 Bonnie L.J. Alberry
Bonnie L.J. Alberry Shiva M. Singh
Shiva M. Singh- Department of Biology, Western University, London, ON, Canada
Fetal alcohol spectrum disorder (FASD) is characterized by developmental and behavioral deficits caused by maternal drinking during pregnancy. Children born with FASD often face additional stresses, including maternal separation, that add yet additional deficits. The mechanism associated with this interaction is not known. We have used a mouse model for prenatal ethanol exposure and maternal separation to demonstrate that the combination of the two treatments results in more than additive deficits. Furthermore, the behavioral deficits are associated with changes in hippocampal gene expression that persist into adulthood. What initiates and maintains these changes remains to be established and forms the focus of this report. Specifically, MeDIP-Seq was used to assess if changes in promoter DNA methylation are affected by exposure to prenatal ethanol and maternal separation including its relationship to gene expression. The novel results show that different sets of genes implicated by promoter DNA methylation are affected by both treatments independently, and a relatively unique set of genes are affected by the combination of the two treatments. Prenatal ethanol exposure leads to altered promoter DNA methylation at genes important for transcriptional regulation. Maternal separation leads to changes at genes important for histone methylation and immune response, and the combination of two treatments results in DNA methylation changes at genes important for neuronal migration and immune response. Our dual results from the same hippocampal samples suggest there is minimal complementarity between changes in promoter DNA methylation and gene expression, although genes involved tend to be critical for brain development and function. While remaining to be validated, such results argue that mechanisms beyond promoter DNA methylation must be involved in lasting gene expression alterations leading to behavioral deficits implicated in FASD. They may facilitate early and reliable diagnosis, as well as novel strategies for the amelioration of FASD-related deficits.
Introduction
Fetal alcohol spectrum disorder (FASD) is a serious neurodevelopmental disorder that begins in utero, manifests during childhood, and lasts a lifetime. It is caused by prenatal ethanol exposure primarily by maternal gestational alcohol consumption and consists of a collection of conditions including developmental delays, restrictions, abnormalities, as well as intellectual and behavioral impairments (Chudley et al., 2005). The incidence of FASD varies across different populations, with the estimated prevalence in 7- to 9-year-old Canadians is between 2% and 3% (Popova et al., 2018), and between 1% and 5% in the United States (May et al., 2018). This incidence is expected to increase given a recent Canadian report that alcohol-related emergencies between 2003 and 2016 rose 4.4 times more than overall emergency visits (Myran et al., 2019). Specifically, this increase is greater for women (86.5%) than men (53.2%). Among women, 15- to 24-year-olds have the highest rate. Of the medical harms identified, suspected fetal damage including fetal alcohol syndrome rose 2133.3% from 2003 to 2016. As it may extend to other populations, this trend is concerning for more than just Ontario and does not bode well for a focus on FASD prevention via alcohol avoidance. The next best strategy is to offer interventions following early diagnosis, which remains problematic given the heterogeneity of manifestation and lack of reliable biomarker. As such, FASD continues to represent a major health and social burden in alcohol-friendly societies.
Given that prenatal alcohol exposure is the cause of FASD, and neurodevelopment is long-lasting, spanning decades, research must also focus on postnatal factors that may determine the manifestation of the disorder. Furthermore, this understanding may offer novel strategies for the remediation of FASD deficits and severity. Molecular brain research in humans is restrictive and must rely on animal models. Our lab has developed a mouse model of FASD using C57BL/6J (B6) mice (Kleiber et al., 2011), establishing that FASD is like an iceberg. Ethanol-exposed pups develop learning deficits, anxiety-like behaviors, and altered activity patterns (Allan et al., 2003; Kaminen-Ahola et al., 2010; Kleiber et al., 2011; Marjonen et al., 2015). What we see as deficit(s) is just the tip—most underlying defects are not visible and are reflected in gene expression alterations (Kleiber et al., 2012).
Mammalian neurodevelopment involves the orchestration of cellular processes that are sensitive to prenatal and postnatal environmental stresses over time (Tau and Peterson, 2010). One common postnatal stress that children born with FASD face is maternal separation. Children entering childcare systems, such as foster care or orphanages, represent a particularly vulnerable population, with FASD considerably overrepresented in such individuals (Lange et al., 2013). In Canada, the prevalence of FASD is 5 to 67 times higher than a global estimate (Lange et al., 2017). In addition to the initial gestational alcohol exposure, exposure to early life stress increases the risk of behavioral deficits, including adult psychiatric disorders (Kisely et al., 2018). Early life stress has a negative effect on learning and memory processes governed by the hippocampus in rodents (Rice et al., 2008; Oomen et al., 2010; Pillai et al., 2018). Little is known about how early life stress may affect children born with FASD (Price et al., 2017), although following gestational alcohol exposure and abuse or neglect during early development, children are more likely to have behavioral deficits, including impaired learning and memory (Coggins et al., 2007; Henry et al., 2007; Koponen et al., 2009; Koponen et al., 2013).
Since children with FASD often face additional early life stress, we have extended the model to explore how postnatal maternal separation may compound developmental and behavioral deficits following prenatal ethanol exposure (Alberry and Singh, 2016). The behaviors affected by prenatal alcohol exposure and maternal separation include anxiety-like behavior as well as learning and memory. While many brain regions may be relevant, the hippocampus forms the focus of this study for its essential role in spatial learning (Jarrard, 1993). To this end, we have also reported changes in hippocampal gene expression that persist into adulthood following prenatal ethanol exposure combined with postnatal maternal separation in B6 mice (Alberry et al., 2019). Given that the initiation and maintenance of aberrant gene expression in response to prenatal ethanol exposure includes epigenetic mechanisms, primarily DNA methylation, noncoding RNA, and histone modifications (Chen et al., 2013; Laufer et al., 2013; Chater-Diehl et al., 2016), these processes are expected to be critical in other models of FASD. In fact, FASD-related human research in epigenetic mechanisms has begun to focus on DNA methylation (Laufer et al., 2015; Portales-Casamar et al., 2016). While there are many epigenetic mechanisms that may alter transcription, here we focus on promoter DNA methylation that may repress transcription via blocked transcription factor binding to explore the canonical relationship between gene expression and promoter DNA methylation (Jaenisch and Bird, 2003).
This research is novel in that it combines the effect of prenatal ethanol exposure and maternal separation on the FASD iceberg using B6 mice at three levels—behavior, hippocampal gene expression and hippocampal DNA methylation on the same samples. Although the behavioral results are compatible with molecular changes, changes in gene expression are rarely related to changes in promoter-specific methylation. It may represent a feature of the experimental design or reflect a general feature of the development of FASD. Although one is tempted to suggest that epigenetic changes other than DNA methylation must represent important underlying mechanisms involved in the development of FASD, the issue deserves further experimentation and comprehensive evaluation. Such clarifications will be needed in the use of these results, including the development of effective measures of remediation.
Materials and Methods
Animals
C57BL/6J mice originally obtained from Jackson Laboratories (Bar Harbor, ME) were bred and housed in same-sex colonies of up to four individuals with unrestricted food and water access at the Animal Care Facility at Western University (London, Ontario, Canada). Temperature and humidity were maintained at 21°C–24°C and 40%–60%, respectively, with lighting on a 14:10-h light-dark cycle. All housing, bedding, and nest material were consistent between cages. All treatment protocols complied with ethical standards established by the Canadian Council on Animal Care and were approved by the Animal Use Subcommittee at Western University (London, Ontario, Canada).
Continuous Preference Drinking Model
Females (10 weeks old) were housed individually and randomly assigned to ethanol or control groups. Both groups had free access to water, with the ethanol group additionally provided free access to 10% ethanol in water solution (Kleiber et al., 2011; Alberry and Singh, 2016). Ethanol was introduced with increasing concentrations of 2%, 5%, and 10% after 48 h with the previous concentration. After 10 days of continuous 10% ethanol availability, females were mated with males (15 weeks old) with only water available. Males were removed after a 24-h period and 10% ethanol was returned, which represented gestational day 0. Until postnatal day 10, 10% ethanol was freely available, with only water available for the remainder of this study. Blood alcohol levels were not taken to minimize maternal stress, voluntary consumption of 10% ethanol at 14 g ethanol per kg body weight per day produces peak blood alcohol levels of 120 mg dl-1 (Allan et al., 2003). This study represents a modest amount of ethanol exposure, with experimental mice consuming 8 g ethanol per kg body weight per day on average, sufficient to produce behavioral deficits in their offspring, including hyperactivity in a novel environment, hypoactivity in a home cage environment, and learning deficits (Alberry and Singh, 2016).
Early Life Stress Via Maternal Separation and Isolation
Postnatal maternal separation and isolation were used to induce early life stress, as described previously (Savignac et al., 2011; Benner et al., 2014; Alberry and Singh, 2016). On postnatal day 2, half of each litter was randomly selected for stress, tail coloring with permanent marker was used to distinguish pups. From postnatal days 2 to 14, stress group pups were removed and isolated for 3 hours per day during the light phase in 8 oz. Dixie cups with bedding and nest material. Pups not selected for maternal separation remained with the dam and other littermates during this time. On postnatal day 21, pups were weaned and housed with same-sex littermates with 2–4 individuals per cage. Following treatments, mice belonged to one of four groups—control, ethanol, stress, or ethanol + stress.
Hippocampal Dissection and DNA Isolation
Male mice were sacrificed via carbon dioxide asphyxiation and cervical dislocation on postnatal day 70. The hippocampus was dissected from the whole brain as described previously (Spijker, 2011). With samples stored in phosphate-buffered saline (PBS), snap-frozen in liquid nitrogen, and kept at −80°C. Samples were ground by pestle over liquid nitrogen to create a powder. Stages of buffer RLT (Qiagen, Valencia, CA) were added and mixed by pipetting. Sampled were centrifuged after a 10-min incubation period. The supernatant was loaded onto AllPrep DNA spin columns and the AllPrep DNA/RNA Mini Kit Protocol (Qiagen, Valencia, CA) was followed to isolate DNA and RNA from the same sample. DNA quantification was determined by NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE).
MeDIP-Seq
Three DNA samples were randomly selected from each treatment group (12 total DNA samples from individual mice processed separately) and sent on dry ice to Arraystar Inc. (Rockville, MD), with one sample per group as an input sample for sequence control. DNA was quantified by NanoDrop 1000 (Thermo Fisher Scientific, Wilmington, DE), then fragmented to a range of 200–1,500 bp using a Diagenode Bioruptor, end-repaired, and 3' adenylated for ligation of genomic adapters. Fragments were immunoprecipitated by anti-5-methylcytosine antibody, then PCR amplified. AMPure XP beads were used to select fragments from 300–800 bp, then quantified by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Following denaturation with 0.1 M NaOH, single-stranded DNA molecules were captured and amplified in situ on an Illumina flow cell. Libraries were sequenced on the Illumina HiSeq 4000 platform using the HiSeq 3000/4000 SBS Kit (300 cycles) protocol, then Off-Line Basecaller (OLB V1.8) was used for base calling. Reads were aligned to the mouse genome (UCSC MM10) using HISAT2 (V2.1.0) (Kim et al., 2015) after passing the Solexa CHASTITY quality filter and resulting BED files were used for differential methylation analysis. Sequences have been made available at GEO accession number GSE137984.
Differential Methylation Analysis
Aligned reads from HISAT2 were used for peak calling using Model-based analysis for ChIP-Seq (MACS) (version 1.4.2) (Zhang et al., 2008), where enriched regions (peaks) for each sample were identified by comparison to input background samples for each group using a dynamic Poisson distribution (q < 10-5). MeDIP enriched regions were annotated to the nearest gene using UCSC RefSeq, with differentially methylated regions (DMRs) in the promoter of known genes for Ethanol vs. Control, Stress vs. Control, as well as Ethanol + stress vs. Control identified by diffReps (cut-off: log2FC = 1.0, p = 10-4) (Shen et al., 2013). Promoter regions are defined here as 2,000 bp upstream and downstream from the transcription start site of a gene. Generalized hypergeometric tests for enrichment of gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathways were used for genes represented by promoter DMRs for each treatment group using goana and kegga functions in the limma software package (Ritchie et al., 2015) in R, filtered by significance (p < 0.05).
Comparison to Transcriptome Analysis
Genes represented by promoter DMRs for each treatment group were compared to previously reported transcript-level expression analysis from an RNA-Seq experiment using RNA isolated from the same samples (Alberry et al., 2019). Briefly, RNA-Seq was carried out by The Centre for Applied Genomics (The Hospital for Sick Children, Toronto, Ontario, Canada). Paired-end reads were pseudo-aligned to version 38 of the Ensembl mouse transcriptome via kallisto (Bray et al., 2016) and differential expression analysis was determined using sleuth (Pimentel et al., 2017; Yi et al., 2018). Gene lists used here were filtered by significance (p < 0.01), resulting in a list of 164 unique transcripts altered following ethanol exposure, 116 for stress, and 217 for the combination of ethanol + stress. Sequence data for the RNA-Seq experiment is available at the Gene Expression Omnibus (GEO), accession number GSE133369.
Results
Hippocampal DNA samples from adult males (postnatal day 70) for each of four groups of mice representing a control group with no experimental interventions, an ethanol group that faced prenatal ethanol exposure, a stress group that faced postnatal maternal separation stress, and an ethanol + stress group subject to both treatments (see methods for specific details). DNA methylation profiles were assessed via MeDIP-Seq to determine how treatment groups differ from controls.
DNA Methylation Changes Following Different Treatments
DMRs (DMRs) were determined for each experimental treatment group compared to controls, then associated with annotated genes via proximity to transcription start sites. Promoter regions are defined here as 2 kb upstream or downstream from the transcription start site of a gene, with genes with promoter DMRs considered for this analysis. In this way, several DMRs considered spatially distinct can be present in the promoter region of a single or multiple gene(s). A gene implicated by more than one DMR is considered separately when the DMRs are in opposition, but only once if the DMRs are in the same direction. Using promoter DMRs, 1,264 genes are implicated by prenatal ethanol treatment (Figure 1A, details in Supplementary Table 1). Following postnatal maternal separation stress, 1,472 genes are represented by promoter DMRs (Figure 1B, details in Supplementary Table 2). When mice faced both ethanol + stress, 958 genes are implicated by promoter DMRs (Figure 1C, details in Supplementary Table 3). Interestingly, while the ethanol group has a similar number of genes implicated by hypo- and hypermethylated promoter DMRs, the stress group genes are represented by 73% hypomethylated promoter DMRs. Genes implicated by promoter DMRs in the ethanol + stress group are also more evenly distributed between hypo- and hypermethylated. While a single DMR is either hypo- or hypermethylated, a single gene may be represented by multiple DMRs with different states. There are between 16 and 29 genes in each comparison that are implicated by both hypo- and hypermethylation of promoter DMRs.
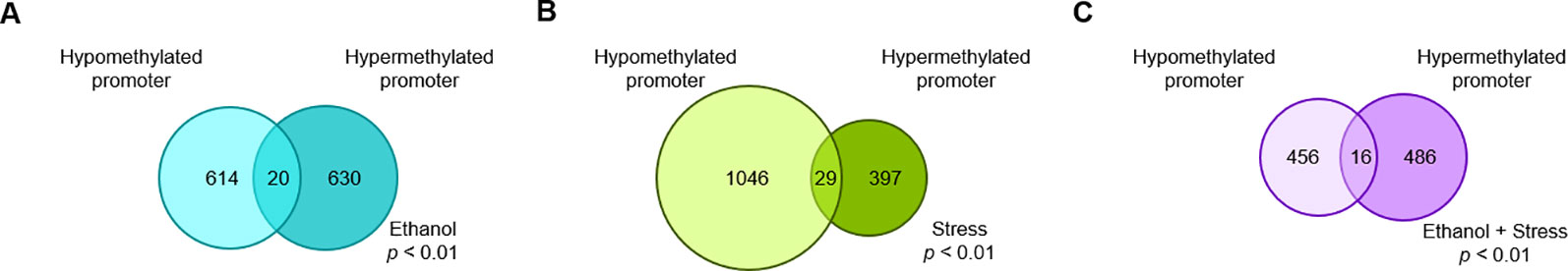
Figure 1 The number of genes with significant (p < 0.01) promoter DNA methylation changes following (A) prenatal ethanol treatment, (B) early life stress treatment, or (C) the combination of prenatal ethanol treatment and early life stress as compared to controls.
Hypo- and hypermethylated DMR-associated genes for each treatment group were analyzed separately for GO and KEGG pathway enrichment. In the ethanol group, genes related to voltage-gated calcium channel activity tended to be hypomethylated at their promoter regions (p < 0.007) (Table 1). In addition, brain-related terms including NMDA receptor activity (p = 0.001), positive regulation of behavior (p = 0.002), circadian rhythm (p = 0.003), and apical dendrite (p = 0.03) were all biological processes overrepresented by genes with hypomethylated promoters following ethanol exposure. Interestingly, components responsible for transcriptional regulation are also implicated by genes with hypomethylated promoters, including PRC1 complex (p = 4.04 x 10-4), transcription factor TFIID complex (p = 0.03), and RNA polymerase II transcription factor complex (p = 0.046). Genes related to phosphoinositide kinase (p < 0.02) and cytoskeleton regulation (p = 0.005) were hypermethylated at their promoter regions following prenatal ethanol exposure. In the stress group, genes important for histone modifications (p < 0.01) and neuronal organization (p = 0.007) were overrepresented by genes with hypomethylated promoters (Table 2). Conversely, genes with hypermethylated promoters following stress are important for cytokine interactions (p < 0.006). When mice faced both ethanol + stress, genes important for neuron migration (p < 0.005) were hypomethylated at their promoters (Table 3). As with stress alone, the combination of ethanol + stress results in promoter hypermethylation for genes responsible for cytokine interactions (p < 0.01).
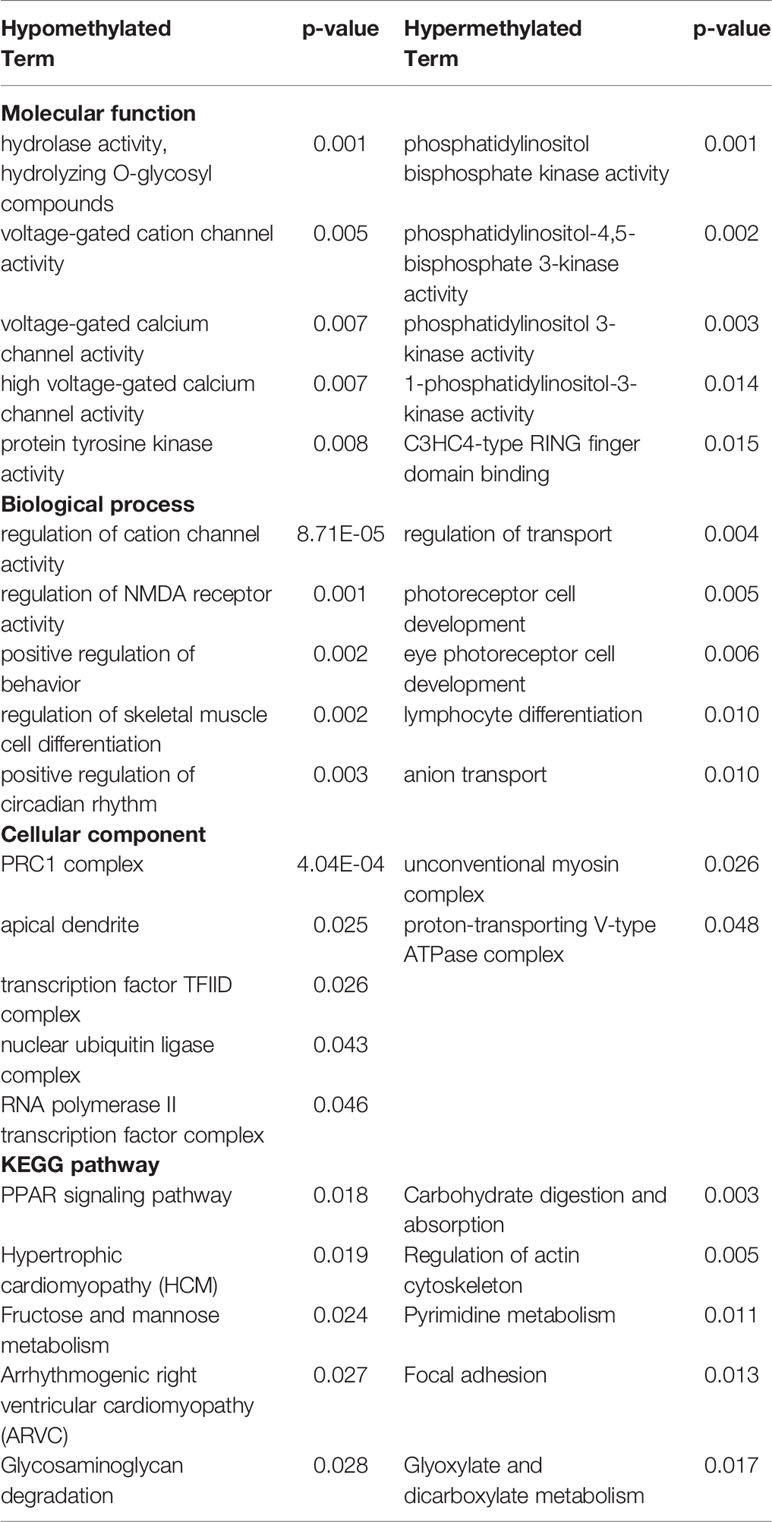
Table 1 Top five most significantly (p < 0.05) overrepresented gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathways represented by either hypo- or hypermethylated differentially methylated region (DMR)-associated genes implicated by Ethanol treatment compared to control (p < 0.01).
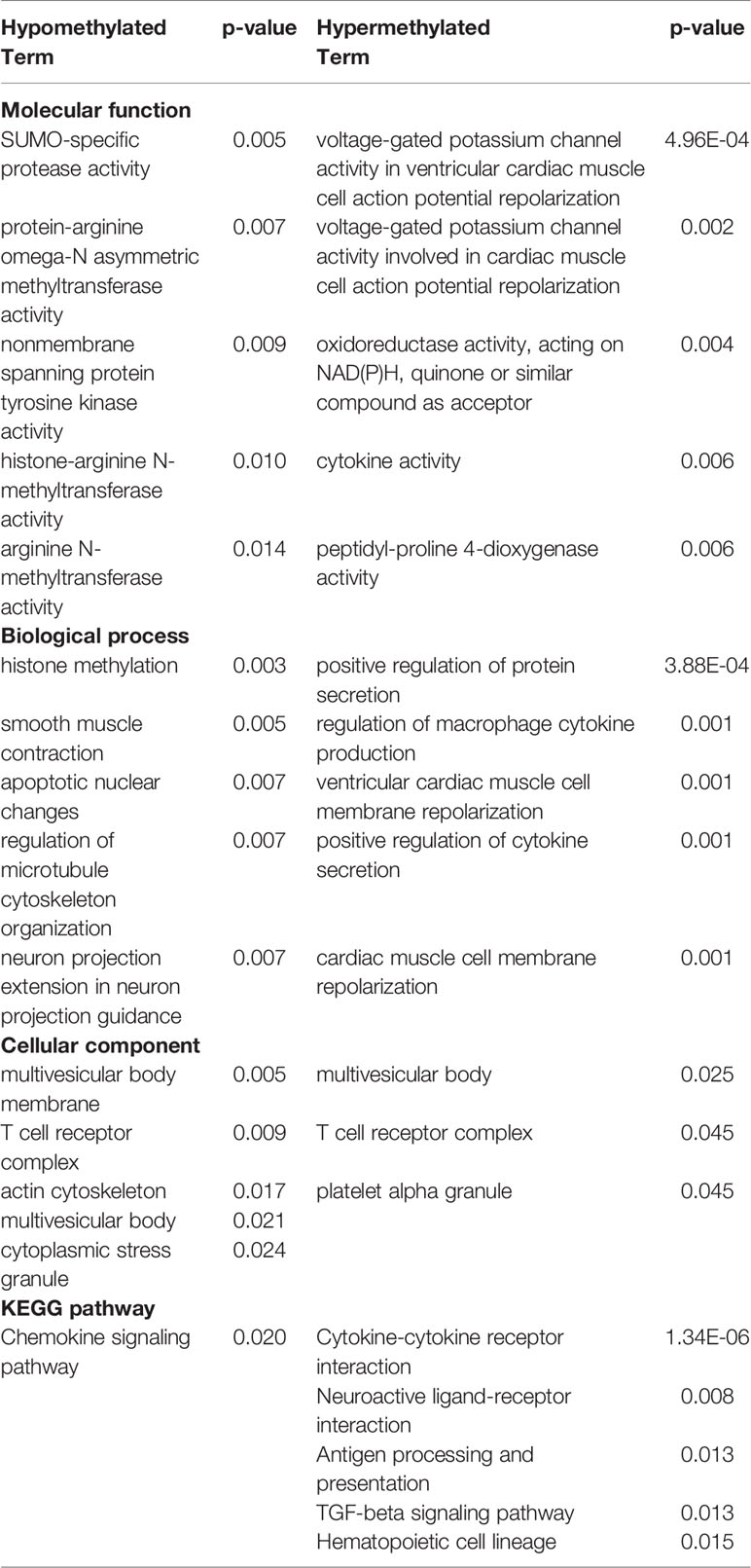
Table 2 Top five most significantly (p < 0.05) overrepresented gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathways represented by either hypo- or hypermethylated differentially methylated region (DMR)-associated genes implicated by Stress treatment compared to control (p < 0.01).
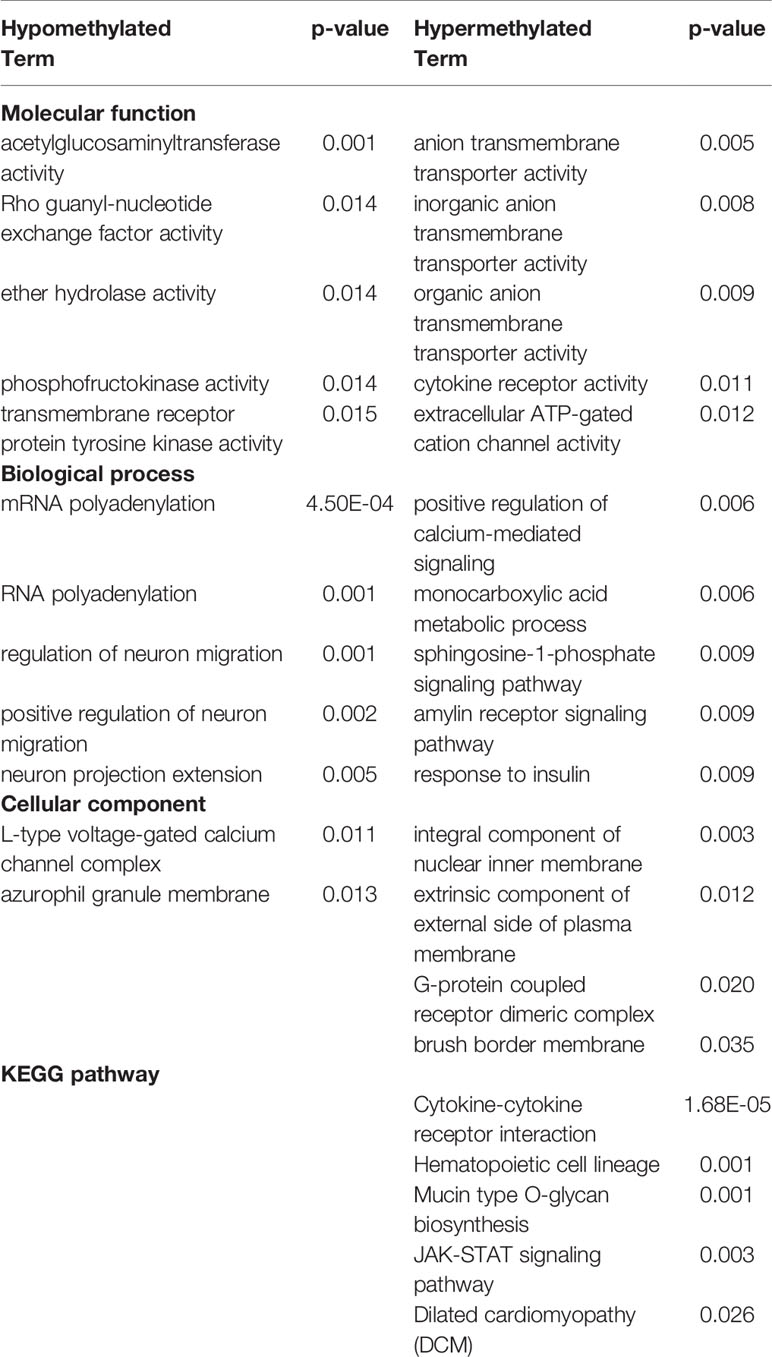
Table 3 Top five most significantly (p < 0.05) overrepresented gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathways represented by either hypo- or hypermethylated differentially methylated region (DMR)-associated genes implicated by combined Ethanol + Stress treatments compared to control (p < 0.01).
DNA Methylation Patterns Are Shared by Different Treatments
While genes implicated by promoter DMRs are unique to each treatment, there are regions of overlap between treatments (Figure 2). 120 genes are implicated by promoter DMRs following ethanol, stress, as well as ethanol + stress (Figure 2A). When directionality is considered, there are 85 genes implicated by hypomethylated promoter DMRs in all three treatment groups (Figure 2B). Finally, there are 19 genes implicated by hypermethylated promoter DMRs in all three treatment groups (Figure 2C). Genes consistently implicated by promoter DMR hypomethylation tend to be important for histone methylation (p < 0.04) and cyclic adenosine monophosphate (cAMP) protein kinase activity (p < 0.04) (Table 4). Conversely, genes implicated by promoter DMR hypermethylation are important for ion channel activity (p < 0.007) as well as neuroactive ligand-receptor interactions (p = 0.04) and chemokine signaling (p < 0.003).
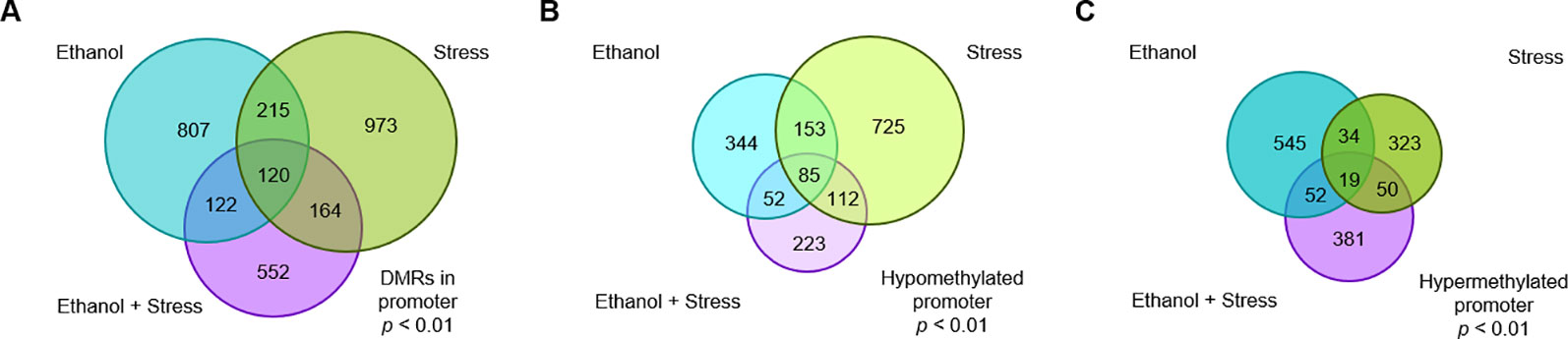
Figure 2 The number of genes with significant promoter DNA methylation changes shared following prenatal ethanol treatment, early life stress, or the combination of ethanol + stress with (A) all genes grouped together, (B) genes with hypomethylated promoters grouped together, and (C) genes with hypermethylated promoters grouped together.
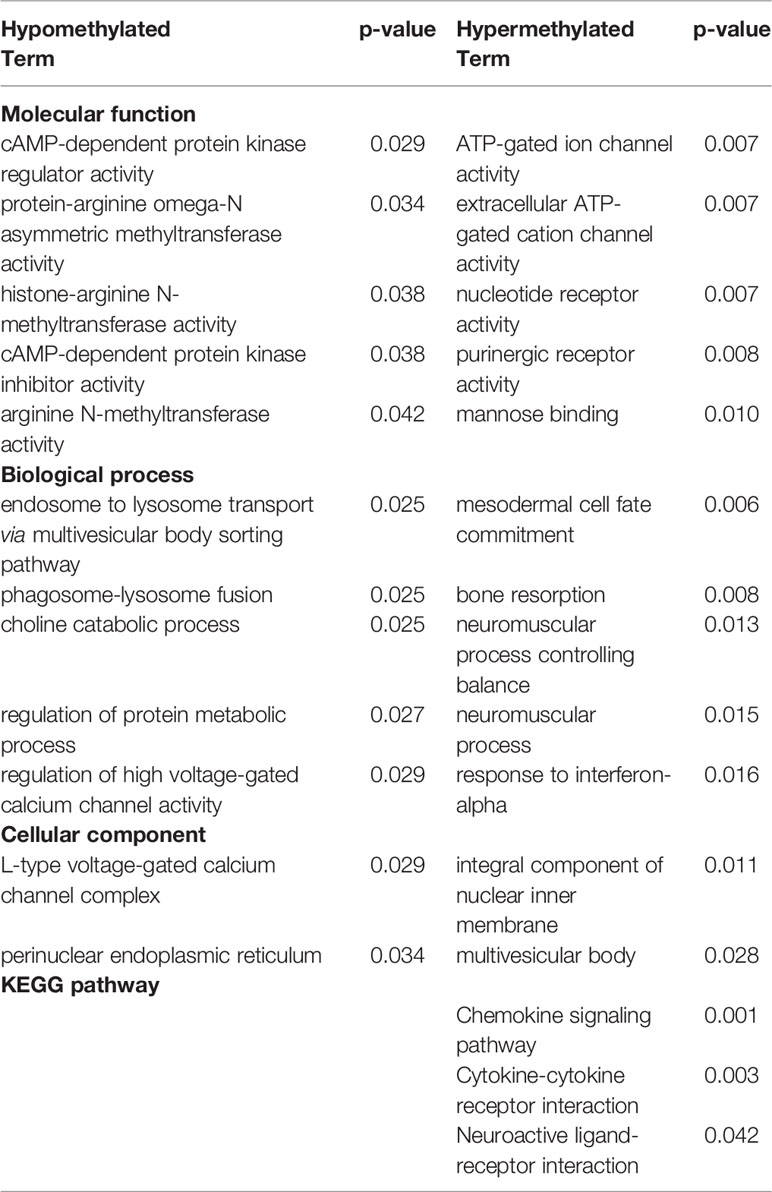
Table 4 Top five most significantly (p < 0.05) overrepresented gene ontology (GO) terms and Kyoto encyclopedia of genes and genomes (KEGG) pathways represented by either hypo- or hypermethylated differentially methylated region (DMR)-associated genes implicated by all three treatment categories—Ethanol, Stress, and combined Ethanol + Stress compared to control (p < 0.01).
DNA Methylation Alterations in Relation to Gene Expression
Since hippocampal transcriptome profiles exist for these mice, genes implicated by altered promoter methylation could be compared to genes with altered transcription. The number of genes represented by altered promoter methylation following each treatment is represented by the direction of transcriptional change following the RNA-Seq study previously reported are presented here (Table 5). While many genes are included for comparison, the overlap between genes implicated by altered DNA methylation and transcription is minimal. For ethanol mice, 1,264 genes with promoter DMRs were compared to the previously reported 164 unique transcripts altered in these samples. There are two corresponding transcripts with hypomethylated promoters and increased expression, Slc2a9, solute carrier family 2 (facilitated glucose transporter), member 9, and Blvrb, biliverdin reductase B (flavin reductase (NADPH)). Conversely, for hypermethylated promoters, there are two transcripts with decreased expression following PAE, Eif4g1, eukaryotic translation initiation factor 4, gamma 1, and Myo6, myosin VI.
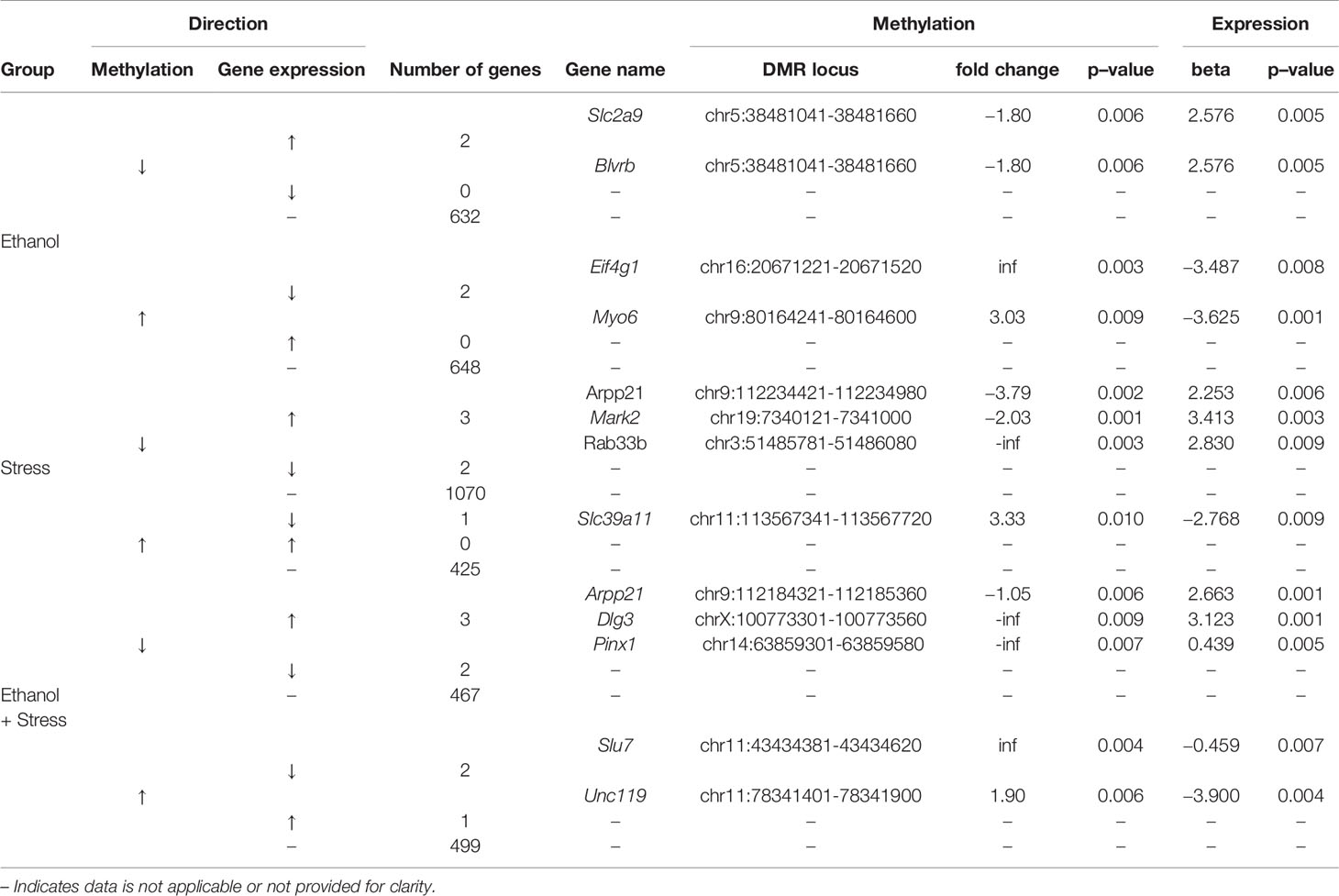
Table 5 Relevant genes through the complementary direction, increase (↑) or decrease (↓) of methylation and gene expression changes compared to control for genes differentially expressed with promoter differentially methylated regions (DMRs).
In the stress group, 1,472 genes with promoter DMRs were compared to the 116 unique transcripts altered in these samples. There are three genes with hypomethylated promoters and increased transcript expression, Arpp21, cyclic AMP-regulated phosphoprotein, 21, Mark2, microtubule affinity regulating kinase 2, and Rab33b, RAB33B, member RAS oncogene family. One gene, Slc39a11, solute carrier family 39 (metal ion transporter), member 11, is hypermethylated at its promoter and has reduced transcript expression following maternal separation.
For ethanol + stress mice, 958 genes with promoter DMRs were compared to the 217 unique transcripts altered in these samples. Interestingly, Arpp21 is also hypomethylated at its promoter and overexpressed following ethanol + stress treatment alongside two additional genes, Dlg3, discs large MAGUK scaffold protein 3, and Pinx1, PIN2/TERF1 interacting, telomerase inhibitor 1. Conversely, two genes share increased promoter methylation and decreased gene expression, Slu7, pre-mRNA-splicing factor SLU7, and Unc119, unc-119 lipid binding chaperone. While the number of genes with altered promoter methylation and expression resulting from each treatment or combination of treatments is minimal, most of these genes have known brain functions in the literature, ranging from neuronal migration to synapse formation and plasticity (Table 6).
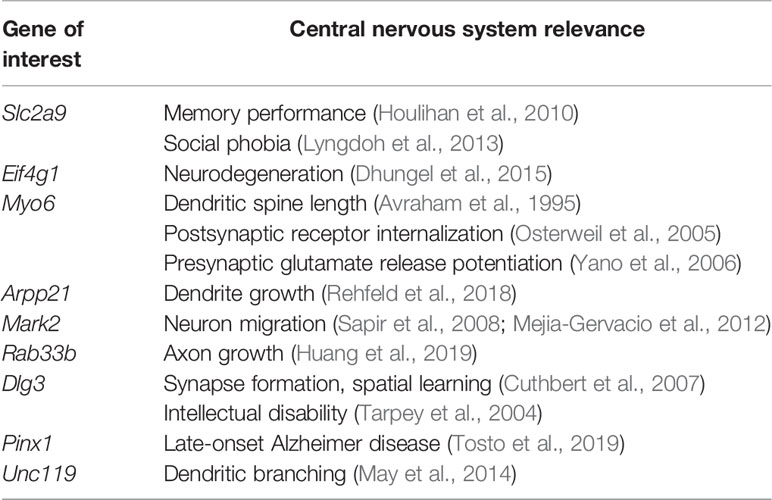
Table 6 Genes of interest identified by altered promoter methylation and gene expression with literature-based evidence of central nervous system function.
Discussion
FASD is a prevalent societal burden that has remained complex to understand. Children born with FASD are often exposed to stressful postnatal environments that include maternal separation. Our main objective was to understand how early life stresses may complicate molecular changes underlying behavioral deficits that arise following prenatal ethanol exposure. Previously, behavioral changes resulting from prenatal ethanol exposure and postnatal maternal separation stress have been characterized (Alberry and Singh, 2016), with lasting changes to the hippocampal transcriptome also being reported (Alberry et al., 2019). Here we focus on alterations to promoter DNA methylation in the same mice. The behavioral deficits originally reported were subtle in nature, a direct result of the subtlety of the treatment paradigms used. In addition, gene expression alterations were small in fold change, detected at postnatal day 70, long after the final experimental interventions.
Ethanol-Induced Alterations in DNA Methylation
Prenatal ethanol exposure results in specific changes in promoter DNA methylation. We report promoter hypomethylation of genes important for brain function and formation as well as for transcriptional regulation following prenatal ethanol exposure. We also found promoter hypermethylation of genes important for phosphoinositide kinase activity as well as cytoskeleton regulation. These alterations may be lasting evidence of altered control of gene expression earlier during development that persists into adulthood. The ethanol exposure model employed here has been used previously, with lasting alterations to whole-brain DNA methylation profiles (Laufer et al., 2013). While we report comparable amounts of hypo- and hypermethylated gene promoters following continuous gestational ethanol exposure, previous work using targeted binge exposure trimester three equivalent models have found alcohol-induced hypermethylation in the hippocampus in both mice (Chater-Diehl et al., 2016) and rats (Otero et al., 2012). Conversely, a model of ethanol exposure more representative of trimester two equivalent exposure in mice found global hypomethylation in the neocortex, but this pattern is more complex when brain structures are independently assessed (Öztürk et al., 2017). Our work uses a continuous exposure model and is aimed at investigating promoter methylation, highlighting the importance of dose and timing of ethanol exposure as well as specific methylation patterns being observed.
Maternal Separation-Induced Alterations in DNA Methylation
We report more genes with hypo- than hypermethylated promoters following early life stress via maternal separation. Of these, hypomethylated genes tend to be important for histone methylation and chemokine activity. Meanwhile, hypermethylated promoters tend to be of genes important for cytokine interactions. Early life stress results in lasting changes to DNA methylation in the hippocampus of rats (Kosten et al., 2014). In humans, child abuse has been associated with hypermethylation of several genomic loci in the hippocampus (McGowan et al., 2009). Meanwhile, maternal separation in rats induces an inflammatory immune response in the hippocampus (Roque et al., 2016). Interestingly, early life stress has been associated with reduced serum cytokine levels, suggesting that hypermethylation of related genes may not be hippocampal specific, rather playing a more ubiquitous role in the body (Raineki et al., 2017).
Ethanol + Maternal Separation-Induced Alterations in DNA Methylation
The combination of prenatal ethanol exposure and maternal separation stress resulted in the hypomethylation of genes important for neuronal migration and hypermethylation of genes related to cytokine interactions. Similarly, genes that were implicated by hypermethylated promoters following any of the treatments were also important for chemokine and cytokine interactions. It is apparent from Figure 1 that the addition of early life stress to individuals that faced prenatal ethanol exposure does not result in an additive DNA methylation response. Few genes are shared between ethanol only or stress only and the combination of treatments as implicated by promoter DMRs. Prenatal ethanol exposure followed by early life stress has been associated with altered immune system development, with early life adversity differentially affecting peripheral and central immune response depending on previous ethanol exposure (Raineki et al., 2017).
Overlaps Between DNA Methylation and Gene Expression Changes Are Scarce
We previously reported changes in gene expression in association with behavioral deficits following prenatal ethanol exposure and maternal separation in these mice (Alberry et al., 2019). Interestingly the module of genes most closely associated with treatment and anxiety-like behavior includes genes important for transcriptional regulation and neurodevelopment. With these results in mind, we examined what overlap exists between genes with altered expression and altered promoter methylation that is complementary.
Following prenatal ethanol exposure, two genes have decreased promoter methylation and corresponding increased expression—Slc2a9 and Blvrb. Slc2a9 codes for a fructose transporter that acts as a uric acid transporter (Le et al., 2008). While not much is known regarding its role in the brain, SLC2A9 SNPs have been associated with better memory performance (Houlihan et al., 2010), and social phobia (Lyngdoh et al., 2013). Blvrb codes for an oxidoreductase important for heme degradation, with no clear link to brain function. Prenatal ethanol exposure results in promoter hypermethylation and decreased transcript expression for two genes, Eif4g1 and Myo6. Eif4g1 produces a translation initiation factor, eIF4G, with mutations present in familial Parkinson Disease (Chartier-Harlin et al., 2011). In fact, this gene acts as a scaffold protein with others in the hippocampus in ways that are perturbed in neurodegeneration likely through protein misfolding (Dhungel et al., 2015). Myo6 is an actin-based motor protein that has been previously linked to stress response in the mouse hippocampus (Tamaki et al., 2008). Mice lacking this gene have fewer synapses and shorter dendritic spines (Avraham et al., 1995). Myo6 is important for postsynaptic stimulus-dependent α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid-type glutamate receptor (AMPAR) internalization (Osterweil et al., 2005), as well as for brain-derived neurotrophic factor (BDNF)-mediated glutamate release from presynaptic terminals in long-term potentiation (Yano et al., 2006). Together, these results suggest the combined accumulation of DNA methylation, followed by reduced expression of these transcripts may be important for the altered brain-related phenotypes observed in these mice following prenatal ethanol exposure.
There are three genes with promoter hypomethylation and a corresponding increase in transcript expression following maternal separation stress, Arpp21, Mark2, and Rab33b. Arpp21 is also implicated by the combination of ethanol + stress through promoter hypomethylation and increased expression. The ARPP21 protein product is composed of conserved domains for RNA-binding, with the ability to interact with eIF4G and preferentially bind to the 3' untranslated regions of mRNAs. Additionally, overexpression and knockdown experiments of ARPP21 show it positively regulates dendrite growth (Rehfeld et al., 2018). While we report increased Arpp21 expression following stress or ethanol + stress, stress-induced decreases have also been reported (Constantinof et al., 2019). Mark2 produces a serine/threonine-protein kinase involved in cell polarity and microtubule dynamics, as well as in neuron migration (Sapir et al., 2008; Mejia-Gervacio et al., 2012). Rab33b is a member of Rab family proteins that regulate intracellular vesicular trafficking, particularly at the Golgi apparatus (Zheng et al., 1998). Recently, the zebrafish ortholog of Rab33b, rab33ba, has been shown to mediate axon outgrowth in the forebrain (Huang et al., 2019). Slc39a11 is the only gene that has promoter hypermethylation and overexpression in the stress group. It codes for a Zrt-, Irt-like protein (ZIP) for zinc transport (Yu et al., 2013). Taken together, these results suggest that the loss of methylation and subsequent overexpression of these genes following postnatal maternal separation stress may lead to observed changes in behavioral phenotypes mediated by improper neuronal migration and growth.
The combination of ethanol + stress treatments results in three genes with increased expression and corresponding promoter hypomethylation, Arpp21, Dlg3, and Pinx1. Arpp21 was introduced previously as it was also implicated by stress alone. Of these, Dlg3 is one of the most significantly altered transcripts in response to Stress and Ethanol + Stress treatments that we previously reported (Alberry et al., 2019). It codes for synapse-associated protein 102, a protein important for synapse formation. Knockout mice have impaired synaptic plasticity and spatial learning (Cuthbert et al., 2007), while human DLG3 mutations result in nonsyndromic X-linked intellectual disability (Tarpey et al., 2004). Pinx1 is a telomerase inhibitor with little known brain function, although variants have been associated with late-onset Alzheimer disease (Tosto et al., 2019). Two genes share decreased promoter methylation and increased expression following the combination of treatments, Slu7 and Unc119. Slu7 is a pre-mRNA splicing factor that forms part of the spliceosome and is important for genome stability (Jiménez et al., 2019). Unc119 codes for a lipid binding chaperone required for G protein localization in sensory neurons (Zhang et al., 2011). In fact, knockdown of Unc119 results in neurotoxicity and decreased dendritic branching (May et al., 2014). These results suggest that dysregulation of genes important for neuronal migration and synapse formation as well as genome integrity via altered promoter methylation following the combination of ethanol + stress may underlie the complex and heterogeneous behavioral changes observed in these mice.
Beyond Promoter DNA Methylation
Promoter DNA methylation is an essential mechanism of epigenetic control for the expression of genes. It is affected by environmental factors, including ethanol and stress. Changes in promoter DNA methylation status can lead to the initiation and maintenance of gene expression over time. Promoter DNA methylation has been the research focus in FASD, however, DNA methylation does not act in isolation. While we focus on DMRs in gene promoters, methylation status at a single CpG site may be sufficient to influence gene expression. In this analysis, we may miss the influence of a single CpG that has this type of influence without resulting in a significantly altered region of DNA methylation. Similarly, we have focused on promoter DNA methylation for its known influence on gene expression via altered transcription factor binding. Gene body or other intergenic changes in methylation would not be captured in this analysis and may be important for gene expression. Phenotypic outcomes are often the result of interrelated epigenetic mechanisms. For example, noncoding RNAs are also important for the regulation of gene expression at the transcriptional and posttranscriptional levels. They include long noncoding RNAs (lncRNAs) (Lee, 2012), microRNAs (miRNAs) (Chuang and Jones, 2007), short interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs) (Holoch and Moazed, 2015). In FASD research, the noncoding RNA research has been focused on the relationship between microRNAs and their mRNA gene targets. Several microRNAs are altered in reciprocal direction to their gene targets following prenatal alcohol exposure (Balaraman et al., 2013; Laufer et al., 2013), offering another potential avenue for control. Additionally, modifications to histone tails at specific genomic loci are the foundation of epigenetic mechanisms, with a wide variety of known potential modifications (Allis and Jenuwein, 2016). In FASD mouse models, altered histone modifications (Veazey et al., 2015; Chater-Diehl et al., 2016), as well as enhanced lysine dimethyltransferase activity resulting in increased histone H3 lysine 9 and 27 dimethylation and neurodegeneration have been reported (Subbanna et al., 2013). Such results suggest changes in gene-specific expression in response to prenatal ethanol exposure must be the result of a variety of epigenetic mechanisms. Although DNA methylation, noncoding RNAs, and histone modifications have all been implicated, their inter-relationship is not known.
The promoter DNA methylation results included in this report are novel in that they were obtained from the same mice that were used to assess behavioral changes (Alberry and Singh, 2016) and hippocampal gene expression (Alberry et al., 2019). This allows for assessment of the relationship between promoter DNA methylation and expression of that gene in response to prenatal ethanol exposure, postnatal maternal separation, and the combination of both treatments. Our results implicate many genes via altered expression or promoter DNA methylation, and these genes are relevant in the context of their involvement in ethanol or stress response. In fact, many genes implicated are important for brain development and function. What is apparent is that changes in expression of only a small subset of genes is compatible with changes in promoter DNA methylation. The results argue that promoter methylation is only partly involved in the environmentally induced changes in gene expression we have modeled. Epigenetic mechanisms other than promoter DNA methylation therefore are likely responsible for the lasting gene expression alterations and behavioral abnormalities seen in pups subjected to prenatal ethanol exposure, maternal separation, and the combination of the two treatments. Although logical, such conclusions must be assessed experimentally, as the hippocampus represents a heterogenous cell population. Also, the interventions used are relatively mild, applied during neurodevelopment, with methylation status assessed in adulthood. Not surprisingly, this experimental paradigm has resulted in subtle changes in both gene expression and DNA methylation. Future research must focus on experimentally establishing mechanisms involved in altered gene expression and maintenance of these changes in different cell types within the brain. This will require modified experimental protocols and the use of single cell sequencing. Greater understanding into how these changes occur and persist has the potential to lead to the development of novel strategies for the amelioration of FASD-related deficits for children born with this common and complex disorder.
Data Availability Statement
The datasets generated for this study can be found at the Gene Expression Omnibus (GEO), accession numbers GSE137984 and GSE133369 for MeDIP-Seq and RNA-Seq experiments, respectively.
Ethics Statement
The animal study was reviewed and approved by Animal Use Subcommittee at Western University.
Author Contributions
BA and SS have made substantial contributions to the conception and design of the work. BA was responsible for data acquisition and analysis. All authors completed data interpretation. BA created the first manuscript draft. All authors substantively revised it and have approved the submitted version.
Funding
This work has been supported by a Discovery Research Grant from the Natural Sciences and Engineering Research Council of Canada awarded to SS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Christina Castellani, Eric Chater-Diehl, and Benjamin Laufer for assistance with experimental design, training, and manuscript guidance, as well as Shruthi Rethi, Ali Pensamiento, David Seok, and Yuchen Li for data collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00070/full#supplementary-material
Supplementary Table 1 | Hypo- and hypermethylated promoter DMRs from adult hippocampus following prenatal ethanol exposure compared to untreated controls filtered by significance (p < 0.01).
Supplementary Table 2 | Hypo- and hypermethylated promoter DMRs from adult hippocampus following postnatal maternal separation stress treatment compared to controls filtered by significance (p < 0.01).
Supplementary Table 3 | Hypo- and hypermethylated promoter DMRs from adult hippocampus following prenatal alcohol exposure and postnatal maternal separation stress as compared to controls, filtered by significance (p < 0.01).
References
Alberry, B., Singh, S. M. (2016). Developmental and behavioral consequences of early life maternal separation stress in a mouse model of fetal alcohol spectrum disorder. Behav. Brain Res. 308, 94–103. doi: 10.1016/j.bbr.2016.04.031
Alberry, B. L., Castellani, C. A., Singh, S. M. (2019). Hippocampal transcriptome analysis following maternal separation implicates altered RNA processing in a mouse model of fetal alcohol spectrum disorder. bioRxiv, 685586. doi: 10.1101/685586
Allan, A. M., Chynoweth, J., Tyler, L. A., Caldwell, K. K. (2003). A mouse model of prenatal ethanol exposure using a voluntary drinking paradigm. Alcohol. Clin. Exp. Res. 27, 2009–2016. doi: 10.1097/01.ALC.0000100940.95053.72
Allis, C. D., Jenuwein, T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500. doi: 10.1038/nrg.2016.59
Avraham, K. B., Hasson, T., Steel, K. P., Kingsley, D. M., Russell, L. B., Mooseker, M. S., et al. (1995). The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 11, 369–375. doi: 10.1038/ng1295-369
Balaraman, S., Tingling, J. D., Tsai, P. C., Miranda, R. C. (2013). Dysregulation of microRNA expression and function contributes to the etiology of fetal alcohol spectrum disorders. Alcohol Res. 35, 18–24.
Benner, S., Endo, T., Endo, N., Kakeyama, M., Tohyama, C. (2014). Early deprivation induces competitive subordinance in C57BL/6 male mice. Physiol. Behav. 137, 42–52. doi: 10.1016/j.physbeh.2014.06.018
Bray, N. L., Pimentel, H., Melsted, P., Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Chartier-Harlin, M.-C., Dachsel, J. C., Vilariño-Güell, C., Lincoln, S. J., Leprêtre, F., Hulihan, M. M., et al. (2011). Translation Initiator EIF4G1 Mutations in Familial Parkinson disease. Am. J. Hum. Genet. 89, 398–406. doi: 10.1016/J.AJHG.2011.08.009
Chater-Diehl, E. J., Laufer, B. I., Castellani, C. A., Alberry, B. L., Singh, S. M. (2016). Alteration of gene expression, DNA methylation, and histone methylation in free radical scavenging networks in adult mouse hippocampus following fetal alcohol exposure. PloS One 11, e0154836. doi: 10.1371/journal.pone.0154836
Chen, Y., Ozturk, N. C., Zhou, F. C. (2013). DNA methylation program in developing hippocampus and its alteration by Alcohol. PloS One 8, e60503. doi: 10.1371/journal.pone.0060503
Chuang, J. C., Jones, P. A. (2007). Epigenetics and MicroRNAs. Pediatr. Res. 61, 24R–29R. doi: 10.1203/pdr.0b013e3180457684
Chudley, A. E., Conry, J., Cook, J. L., Loock, C., Rosales, T., LeBlanc, N., et al. (2005). Fetal alcohol spectrum disorder: Canadian guidelines for diagnosis. Can. Med. Assoc. J. 172, S1–S21. doi: 10.1503/cmaj.1040302
Coggins, T. E., Timler, G. R., Olswang, L. B. (2007). A state of double jeopardy: impact of prenatal alcohol exposure and adverse environments on the social communicative abilities of school-age children with fetal alcohol spectrum disorder. Lang. Speech. Hear. Serv. Sch. 38, 117–127. doi: 10.1044/0161-1461(2007/012)
Constantinof, A., Moisiadis, V. G., Kostaki, A., Szyf, M., Matthews, S. G. (2019). Antenatal glucocorticoid exposure results in sex-specific and transgenerational changes in prefrontal cortex gene transcription that relate to behavioural outcomes. Sci. Rep. 9, 764. doi: 10.1038/s41598-018-37088-3
Cuthbert, P. C., Stanford, L. E., Coba, M. P., Ainge, J. A., Fink, A. E., Opazo, P., et al. (2007). Synapse-Associated Protein 102/dlgh3 couples the NMDA receptor to specific plasticity pathways and learning strategies. J. Neurosci. 27, 2673–2682. doi: 10.1523/jneurosci.4457-06.2007
Dhungel, N., Eleuteri, S., Li, L., Kramer, N. J., Chartron, J. W., Spencer, B., et al. (2015). Parkinson's disease genes VPS35 and EIF4G1 interact genetically and Converge on α-Synuclein. Neuron 85, 76–87. doi: 10.1016/J.NEURON.2014.11.027
Henry, J., Sloane, M., Black-Pond, C. (2007). Neurobiology and neurodevelopmental impact of childhood traumatic stress and prenatal alcohol exposure. Lang. Speech. Hear. Serv. Sch. 38, 99–108. doi: 10.1044/0161-1461(2007/010)
Holoch, D., Moazed, D. (2015). RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71. doi: 10.1038/NRG3863
Houlihan, L. M., Wyatt, N. D., Harris, S. E., Hayward, C., Gow, A. J., Marioni, R. E., et al. (2010). Variation in the uric acid transporter gene (SLC2A9) and memory performance. Hum. Mol. Genet. 19, 2321–2330. doi: 10.1093/hmg/ddq097
Huang, L., Urasaki, A., Inagaki, N. (2019). Rab33a and Rab33ba mediate the outgrowth of forebrain commissural axons in the zebrafish brain. Sci. Rep. 9, 1799. doi: 10.1038/s41598-018-38468-5
Jaenisch, R., Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33 Suppl, 245–254. doi: 10.1038/ng1089
Jarrard, L. E. (1993). On the role of the hippocampus in learning and memory in the rat. Behav. Neural Biol. 60, 9–26. doi: 10.1016/0163-1047(93)90664-4
Jiménez, M., Urtasun, R., Elizalde, M., Azkona, M., Latasa, M. U., Uriarte, I., et al. (2019). Splicing events in the control of genome integrity: role of SLU7 and truncated SRSF3 proteins. Nucleic Acids Res. 47, 3450–3466. doi: 10.1093/nar/gkz014
Kaminen-Ahola, N., Ahola, A., Flatscher-Bader, T., Wilkins, S. J., Anderson, G. J., Whitelaw, E., et al. (2010). Postnatal growth restriction and gene expression changes in a mouse model of fetal alcohol syndrome. Birth defects Res. A Clin. Mol. Teratol. 88, 818–826. doi: 10.1002/bdra.20729
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kisely, S., Abajobir, A. A., Mills, R., Strathearn, L., Clavarino, A., Najman, J. M. (2018). Child maltreatment and mental health problems in adulthood: birth cohort study. Br. J. Psychiatry 213, 698–703. doi: 10.1192/bjp.2018.207
Kleiber, M. L., Wright, E., Singh, S. M. (2011). Maternal voluntary drinking in C57BL/6J mice: advancing a model for fetal alcohol spectrum disorders. Behav. Brain Res. 223, 376–387. doi: 10.1016/j.bbr.2011.05.005
Kleiber, M. L., Laufer, B. I., Wright, E., Diehl, E. J., Singh, S. M. (2012). Long-term alterations to the brain transcriptome in a maternal voluntary consumption model of fetal alcohol spectrum disorders. Brain Res. 1458, 18–33. doi: 10.1016/j.brainres.2012.04.016
Koponen, A. M., Kalland, M., Autti-Rämö, I. (2009). Caregiving environment and socio-emotional development of foster-placed FASD-children. Child. Youth Serv. Rev. 31, 1049–1056. doi: 10.1016/j.childyouth.2009.05.006
Koponen, A. M., Kalland, M., Autti-Rämö, I., Laamanen, R., Suominen, S. (2013). Socio-emotional development of children with foetal alcohol spectrum disorders in long-term foster family care: a qualitative study. Nord. Soc. Work Res. 3, 38–58. doi: 10.1080/2156857x.2013.766234
Kosten, T. A., Huang, W., Nielsen, D. A. (2014). Sex and litter effects on anxiety and DNA methylation levels of stress and neurotrophin genes in adolescent rats. Dev. Psychobiol. 56, 392–406. doi: 10.1002/dev.21106
Lange, S., Shield, K., Rehm, J., Popova, S. (2013). Prevalence of fetal alcohol spectrum disorders in child care settings: a meta-analysis. Pediatrics 132, e980–e995. doi: 10.1542/peds.2013-0066
Lange, S., Probst, C., Gmel, G., Rehm, J., Burd, L., Popova, S. (2017). Global prevalence of fetal alcohol spectrum disorder among children and youth: A systematic review and meta-analysis. JAMA Pediatr. 171, 948–956. doi: 10.1001/jamapediatrics.2017.1919
Laufer, B. I., Mantha, K., Kleiber, M. L., Diehl, E. J., Addison, S. M., Singh, S. M. (2013). Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Model. Mech. 6, 977–992. doi: 10.1242/dmm.010975
Laufer, B. I., Kapalanga, J., Castellani, C. A., Diehl, E. J., Yan, L., Singh, S. M. (2015). Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics 7, 1259–1274. doi: 10.2217/epi.15.60
Le, M. T., Shafiu, M., Mu, W., Johnson, R. J. (2008). SLC2A9–a fructose transporter identified as a novel uric acid transporter. Nephrol. Dial. Transplant. 23, 2746–2749. doi: 10.1093/ndt/gfn349
Lee, J. T. (2012). Epigenetic Regulation by Long Noncoding RNAs. Sci. (80-) 338, 1435–1439. doi: 10.1126/science.1231776
Lyngdoh, T., Bochud, M., Glaus, J., Castelao, E., Waeber, G., Vollenweider, P., et al. (2013). Associations of serum uric acid and SLC2A9 variant with depressive and anxiety disorders: a population-based study. PloS One 8, e76336. doi: 10.1371/journal.pone.0076336
Marjonen, H., Sierra, A., Nyman, A., Rogojin, V., Gröhn, O., Linden, A.-M. M., et al. (2015). Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PloS One 10, e0124931. doi: 10.1371/journal.pone.0124931
May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503. doi: 10.1007/s00401-014-1329-4
May, P. A., Chambers, C. D., Kalberg, W. O., Zellner, J., Feldman, H., Buckley, D., et al. (2018). Prevalence of fetal alcohol spectrum disorders in 4 US communities. JAMA - J. Am. Med. Assoc. 319, 474–482. doi: 10.1001/jama.2017.21896
McGowan, P. O., Sasaki, A., D'Alessio, A. C., Dymov, S., Labonte, B., Szyf, M., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348. doi: 10.1038/nn.2270
Mejia-Gervacio, S., Murray, K., Sapir, T., Belvindrah, R., Reiner, O., Lledo, P.-M. (2012). MARK2/Par-1 guides the directionality of neuroblasts migrating to the olfactory bulb. Mol. Cell. Neurosci. 49, 97–103. doi: 10.1016/J.MCN.2011.10.006
Myran, D. T., Hsu, A. T., Smith, G., Tanuseputro, P. (2019). Rates of emergency department visits attributable to alcohol use in Ontario from 2003 to 2016: a retrospective population-level study. CMAJ 191, E804–E810. doi: 10.1503/cmaj.181575
Oomen, C. A., Soeters, H., Audureau, N., Vermunt, L., van Hasselt, F. N., Manders, E. M. M., et al. (2010). Severe early life stress hampers spatial learning and neurogenesis, but improves hippocampal synaptic plasticity and emotional learning under high-stress conditions in adulthood. J. Neurosci. 30, 6635–6645. doi: 10.1523/JNEUROSCI.0247-10.2010
Osterweil, E., Wells, D. G., Mooseker, M. S. (2005). A role for myosin VI in postsynaptic structure and glutamate receptor endocytosis. J. Cell Biol. 168, 329–338. doi: 10.1083/jcb.200410091
Otero, N. K. H. H., Thomas, J. D., Saski, C. A., Xia, X., Kelly, S. J. (2012). Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol During Development. Alcohol. Clin. Exp. Res. 36, 1701–1709. doi: 10.1111/j.1530-0277.2012.01784.x
Öztürk, N. C., Resendiz, M., Öztürk, H., Zhou, F. C. (2017). DNA Methylation program in normal and alcohol-induced thinning cortex. Alcohol 60, 135–147. doi: 10.1016/j.alcohol.2017.01.006
Pillai, A. G., Arp, M., Velzing, E., Lesuis, S. L., Schmidt, M. V., Holsboer, F., et al. (2018). Early life stress determines the effects of glucocorticoids and stress on hippocampal function: Electrophysiological and behavioral evidence respectively. Neuropharmacology 133, 307–318. doi: 10.1016/j.neuropharm.2018.02.001
Pimentel, H., Bray, N. L., Puente, S., Melsted, P., Pachter, L. (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. doi: 10.1038/nmeth.4324
Popova, S., Lange, S., Chudley, A. E., Reynolds, J. N., Rehm, J., May, P. A., et al. (2018). World Health Organization International study on the Prevalence of fetal alcohol spectrum disorder (FASD). Cent. Addit. Ment. Heal. Available at: www.camh.ca [Accessed April 25, 2019].
Portales-Casamar, E., Lussier, A. A., Jones, M. J., MacIsaac, J. L., Edgar, R. D., Mah, S. M., et al. (2016). DNA methylation signature of human fetal alcohol spectrum disorder. Epigenet. Chromatin 9, 25. doi: 10.1186/s13072-016-0074-4
Price, A., Cook, P. A., Norgate, S., Mukherjee, R. (2017). Prenatal alcohol exposure and traumatic childhood experiences: a systematic review. Neurosci. Biobehav. Rev. 80, 89–98. doi: 10.1016/j.neubiorev.2017.05.018
Raineki, C., Bodnar, T. S., Holman, P. J., Baglot, S. L., Lan, N., Weinberg, J. (2017). Effects of early-life adversity on immune function are mediated by prenatal environment: Role of prenatal alcohol exposure. Brain Behav. Immun. 66, 210–220. doi: 10.1016/j.bbi.2017.07.001
Rehfeld, F., Maticzka, D., Grosser, S., Knauff, P., Eravci, M., Vida, I., et al. (2018). The RNA-binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat. Commun. 9, 1235. doi: 10.1038/s41467-018-03681-3
Rice, C. J., Sandman, C. A., Lenjavi, M. R., Baram, T. Z. (2008). A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology 149, 4892–4900. doi: 10.1210/en.2008-0633
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi: 10.1093/nar/gkv007
Roque, A., Ochoa-Zarzosa, A., Torner, L. (2016). Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav. Immun. 55, 39–48. doi: 10.1016/J.BBI.2015.09.017
Sapir, T., Sapoznik, S., Levy, T., Finkelshtein, D., Shmueli, A., Timm, T., et al. (2008). Accurate balance of the polarity kinase MARK2/Par-1 is required for proper cortical neuronal migration. J. Neurosci. 28, 5710–5720. doi: 10.1523/JNEUROSCI.0911-08.2008
Savignac, H. M., Dinan, T. G., Cryan, J. F. (2011). Resistance to early-life stress in mice: effects of genetic background and stress duration. Front. Behav. Neurosci. 5, 13. doi: 10.3389/fnbeh.2011.00013
Shen, L., Shao, N.-Y., Liu, X., Maze, I., Feng, J., Nestler, E. J. (2013). diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PloS One 8, e65598. doi: 10.1371/journal.pone.0065598
Spijker, S. (2011). “Dissection of rodent brain regions,” in Neuroproteomics Neuromethods. Ed. Li, W. W. K. (Humana Press), 13–26. doi: 10.1007/978-1-61779-111-6_2
Subbanna, S., Shivakumar, M., Umapathy, N. S., Saito, M., Mohan, P. S., Kumar, A., et al. (2013). G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol. Dis. 54, 475–485. doi: 10.1016/J.NBD.2013.01.022
Tamaki, K., Kamakura, M., Nakamichi, N., Taniura, H., Yoneda, Y. (2008). Upregulation of Myo6 expression after traumatic stress in mouse hippocampus. Neurosci. Lett. 433, 183–187. doi: 10.1016/J.NEULET.2007.12.062
Tarpey, P., Parnau, J., Blow, M., Woffendin, H., Bignell, G., Cox, C., et al. (2004). Mutations in the DLG3 gene cause Nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 75, 318–324. doi: 10.1086/422703
Tau, G. Z., Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35, 147–168. doi: 10.1038/npp.2009.115
Tosto, G., Vardarajan, B., Sariya, S., Brickman, A. M., Andrews, H., Manly, J. J., et al. (2019). Association of Variants in PINX1 and TREM2 with late-onset Alzheimer Disease. JAMA Neurol. 76, 942. doi: 10.1001/jamaneurol.2019.1066
Veazey, K. J., Parnell, S. E., Miranda, R. C., Golding, M. C. (2015). Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenet. Chromatin 8, 39. doi: 10.1186/s13072-015-0031-7
Yano, H., Ninan, I., Zhang, H., Milner, T. A., Arancio, O. (2006). BDNF-mediated neurotransmission relies upon a myosin VI motor complex. Nat. Neurosci. 9, 1009–1018. doi: 10.1038/nn1730
Yi, L., Pimentel, H., Bray, N. L., Pachter, L. (2018). Gene-level differential analysis at transcript-level resolution. Genome Biol. 19, 53. doi: 10.1186/s13059-018-1419-z
Yu, Y., Wu, A., Zhang, Z., Yan, G., Zhang, F., Zhang, L., et al. (2013). Characterization of the GufA subfamily member SLC39A11/Zip11 as a zinc transporter. J. Nutr. Biochem. 24, 1697–1708. doi: 10.1016/J.JNUTBIO.2013.02.010
Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. doi: 10.1186/gb-2008-9-9-r137
Zhang, H., Constantine, R., Vorobiev, S., Chen, Y., Seetharaman, J., Huang, Y. J., et al. (2011). UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci. 14, 874–880. doi: 10.1038/nn.2835
Keywords: fetal alcohol spectrum disorder, prenatal alcohol, hippocampus, maternal separation, DNA methylation, early life stress
Citation: Alberry BLJ and Singh SM (2020) Hippocampal DNA Methylation in a Mouse Model of Fetal Alcohol Spectrum Disorder That Includes Maternal Separation Stress Only Partially Explains Changes in Gene Expression. Front. Genet. 11:70. doi: 10.3389/fgene.2020.00070
Received: 27 September 2019; Accepted: 22 January 2020;
Published: 27 February 2020.
Edited by:
Hehuang Xie, Virginia Tech, United StatesReviewed by:
Silvio Zaina, University of Guanajuato, MexicoHelge Frieling, Hannover Medical School, Germany
Copyright © 2020 Alberry and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiva M. Singh, c3NpbmdoQHV3by5jYQ==