 Elana Vorster
Elana Vorster Fahmida B. Essop
Fahmida B. Essop John L. Rodda
John L. Rodda Amanda Krause
Amanda Krause- 1National Health Laboratory Service and School of Pathology, University of the Witwatersrand, Johannesburg, South Africa
- 2Department of Paediatrics, University of the Witwatersrand, Johannesburg, South Africa
Spinal muscular atrophy (SMA) is a neuromuscular disorder, characterized by muscle atrophy and impaired mobility. A homozygous deletion of survival motor neuron 1 (SMN1), exon 7 is the main cause of SMA in ~94% of patients worldwide, but only accounts for 51% of South African (SA) black patients. SMN1 and its highly homologous centromeric copy, survival motor neuron 2 (SMN2), are located in a complex duplicated region. Unusual copy number variations (CNVs) have been reported in black patients, suggesting the presence of complex pathogenic rearrangements. The aim of this study was to further investigate the genetic cause of SMA in the black SA population. Multiplex ligation-dependent probe amplification (MLPA) testing was performed on 197 unrelated black patients referred for SMA testing (75 with a homozygous deletion of SMN1, exon 7; 50 with a homozygous deletion of SMN2, exon 7; and 72 clinically suggestive patients with no homozygous deletions). Furthermore, 122 black negative controls were tested. For comparison, 68 white individuals (30 with a homozygous deletion of SMN1, exon 7; 8 with a homozygous deletion of SMN2, exon 7 and 30 negative controls) were tested. Multiple copies (>2) of SMN1, exon 7 were observed in 50.8% (62/122) of black negative controls which could mask heterozygous SMN1 deletions and potential pathogenic CNVs. MLPA is not a reliable technique for detecting carriers in the black SA population. Large deletions extending into the rest of SMN1 and neighboring genes were more frequently observed in black patients with homozygous SMN1, exon 7 deletions when compared to white patients. Homozygous SMN2, exon 7 deletions were commonly observed in black individuals. No clear pathogenic CNVs were identified in black patients but discordant copy numbers of exons suggest complex rearrangements, which may potentially interrupt the SMN1 gene. Only 8.3% (6/72) of clinically suggestive patients had heterozygous deletions of SMN1, exon 7 (1:0) which is lower than previous SA reports of 69.5%. This study emphasizes the lack of understanding of the architecture of the SMN region as well as the cause of SMA in the black SA population. These factors need to be taken into account when counseling and performing diagnostic testing in black populations.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neurological disorder, characterized by the progressive degeneration of anterior horn cells (lower motor neurons) of the spinal cord, causing symmetrical muscle atrophy, weakness and paralysis. Historically, SMA was categorized into four clinical subtypes (SMA I–IV), ranging in severity, maximum muscle activity achieved, and age of onset, although it has been suggested that the SMA phenotype rather spans a continuum (Prior et al., 2004). SMA type I is the most severe form, with onset usually at birth or before six months with an average lifespan of two years. SMA type II is an intermediate form with an onset between 6 and 18 months (Fried and Emery, 1971); SMA type III is a mild form with onset after 18 months (Kugelberg and Welander, 1956) and SMA type IV is the mildest form with adult onset (Pearn et al., 1978).
A previous study suggested that the clinical presentation of SMA in black South African (SA) patients differs from worldwide reports with more frequent involvement of facial muscles in the severe infantile form of SMA leading to an expressionless facies (Moosa and Dawood, 1990). This is supported by clinical observation, but has not been scientifically documented.
SMA has been reported to be the second most common autosomal recessive disorder in Caucasian individuals after cystic fibrosis. The predicted birth incidence of SMA varies between 1 in 6,000 and 1 in 10,000 with a carrier frequency estimated at 1 in 40 to 1 in 60 worldwide (Hendrickson et al., 2009). The birth incidence of SMA in black SA patients has been estimated to be much higher at 1 in 3,574. This indicates that SMA may have a higher birth incidence than albinism (birth incidence: 1 in 3,900) in the black SA population. The carrier rate of SMA was previously estimated to be 1 in 23 in the white SA population and 1 in 50 in the black SA population (Labrum et al., 2007).
SMA is caused by mutations within the survival motor neuron 1 gene (SMN1; OMIM #6003541). A homozygous deletion of SMN1, exon 7 is reported to cause SMA in ~94% of patients with SMA worldwide (Hendrickson et al., 2009). In contrast, only 51% of SA SMA cases have been reported to be caused by a homozygous deletion of SMN1, exon 7 (Stevens et al., 1999; Labrum et al., 2007). An SMN1 deletion in conjunction with a second mutation, results in a compound heterozygote pattern and accounts for an additional 2–5% of patients with SMA worldwide (Wirth, 2000). The heterozygous deletion of SMN1, exon 7 rate in black SA patients with SMA who tested negative for the homozygous SMN1, exon 7 deletion, was previously reported to be as high at 69.5%, supporting the diagnosis of SMA in these patients and suggesting that SMA is probably due to additional unidentified mutations in this region (Labrum et al., 2007).
The SMN1 gene and its highly homologous copy, survival motor neuron 2 (SMN2; OMIM #6016271) are located in the SMN region on chromosome 5q13. The SMN region consists of multiple copy genes, pseudogenes (Selig et al., 1995), repetitive sequences (Bürglen et al., 1996), and retrotransposon-like elements (Francis et al., 1995), resulting in a large 500 kb inverted duplication, containing both a telomeric copy (SMN1) and a centromeric copy (SMN2) of the region. The historic terms, “telomeric” and “centromeric” refer to the relative positions of the SMN1 and SMN2 genes, respectively, within the SMN critical region at chromosome 5q13. As a result of the complexity and hypervariability of the region, there is no current complete and accurate map of the SMN region.
Homozygous deletions of the centromeric SMN2, exon 7 are not thought to be pathogenic (Schwartz et al., 1997), but are commonly encountered in black SA patients referred for SMA testing (Stevens et al., 1999; Labrum et al., 2007). A number of studies have suggested that SMN2 acts as a disease modifying gene as SMA disease severity is inversely correlated with SMN2 copy number (McAndrew et al., 1997; Wirth et al., 1999; Feldkötter et al., 2002; Jedrzejowska et al., 2008). SMA type I patients tend to have two copies of SMN2, type II and type 3b (onset before three years)— three copies, SMA type IIIb (onset after three years)— four copies and SMA type IV— four to six copies (reviewed by Mercuri et al., 2018).
Recombination between SMN1 and SMN2 could potentially interrupt a critical region of SMN1, leading to the loss of full-length functional SMN transcripts. SMA type II and III patients have been shown to have gene conversions from SMN1 to SMN2 rather than deletions, resulting in a higher copy number of SMN2, which has been associated with a milder phenotype (Campbell et al., 1997). A high frequency (31.5%) of black SA patients with SMA were shown to have smaller deletions including SMN1, exon 7, but with exon 8 present, possibly due to gene conversions (Stevens et al., 1999). Additional evidence for this hypothesis was reported by Labrum et al. (2007) who observed a lower frequency of large deletions spanning SMN1, exons 7, 8, and the NLR family, apoptosis inhibitory protein gene (NAIP) in black SA patients (9.8%) when compared to white SA patients (41.7%).
Other genes located in the duplicated SMN region at chromosome 5q13 include NAIP, GTF2H2, and SERF1A and their multiple pseudo copies. The lack of understanding of the physical structure and orientation of these genes in the SMN region, hampers the better understanding of the role of these genes in the SMA disease mechanism.
The SMN protein is present in both the cytoplasm and nucleus of all cells, but is particularly abundant in motor neurons. The SMN protein's main function involves the assembly of small nuclear ribonucleoprotein (snRNP) complexes important for pre-messenger RNA splicing (Markowitz et al., 2012).
SMN1 and SMN2 differ in only 5 nucleotides of sequence, with the critical difference being a silent C to T transition at cDNA position 840 (c.840C > T) in SMN2, resulting in the exclusion of exon 7 during splicing and causing the majority of SMN2 transcripts to be truncated and unstable (Lorson et al., 1999; Monani et al., 1999). Only 20% of the total full-length SMN (FL-SMN) transcript is produced from the SMN2 gene, which partly compensates for the lack of FL-SMN transcript produced from SMN1 in patients with SMA, but does not produce sufficient SMN protein levels in motor neurons for their survival (Zheleznyakova et al., 2011). A milder phenotype (SMA types III and IV) have been associated with four or more copies of SMN2 (Wirth et al., 2006). Recently, Nusinersen, an antisense oligonucleotide drug that modifies splicing of SMN2, has been shown to lead to an increase in the total FL-SMN transcripts of SMN2, leading to improvements in motor function (Finkel et al., 2017).
Approximately 4% of American and Canadian individuals have been found to have heterozygous SMN1 deletions with two SMN1 gene copies on a single chromosome in addition to a chromosome with a deletion of the SMN1 gene (2:0 genotype) (Scheffer et al., 2001). These individuals are SMA carriers since they have the ability to pass on a deletion chromosome to subsequent generations. Carrier testing is compromised since quantitative techniques cannot distinguish between two copies in cis or trans of SMN1, one copy present on each chromosome or two copies of SMN1 present on a single chromosome in conjunction with 0 copies on the second chromosome (McAndrew et al., 1997). It is recommended that potential carriers with multiple copies of SMN1 need to be analyzed in a family context to try and clarify the phase of these copy number variations (CNVs) and to accurately assign carrier status.
Studies performed on various American population groups, showed an unusually high frequency of multiple copies of SMN1 in the African American population when compared to other populations (Hendrickson et al., 2009; Sugarman et al., 2012). A study performed on unaffected individuals from various sub-Saharan African populations (Kenyan, Malian and Nigerian) confirmed this observation and showed a higher frequency of multiple copies of SMN1 and deletions of SMN2 than European populations (Sangaré et al., 2014).
SMA was previously thought to be rare in African populations with limited studies performed in Northern Africa (Tunisia, Egypt, Nigeria, Algeria and Senegal), but this was likely due to an underestimation (Pelleboer et al., 1989; Tazir and Geronimi, 1990; Shawky et al., 2001; Ndiaye et al., 2002; Mrad et al., 2006).
It has been hypothesized that complex population-specific rearrangements of the SMN region could cause SMA in the black SA population (Labrum et al., 2007; Vorster et al., 2011). The main aim of this study was to investigate CNVs of the SMN region using the P021 multiplex ligation-dependent probe amplification (MLPA) probe mix (MRC Holland, Amsterdam, Netherlands), which has multiple probes spanning the SMN region, in an attempt to identify potential pathogenic CNVs which could contribute to the disease mechanism of SMA in the black SA population. A better understanding of potential pathogenic CNVs of the SMN region could improve diagnostic testing for the 49% of black SA patients affected with SMA who currently test negative for the common homozygous SMN1, exon 7 deletion.
Subjects, Materials, and Methods
Subjects: U/Ub Patients
U/Ub patients (Unidentified mutation/Unidentified mutation genotype) represent black patients who presented with symptoms clinically suggestive of SMA and who previously tested negative for a homozygous deletion of SMN1, exon 7 in a diagnostic setting using an in-house PCR and restriction enzyme assay. U/Ub patients were identified and selected in collaboration with the Clinical Section of the Division of Human Genetics, molecular diagnostic laboratory, National Health Laboratory Service Johannesburg (NHLS), and the University of the Witwatersrand (WITS), henceforth referred to as “the Division” and in collaboration with the Departments of Paediatrics of the Chris Hani Baragwanath and Charlotte Maxeke Academic Hospitals.
In total, 72 U/Ub patients were identified, nine of whom had muscle biopsies suggestive of SMA. MLPA analysis was performed on these patients to identify potential pathogenic CNV patterns. DNA samples of family members of U/Ub patients were not available. These patients formed the main focus of this research study. DNA samples of all of these patients are stored in the Division.
Groups Used for Comparison
N/Nb Individuals
N/Nb individuals (Negative/Negative genotype) represent black controls negative for SMA. MLPA analysis was performed on family members of 61 N/Nb families (200 individuals in total). In order to be included, DNA had to be available from two unrelated parents and at least one child. The unaffected parents of these families were used as negative controls in this study and consisted of a total of 122 unrelated N/Nb individuals. Haplotypes were constructed from the MLPA data and family pedigrees were drawn to investigate potential novel CNV events in these families.
N/Nw Individuals
N/Nw individuals (Negative/Negative genotype) represent white controls negative for SMA. To compare the typical non-pathogenic CNV patterns of black and white individuals, 30 random unrelated N/Nw individuals were tested on MLPA.
M1/M1b Patients
M1/M1b patients (Mutation 1: deletion of SMN1, exon 7/Mutation 1: deletion of SMN1, exon 7 genotype) represent black patients who were previously identified to have the common homozygous deletion of SMN1, exon 7 on a diagnostic PCR and restriction enzyme assay designed to detect and distinguish homozygous deletions of SMN1, exon 7 and SMN2, exon 7 (van der Steege et al., 1996). MLPA analysis was performed on 75 M1/M1b patients to investigate the molecular structure of pathogenic CNV patterns, including the extent of homozygous deletions of SMN1, exon 7 and potential gene conversion events. Furthermore, 25 of these patients formed part of families (71 individuals in total). MLPA was performed on all family members and haplotypes were constructed from the MLPA data and family pedigrees to investigate the phase of potential common pathogenic CNV patterns.
M1/M1w Patients
M1/M1w patients (Mutation 1: deletion of SMN1, exon 7/Mutation 1: deletion of SMN1, exon 7 genotype) represent white patients who were previously identified to have a homozygous deletion of SMN1, exon 7 on the diagnostic PCR, and restriction enzyme assay. For comparison, 30 random unrelated M1/M1w patients were tested to compare the molecular structure of pathogenic CNV patterns between M1/M1b and M1/M1w patients.
M2/M2b Patients
M2/M2b patients (Mutation 2: deletion of SMN2, exon 7/Mutation 2: deletion of SMN2, exon 7 genotype) represent black patients who were previously identified to have a homozygous deletion of SMN2, exon 7 on the diagnostic assay. Fifty M2/M2b patients were tested on MLPA to determine the underlying molecular structure of this common CNV and to understand the interaction between the SMN1 and SMN2 genes.
M2/M2w Patients
M2/M2w patients (Mutation 2: deletion of SMN2, exon 7/Mutation 2: deletion of SMN2, exon 7 genotype) represent white patients who were previously identified to have a homozygous deletion of SMN2, exon 7 on the diagnostic assay. For comparison, eight random unrelated M2/M2w patients were tested on MLPA to compare the molecular structure of this CNV between M2/M2b and M2/M2w patients. Only eight M2/M2w patients were included in this group, since they were the only M2/M2w patients available who have been identified in the white SA population.
Methods
Dna Extraction
Genomic DNA was extracted from whole blood using the salting out method (Miller et al., 1988), a commercial DNA extraction kit (High Pure PCR Template Preparation Kit, Roche Diagnostics), or in the case of chorionic villus sampling (CVS) and amniocyte material, the phenol-chloroform extraction method was used (Barker, 2004). All samples were processed and extracted in a diagnostic setting, with stringent quality control. The P021 probe mix was validated on blood, amniocyte material, and CVS samples. A quantity of 50–250 ng of DNA is recommended for MLPA2. All DNA samples were normalized in order to accurately compare probe copy numbers to each other.
MLPA Analysis
The MLPA P021 probe mix (MRC Holland, Amsterdam, Netherlands) is mainly designed to detect SMN1 and SMN2, exon 7 copy numbers. The P021 probe mix consists of a multiplex of 46 probes, consisting of seven DQ (dosage quality) control probes; two sex-chromosome specific probes (for gender determination and to detect sample mix-up); 22 internal reference probes, specific to various chromosomal regions and not associated with SMA; 15 probes specific to the SMN region (eight targeting the SMN1 and SMN2 genes and seven probes targeting neighboring genes). The DQ control probes amplify four Q fragments which determine whether sufficient DNA has been added to the reaction and whether ligation has been successful and two D fragments which determine whether successful denaturation of the DNA sample took place2.
Probes have been designed to target the critical one base pair difference between SMN1 and SMN2 in exon 7 and can therefore distinguish between exon 7 of SMN1 and SMN2. Similarly, probes have been designed to target a one base pair difference between SMN1 and SMN2, exon 8 and can therefore distinguish between exon 8 of SMN1 and SMN2. Probes specific to exons 1, 4, and 6 of the SMN1 and SMN2 genes as well as neighboring genes in the SMN region have been included to assist with determining the extent of deletions (He et al., 2013). Neighbouring genes include the RAD17 checkpoint clamp loader component gene (RAD17) and telomeric as well as centromeric copies of the NAIP genes (NAIP/NAIPΨ), the general transcription factor IIH subunit 2 genes (GTF2H2), and the small EDRK-rich factor 1A genes (SERF1A/1B)3.
MLPA was performed using the Applied Biosystems (ABI) 9700 thermal cycler and fragment separation was performed using the ABI Genetic Analyzer 3130xl (Applied Biosystems, Foster City, CA, USA). Dosage analysis was performed using the freely available Coffalyser software (MRC Holland, Amsterdam, Netherlands) to quantify CNVs. By comparing the copy number of PCR products observed in a patient sample with endogenous reference probes and several external control samples, relative quantitative changes in DNA fragments can be determined (Schouten et al., 2002). The copy number of probe regions was determined using the parameters as set out in Table 1. MLPA results were analyzed by statistical analysis using Statistica (Dell, version 12.7) and Real Statistics Using Excel software4 to compare dosage trends between different patient groups and to determine significant differences among patient and control groups.

Table 1 The relationship of the P021 probe mix dosage quotient with copy number.
MLPA has been shown to be a reliable technique to detect multiple copy numbers in regions of segmental duplication, without the need for repeat testing or replicates (Cantsilieris et al., 2014). Furthermore, the analytical sensitivity and specificity of the P021 probe mix has been reported to be >99%2. The P021 probe mix was validated using samples of individuals whose SMN1 copy number was previously identified using the in-house PCR and restriction enzyme diagnostic assay or through ISO 17043 accredited external quality assessors, the European Molecular Genetics Quality Network (EMQN)3. Furthermore, negative, homozygous SMN1, exon 7 deletion and heterozygous SMN1, exon 7 deletion control samples were included in every experiment to ensure consistency among experiments. MLPA experiments were repeated and/or excluded when results did not adhere to quality requirements.
Haplotype analysis of pedigrees of N/Nb and M1/M1b families were performed to determine common chromosomal CNV patterns/haplotypes in the black SA population. Multiple copies of genes in the SMN region were assumed to be located on chromosome 5 and the gene order was based on current map data from the Ensembl genome browser (genome built: GRCh38)5. Family pedigrees were categorized as informative if a clear pattern of inheritance from parents to their children could be established for each of the MLPA probes. Family pedigrees were categorized as uninformative if the phase of the CNVs could not be correctly determined. Multiple copies of a specific probe region complicate the assignment of phase due to various combinations being possible within a family and apparent discrepant results could arise due to potential novel CNV events, family members who are not related as specified or technical MLPA faults. All discrepant results were repeated on MLPA to ensure the accuracy of these results.
An ethics application was approved unconditionally by the WITS Medical Human Research Ethics Committee (ethics clearance number: M130950).
Results
MLPA Analysis
The results of MLPA analysis of SMN1 and SMN2 of the patient and control groups are summarized in Table 2. All MLPA data is available as Supplementary Material.
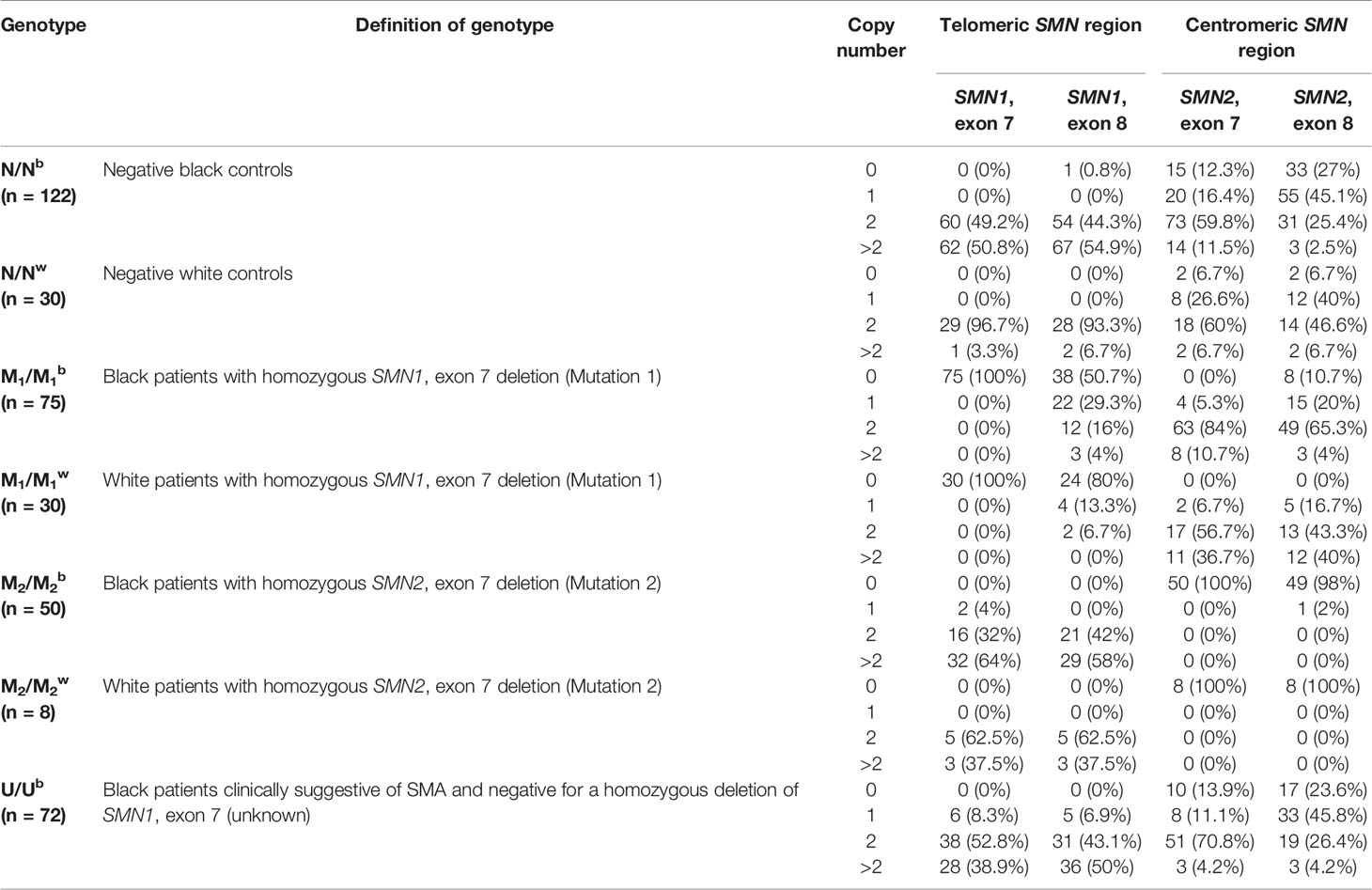
Table 2 Summary of telomeric SMN1, exons 7 and 8 and centromeric SMN2, exons 7 and 8 copy number across various patient and control groups.
Comparative Analysis of N/Nb and N/Nw Individuals
The SMN1, exon 7 copy number was found to differ statistically between N/Nb and N/Nw individuals (Kruskal-Wallis test: H = 32.7; p < 0.0001). In this study, 50.8% (62/122) of N/Nb individuals were found to have multiple copies (3–6) of SMN1, exon 7. These results stand in sharp contrast to trends observed in N/Nw individuals, with only 3.3% (1/30) with multiple copies of SMN1, exon 7. Figure 1 compares the SMN1 copy numbers observed in SA populations with various international population groups.
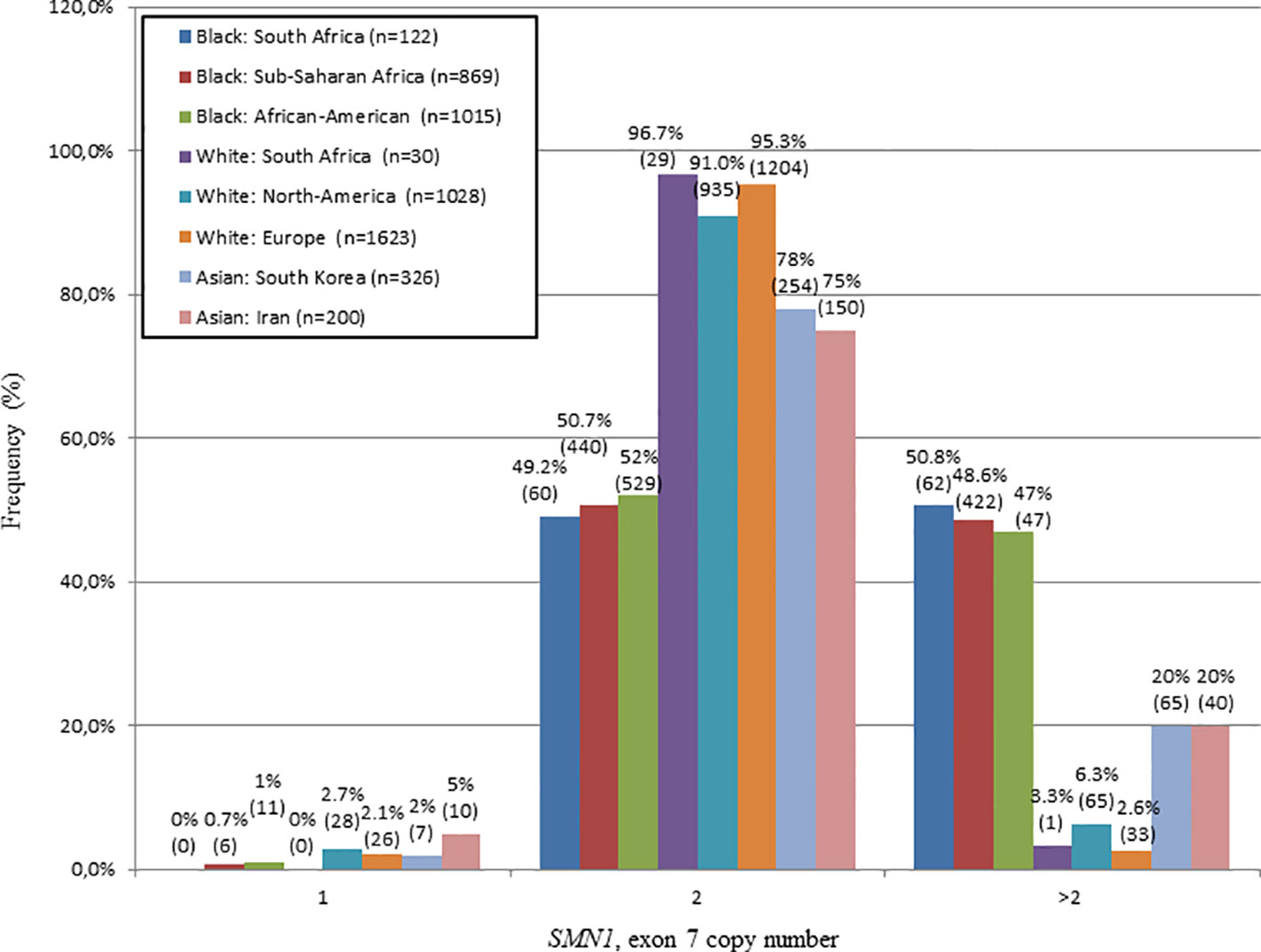
Figure 1 Comparison of the SMN1, exon 7 copy number distribution of N/Nb and N/Nw individuals with various international studies. Graph produced from combined data of this study and a study performed by Sangaré et al., 2014. A similar frequency of white individuals with 2 copies of SMN1, exon 7 was observed in SA, North-America and Europe. A similar frequency of black individuals with multiple copies (>2) of SMN1, exon 7 was observed in SA, Sub-Saharan Africa and North-America.
Similarly, the telomeric SMN1, exon 8 and NAIP, exon 5 copy numbers differed significantly between N/Nb and N/Nw individuals (Kruskal-Wallis test: H = 14.3; p = 0.0002 and H = 7.2; p = 0.0071, respectively). As seen with the SMN1, exon 7 region; 54.9% (67/122) of N/Nb individuals were found to have multiple copies (>2 copies) of SMN1, exon 8 and 37.7% (46/122) were found to have multiple copies of NAIP, exon 5, which are both assumed to co-locate with SMN1 in the telomeric SMN region. Once again, these results stand in contrast to trends observed in N/Nw individuals, with only 6.7% (2/30) of individuals with multiple copies of SMN1, exon 8 and only 13.3% (4/30) with multiple copies of NAIP, exon 5. The SMN1, exon 7 and 8 copy numbers did not correlate fully, suggesting that some of these copies may not be contiguous.
The centromeric SMN2, exon 7 copy number was not found to differ significantly between N/Nb and N/Nw individuals (Kruskal-Wallis test: H = 0.09; p = 0.7652). The majority of N/Nb and N/Nw individuals had two copies of SMN2, exon 7; 59.8% (73/122) and 60% (18/30), respectively. The SMN2, exon 8 copy number was found to differ significantly between N/Nb and N/Nw individuals (Kruskal-Wallis test: H = 11.1; p = 0.0009). N/Nb individuals had a higher rate of homozygous SMN2, exon 8 deletions (27% (33/122) than N/Nw individuals (6.7% (2/30). This finding could be due to gene conversions from centromeric SMN2, exon 8 to telomeric SMN1, exon 8 resulting in hybrid genes consisting of SMN1, exon 7 and SMN2, exon 8. Deletions of these hybrid genes would result in a loss of SMN1, exon 7 in conjunction with SMN2, exon 8.
No N/Nb (0/122) and N/Nw (0/30) individuals appeared to have a detectable heterozygous deletion of SMN1, exon 7 (1:0 genotype), usually accepted to be carriers of SMA. Two N/Nb families had de novo CNV events with a new mutation rate of 3.3% (2/61).
N/Nb individuals had a significantly higher variance in copy number than N/Nw individuals for the majority of MLPA probes. Whereas the copy number of the telomeric SMN1, exons 7, 8 and NAIP, exon 5 of N/Nw individuals seem to cluster at two copies, as expected; the copy number of SMN1, exons 7, 8 and NAIP, exon 5 of N/Nb individuals varies extensively from 1 to 6. Figure 2 shows the difference in variance in copy number of SMN1, exon 7 between N/Nb and N/Nw individuals. In N/Nw individuals the SMN1, exon 7 copy number clusters around integers (one or two copy numbers) whereas the SMN1, exon 7 copy number varies extensively in N/Nb individuals (one to six copy numbers). It is hypothesized that some of these multiple SMN1 gene copies may be partial discontinuous gene copies which may be non-functional due to interruptions in the coding region.
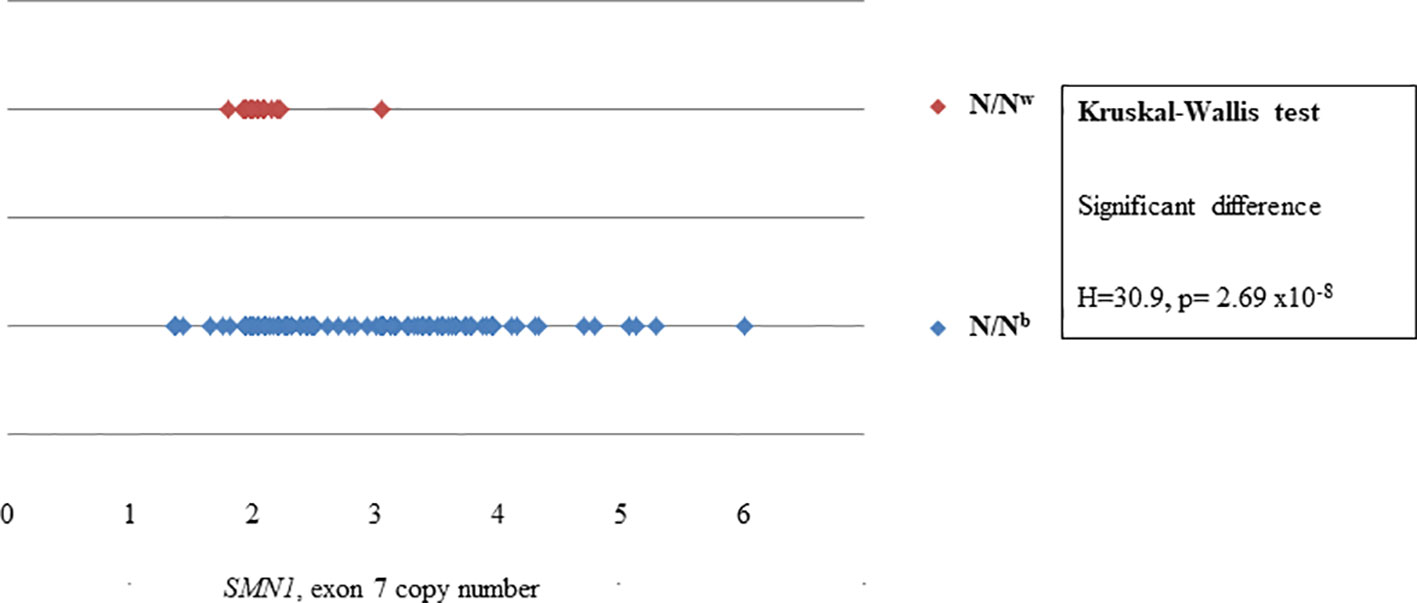
Figure 2 Scatterplot comparison of probe copy numbers of SMN1, exon 7 between N/Nb and N/Nw individuals. Whereas the copy number of SMN1, exon 7 of N/Nw individuals seem to cluster at 2 copies; the copy number of SMN1, exon 7 of N/Nb individuals varies extensively from 1 to 6.
Further, The SERF1B and RAD17 copy numbers were not found to differ significantly between N/Nb and N/Nw individuals (Kruskal-Wallis test: H = 0.80; p = 0.367 and H = 0.00; p = 0.967, respectively). Multiple copies (more than 2) of RAD17 were observed in 14.8% (18/122) of N/Nb individuals in contrast to no CNVs of RAD17 in N/Nw individuals, suggesting that N/Nb individuals have a higher variability of these regions than N/Nw individuals.
Comparative Analysis of M1/M1b and M1/M1w Patients
As expected, MLPA analysis confirmed SMN1, exon 7 homozygous deletions in all 75 M1/M1b patients and all 30 M1/M1w patients. The telomeric SMN1, exon 8 and NAIP, exon 5 copy numbers were found to differ statistically between M1/M1b and M1/M1w patients (Kruskal-Wallis test: H = 7.8, p = 0.0053; H = 5.6, p = 0.0181, respectively). This significant difference could be due to homozygous SMN1, exon 8 deletions being more common in M1/M1w patients [80% (24/30)] than M1/M1b patients [50.7% (38/75)] and heterozygous deletions of NAIP, exon 5 being more common in M1/M1w patients [56.7% (17/30)] than M1/M1b patients [21.3% (16/75)].
The SMN2, exon 7 copy number was found to differ significantly between M1/M1b and M1/M1w patients (Kruskal-Wallis test: H = 9, p = 0.0027), likely due to a higher frequency of multiple copies (> 2) of this region in M1/M1w patients [36.7% (11/30)] when compared to M1/M1b patients [10.7% (8/75)]. Multiple copies of SMN2, exon 7 in conjunction with homozygous deletions of SMN1, exon 7 suggest gene conversion from telomeric SMN1, exon 7 to centromeric SMN2, exon 7 being more common in M1/M1w patients.
Due to identical sequences of exons 1, 4, 6, and another region of exon 8 of the SMN1 and SMN2 genes, the P021 probe mix cannot distinguish between SMN1 and SMN2 for these regions and will give a combined copy number result (representing both the SMN1 and SMN2 genes: SMN1/2). An absence of these probes could therefore represent a deletion in either or both copies of the SMN1 and SMN2, which complicates analysis.
A homozygous deletion of SMN1/2, exons 1, 4, and 6 were more frequently observed in M1/M1b individuals [exon 1: 60% (45/75), exon 4: 61.3% (46/75) and exon 6: 62.7% (47/75) than M1/M1w individuals (exon 1, 4 and 6: 31.3% (10/30)]. The SMN1/2, exons 4 and 6 copy numbers were found to differ statistically between M1/M1b and M1/M1w individuals (Kruskal-Wallis test: H = 4.4, p = 0.0355 and H = 4.2, p = 0.0399, respectively). The SMN1/2, exon 1 copy number was not found to differ significantly between M1/M1b and M1/M1w individuals (Kruskal-Wallis test: H = 1; p = 0.3188). The MLPA probes for exons 1, 4, and 6 cannot distinguish between SMN1 and SMN2 but since these patients have homozygous deletions of SMN1, exons 7 and 8, the exons 1, 4, and 6 deletions are most likely located in SMN1. The SMN1/2, exons 1, 4, and 6 copy numbers did not correlate fully with each other or with the SMN1, exon 7 and 8 copy numbers in M1/M1b or M1/M1w individuals suggesting that these copies may not be contiguous.
The GTF2H2, exon 5 and NAIP/NAIPΨ, exon 13 copy numbers were found to differ statistically between M1/M1b and M1/M1w individuals (Kruskal-Wallis test: H = 10.8, p = 0.001 and H = 13.8, p = 0.0002, respectively). A deletion of GTF2H2, exon 5 and NAIP/NAIPΨ, exon 13 were more frequently observed in M1/M1b individuals [66.7% (50/75) and 61.3% (46/75) respectively] than M1/M1w individuals [50% (15/30) and 23.3% (7/30) respectively].
SERF1B copy numbers were found to differ significantly between M1/M1b and M1/M1w individuals (Kruskal-Wallis test: H = 3.9, p = 0.0480), most likely due to a higher frequency of heterozygous deletions of SERF1B in 34.7% (26/75) of M1/M1b individuals compared to 0% (0/30) in M1/M1w individuals. There was no difference in copy number of RAD17 between M1/M1b and M1/M1w individuals.
A higher frequency of deletions extending into the rest of SMN1/2 (exons 1, 4, and 6), NAIP/NAIPΨ, exon 13, GTF2H2, exon 5 and SERF1B in M1/M1b patients when compared to M1/M1w patients suggest that large deletions are more common in M1/M1b patients than M1/M1w patients, discrepant from results obtained from SMN1, exon 8 and NAIP, exon 5 analysis.
Comparative Analysis of M2/M2b and M2/M2w Patients
From a previous retrospective audit of patients referred to the Division for SMA testing, performed from September 1991 to October 2015, it was shown that homozygous deletions of SMN2, exon 7 were identified in 12.4% (123/991) of black patients, 4.7% (9/192) of white patients, 4% (2/50) of Indian patients and 18.8% (3/16) of patients with mixed ancestry. There is a significantly higher percentage of SMN2, exon 7 deletions in black patients when compared to white patients (Chi-square test: χ2 = 11.64; p = 0.000645).
MLPA analysis confirmed homozygous SMN2, exon 7 deletions in 100% (50/50) of M2/M2b patients and 100% (8/8) of M2/M2w patients identified. Homozygous deletions of SMN2, exon 8 were detected in 98% (49/50) of M2/M2b patients and 100% (8/8) of M2/M2w patients. Only one M2/M2b individual had a smaller deletion of SMN2, exon 7 which did not extend into exon 8. CNVs of SMN2, exons 7 and 8 were not found to differ statistically between M2/M2b and M2/M2w patients (Kruskal-Wallis test: H = 2.4; p = 0.1252 and H = 0.2; p = 0.6921, respectively).
CNVs of NAIP/NAIPΨ, exon 13 were found to differ statistically between M2/M2b and M2/M2w individuals (Kruskal-Wallis test: H = 5.6; p = 0.0177), most likely due to deletions being more frequently observed in M2/M2b [44% (22/50) than M2/M2w individuals (0% (0/8)].
CNVs of SMN1/2, exons 1 (Kruskal-Wallis test: H = 0.07; p = 0.7904), 4 (Kruskal-Wallis test: H = 2.5; p = 0.1107), and 6 (Kruskal-Wallis test: H = 0.8; p = 0.3579), GTF2H2, exon 5 (Kruskal-Wallis test: H = 1.98; p = 0.1596) and SERF1B (Kruskal-Wallis test: H = 0.02; p = 0.8855) were not found to differ statistically between M2/M2b and M2/M2w individuals. There was no difference in copy number of RAD17 between M2/M2b and M2/M2w individuals. The MLPA probes for exons 1, 4, and 6 cannot distinguish between SMN1 and SMN2, but since these patients have homozygous deletions of SMN2, exons 7 and 8, the exons 1, 4, and 6 deletions are most likely located in SMN2.
Comparative Analysis of U/Ub Patients With Black Control Groups (N/Nb, M1/M1b and M2/M2b)
Significant differences (p < 0.05) between U/Ub patients and M1M1B and M2M2B patients were observed, suggesting that hypothesized novel pathogenic CNVs are distinct from the common homozygous deletions of SMN1, exon 7 and SMN2, exon 7. U/Ub patients more closely resembled N/Nb individuals. Multiple copies (>2 copies) were observed for SMN1, exon 7 [38.9% (28/72) and SMN1, exon 8 [50% (36/72)] in U/Ub patients which is similar to that found in N/Nb individuals. A significant difference of the SMN1, exon 7 copy number between U/Ub patients and N/Nb individuals was observed and could be attributed to the presence of heterozygous SMN1, exon 7 deletions in 8.3% (6/72) of U/Ub patients, not observed in any N/Nb individuals [0% (0/122)]. This result stands in contrast to a previous South-African study that reported a heterozygous SMN1, exon 7 deletion rate of 69.5% (16/23) (Labrum et al., 2007).
Haplotype and CNV Pattern Analysis N/Nb Families
Only 31.7% (19/60) of N/Nb families were completely informative where the phase of CNVs could be determined with certainty. For 60% (36/60) of N/Nb families, multiple combinations were possible, due to the presence of multiple copies of one or more probe regions. The exact locations of these multiple copies are uncertain. Discrepant results were observed in 5% (3/60) of N/Nb families possibly due to non-paternity or novel deletion/duplication events in the proband. Two N/Nb families had clear novel results with a new mutation rate of 3.3% (2/60). In total, 19 N/Nb families, consisting of 38 unrelated parents were found to be informative, from which 76 haplotypes were constructed. In total, 35 unique haplotypes were identified, emphasizing the high variability of this region.
M1/M1b Families
Only 44% (11/25) of M1/M1b families were completely informative where the phase of CNVs could be assigned with certainty. For 48% (12/25) of these M1/M1b families, multiple combinations of CNVs were possible, due to the presence of multiple copies of one or more probes. Discrepant results were observed in 8% (2/25) of M1/M1b families which could be due to a variety of causes such as non-paternity or novel deletion or duplication events in the proband. In total, 22 pathogenic haplotypes were constructed from probands from M1/M1b families. Of these, 17 unique haplotypes were identified, once again emphasizing the high variability of this region.
Discussion
The SMN1 gene is the key gene associated with SMA with the SMN2 gene thought to have a disease-modifying effect. Current drug therapies are aimed at increasing the FL-SMN transcripts produced from SMN2. Potential large complex rearrangements of the SMN region may play a role in the SMA disease mechanism in the black SA population and may influence diagnosis and potentially the effect of drug therapies. Therefore it is valuable to investigate the genetic CNV background of the black SA population.
A major limitation of previous quantitative studies of the SMN region performed in African-American (Hendrickson et al., 2009; Sugarman et al., 2012) and sub-Saharan African populations (Sangaré et al., 2014) was that CNV analysis was performed in unaffected individuals, with the exception of prenatal screening performed by Sugarman et al. This study focuses on comparing CNVs in black individuals who are negative for SMA (N/Nb) to identify non-pathogenic CNVs as well as patients with known homozygous SMN1 and SMN2, exon 7 deletions (M1/M1b and M2/M2b, respectively) and patients who are clinically suggestive of SMA (U/Ub) to delineate potential pathogenic CNVs.
Multiple Copies of the Telomeric Region (SMN1, Exons 7 and 8 and NAIP, Exon 5) Were Observed in N/Nb Individuals and Could Complicate Analysis
In this study, 50.8% of N/Nb individuals were found to have multiple (3–6) copies of SMN1, exon 7, which is similar to previous reports of 46.8% (Hendrickson et al., 2009) and 47.1% (Sugarman et al., 2012) in African-American individuals and a combined percentage of 48.6% in sub-Saharan African populations (Mali, Nigeria and Kenya) (adapted from Sangaré et al., 2014). In contrast, N/Nw individuals have a much lower percentage of multiple SMN1, exon 7 copies of 3.3%, which is comparable to previous reports of 6.3% in white North-American populations (Hendrickson et al., 2009) and a combined percentage of 2.6% in European populations (Germany, France and Sweden) (summarized in Figure 1, Feldkötter et al., 2002; Corcia et al., 2012).
Similarly, multiple copies of SMN1, exon 8 and NAIP, exon 5 were more frequently observed in N/Nb individuals (54.9% and 37.7%, respectively) when compared to N/Nw individuals (6.7% and 13.3%, respectively).
No SMA carriers (individuals with a heterozygous deletions of SMN1, exon 7) were identified in either N/Nb or N/Nw individuals in this study in contrast to the previously predicted SA carrier rate of 1/50 in the black population and 1/23 in the white population (Labrum et al., 2007). Small sample sizes could have caused this discrepancy in both N/Nb (n = 122) and N/Nw (n = 30) individuals in this study.
Further, the discrepancy in N/Nb individuals could also be due to two additional reasons. Firstly, MLPA analysis is a very robust technique, which has built-in statistical tests and extensive normalization to multiple exogenous regions, which are likely to yield more reliable and accurate results than the previously used in-house dosage system, which normalized results against a single exogenous region (Labrum et al., 2007). Secondly, it is likely that there is a higher frequency of heterozygous SMN1 deletion carriers (2:0, 3:0, 4:0, 5:0, 6:0, etc.) in the black SA population, not detectable by either of these two assays. These results are supported by a previous study performed by Sugarman et al. (2012) who reported heterozygous SMN1 deletions (2:0) to be more common in African-American individuals (27%, n = 4 883) when compared to white individuals (3.6%, n = 24 471). As a result, the carrier detection rate in African-American individuals was lower at 70% versus 91% in other population groups.
MLPA cannot provide information on the location or phase of multiple SMN1, exon 7 copies on an individual's chromosomes and therefore these multiple copies could be located on a single chromosome, resulting in a heterozygous SMN1 deletion carrier profile. MLPA is therefore not a reliable technique to detect SMA carriers in the black SA population.
Large Deletions Extending Into the Rest of the SMN Region Appear to Be More Common in M1/M1b Patients
SMN1, exon 8 has been reported to be deleted together with SMN1, exon 7 in 93% of positive SMA cases (Lefebvre et al., 1995). Furthermore, NAIP deletions have been associated with SMN1 deletions in 67% of SMA type I patients (Roy et al., 1995). A previous SA study proposed that M1/M1w patients had larger homozygous deletions of SMN1, exon 7 also encompassing the telomeric SMN1, exon 8 and NAIP more often than M1/M1b patients (Labrum et al., 2007). Furthermore, M1/M1b patients were previously reported to have a homozygous SMN1, exon 7 deletion in conjunction with a homozygous NAIP deletion (SMN1, exon 8 is present), suggestive of a gene conversion from SMN1, exon 7 to SMN2, exon 7, more often than M1/M1w patients (Stevens et al., 1999; Labrum et al., 2007). CNV results of SMN1, exon 7, 8 and NAIP from this study did not differ significantly from results from the previous study in either M1/M1w (Chi-square test: χ2 = 0.9, p = 0.8) or M1/M1b patients (Chi-square test: χ2 = 1.6, p = 0.7).
MLPA analysis using the P021 probe mix offers a more extensive look into the rest of the SMN region, suggesting a different hypothesis. NAIP and GTF2H2 deletions have been observed in patients with SMA (Roy et al., 1995; Carter et al., 1997)) and could therefore provide some information on the extent of SMN1, exon 7 deletions. A higher frequency of homozygous and heterozygous deletions extending into the rest of SMN1/2 (exons 1, 4, 6, and 8), NAIP/NAIPΨ, exon 13, GTF2H2, exon 5 and SERF1B were observed in M1/M1b patients compared to M1/M1w patients. These observations suggest that large deletions are more common in M1/M1b patients than M1/M1w patients contrasting results from only analyzing SMN1, exon 8 and NAIP, exon 5.
Hybrid genes could mask larger deletions of the SMN1 gene and could have confounded previous SA reports (Stevens et al., 1999; Labrum et al., 2007), creating the impression of smaller deletions in M1/M1b patients when compared to M1/M1w patients. In support of this hypothesis, the CNVs of the centromeric SMN2, exons 7 and 8 do not correlate in N/Nb, M1/M1b, and N/Nb individuals, suggesting that these two regions may not be contiguous potentially due to gene conversions or other rearrangements.
In contrast, M1/M1w patients had a higher frequency of multiple copies of SMN2, exon 7 (3 copies: 33.3%, 4 copies: 3.3%) when compared to M1/M1b patients (3 copies: 6.7%, 4 copies: 4%), suggesting that gene conversion from telomeric SMN1, exon 7 to centromeric SMN2, exon 7 might be more common in M1/M1w patients.
Homozygous SMN2, Exons 7 and 8 Deletions Could Form Part of the Normal Variation in N/Nb Individuals
The high frequency of homozygous SMN2, exon 7 deletions in N/Nb individuals (27%) when compared to N/Nw individuals (6.7%) suggests that these deletions form part of the general variation in the black SA population. These frequencies are similar to international reports of ~10% (Corcia et al., 2002; Gamez et al., 2002).
Primates only have one copy of the SMN1 gene. It has been hypothesized that the SMN region in early humans consisted of only the SMN1 gene. Due to the hypervariable nature of the SMN region, duplications of the SMN region resulted in multiple copies of the SMN1 gene, often observed in individuals of African descent (Dennis et al., 2017). This scenario is supported by the observation of multiple copies of SMN1 in conjunction with the lack of SMN2 in black SA individuals (N/Nb and U/Ub individuals). The duplicated SMN1 gene diverged into the SMN2 gene due to mutations (more specifically, the critical c.840C > T change in exon 7). A CNV containing both the SMN1 and SMN2 genes is more commonly observed in individuals of European descent (Kelter et al., 2000). A loss of SMN1 could take place as a result of a deletion of SMN1 or a gene conversion from SMN1 to SMN2, an arrangement observed more frequently in individuals of European descent (van der Steege et al., 1996). The higher rate of gene conversion in white SA individuals supports this hypothesis (M1/M1w). Figure 3 summarizes the different CNVs of the SMN region and their evolution.
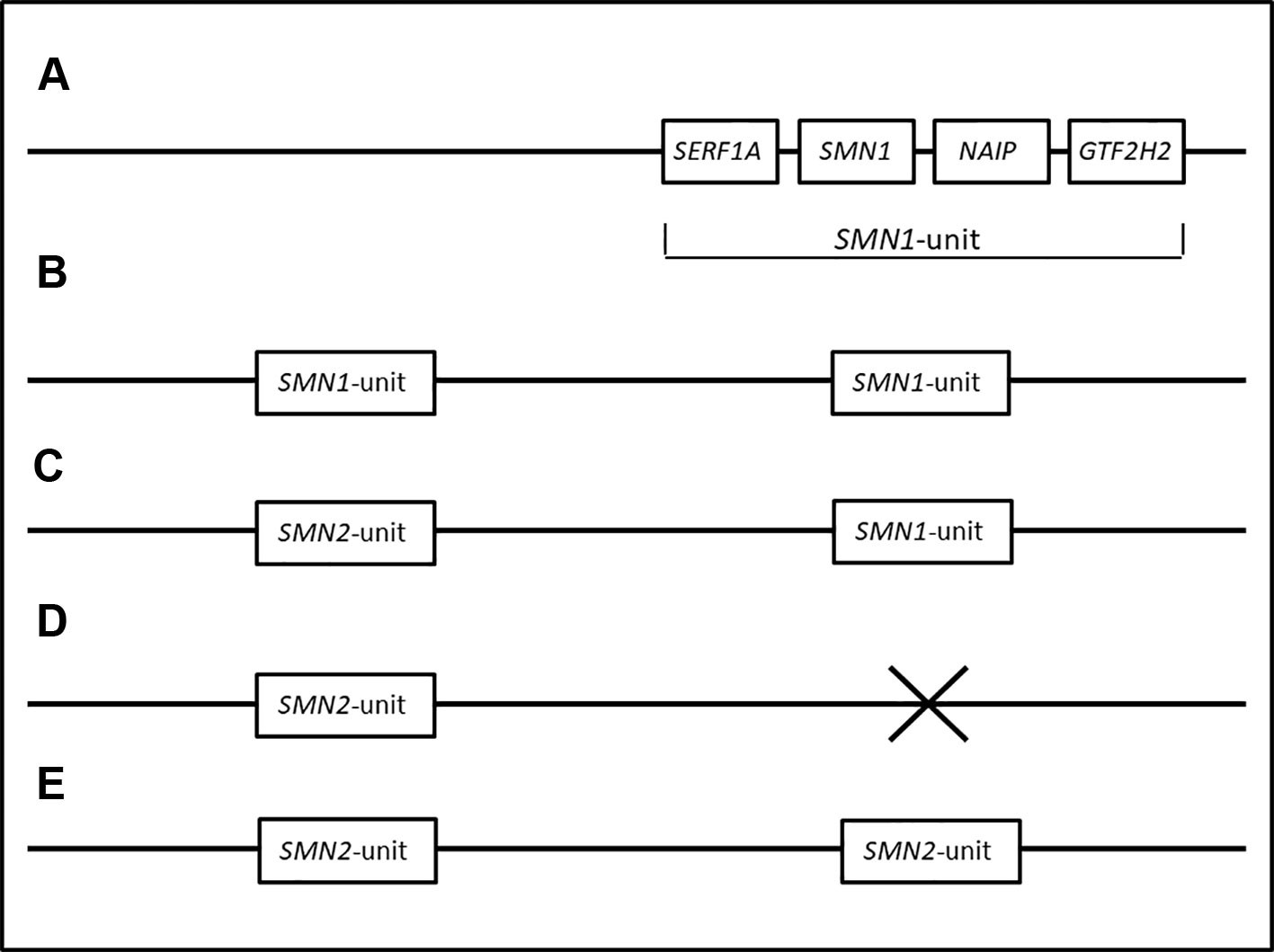
Figure 3 Hypothetical haplotypes representing the transformation of the SMN region from ancestral to modern populations. (A) represents the proposed order of genes in the SMN1 region. The SMN region in primates and early humans are thought to have consisted of only one copy of the SMN1 gene. (B) Duplications of the SMN1 region resulted in multiple copies of the SMN1 gene, frequently observed in individuals of African descent (Dennis et al., 2017). This is supported by the observation of multiple copies of SMN1 in conjunction with the lack of SMN2 in black SA individuals (N/Nb and U/Ub individuals). (C) Mutations in the duplicated SMN1 gene resulted in the SMN2 gene. A chromosome consisting of one SMN1 and one SMN2 gene.is thought to be the most common genotype seen in populations of European descent (N/Nw individuals). (D, E) represent individuals with a deletion of SMN1. A homozygous deletion of SMN1, exon 7 causes SMA (M1/M1w and M1/M1b). (E) Deletions of SMN1 in M1/M1w individuals are commonly caused by gene conversions from SMN1 to SMN2, resulting in multiple copies of SMN2.
Human-specific segmental duplication of the SMN region resulting in the inverted centromeric SMN duplication (including SMN2) has been estimated to have taken place 0.3 mya. The exact method of further duplication of the SMN ancestral structure to the structure of the human reference today has been difficult to determine due to polymorphic, palindromic duplications of the region (Dennis et al., 2017).
No Novel Pathogenic CNVs Were Identified in U/Ub Patients
In contrast to previous SA reports of heterozygous SMN1, exon 7 deletions being present in 69.5% of U/Ub patients (Labrum et al., 2007), only 8.3% of U/Ub patients were confirmed to have heterozygous deletions of SMN1, exon 7 in this study. This discrepancy could firstly be due to MLPA analysis being a very robust technique, which has built-in statistical tests and extensive normalization, which are likely to yield more reliable and accurate results than the previously used in-house dosage assay (Labrum et al., 2007). In support of this hypothesis, seven individuals previously reported to have heterozygous deletions of SMN1, exon 7 on the in-house dosage assay were retested on MLPA of which four individuals had discrepant results. Two of these individuals had two copies and two individuals had three copies of SMN1, exon 7, which were mistaken for one copy on the in-house dosage assay.
Secondly the presence of multiple copies of SMN1, exon 7 could mask the actual heterozygous SMN1, exon 7 deletion rate in U/Ub patients.
No novel pathogenic CNVs were identified in U/Ub patients. The presence of potential large complex rearrangements in the black SA population not detectable by current standard diagnostic techniques is supported by the high variability and lack of correlation of copy number between different genes and exons seen in black SA individuals. SMN genes and exons may not have contiguous coding regions and the relationship between these complex rearrangements and the effect on SMN protein expression needs to be further investigated.
Haplotype and CNV Pattern Analysis
As part of a previous study performed in the Division, linkage analysis, using two intragenic and six extragenic microsatellite markers across the SMN1 gene, was performed to see if a common chromosomal background could be established in U/Ub patients. No clear haplotype or common allele was identified and it was reported that it was particularly difficult to construct haplotypes (Labrum et al., 2007).
Similarly, in this study, multiple gene and exon copies in the black SA population complicated haplotype construction. Only 44% of M1/M1b families and 31.7% of N/Nb families were completely informative where the phase of the haplotype could be determined with certainty. The orientation of genes in the SMN region is not known and this study could not predict the arrangement of genes or exons even though the phase could be determined.
Potential novel events (sporadic deletions or duplications) were observed in 3.3% (2/61) of N/Nb families. A new mutation rate of 3.3% is not unexpected as novel mutations have been reported at a rate of 2% due to the high instability of this region (Wirth et al., 1997). Moosa and Dawood reported a paucity of family history in black SA families affected by SMA potentially due to SMA being more sporadic in this population (Moosa and Dawood, 1990) although this may be due to poor ascertainment. Sporadic mutations could be explained as a consequence of novel gene conversion and rearrangement events.
Two variants, c.885+83T > G and c.885+667delAT in exon 8 of the SMN1 gene have been described to be associated with multiple SMN1 copies on a single chromosome in combination with a SMN1, exon 7 deletion on the other chromosome in African American and Ashkenazi Jewish population groups. It was suggested that these two variants are associated with heterogous SMN1 deletions (2:0) and could be useful in refining the carrier risk in individuals who have multiple copies of SMN1, exon 7 (Luo et al., 2014). The association of the c.885+83T > G and c.885+667delAT variants to heterozygous SMN1 deletion (2:0) haplotypes in the black SA population was investigated as part of a previous unpublished study in the Division. Both the c.885+83T > G and c.885+667delAT variants were observed in 60% (3/5) of individuals with known heterozygous SMN1 deletions (2:0) compared to 0% (0/7) of individuals known to have two copies of SMN1, exon 7 copy, one copy on each of their two chromosomes (1:1), suggesting that the two variants are associated with duplicated SMN1, exon 7 alleles. These two variants could be useful in refining the carrier risk of black SA individuals who have multiple copies of SMN1, exon 7.
Implications for Diagnostic Testing
With the advent of new sequencing technologies, pan-ethnic population-based expanded carrier screening has been gaining momentum internationally. As an example, Israel has implemented a genetic screening program including carrier testing for SMA for couples of reproductive age (Zlotogora et al., 2016). The American College of Medical Genetics (ACMG) supports the inclusion of SMA into expanded carrier screening tests (Prior and Professional Practice and Guidelines Committee, 2008). Caution is advised against population screening in the black SA population, due to the presence of multiple copies of SMN1, exon 7 which could significantly impair accurate carrier detection and lead to false negative carrier results. MLPA may be useful in detecting the carrier risk in members of M1/M1b families but it is highly recommended that MLPA results of samples referred for prenatal and carrier testing should be analyzed within a family context to identify the phase of multiple SMN1 gene copies in all SA populations.
Challenges and Limitations of the Study
The SMN region is extremely complex containing multiple pseudogenes (Selig et al., 1995) and repetitive sequences (Bürglen et al., 1996) within a large inverted segmental duplication. Due to this complexity, there is limited understanding of the exact order and location of genes in the SMN region. This is complicated even further since CNV trends observed in the various patient and control groups tested on MLPA, suggest that large rearrangements in the SMN region form part of the general variation within the black SA population.
It is well established that African populations have a higher level of genetic diversity than any other population (Tishkoff and Williams, 2002; Tishkoff and Kidd, 2004; Conrad et al., 2006; Pickrell et al., 2014). A local group of researchers who investigated CNVs in SA populations found that haplotype block lengths are significantly smaller in African populations when compared to non-African populations. These regions seem to coincide with recombination hotspots (Chimusa et al., 2015). Very few of these recombination hotspots seem to be shared between African and other populations (Choudhury et al., 2014; Chimusa et al., 2015). Perhaps the high variability of the SMN region in the black SA population could be due to frequent recombination events in the SMN region. This hypothesis is supported by a new mutation rate of 3.3% in this study which is comparable to the high new mutation rate seen in other populations (Wirth et al., 1997). Novel events may also influence the recurrence risk in black SA SMA families.
We need to identify and comprehend non-pathogenic CNVs in the general SA population to fully understand disease mechanisms overlaying these variations, specifically in the SMN region. The Southern African Human Genome Project (SAHGP) shows some promise in creating a better understanding of the baseline CNVs in the general black SA population (Pepper, 2011) although it is unlikely to provide detailed information on the architecture of the SMN region.
The high sequence homology of the SMN1 and SMN2 genes, with only a five nucleotide difference between the two genes and the highly variable CNVs of these genes make molecular diagnosis extremely challenging. This limitation complicates and restricts the design of primers and probes in this region and limits the choice of laboratory techniques that can be used to understand this region better. The P021-A2 MLPA kit was mainly designed to distinguish between exons 7 and 8 of the SMN1 and SMN2 gene, but cannot distinguish between exons 1, 4, and 6 of the SMN1 and SMN2 genes. This means that a combined result was observed for these probes. This makes it difficult to assign multiple copies of specific exons to either SMN1 or SMN2. Similarly, the NAIP/NAIPΨ, exon 13 probe was designed to detect the combined copy number of the NAIP gene and its centromeric copy, NAIPΨ and the GTF2H2, exon 5 was designed to detect the combined copy number of telomeric-GTF2H2 and centromeric-GTF2H2.
The copy numbers of SMN1/2 exons 1, 4, 6, and 8 do not correlate fully with each other or with the SMN1, exons 7 and 8 copy numbers in any of the groups suggesting that CNVs of the SMN1 and SMN2 genes do not consist of complete gene copies and that the exons may be non-contiguous. This is further complicated by gene conversion events between SMN1 and SMN2. Other factors which could potentially influence within- and between sample variance, is sample quality and experimental design. Result interpretation is therefore incredibly difficult and it is not possible to construct accurate CNV patterns using MLPA.
Future Studies
MLPA testing cannot give us information about the functionality of potential multiple, partial copies. RNA expression studies may be able to quantify the expression of FL-SMN transcripts which may be a more accurate indication of the amount of SMN protein produced in U/Ub patients even in the presence of multiple SMN1, exon 7 copies. If multiple copies of the SMN1 gene are present on MLPA, but there is no corresponding FL-SMN transcript, it could be indicative of partial/interrupted non-functional SMN1 copies. The expression of SMN transcripts using real-time reverse transcription PCR (qRT-PCR) is being investigated as part of a current study in the Division.
Sixteen additional genes with overlapping phenotypes to SMA have been shown to be associated with non-5q forms of SMA (Peeters et al., 2014). Due to the lack of clinical information on patients referred for SMA testing to the Division it may be more practical to perform testing by an NGS neuromuscular panel first to exclude other related neuromuscular diseases and other causes of SMA before continuing SMA testing in individuals who test negative for the homozygous SMN1, exon 7 deletion.
A previous SA study sequenced the SMN1 gene in patients found to have heterozygous SMN1, exon 7 deletions on the previously used in-house dosage assay to look for additional mutations in SMN1 (Labrum et al., 2007). No pathogenic mutations were identified. Since the accuracy of the previously used in-house dosage assay has been questioned by this study, all U/Ub and M2/M2b individuals who were found to have heterozygous deletions of SMN1, exon 7 should be sequenced to try and find a potential second pathogenic mutation. The high homology of the SMN1 and SMN2 genes complicates sequencing analysis however this challenge can be overcome with long range PCR targeting the SMN1 gene, followed by nested PCR and Sanger sequencing of exons 1–8 (Kubo et al., 2015).
PacBio single molecule, real-time sequencing (SMRT) technology6 has shown some promise with resolving large CNVs. This technology is currently limited to 20 kb reads, which may still be too small to detect the full sequence of the SMN region, which is at least 500kb. The MinION (Oxford Nanoppore technologies) nanopore sequencer generate ultra-long sequencing reads of up to 800kb and have been shown to improve the accuracy and to close gaps in the reference human genome (Jain et al., 2018). These long range sequencing technologies could be investigated to try and further define the structure of the SMN region in the black SA population.
Conclusion
This is the first report summarizing CNV patterns of the SMN region in African patients with known homozygous SMN1 and SMN2, exon 7 deletions (M1/M1b, M2/M2b) and patients who have features clinically suggestive of SMA (U/Ub). This is also the first report of CNVs patterns of the SMN region in the general black SA population.
Multiple copies of SMN1, exon 7 were observed as evidence of the marked hypervariability of the SMN region in the black SA population. These multiple copies potentially confound diagnostic and carrier testing and could potentially consist of partial, non-contiguous copies. Future studies investigating the expression of these multiple gene copies may provide information on their functional effect. No clear additional pathogenic CNV patterns were identified in U/Ub patients. This study emphasizes the lack of understanding of the architecture of the SMN region and the composition of CNVs in the black SA population. These factors need to be taken into account when counselling and performing diagnostic, carrier and prenatal testing in the black SA population.
Data Availability Statement
All datasets generated for this study are included in the article and the supplementary files.
Ethics Statement
This study was carried out in accordance with the recommendations of the 'WITS Medical Human Research Ethics Committee. Written informed consent of patients was not required as all DNA samples were referred and banked in a diagnostic setting and have been anonymised for the purpose of this study. The protocol was approved by the 'WITS Medical Human Research Ethics Committee (ethics clearance number: M130950).
Author Contributions
EV, FE, and AK contributed to the conception and design of the study. JR assisted in identifying appropriate patients for this study. EV performed all laboratory work, MLPA analysis, haplotype analysis, and statistical analysis as part of her MSc (Medicine) Human Genetics degree (obtained with distinction). EV wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
National Health Laboratory Service (NHLS) Research Trust Grant (2014-2DEV41-EVO1). WITS Faculty Research Council Grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The National Health Laboratory Service (NHLS) for the use of their facilities for this project. MRC Holland who has generously sponsored 800 MLPA probe mix reactions for this project. Ms Haseena Khan who investigated the association of the c.885+83T > G and c.885+667delAT variants to heterozygous SMN1 deletion (2:0) carrier haplotypes in the black SA population as part of her BSc (Hons) project. This manuscript is based on findings reported in a Masters dissertation: Vorster (2017) Determining the molecular basis of spinal muscular atrophy in the black South African population (University of the Witwatersrand, Johannesburg, South Africa): http://wiredspace.wits.ac.za/handle/10539/25810.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00054/full#supplementary-material
Footnotes
- ^ Online Mendelian Inheritance in Man (OMIM), OMIM accession numbers: 600354, 601627, http://www.omim.org.
- ^ MRC Holland MLPA general protocol and P021 product description, http://www.mlpa.com/WebForms/.
- ^ EMQN, https://www.emqn.org.
- ^ Real Statistics Using Excel, http://www.real-statistics.com.
- ^ Ensembl Genome Browser, https://www.ensembl.org
- ^ PacBio, https://www.pacb.com/smrt-science/smrt-sequencing/
References
Bürglen, L., Lefebvre, S., Clermont, O., Burlet, P., Viollet, L., Cruaud, C., et al. (1996). Structure and organization of the human survival motor neurone (SMN) gene. Genomics 32, 479–482. doi: 10.1006/geno.1996.0147
Barker, K. (2004). At the Bench: A Laboratory Navigator, Updated Edition. 2nd edition (Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press).
Campbell, L., Potter, A., Ignatius, J., Dubowitz, V., Davies, K. (1997). Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am. J. Hum. Genet. 61, 40–50. doi: 10.1086/513886
Cantsilieris, S., Western, P. S., Baird, P. N., White, S. J. (2014). Technical considerations for genotyping multi-allelic copy number variation (CNV), in regions of segmental duplication. BMC Genomics 15, 329. doi: 10.1186/1471-2164-15-329
Carter, T. A., Bönnemann, C. G., Wang, C. H., Obici, S., Parano, E., De Fatima Bonaldo, M., et al. (1997). A multicopy transcription-repair gene, BTF2p44, maps to the SMA region and demonstrates SMA associated deletions. Hum. Mol. Genet. 6, 229–236. doi: 10.1093/hmg/6.2.229
Chimusa, E. R., Meintjies, A., Tchanga, M., Mulder, N., Seoighe, C., Seioghe, C., et al. (2015). A genomic portrait of haplotype diversity and signatures of selection in indigenous southern African populations. PloS Genet. 11, e1005052. doi: 10.1371/journal.pgen.1005052
Choudhury, A., Hazelhurst, S., Meintjes, A., Achinike-Oduaran, O., Aron, S., Gamieldien, J., et al. (2014). Population-specific common SNPs reflect demographic histories and highlight regions of genomic plasticity with functional relevance. BMC Genomics 15, 437. doi: 10.1186/1471-2164-15-437
Conrad, D. F., Jakobsson, M., Coop, G., Wen, X., Wall, J. D., Rosenberg, N. A., et al. (2006). A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 38, 1251–1260. doi: 10.1038/ng1911
Corcia, P., Mayeux-Portas, V., Khoris, J., de Toffol, B., Autret, A., Muh, J.-P., et al. (2002). Abnormal SMN1 gene copy number is a susceptibility factor for amyotrophic lateral sclerosis. Ann. Neurol. 51, 243–246. doi: 10.1002/ana.10104
Corcia, P., Ingre, C., Blasco, H., Press, R., Praline, J., Antar, C., et al. (2012). Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur. J. Hum. Genet. 20, 588–591. doi: 10.1038/ejhg.2011.255
Dennis, M. Y., Harshman, L., Nelson, B. J., Penn, O., Cantsilieris, S., Huddleston, J., et al. (2017). The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol. 1, 692–693. doi: 10.1038/s41559-016-0069
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T. F., Wirth, B. (2002). Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368. doi: 10.1086/338627
Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Eng. J. Med. 377 (18), 1723–1732. doi: 10.1056/NEJMoa1702752
Francis, M. J., Nesbit, M. A., Theodosiou, A. M., Rodrigues, N. R., Campbell, L., Christodoulou, Z., et al. (1995). Mapping of retrotransposon sequences in the unstable region surrounding the spinal muscular atrophy locus in 5q13. Genomics 27, 366–369. doi: 10.1006/geno.1995.1059
Fried, K., Emery, A. E. (1971). Spinal muscular atrophy type II. A separate genetic and clinical entity from type I (Werdnig-Hoffmann disease) and type 3 (Kugelberg-Welander disease). Clin. Genet. 2, 203–209. doi: 10.1111/j.1399-0004.1971.tb00278.x
Gamez, J., Barcelo, M. J., Munoz, X., Carmona, F., Cusco, I., Baiget, M., et al. (2002). Survival and respiratory decline are not related to homozygous SMN2 deletions in ALS patients. Neurology 59, 1456–1460. doi: 10.1212/01.WNL.0000032496.64510.4E
He, J., Zhang, Q.-J., Lin, Q.-F., Chen, Y.-F., Lin, X.-Z., Lin, M.-T., et al. (2013). Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene 518, 325–329. doi: 10.1016/j.gene.2012.12.109
Hendrickson, B. C., Donohoe, C., Akmaev, V. R., Sugarman, E. A., Labrousse, P., Boguslavskiy, L., et al. (2009). Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 46, 641–644. doi: 10.1136/jmg.2009.066969
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi: 10.1038/nbt.4060
Jedrzejowska, M., Borkowska, J., Zimowski, J., Kostera-Pruszczyk, A., Milewski, M., Jurek, M., et al. (2008). Unaffected patients with a homozygous absence of the SMN1 gene. Eur. J. Hum. Genet. EJHG 16, 930–934. doi: 10.1038/ejhg.2008.41
Kelter, A. R., Herchenbach, J., Wirth, B. (2000). The transcription factor-like nuclear regulator (TFNR) contains a novel 55-amino-acid motif repeated nine times and maps closely to SMN1. Genomics 70, 315–326. doi: 10.1006/geno.2000.6396
Kubo, Y., Nishio, H., Saito, K. (2015). A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J. Hum. Genet. 60, 233–239. doi: 10.1038/jhg.2015.16
Kugelberg, E., Welander, L. (1956). Heredofamilial juvenile muscular atrophy simulating muscular dystrophy. AMA Arch. Neurol. Psychiatry 75, 500–509. doi: 10.1001/archneurpsyc.1956.02330230050005
Labrum, R., Rodda, J., Krause, A. (2007). The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul. Disord. NMD 17, 684–692. doi: 10.1016/j.nmd.2007.05.005
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lorson, C. L., Hahnen, E., Androphy, E. J., Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Luo, M., Liu, L., Peter, I., Zhu, J., Scott, S. A., Zhao, G., et al. (2014). An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. Off. J. Am. Coll. Med. Genet. 16, 149–156. doi: 10.1038/gim.2013.84
Markowitz, J. A., Singh, P., Darras, B. T. (2012). Spinal muscular atrophy: a clinical and research update. Pediatr. Neurol. 46, 1–12. doi: 10.1016/j.pediatrneurol.2011.09.001
McAndrew, P. E., Parsons, D. W., Simard, L. R., Rochette, C., Ray, P. N., Mendell, J. R., et al. (1997). Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422. doi: 10.1086/515465
Mercuri, E., Finkel, R. S., Muntoni, F., Wirth, B., Montes, J., Main, M., et al. (2018). Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 28, 103–115. doi: 10.1016/j.nmd.2017.11.005
Miller, S. A., Dykes, D. D., Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215. doi: 10.1093/nar/16.3.1215
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Moosa, A., Dawood, A. (1990). Spinal muscular atrophy in African children. Neuropediatrics 21, 27–31. doi: 10.1055/s-2008-1071453
Mrad, R., Dorboz, I., Ben Jemaa, L., Maazoul, F., Trabelsi, M., Chaabouni, M., et al. (2006). Molecular analysis of the SMN1 and NAIP genes in 60 Tunisian spinal muscular atrophy patients. Tunis. Médicale 84, 465–469.
Ndiaye, O., Sall, G., Sylla, A., Diouf, S., Diagne, I., Kuakuvi, N. (2002). Progressive spinal amyotrophy type I or Werdnig-Hoffman disease. Apropos of 5 cases in Dakar (Senegal). Bull. Société Pathol. Exot. 1990 95, 81–82.
Pearn, J. H., Hudgson, P., Walton, J. N. (1978). A clinical and genetic study of spinal muscular atrophy of adult onset: the autosomal recessive form as a discrete disease entity. Brain J. Neurol. 101, 591–606. doi: 10.1093/brain/101.4.591
Peeters, K., Chamova, T., Jordanova, A. (2014). Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain J. Neurol. 137, 2879–2896. doi: 10.1093/brain/awu169
Pelleboer, R. A., Maaswinkel-Mooy, P. D., Gelderen, H. H. (1989). Werdnig-Hoffmann disease in a Nigerian infant. Trop. Geogr. Med. 41, 282–284.
Pepper, M. S. (2011). Launch of the Southern African human genome programme. South Afr. Med. J. Suid-Afr. Tydskr. Vir Geneeskd. 101, 287–288. doi: 10.7196/SAMJ.4860
Pickrell, J. K., Patterson, N., Loh, P.-R., Lipson, M., Berger, B., Stoneking, M., et al. (2014). Ancient west Eurasian ancestry in southern and eastern Africa. Proc. Natl. Acad. Sci. U. S. A. 111, 2632–2637. doi: 10.1073/pnas.1313787111
Prior, T. W., Swoboda, K. J., Scott, H. D., Hejmanowski, A. Q. (2004). Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A. 130A, 307–310. doi: 10.1002/ajmg.a.30251
Prior, T. W., Professional Practice and Guidelines Committee. (2008). Carrier screening for spinal muscular atrophy. Genet Med. 10(11), 840–2. doi: 10.1097/GIM.0b013e318188d069
Roy, N., Mahadevan, M. S., McLean, M., Shutler, G., Yaraghi, Z., Farahani, R., et al. (1995). The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80, 167–178. doi: 10.1016/0092-8674(95)90461-1
Sangaré, M., Hendrickson, B., Sango, H. A., Chen, K., Nofziger, J., Amara, A., et al. (2014). Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann. Neurol. 75, 525–532. doi: 10.1002/ana.24114
Scheffer, H., Cobben, J. M., Matthijs, G., Wirth, B. (2001). Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur. J. Hum. Genet. 9, 484–491. doi: 10.1038/sj.ejhg.5200667
Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D., Diepvens, F., Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30, e57. doi: 10.1093/nar/gnf056
Schwartz, M., Sørensen, N., Hansen, F. J., Hertz, J. M., Nørby, S., Tranebjaerg, L., et al. (1997). Quantification, by solid-phase minisequencing, of the telomeric and centromeric copies of the survival motor neuron gene in families with spinal muscular atrophy. Hum. Mol. Genet. 6, 99–104. doi: 10.1093/hmg/6.1.99
Selig, S., Bruno, S., Scharf, J. M., Wang, C. H., Vitale, E., Gilliam, T. C., et al. (1995). Expressed cadherin pseudogenes are localized to the critical region of the spinal muscular atrophy gene. Proc. Natl. Acad. Sci. U. S. A. 92, 3702–3706. doi: 10.1073/pnas.92.9.3702
Shawky, R. M., Abd el-Aleem, K., Rifaat, M. M., Moustafa, A. (2001). Molecular diagnosis of spinal muscular atrophy in Egyptians. East Mediterr. Health J. Rev. Santé Méditerranée Orient 7, 229–237. Al-Majallah Al-Ṣiḥḥīyah Li-Sharq Al-Mutawassit.
Stevens, G., Yawitch, T., Rodda, J., Verhaart, S., Krause, A. (1999). Different molecular basis for spinal muscular atrophy in South African black patients. Am. J. Med. Genet. 86, 420–426. doi: 10.1002/(SICI)1096-8628(19991029)86:5<420::AID-AJMG5>3.0.CO;2-S
Sugarman, E. A., Nagan, N., Zhu, H., Akmaev, V. R., Zhou, Z., Rohlfs, E. M., et al. (2012). Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. EJHG 20, 27–32. doi: 10.1038/ejhg.2011.134
Tazir, M., Geronimi, C. (1990). Chronic childhood spinal muscular atrophies in Algeria. A Genet. Study J. Neurol. Sci. 96, 89–101. doi: 10.1016/0022-510X(90)90059-V
Tishkoff, S. A., Kidd, K. K. (2004). Implications of biogeography of human populations for “race” and medicine. Nat. Genet. 36, S21–S27. doi: 10.1038/ng1438
Tishkoff, S. A., Williams, S. M. (2002). Genetic analysis of African populations: human evolution and complex disease. Nat. Rev. Genet. 3, 611–621. doi: 10.1038/nrg865
van der Steege, G., Grootscholten, P. M., Cobben, J. M., Zappata, S., Scheffer, H., den Dunnen, J. T., et al. (1996). Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus on chromosome 5. Am. J. Hum. Genet. 59, 834–838.
Vorster, E., Essop, F., Krause, A. (2011). “MLPA as a diagnostic tool to detect spinal muscular atrophy (SMA) in South African populations (Poster),” SASHG (South African Society of Human Genetics), 2010 Joint International Conference of the African and Southern African Societies of Human Genetics (SASHG)Cape Town, South Africa6-9 March 2011(NHLS & WITS: Johannesburg).
Vorster, E. (2017). Determining the molecular basis of spinal muscular atrophy in the black south African population. [dissertation/master"s thesis] (Johannesburg, South Africa: University of the Witwatersrand).
Wirth, B., Schmidt, T., Hahnen, E., Rudnik-Schöneborn, S., Krawczak, M., Müller-Myhsok, B., et al. (1997). De Novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am. J. Hum. Genet. 61, 1102–1111. doi: 10.1086/301608
Wirth, B., Herz, M., Wetter, A., Moskau, S., Hahnen, E., Rudnik-Schöneborn, S., et al. (1999). Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 64, 1340–1356. doi: 10.1086/302369
Wirth, B., Brichta, L., Schrank, B., Lochmuller, H., Blick, S., Baasner, A., et al. (2006). Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 119, 422–428. doi: 10.1007/s00439-006-0156-7
Wirth, B. (2000). An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 15, 228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9
Zheleznyakova, G. Y., Kiselev, A. V., Vakharlovsky, V. G., Rask-Andersen, M., Chavan, R., Egorova, A. A., et al. (2011). Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med. Genet. 12, 96. doi: 10.1186/1471-2350-12-96
Keywords: spinal muscular atrophy, survival motor neuron 1, survival motor neuron 2, multiplex ligation-dependent probe amplification, copy number variations, rearrangement, South Africa
Citation: Vorster E, Essop FB, Rodda JL and Krause A (2020) Spinal Muscular Atrophy in the Black South African Population: A Matter of Rearrangement? Front. Genet. 11:54. doi: 10.3389/fgene.2020.00054
Received: 16 November 2018; Accepted: 17 January 2020;
Published: 13 February 2020.
Edited by:
Nicola Mulder, University of Cape Town, South AfricaReviewed by:
Brunhilde Wirth, University of Cologne, GermanyZeljka Pezer, Rudjer Boskovic Institute, Croatia
Copyright © 2020 Vorster, Essop, Rodda and Krause. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elana Vorster, dm9yc3RlcmVAYW1wYXRoLmNvLnph