phylogenetic analysis. Resulting amino acid sequence alignments were manually edited in MEGA 7.0 (Kumar et al., 2016) for further improvement. Conserved protein motifs were analyzed by the online programs MEME (Multiple Expectation Maximization for Motif Elicitation) and WebLogo (Crooks et al., 2004; Bailey et al., 2006) with default parameter settings.
Phylogenetic Analysis and Reconciliation of Gene Loss/Duplication Events
In order to explore the relationships of NBS-LRR genes in the four orchids, a phylogenetic tree was reconstructed based on the aligned amino acid sequences of the conserved NBS domains. To avoid interference from “noisy characters,” too short or extremely divergent sequences were excluded from the phylogenetic analysis. Phylogenetic analyses were conducted using IQ-TREE and the maximum likelihood method (Nguyen et al., 2015). The best-fit model was estimated by ModelFinder (Kalyaanamoorthy et al., 2017). Branch support values were assessed with UFBoot2 tests (Minh et al., 2013). The scale bar indicated the genetic distance. TNL genes from the basal angiosperm, Amborellla trichopoda, were used as outgroups. Additionally, gene loss/duplication events during the speciation of the four orchid taxa were recovered by reconciling the NBS-LRR gene phylogenetic tree with the real species tree using Notung software (Stolzer et al., 2012). The phylogenetic analysis of RNL genes used the full length amino acid sequences of RNL proteins of 45 seed plants downloaded from Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html).
Syntenic Analyses Within and Across the Four Orchid Genomes
A synteny network approach was employed in this study (Zhao et al., 2017; Zhao and Schranz, 2019). Briefly, pair-wise all-against-all blast of protein sequences from the four genomes (Apostasia, Gastrodia, Phalaenopsis and Dendrobium) was performed. The obtained results and gff annotation files were then subjected to MCScanX for intra- and interspecies microsynteny detection and gene duplication type determination (Wang et al., 2012). Microsynteny relationship was displayed by TBtools (https://github.com/CJ-Chen/TBtools).
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
J-YX, Z-QS, S-ZZ and G-CZ conceived and designed the project. JYX, G-CZ, TZ and Z-QS obtained and analyzed the data. YL (3rd Author), YL (4th Author), Y-XZ, G-QZ, HC and S-ZZ participated in the data analysis and discussion. J-YX drafted the initial manuscript. Z-QS and YL (3rd Author) complemented the writing. All authors contributed to discussion of the results, reviewed the manuscript and approved the final article.
Funding
This work was supported by grants from the Shenzhen Key Laboratory of Southern Subtropical Plant Diversity (SLPD-2018-3 to J-YX), the Jiangsu Key Laboratory for the Research and Uti1ization of Plant Resources (Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, KSPKLB201835 to J-YX), and the Strategic Priority Research Program of Chinese Academy of Sciences (XDA13020603 to HC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We also thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01286/full#supplementary-material
Figure S1 | A detailed ML phylogenetic tree with all sequence names and branch support values. The tree was reconstructed based on the NBS domain sequences of the NBS-LRR genes from the four genomes and seven transcriptomes.
Figure S2 | Reconciled NBS-LRR gene tree with real species phylogeny and various loss and duplication events restored. “n3014” indicates a loss event that occurred in the common ancestor of Epidendroideae, Orchidoeae, Cypripedioideae, and Vanilloideae.
Figure S3 | A detailed ML phylogenetic tree based on the full length sequences of RNL proteins from 45 seed plants.
Table S1 | A list of NBS-LRR genes identified from seven orchid transcriptomes.
Table S2 | Detailed information of the two synteny blocks.
References
Andolfo, G., Sanseverino, W., Rombauts, S., Van de Peer, Y., Bradeen, J. M., Carputo, D., et al. (2013). Overview of tomato (Solanum lycopersicum) candidate pathogen recognition genes reveals important Solanum R locus dynamics. New Phytol. 197, 223–237. doi: 10.1111/j.1469-8137.2012.04380.x
Bai, J., Pennill, L. A., Ning, J., Lee, S. W., Ramalingam, J., Webb, C. A., et al. (2002). Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 12, 1871–1884. doi: 10.1101/gr.454902
Bailey, T. L., Williams, N., Misleh, C., Li, W. W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373. doi: 10.1093/nar/gkl198
Bonardi, V., Tang, S. J., Stallmann, A., Roberts, M., Cherkis, K., Dangl, J. L. (2011). Expanded functions for a family of plant intracellular immune receptors beyond specific recognition of pathogen effectors. Proc. Natl. Acad. Sci. United States America 108, 16463–16468. doi: 10.1073/pnas.1113726108
Castel, B., Ngou, P. M., Cevik, V., Redkar, A., Kim, D. S., Yang, Y., et al. (2019). Diverse NLR immune receptors activate defence via the RPW8-NLR NRG1. New Phytol. 222, 966–980. doi: 10.1111/nph.15659
Collier, S. M., Hamel, L. P., Moffett, P. (2011). Cell death mediated by the N-terminal domains of a unique and highly conserved class of NB-LRR protein. Mol. Plant Microbe Interact. 24, 918–931. doi: 10.1094/MPMI-03-11-0050
Crooks, G. E., Hon, G., Chandonia, J.-M., Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
DeYoung, B. J., Innes, R. W. (2006). Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 7, 1243–1249. doi: 10.1038/ni1410
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Friedman, A. R., Baker, B. J. (2007). The evolution of resistance genes in multi-protein plant resistance systems. Curr. Opin. Genet. Dev. 17, 493–499. doi: 10.1016/j.gde.2007.08.014
Jia, Y., Yuan, Y., Zhang, Y., Yang, S., Zhang, X. (2015). Extreme expansion of NBS-encoding genes in Rosaceae. BMC Genet. 16, 48. doi: 10.1186/s12863-015-0208-x
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/Nmeth.4285
Kourelis, J., van der Hoorn, R. A. L. (2018). Defended to the Nines: 25 Years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 30, 285–299. doi: 10.1105/tpc.17.00579
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, J., Ding, J., Zhang, W., Zhang, Y., Tang, P., Chen, J. Q., et al. (2010a). Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol. Genet. Genomics 283, 427–438. doi: 10.1007/s00438-010-0527-6
Li, J., Ding, J., Zhang, W., Zhang, Y., Tang, P., Chen, J. Q., et al. (2010b). Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol. Genet. genomics: MGG 283, 427–438. doi: 10.1007/s00438-010-0527-6
Lin, X., Zhang, Y., Kuang, H. H., Chen, J. J. (2013). Frequent loss of lineages and deficient duplications accounted for low copy number of disease resistance genes in Cucurbitaceae. BMC Genomics 14, 335. doi: 10.1186/1471-2164-14-335
Lozano, R., Ponce, O., Ramirez, M., Mostajo, N., Orjeda, G. (2012). Genome-wide identification and mapping of NBS-encoding resistance genes in Solanum tuberosum group phureja. PloS One 7, e34775. doi: 10.1371/journal.pone.0034775
Luo, S., Zhang, Y., Hu, Q., Chen, J., Li, K., Lu, C., et al. (2012). Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol. 159, 197–210. doi: 10.1104/pp.111.192062
McHale, L., Tan, X., Koehl, P., Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212. doi: 10.1186/gb-2006-7-4-212
Meyers, B. C., Kozik, A., Griego, A., Kuang, H., Michelmore, R. W. (2003a). Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15, 809–834. doi: 10.1105/tpc.009308
Meyers, B. C., Kozik, A., Griego, A., Kuang, H. H., Michelmore, R. W. (2003b). Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15, 809–834. doi: 10.1102/tpc.009308
Meyers, B. C., Kaushik, S., Nandety, R. S. (2005). Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134. doi: 10.1016/j.pbi.2005.01.002.S1369-5266(05)00006-3
Minh, B. Q., Nguyen, M. A. T., von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Peart, J. R., Mestre, P., Lu, R., Malcuit, I., Baulcombe, D. C. (2005). NRG1, a CC-NB-LRR protein, together with N, a TIR-NB-LRR protein, mediates resistance against tobacco mosaic virus. Curr. Biol. 15, 968–973. doi: 10.1016/j.cub.25005.04.053
Porter, B. W., Paidi, M., Ming, R., Alam, M., Nishijima, W. T., Zhu, Y. J. (2009). Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol. Genet. Genomics 281, 609–626. doi: 10.1007/s00438-009-0434-x
Qi, T., Seong, K., Thomazella, D. P. T., Kim, J. R., Pham, J., Seo, E., et al. (2018). NRG1 functions downstream of EDS1 to regulate TIR-NLR-mediated plant immunity in Nicotiana benthamiana. Proc. Natl. Acad. Sci. U.S.A. 115, E10979–E10987. doi: 10.1073/pnas.1814856115
Qian, L. H., Zhou, G. C., Sun, X. Q., Lei, Z., Zhang, Y. M., Xue, J. Y., et al. (2017). Distinct patterns of gene gain and loss: diverse evolutionary modes of NBS-encoding genes in three solanaceae crop species. G3-. Genes Genomes Genet. 7, 1577–1585. doi: 10.1534/g3.117.040485
Shao, Z.-Q., Zhang, Y.-M., Hang, Y.-Y., Xue, J.-Y., Zhou, G.-C., Wu, P., et al. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol. 166, 217–234. doi: 10.1104/pp.114.243626
Shao, Z. Q., Wang, B., Chen, J. Q. (2016a). Tracking ancestral lineages and recent expansions of NBS-LRR genes in angiosperms. Plant Signal Behav. 11, e1197470. doi: 10.1080/15592324.2016.1197470
Shao, Z. Q., Xue, J. Y., Wu, P., Zhang, Y. M., Wu, Y., Hang, Y. Y., et al. (2016b). Large-scale analyses of angiosperm nucleotide-binding site-Leucine-rich repeat Genes Reveal Three Anciently Diverged Classes with Distinct Evolutionary Patterns. Plant Physiol. 170, 2095–2109. doi: 10.1104/pp.15.01487
Shao, Z. Q., Xue, J. Y., Wang, Q., Wang, B., Chen, J. Q. (2019). Revisiting the origin of plant NBS-LRR Genes. Trends Plant Sci. 24, 9–12. doi: 10.1016/j.tplants.2018.10.015
Stolzer, M., Lai, H., Xu, M., Sathaye, D., Vernot, B., Durand, D. (2012). Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28, i409–i415. doi: 10.1093/bioinformatics/bts386
Sun, G., Xu, Y., Liu, H., Sun, T., Zhang, J., Hettenhausen, C., et al. (2018). Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis. Nat. Commun. 9, 2683. doi: 10.1038/s41467-018-04721-8
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Tarr, D. E., Alexander, H. M. (2009). TIR-NBS-LRR genes are rare in monocots: evidence from diverse monocot orders. BMC Res. Notes 2, 197. doi: 10.1186/1756-0500-2-197
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wu, P., Shao, Z. Q., Wu, X. Z., Wang, Q., Wang, B., Chen, J. Q., et al. (2014). Loss/retention and evolution of NBS-encoding genes upon whole genome triplication of Brassica rapa. Gene 540, 54–61. doi: 10.1016/j.gene.2014.01
Wu, Z., Li, M., Dong, O. X., Xia, S., Liang, W., Bao, Y., et al. (2019). Differential regulation of TNL-mediated immune signaling by redundant helper CNLs. New Phytol. 222, 938–953. doi: 10.1111/nph.15665
Xue, J. Y., Wang, Y., Wu, P., Wang, Q., Yang, L. T., Pan, X. H., et al. (2012). A primary survey on bryophyte species reveals two novel classes of nucleotide-binding site (NBS) genes. PloS One 7, e36700. doi: 10.1371/journal.pone
Yang, S., Zhang, X., Yue, J. X., Tian, D., Chen, J. Q. (2008). Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genomics 280, 187–198. doi: 10.1007/s00438-008-0355-0
Yuan, Y., Jin, X. H., Liu, J., Zhao, X., Zhou, J. H., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9, 1615. doi: 10.1038/s41467-018-03423-5
Zhang, M. P., Wu, Y. H., Lee, M. K., Liu, Y. H., Rong, Y., Santos, T. S., et al. (2010). Numbers of genes in the NBS and RLK families vary by more than four-fold within a plant species and are regulated by multiple factors. Nucleic Acids Res. 38, 6513–6525. doi: 10.1093/nar/gkq524
Zhang, Y. M., Shao, Z. Q., Wang, Q., Hang, Y. Y., Xue, J. Y., Wang, B., et al. (2016). Uncovering the dynamic evolution of nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in Brassicaceae. J. Integr. Plant Biol. 58, 165–177. doi: 10.1111/jipb.12365
Zhang, G. Q., Liu, K. W., Li, Z., Lohaus, R., Hsiao, Y. Y., Niu, S. C., et al. (2017a). The Apostasia genome and the evolution of orchids. Nature 549, 379–383. doi: 10.1038/nature23897
Zhang, Y. M., Xue, J. Y., Liu, L. W., Sun, X. Q., Zhou, G. C., Chen, M., et al. (2017b). Divergence and conservative evolution of XTNX genes in land plants. Front. Plant Sci. 8, 1844. doi: 10.3389/fpls.2017.01844
Zhao, T., Schranz, M. E. (2019). Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc. Natl. Acad. Sci. U. S. A. 116, 2165–2174. doi: 10.1073/pnas.1801757116
Zhao, T., Holmer, R., de Bruijn, S., Angenent, G. C., van den Burg, H. A., Schranz, M. E. (2017). Phylogenomic synteny network analysis of MADS-Box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell 29, 1278–1292. doi: 10.1105/tpc.17.00312
Keywords: orchids, plant resistance genes, evolution, phylogeny, synteny
Citation: Xue J-Y, Zhao T, Liu Y, Liu Y, Zhang Y-X, Zhang G-Q, Chen H, Zhou G-C, Zhang S-Z and Shao Z-Q (2020) Genome- Wide Analysis of the Nucleotide Binding Site Leucine-Rich Repeat Genes of Four Orchids Revealed Extremely Low Numbers of Disease Resistance Genes. Front. Genet. 10:1286. doi: 10.3389/fgene.2019.01286
Received: 02 July 2019; Accepted: 22 November 2019;
Published: 08 January 2020.
Edited by:
Horacio Naveira, University of A Coruña, SpainReviewed by:
Serena Aceto, University of Naples Federico II, ItalyZhonghua Zhang, Chinese Academy of Agricultural Sciences, China
Copyright © 2020 Xue, Zhao, Liu, Liu, Zhang, Zhang, Chen, Zhou, Zhang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Can Zhou, emdjYW4yMDA5QDE2My5jb20=; Shou-Zhou Zhang, c2hvdXpob3V6QHN6YmcuYWMuY24=; Zhu-Qing Shao, emh1cWluZ3NoYW9AMTI2LmNvbQ==; emh1cWluZ3NoYW9Abmp1LmVkdS5jbg==