 Kerong Shi*
Kerong Shi* Fugui Niu
Fugui Niu Qin Zhang
Qin Zhang Chao NingShujian YueChengzhang HuZhongjin XuShengxuan Wang
Chao NingShujian YueChengzhang HuZhongjin XuShengxuan Wang Ranran Li
Ranran Li Qiuling Hou*Zhonghua Wang*
Qiuling Hou*Zhonghua Wang*- College of Animal Science and Technology, Shandong Key Laboratory of Animal Bioengineering and Disease Prevention, Shandong Agricultural University, Taian, China
A genome-wide association study (GWAS) was conducted on 23 serum biochemical traits in Chinese Holstein cattle. The experimental population consisted of 399 cattle, each genotyped by a commercial bovine 50K SNP chip, which had 49,663 SNPs. After data cleaning, 41,092 SNPs from 361 Holstein cattle were retained for GWAS. The phenotypes were measured values of serum measurements of these animals that were taken at 11 days after parturition. Two statistical models, a fixed-effect linear regression model (FLM) and a mixed-effect linear model (MLM), were used to estimate the association effects of SNPs. Genome-wide significant and suggestive thresholds were set up to be 1.22E−06 and 2.43E−06, respectively. In the Chinese Holstein population, FLM identified 81 genome-wide significant (0.05/41,092 = 1.22E−06) SNPs associated with 11 serum traits. Among these SNPs, five SNPs (BovineHD0100005950, ARS-BFGL-NGS-115158, BovineHD1500021175, BovineHD0800028900, and BTB-00442438) were also identified by the MLM to have genome-wide suggestive effects on CHE, DBIL, and LDL. Both statistical models pinpointed two SNPs that had significant effects on the Holstein population. The SNP BovineHD0800028900 (located near the gene LOC101903458 on chromosome 8) was identified to be significantly associated with serum high- and low-density lipoprotein (HDL and LDL), whereas BovineHD1500021175 (located in 73.4Mb on chromosome 15) was an SNP significantly associated with total bilirubin and direct bilirubin (TBIL and DBIL). Further analyses are needed to identify the causal mutations affecting serum traits and to investigate the correlation of effects for loci associated with fatty liver disease in dairy cattle.
Genome-wide association study was proven to be a powerful tool for detecting genetic variants associated with economically important traits, such as production (Jung et al., 2013; Yue et al., 2017; Yan et al., 2019), reproduction (Sahana et al., 2011), and disease traits (Pant et al., 2010). This study was to identify SNPs with significant association effects on serum traits in Chinese Holstein and Jersey cattle through the use of GWAS.
The experimental population consisted of 399 Chinese Holstein dairy cows, all of which were raised on the same farm. The phenotypes were the measured values for 23 serum traits with the serum being sampled from each cow at 11 days after parturition within a month (between September and October). The serum traits were adenosine deaminase (ADA), serum albumin (ALB), alkaline phosphatase (ALP), alanine transaminase (ALT), aspartate aminotransferase (AST), β-hydroxybutyric acid (BHB), cholinesterase (CHE), creatine kinase (CK), serum creatinine (CR), direct bilirubin (DBIL), glucose (GLU), high density lipoprotein (HDL), L-lactate dehydrogenase (LDHL), low density lipoprotein (LDL), non-esterified fatty acid (NEFA), serum urea nitrogen (SUN), total bilirubin (TBIL), total cholesterol (TCHO), triglyceride (TG), total protein (TP), urea acid (UA), very low density lipoprotein (VLDL), and γ-glutamyltransferase (γ-GT). The statistical summary of these phenotypes is listed in Supplementary Table S1.
All animals were genotyped with a bovine 50K SNP chip (49,663 SNPs). SNPs from the X chromosome were counted due to the overall majority of female individuals in the study population. After the data quality control procedure (Yue et al., 2017; Yan et al., 2019), 361 animals with 41,092 SNP genotypes were finally retained for the subsequent GWAS analysis. Physical map length, the number of SNPs, and the SNP density on each chromosome, before and after the data cleaning procedure, are shown in Supplementary Table S2.
A pair-wise linkage disequilibrium (LD) analysis was conducted for the Holstein population. The results showed high genome-wide similarity of LD patterns among the cattle populations (Supplementary Figure S1). The similarity might reflect the sharing of breeding histories among the cattle. Multi-dimensional scaling (MDS) analysis of 12,380 independent SNP markers (Purcell et al., 2007; Yue et al., 2017; Yan et al., 2019) with r2 < 0.2 (Wang et al., 2009), using the first and the second components, indicating that there was slight population stratification (Supplementary Figure S2). To better correct cryptic population stratification, the first MDS component was used to be the covariate in the following genome-wide association analysis (Supplementary Figure S3).
According to the previous method (Yue et al., 2017), a GWAS analysis was carried out by two statistical models, a fixed-effect linear model (FLM) and a mixed-effect linear model (MLM), implemented by the PLINK software package V1.07 (Purcell et al., 2007) and the GCTA (v1.2.4) software package (Yang et al., 2011), respectively. FLM is of the form:
where y is a vector of phenotypic values; α is a vector of fixed effects including the population mean and the first MDS component; W is the designed matrix for fixed effects; β is the marker effect; x a vector of marker genotypes; and e is the random errors with distribution of . Here, is the residual variances. For MLM, an additive genomic relatedness matrix is included to control the type I error, which is of the form
where Z is the designed matrix, and u is the vector of random effects with the distribution of . Here, is the additive genetic variances and K is the additive genomic relatedness matrix. The other symbols are the same as the FLM. Bonferroni corrections for the genome-wide significance and suggestive thresholds (Mapholi et al., 2016; Kerr et al., 2017) were computed to be 1.22E−06 (=0.05/41,092) and 2.43E−06 (=0.1/41,092), respectively.
A GWAS based on the FLM identified 81 SNPs with genome-wide significant (1.22E−06) association effects on 11 serum traits (Table 1) in the Holstein cattle population. A GWAS based on the MLM identified 15 SNPs as having genome-wide suggestive effects on 11 serum traits (Table 2). Among these SNPs, five SNPs (BovineHD0100005950, ARS-BFGL-NGS-115158, BovineHD1500021175, BovineHD0800028900 and BTB-00442438) were identified by both the FLM and MLM to have genome-wide suggestive effects on CHE, DBIL, and LDL.
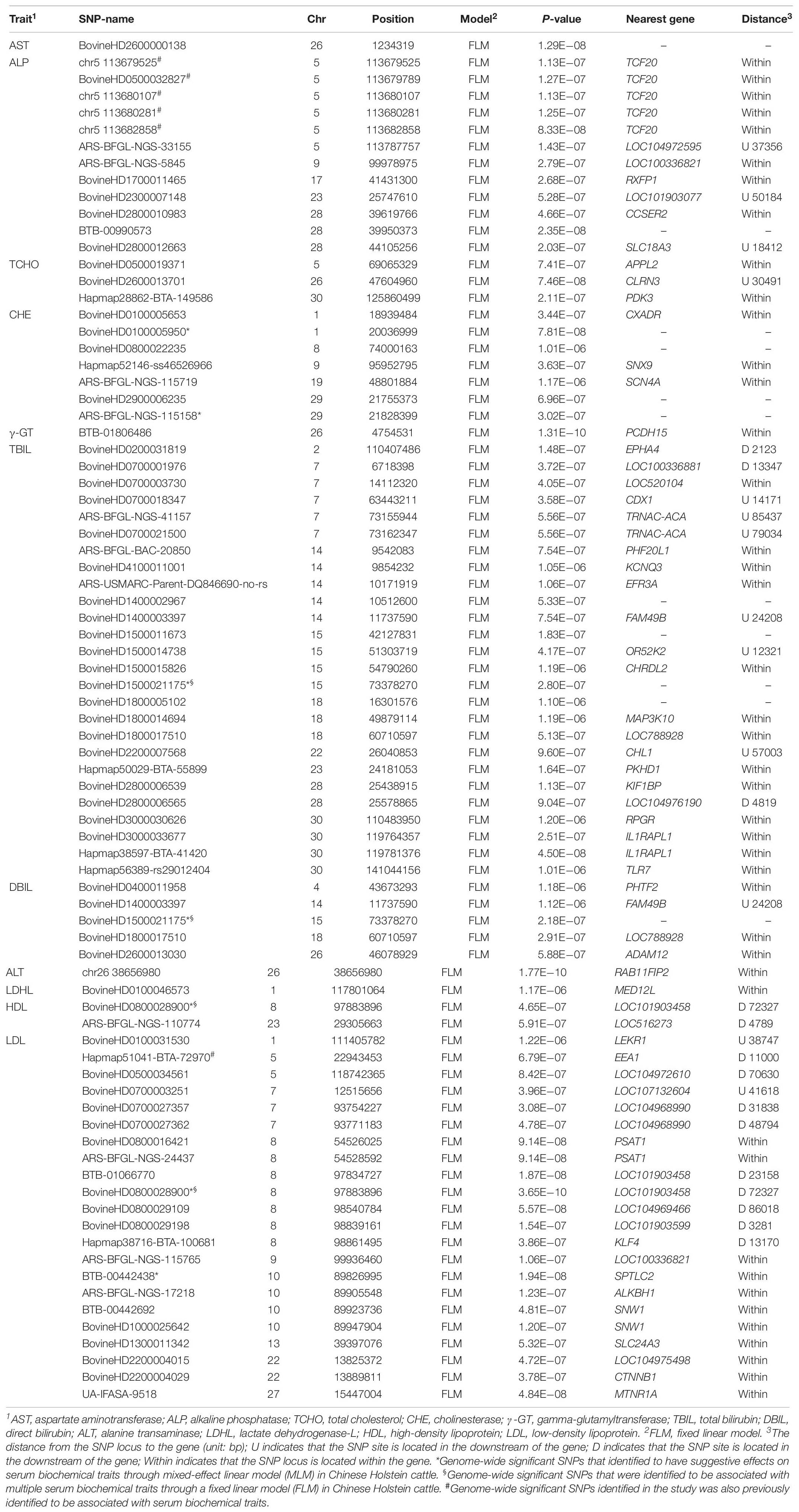
Table 1. Genome-wide significant SNPs that were identified to be associated with serum indexes in Chinese Holstein cattle using a fixed linear model.
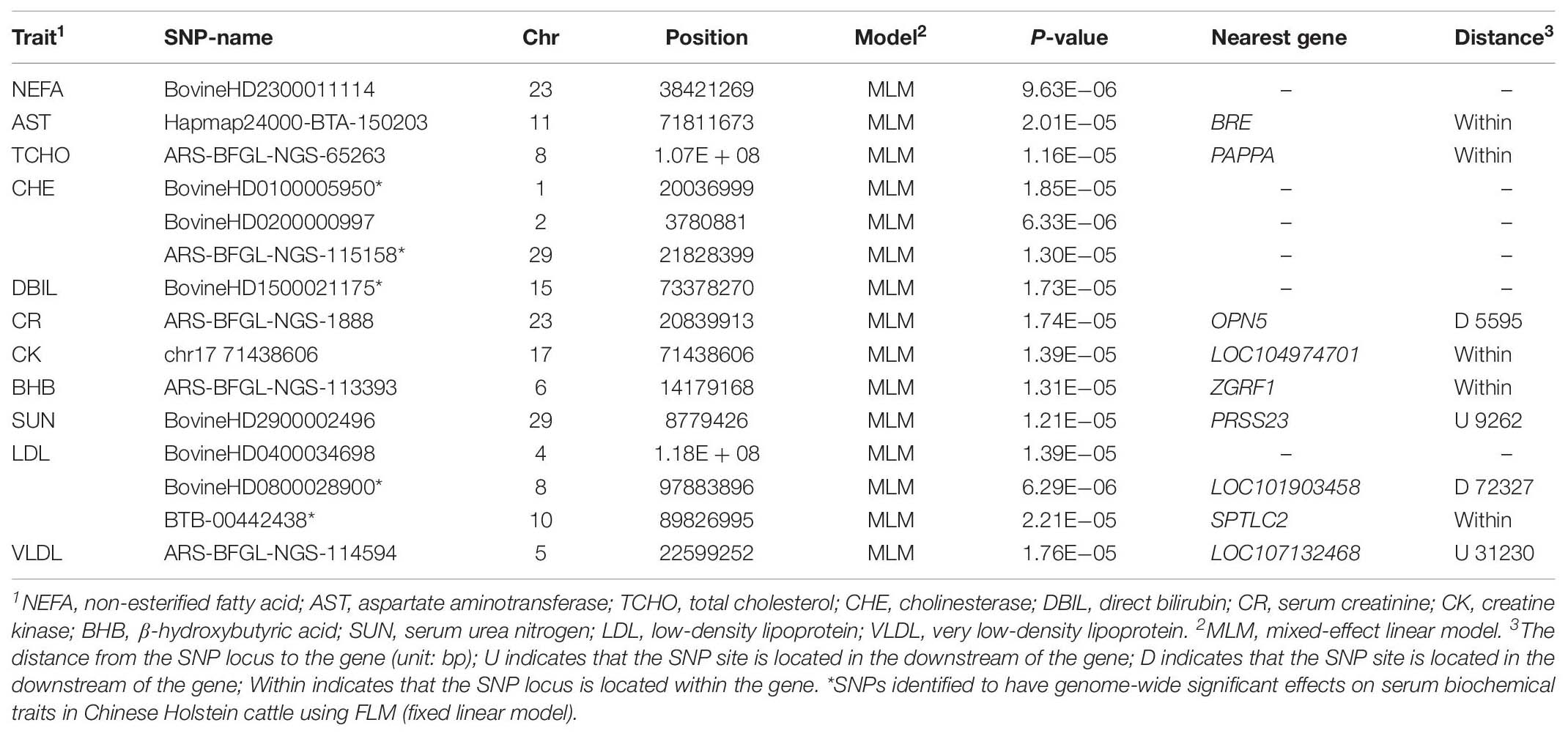
Table 2. SNPs identified to have genome-wide suggestive effects on serum biochemical traits in Holstein cattle using a mixed-effect linear model.
The SNPs identified through the MLM displayed lower overlapping than those identified through the FLM. However, the set of significant SNPs from the MLM in the study was almost a subset of SNPs from the FLM. The SNPs identified through the MLM were more conservative because the MLM took into account the additive genetic effects of each animal, and the false positive rate was expected to be lower than with the FLM. In the GWAS, the FLM with the population structure fitted as covariates may not control the type I error well, while the MLM can lead to false negatives, thus missing some potentially important discoveries (Liu et al., 2016; Supplementary Figure S3). The FLM and MLM are the most popular models in the field of GWAS (Yu et al., 2006; Purcell et al., 2007; Kang et al., 2008, 2010). On the other hand, the low overlapping genome-wide significant SNPs identified from the FLM and MLM also suggest low heritability (h2) of biochemical serum traits, which could be genetically affected by minor genes.
Interestingly, both statistical models pinpointed two SNPs (BovineHD0800028900 and BovineHD1500021175) that displayed genome-wide significant (1.22E−06) association effects on serum traits in the Holstein population. The SNP BovineHD0800028900, located at the downstream of LOC101903458 gene on chromosome 8, was identified to be significantly associated with serum high- and low-density lipoprotein (HDL and LDL). The SNP of BovineHD1500021175 on chromosome 15 was found to have significant association effects on serum bilirubin (TBIL and DBIL). Further analyses are needed to understand the mechanism for the association effects of these SNPs on serum biochemical traits (Du et al., 2013; Hu et al., 2015).
Additionally, several candidate genes or DNA regions that we found to be significantly associated with serum biochemical traits in Holstein cattle coincided with reported association effects on other traits in the literature. For example, six SNPs at the DNA region from 113.6 to 113.7 cM of chromosome 5, closely associated with TCF20 gene, were identified to have a significant effect on the serum ALP level (Table 1). The same DNA region was reported to have a QTL associated with blood triglyceride (TAG) levels (Wu et al., 2014). As another example, Hapmap51041-BTA-72970, located at the downstream region of EEA1 (early endosome antigen 1), was identified to be significantly associated with serum low-density lipoprotein (LDL) level in both Holstein and Jersey cattle in the study. The same region was found to be a QTL, having an effect on abomasum displacement in German Holstein cattle (Mömke et al., 2013). MNTR1A (melatonin receptor 1A) was previously found associated with intramuscular fat and subcutaneous fat (Yang et al., 2015) in Qinchuan beef cattle, and it was also found to be a candidate gene of serum LDL in our study.
In summary, GWAS was conducted using two statistical models on 23 serum biochemical traits in a Chinese Holstein cattle population. Eighty-one genome-wide significant (1.22E−06) SNPs were identified to have association effects on 11 serum biochemical traits through FLM. Among these SNPs, five SNPs were also identified by the MLM to have genome-wide suggestive effects on CHE, DBIL, and LDL. There were two SNPs, BovineHD0800028900 and BovineHD1500021175, that were found to be associated with multiple serum lipoprotein levels and serum bilirubin traits, respectively. The role of these identified SNPs associated with serum biochemical traits remains to be further investigated and validated in future studies. Understand their roles may increase our understanding of the underlying molecular biology of perinatal metabolic disorder, such as fatty liver disease, in dairy cows.
Data Availability Statement
The dataset generated in this study has been deposited into the Animal QTLdb (https://www.animalgenome.org/cgi-bin/QTLdb/BT/pubtails?PUBMED_ID=ISU0115).
Ethics Statement
All experiments were carried out according to the Regulations for the Administration of Affairs Concerning Experimental Animals published by the Ministry of Science and Technology, China (2004) and approved by the Animal Care and Use Committee in Shandong Agricultural University, Shandong, China.
Author Contributions
KS, QZ, and ZW conceived and designed the experiments. QH, FN, CH, ZX, SW, and RL performed the experiments. KS, CN, and SY analyzed the data. ZW, CH, SW, FN, and RL contributed the reagents, materials, and analysis tools. KS, SY, and CN wrote the manuscript.
Funding
This work was financially supported by the Key Project of Agricultural Fine Breeding of Shandong Province (2016LZGC030 and 2019LZGC011), the National Natural Science Foundation of China (31402054), the Natural Science Foundation of Shandong (ZR2013CM013), the Funds of Shandong “Double Tops” Program (SYL2017YSTD08), the Modern Agricultural Industry Technology System (CARS-36), and the Tai Mountain Scholar Innovation Team.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the farm owners who generously allowed us to sample blood from their cattle. We are also thankful to Ms. Zijuan Qin and Ms. Mei Zhao in the Animal Husbandry Laboratory platform of Shandong Agricultural University for their kind laboratory support on the serum biochemical tests.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00163/full#supplementary-material
References
Du, H. T., Wang, C. Y., Wang, X. P., Ma, M. W., and Li, F. C. (2013). The effects of dietary α-linolenic acid on growth performance, meat quality, fatty acid composition, and liver relative enzyme mRNA expression of growing meat rabbits. J. Anim. Feed Sci. 22, 122–129. doi: 10.22358/jafs/66002/2013
Hu, Z. Y., Yin, Z. Y., Lin, X. Y., Yan, Z. G., and Wang, Z. H. (2015). Effects of feeding fatty acid calcium and the interaction of forage quality on production performance and biochemical indexes in early lactation cow. J. Anim. Physiol. Anim. Nutr. 99, 899–904. doi: 10.1111/jpn.12302
Jung, E. J., Park, H. B., Lee, J. B., Yoo, C. K., Kim, B. M., Kim, H. I., et al. (2013). Genome-wide association analysis identifies quantitative trait loci for growth in a Landrace purebred population. Anim. Genet. 45, 442–444. doi: 10.1111/age.12117
Kang, H. M., Sui, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckerman, D., Daly, M. J., et al. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. doi: 10.1534/genetics.107.080101
Kerr, K. F., Avery, C. L., Lin, H. J., Raffield, L. M., Zhang, Q. S., Browning, B. L., et al. (2017). Genome-wide association study of heart rate and its variability in Hispanic/Latino cohorts. Heart Rhythm. 14, 1675–1684. doi: 10.1016/j.hrthm.2017.06.018
Liu, X., Huang, M., Fan, B., Buckler, E. S., and Zhang, Z. (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12:e1005767. doi: 10.1371/journal.pgen.1005767
Mapholi, N. O., Maiwashe, A., Matika, O., Riggio, V., Bishop, S. C., MacNeil, M. D., et al. (2016). Genome-wide association study of tick resistance in South African Nguni cattle. Ticks Tick Borne Dis. 7, 487–497. doi: 10.1016/j.ttbdis.2016.02.005
Mömke, S., Sickinger, M., Lichtner, P., Doll, K., Rehage, J., and Distl, O. (2013). Genome-wide association analysis identifies loci for left-sided displacement of the abomasum in German Holstein cattle. J. Dairy Sci. 96, 3959–3964. doi: 10.3168/jds.2012-5679
Pant, S. D., Schenkel, F. S., Verschoor, C. P., You, Q., Kelton, D. F., Moore, S. S., et al. (2010). A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in Holstein cattle. Genomics 95, 176–182. doi: 10.1016/j.ygeno.2010.01.001
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Sahana, G., Guldbrandtsen, B., and Lund, M. S. (2011). Genome-wide association study for calving traits in Danish and Swedish Holstein cattle. J. Dairy Sci. 94, 479–486. doi: 10.3168/jds.2010-3381
Wang, D., Sun, Y., Stang, P., Berlin, J. A., Wilcox, M. A., Li, Q., et al. (2009). Comparison of methods for correcting population stratification in a genome-wide association study of rheumatoid arthritis: principal-component analysis versus multidimensional scaling. BMC Proc. 3, (Suppl. 7):S109. doi: 10.1186/1753-6561-3-s7-s109
Wu, Y., Fan, H., Wang, Y., Zhang, L., Gao, X., Chen, Y., et al. (2014). Genome-wide association studies using haplotypes and individual SNPs in Simmental cattle. PLoS One 9:e109330. doi: 10.1371/journal.pone.0109330
Yan, Z. G., Wang, Z. H., Zhang, Q., Yue, S., Yin, B., Jiang, Y., et al. (2019). Identification of whole-genome significant single nucleotide polymorphisms in candidate genes associated with body conformation traits in Chinese Holstein cattle. Anim. Genet. 51, 141–146. doi: 10.1111/age.12865
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yang, W., Wang, Y., Fu, C., and Zan, L. S. (2015). Association study and expression analysis of MTNR1A as a candidate gene for body measurement and meat quality traits in Qinchuan cattle. Gene 570, 199–204. doi: 10.1016/j.gene.2015.06.012
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Keywords: GWAS, serum biochemical traits, cattle, Chinese Holstein, SNPs, QTL
Citation: Shi K, Niu F, Zhang Q, Ning C, Yue S, Hu C, Xu Z, Wang S, Li R, Hou Q and Wang Z (2020) Identification of Whole-Genome Significant Single Nucleotide Polymorphisms in Candidate Genes Associated With Serum Biochemical Traits in Chinese Holstein Cattle. Front. Genet. 11:163. doi: 10.3389/fgene.2020.00163
Received: 06 September 2019; Accepted: 12 February 2020;
Published: 04 March 2020.
Edited by:
Fabyano Fonseca Silva, Universidade Federal de Viçosa, BrazilReviewed by:
Xiangdong Ding, China Agricultural University, ChinaHayrettin Okut, University of Kansas Hospital, United States
Copyright © 2020 Shi, Niu, Zhang, Ning, Yue, Hu, Xu, Wang, Li, Hou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kerong Shi, a3JzaGlAc2RhdS5lZHUuY24=; Qiuling Hou, aG91cWxAc2RhdS5lZHUuY24=; Zhonghua Wang, emh3YW5nQHNkYXUuZWR1LmNu