 Lianmei Zhang1,2Jingliang Cheng2Qi Zhou3
Lianmei Zhang1,2Jingliang Cheng2Qi Zhou3 Md. Asaduzzaman Khan2Jiewen Fu2Chengxia Duan3Suan Sun1*Hongbin Lv3*
Md. Asaduzzaman Khan2Jiewen Fu2Chengxia Duan3Suan Sun1*Hongbin Lv3* Junjiang Fu2*
Junjiang Fu2*- 1Department of Pathology, The Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University, Huai’an, China
- 2Key Laboratory of Epigenetics and Oncology, The Research Center for Preclinical Medicine, Southwest Medical University, Luzhou, China
- 3Department of Ophthalmology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Usher syndrome includes a group of genetically and clinically heterogeneous autosomal recessive diseases, such as retinitis pigmentosa (RP) and sensorineural hearing loss. Usher syndrome type I (USHI) is characterized by profound hearing impairment beginning at birth, vestibular dysfunction, and unintelligible speech in addition to RP. The relationships between the Usher syndrome causing genes and the resultant phenotypes of Usher syndrome have not yet been fully elucidated. In the present study, we recruited a Chinese family with Usher syndrome and conducted paneled next-generation sequencing, Sanger sequencing, segregation analysis, and expression profile analysis. The functional effects of the identified cadherin-related 23 (CDH23) pathogenic variants were analyzed. The M101 pedigree consisted of a proband and seven family members, and the proband was a 39-year-old Chinese male who claimed that he first began to experience night blindness 11 years ago. We revealed novel, missense compound heterozygous variants c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) in the CDH23 gene, which co-segregated with the disease phenotype causing Usher syndrome type ID (USH1D) in this Chinese pedigree. CDH23 mRNA was highly expressed in the retina, and this protein was highly conserved as revealed by the comparison of Homo sapiens CDH23 with those from nine other species. This is the first study to identify the novel, missense compound heterozygous variants c. 2572G > A (p.V858I) and c.2891G > A (p.R964Q) of CDH23, which might cause USH1D in the studied Chinese family, thereby extending CDH23 mutation spectra. Identifying CDH23 pathogenic variants should help in the detailed phenotypic characterization of USH1D.
Introduction
Usher syndrome is a group of genetically and clinically heterogeneous autosomal recessive diseases, such as progressive retinitis pigmentosa (RP) and sensorineural hearing loss (Boughman et al., 1983). Clinical differences have led to the categorization of three types of Usher syndrome: type I, II, and III (Keats and Corey, 1999; Mathur and Yang, 2015; Wei et al., 2018; Okano et al., 2019). Usher syndrome type I (USHI) is characterized by profound hearing impairment beginning at birth, vestibular dysfunction, and unintelligible speech in addition to RP (Okano et al., 2019).
Genetically, USHI is an autosomal recessive heterogeneous disorder causing mutations in at least eight genes that produces a similar disease or phenotype. These genes are: MYO7A (OMIM: 276903) at 11q13.5 causing USH1B (USH1A is considered to be the same; OMIM: 276900), harmonin or USH1C (OMIM: 605242) at 11p15.1 causing USH1C (OMIM: 276904), CDH23 (OMIM: 605516) at 10q21-q22, causing USH1D (OMIM: 601067), PCDH15 (OMIM: 605514) at 10q21.1 causing USH1F (OMIM: 602083), SANS or USH1G (OMIM: 607696) at 17q24-25 causing USH1G (OMIM: 606943), and CIB2 (605564) at 15q24 causing USH1J (OMIM: 614869). Mutations in as yet unnamed genes in loci at 21q21 (USH1E; OMIM: 602097), 10p11.21-q21.1 (USH1K, OMIM: 614990), and 15q22-q23 (USH1H; OMIM: 612632) may also cause this type I phenotype. The clinical features of each are indistinguishable but are caused by different genes. Herein, Usher syndrome type ID (USH1D) is caused by homozygous or compound heterozygous mutation in the gene CDH23, a member of the cadherin superfamily which comprises calcium-dependent cell–cell adhesion glycoproteins. The CDH23 gene locates on chromosome 10q22.1 and encodes a predicted protein of 3,354 amino acids with a single transmembrane domain and 27 cadherin repeats. During late embryonic or early postnatal development, the CDH23 protein was required for establishing and maintaining the proper organization of the stereocilia bundle of hair cells in the cochlea and the vestibule. However, the expression levels of CDH23 in different tissues were not fully described.
The relationships between the variations in the Usher syndrome-caused genes and the resultant different Usher syndrome types/phenotypes in the patients are highly variable. Novel CDH23 mutations in patients with USH1D and their relationships with genotypes/phenotypes are not well-documented in the Chinese population. Next-generation sequencing (NGS) provides a useful genetic diagnosis approach (Adams and Eng, 2018; Fu et al., 2019; Rahbaran et al., 2019). In the present study, novel missense compound heterozygous mutations of CDH23 are revealed in Chinese pedigree links with USH1D.
Case Presentation
The M101 pedigree consisted of a proband and seven family members (Figure 1A). The proband (Figure 1, II: 2) was a 39-year-old Chinese male who claimed that he first began to experience night blindness 11 years ago. Fundus examinations showed macular changes in both eyes (Figure 1B). The retinal pigment epitheliums (RPEs) were atrophied (data not shown). The proband was first characterized as having RP and currently claims that they are presently experiencing hearing loss. Pure tone audiometry subsequently confirmed the presence of sensorineural hearing loss (data not shown). His parents and his daughter tested negatively for hearing loss. Therefore, the most likely diagnosis for the proband was Usher syndrome (type I).
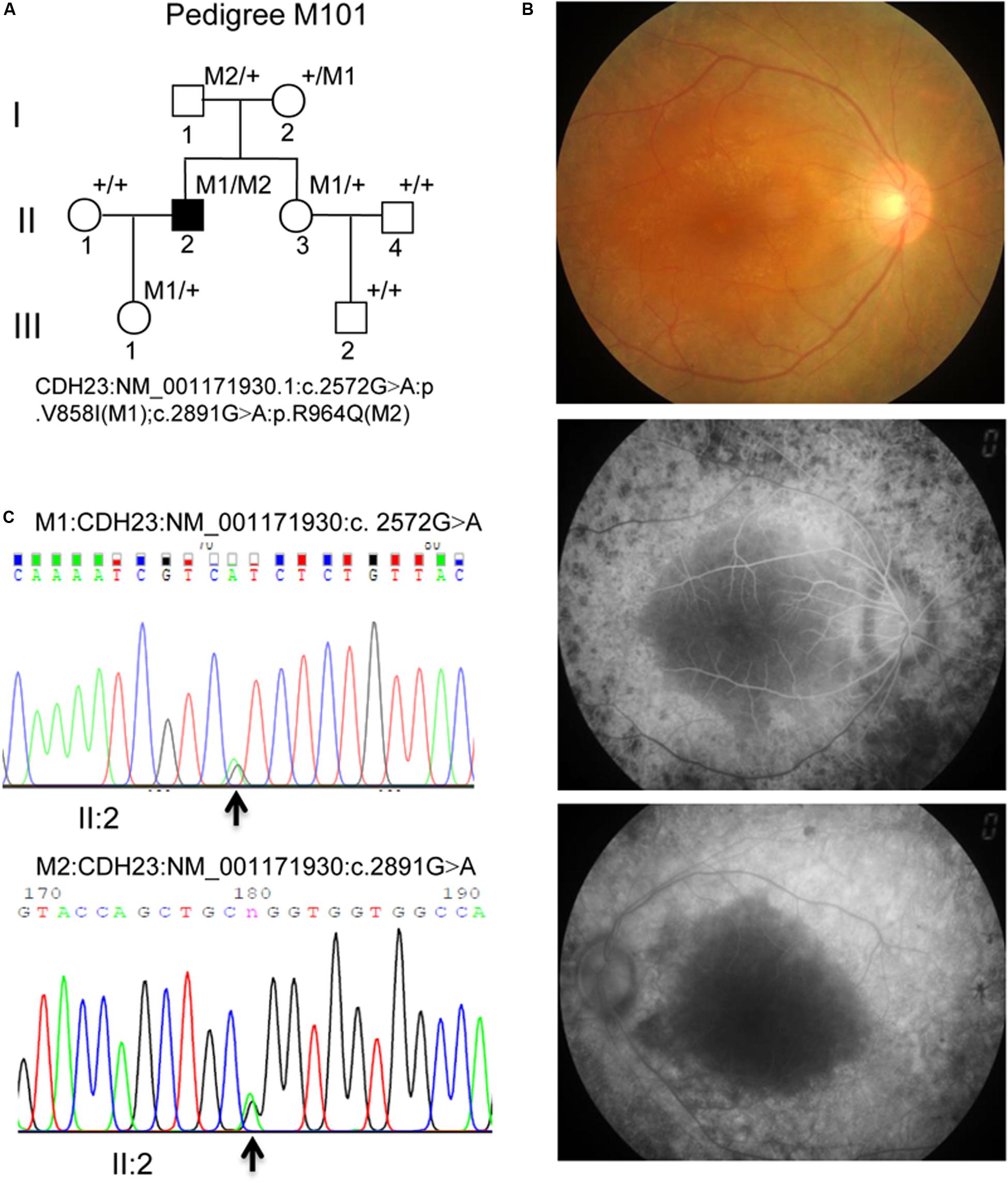
Figure 1. (A) M101 pedigree with Usher syndrome type ID. Normal individuals are shown as clear circles (females) or squares (male). The filled square indicates the proband (II: 2, arrow) with the compound heterozygous mutation of the CDH23 gene: NM_001171930.1:exon22:c.2572G > A:p.V858I (M1); exon24: c. 2891G > A: p.R964Q(M2). “+,” wild-type allele. (B) Representative retinal phenotypes of proband II: 2. Top panel. Representatively FP in patient II: 2 of the right eye. Middle and bottom panel. Representatively FFA in patient II: 2 of both right and left eyes, respectively. (C) Pyrogram profiles for variant verification by Sanger sequencing, indicating the sequenced results in II: 2 (missense c. 2572G > A mutant type) (top panel), II: 2 (missense c.2891G > A mutant type) (bottom panel). The arrows show the mutation position NM_001171930.1: c.2572G > A or c. 2891G > A in the CDH23 gene, respectively.
The study has been approved by the Ethics Committee of Southwest Medical University/the Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University, and with the Helsinki Declaration of 1975, as revised in 2013. The protocol and procedures employed for the mouse experiments were ethically reviewed and approved by the Ethics Committee of Southwest Medical University/the Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University. The patient and the family members gave written informed consent to participate in the study, and for the publication of their clinical cases. All experimental works were carried out following the approved protocols and procedures.
Methods
Clinical Assessment, Blood Collection, and DNA Isolation
Clinical ophthalmic examinations and audiometric testing of the proband were performed as described previously (Wei et al., 2018; Huang et al., 2019). Blood samples from the family being studied were retrieved, and DNA was isolated from these samples. Healthy control blood samples in Luzhou were also collected (Fu et al., 2002; Cheng et al., 2019).
Targeted NGS, Quality Control, and Bioinformatics Analysis
Targeted next-generation sequencing (TGS) analyses with 256 disease-causing genes panel were performed on an M101 DNA sample, according to the manual from Illumina as previously described (Zhang et al., 2016; Fu et al., 2017, 2018; Soens et al., 2017; An et al., 2019). Library construction, TGS, bioinformatic computational framework and data analysis were executed according to the manufacturer’s protocols (Koenekoop et al., 2012; Soens et al., 2016; Fu et al., 2017; Adams and Eng, 2018; An et al., 2019). Ninety-six percent of the targeted regions have coverage >20× and 91.1% of the targeted regions have coverage >40×. In all, more than 10 million bases of the sequence with 100-bp read length, 40,000 SNPs and 11,400 INDELs were generated. After quality assessment, more than 97% of billion bases were aligned to the human reference sequences (hg19) by BWA-MEM, duplicate reads were removed by Picard, local realignments were conducted by GATK, and variants were called by using Atlas2. A population frequency threshold of 0.5% was applied to filter out common variants which occur too frequently to be the cause of a rare Mendelian disease. Among those, billions of bases covered with a 10-fold coverage target region. Four NGS cohort databases were used to determine allele frequencies (Soens et al., 2016). The functional consequence of remaining rare variants was annotated using ANNOVAR, and dbNSFP was used to compile in silico predictions about the deleteriousness of non-synonymous variants. UGENE was used to perform the multiple sequence alignment using the MUSCLE alignment algorithm (Soens et al., 2016). The comprehensive and method of variant interpretation in detail has been described in An’s study (An et al., 2019).
The pathogenicity of each variant was assessed with the following databases: PolyPhen-2, Sorting Intolerant From Tolerant (SIFT), MutationTaster, and I-Mutant2.0 (Fu et al., 2018; Cheng et al., 2020). Variants, considered as pathogenic candidates, were searched for in the Human Gene Mutation Database (HGMD) and The Exome Aggregation Consortium (ExAC) to determine whether they had been reported previously.
Sanger Sequencing
Primer pairs M101-CDH23-22 and M101-CDH23-24 were designed using the Primer3 program with genomic sequences containing NM_001171930.1: c.G2572A in exon 22 and c.G2891A in exon 24, respectively, in the CDH23 gene (Supplementary Table 1). PCR amplification was conducted using gDNA as a template by above designed primer pairs. Purified the PCR products and then the products were used for Sanger sequencing (ABI-3500DX Genetic Analyzer, Thermo Fisher Scientific) using M101-CDH23-22L and M101-CDH23-24L, which are shown in Supplementary Table 1.
Co-segregation analysis of the M101 family was conducted based on Sanger sequencing results.
Prediction of Protein Structure and Bioinformatics Analysis
The homologs of the cdh23 gene (NM_001171930.1) in humans were analyzed as previously described. The conserved domains of CDD in CDH23 protein (NP_071407.4) were searched using the NCBI (Marchler-Bauer et al., 2017; Imani et al., 2018, 2019; Cheng et al., 2020).
RNA-Sequencing and Reverse Transcription-PCR Profiles
The CDH23 gene expression profiles were analyzed from RNA-sequencing (RNA-seq) data, which were retrieved from healthy human samples of 95 individuals representing 27 different tissues from the NCBI. The expression values of CDH23 mRNA in different human tissues were retrieved from the NCBI.
RNA was isolated, and semi-quantitative RT-PCR was executed as previously reported (Fu et al., 2018). The primer pair for RT-cdh23 (RT-cdh23-nL and RT-cdh23-nR) targeted the mouse Cdh23 gene and is listed in Supplementary Table 1 (Fu et al., 2018). The amplified PCR product was separated on 1.2% agarose gel three times (Cheng et al., 2019). The mouse β-actin gene served as a control.
Results
NGS Results and Co-segregation Analysis
Compound heterozygous variants (c. 2572G > A) at exons 22 and (c. 2891G > A) 24 in the CDH23 gene (NM_001171930.1) with high confidence were identified in the proband, leading to the production of Isoleucine from Valine, and Glutamine from Arginine at amino acid positions 858 (V858I) and 964 (p.R964Q), respectively, in the CDH23 protein (NP_001165401) (Figure 1A, II: 2). Both variants c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) were proven to be novel by searching ExAC and HGMD databases (Table 1).
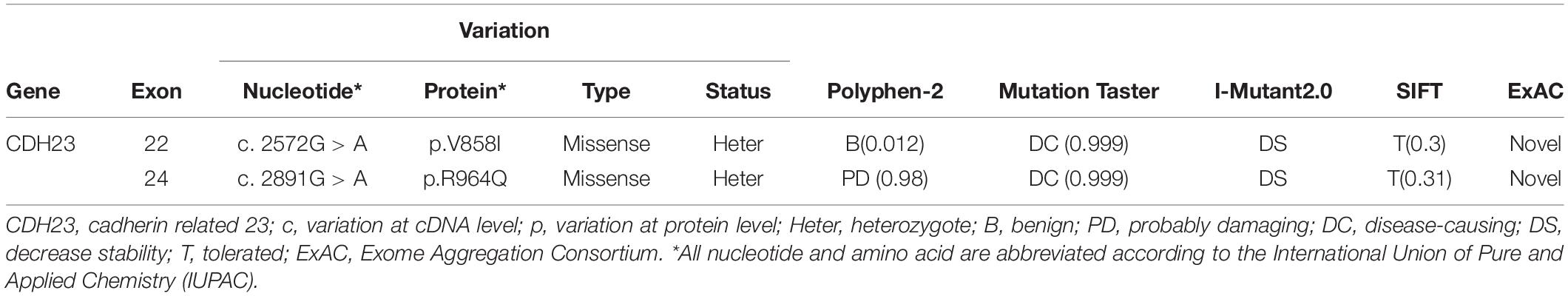
Table 1. Characteristics of CDH23 variants and the analysis of disease-causing effects.
The mutations c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) in the CDH23 gene were validated as compound heterozygous in the proband (Figure 1C; pedigree II: 1), with c. 2891G > A being inherited from his father (Supplementary Figure 1; pedigree I: 1) and c. 2572G > A being inherited from his mother (Supplementary Figure 1; pedigree I: 2), by Sanger sequencing. The proband’s daughter was found to be heterozygous c. 2891G > A with a normal phenotype (pedigree III: 1; Supplementary Figure 1), and the proband’s wife had a normal phenotype with alleles of wild-type (data not shown; pedigree II: 1). Therefore, it was ascertained that these variants in the CDH23 gene in the proband and the variants were co-segregated with this clinical phenotype in the family members. Neither variant was identified in either 1000 Genomes or the ExAC Browser. Altogether, this finding presents co-segregation of the variants in this family and pinpoints possible roles in pathogenesis of USH1D.
Functional Effects of the CDH23 Mutations c.G2572A (p.V858I) and c.G2891A (p.R964Q) for USH1D Disease
The characteristics of CDH23 mutations and the disease-causing effects on the proband are shown in Table 1. From Table 1, it can be observed that PolyPhen-2 analysis shows probable damage for p.R964Q change (score 0.98) and is benign for p.V858I change (score 0.012), respectively. MutationTaster revealed these changes to be caused by disease, with a score of 0.999. I-Mutant 2.0 indicated that both p.R964Q and p.V858I were damaged (score 0). Sorting Intolerant From Tolerant discovered tolerance for the p.R964Q change with a score of 0.31, and for the p.V858I change with a score of 0.30. Therefore, these missense mutations (c.2572G > A, p.V858I and c.2891G > A, p.R964Q) in the CDH23 gene damage protein function resulting in a diagnosis of USH1D in the studied Chinese family based on the gene mutations and clinical phenotypes. By comparing Homo sapiens CDH23 protein to nine other species, including Pan troglodytes, Macaca mulatta, Canis lupus, Bos taurus, Mus musculus, Rattus norvegicus, Gallus gallus, Xenopus tropicalis, and Danio rerio, it was revealed that CDH23 is a highly conserved ortholog (Figure 2A). Valine and Arginine at amino acid positions 858 and 964, respectively, were also found to be highly conserved (Figures 2B,C, highlighted in green). Taken together, our study reveals that the CDH23 missense compound heterozygous variants c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) might cause USH1D disease in the studied Chinese family.

Figure 2. The CDH23 comparison and conserved mutant positions. (A) The conservation analysis of CDH23 in indicated species. (B) The conserved mutant position of p.V858 (highlighted in green). (C) The conserved mutant position of p.R964 (highlighted in green). Variants p.V858I and p.R964Q of CDH23 are indicated in Figures 3B,C, respectively.
CDH23 and Cdh23 Expression Profiles
CDH23 expression indicated that CDH23 mRNA is highly expressed in the ovaries, with a value of RPKM 44.218 ± 0.225, followed by expression in fat and the testes, with the lowest expression in kidneys with a value of RPKM 0.072 ± 0.026 (Figure 3A and Supplementary Table 2). The expression values of RNA-seq in 27 different tissues are presented in Supplementary Table 2. No human eye tissues or developmental retinal stages were available. Therefore, Cdh23 expression in mice was studied. From Figure 3B, it was found that Cdh23 mRNA is highly expressed in the retina and testes, as well as in the sclera eye tissue and tissues of the ovaries, brain, and skeletal muscles. No or very low expression was identified in all other indicated tissues (Figure 3B). Cdh23 is also expressed at the different developmental retinal stages and is highly expressed at the latter two stages (one and two months for the retina) (Figure 3C), indicating a role in retina maturation. Therefore, a vital role in the retina and other tissues for CDH23 was revealed through CDH23 and Cdh23 expression profiles.
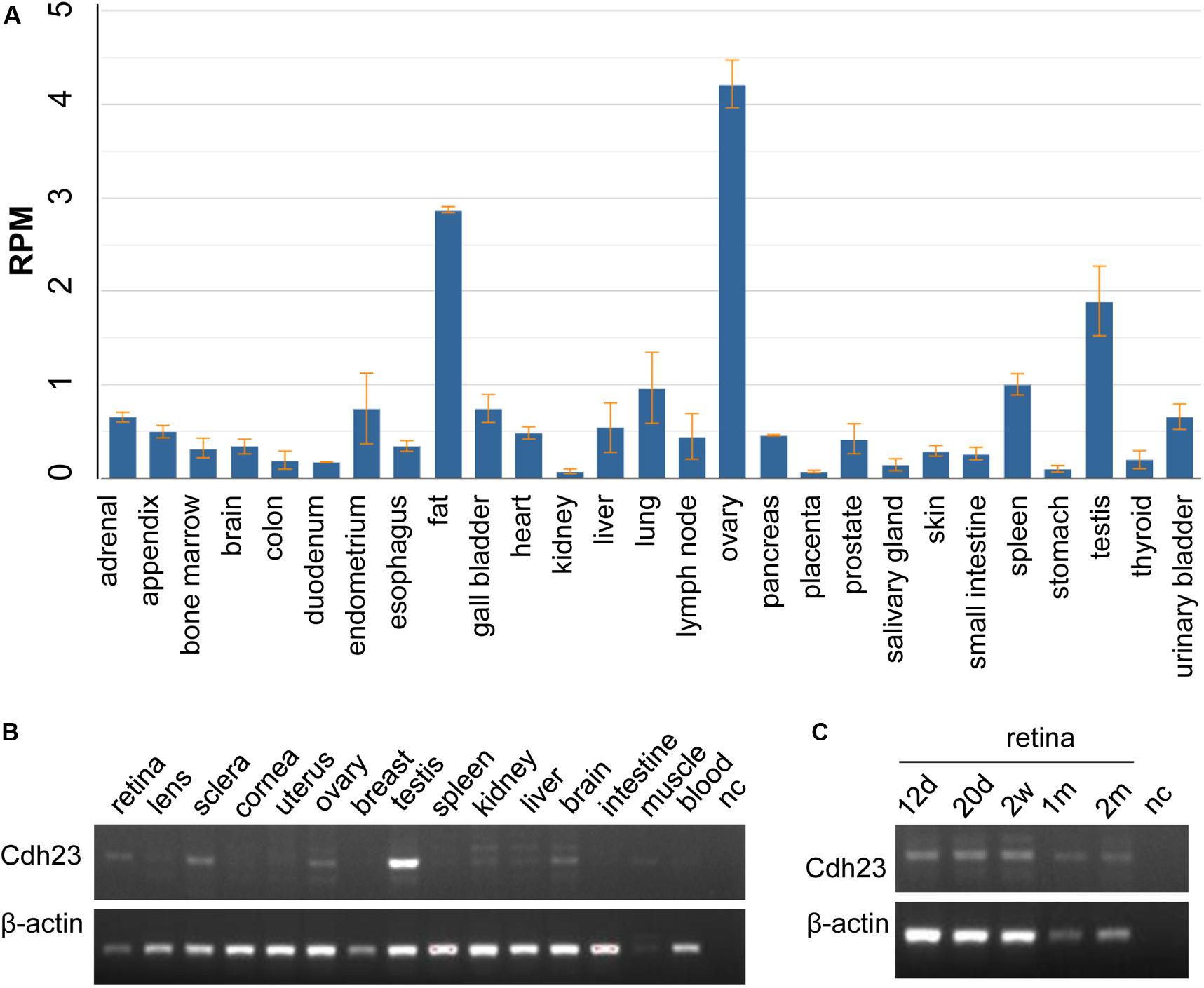
Figure 3. The mRNA expression in human CDH23 and mouse Cdh23 tissues. (A) The CDH23 mRNA expression in human tissues from RNA-seq data. (B) The Cdh23 mRNA levels in the indicated tissues. (C) The Cdh23 mRNA levels in the indicated development times in retinal tissue in mice. d, days; w, weeks; m, month(s); nc, no DNA template; muscle, skeletal muscle. Whole embryo eyeballs at 12.5 days (12 d) and 20.5 days (20 d) before birth, in panel (C), respectively.
Discussion
Mutations in the CDH23 gene cause USH1D (Bolz et al., 2001; Bork et al., 2001). Germline mutations in CDH23 have also recently been identified and associated with both familial and sporadic pituitary adenomas (Zhang et al., 2017). The upregulation of this gene may also be associated with breast cancer. However, the relationships between the variations in the Usher syndrome-caused genes and the resultant different types of Usher syndrome diseases or phenotypes in patients are highly variable, and the genotype/phenotype correlations are divergent. Patients with mutations in less frequently mutated genes, including CDH23, have not been well-reported for Chinese patients with USH (Jiang et al., 2015; Sun et al., 2018). In the present study, by using TGS, Sanger sequencing, and co-segregation analysis, missense compound heterozygous pathogenic variants c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) were successfully identified in the CDH23 gene in a Chinese USH1D pedigree, thereby elucidating the genetic roles of the CDH23 mutant alleles and confirming USH1D disease in this family. Therefore, CDH23 mutations with USH1D and the relationship with the genotype/phenotype were successfully linked for the proband of this studied family. By searching the HGMD1 (access date, February 22, 2020), 275 pathogenic variants have been reported (Supplementary Table 3). Disease/phenotype and CDH23 mutation relationship was provided in Supplementary Table 4. To the best of our knowledge, the CDH23 variants of c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) are novel, thereby extending the CDH23 mutation spectra.
By comparing H. sapiens CDH23 to nine other species, it was revealed that the CDH23 protein is a highly conserved ortholog. Cdh23 mRNA was only highly expressed in the retina, indicating its role in retinal functions. Taken together, this study revealed that the CDH23 compound heterozygous variants c.2572G > A (p.V858I) and c.2891G > A (p.R964Q) cause disease linked to USH1D.
As a rare disease with common inherited forms resulting in combined visual and hearing impairment, up to 14 genes, including MYO7A, CDH23, USH1C, PCDH15, USH1G, and CIB2 for USH type I, USH2A, ADGRV1, and WHRN for USH type II, CLRN1 and HARS for USH type III, and PDZD7, CEP250, and C2orf71, are associated with Usher syndrome, with CDH23 mutation identification in this study. Therefore, identifying CDH23 pathogenic variants should help to fine map and characterize the clinical phenotype with USH1D.
Genetic screening and the accurate diagnosis of Usher syndrome in different populations is necessary (Ben-Rebeh et al., 2016; Sun et al., 2018; Santana et al., 2019). Efficient molecular diagnosis of Usher syndrome in a patient’s early childhood is of utmost importance, allowing better genetic counseling and therapeutic management (Ben-Rebeh et al., 2016; Ivanova et al., 2018; Maddalena et al., 2018; Jouret et al., 2019). With accurate gene diagnosis for Usher syndrome, the repair of specific mutations using a CRISPR/Cas9 editing system may be possible. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology-directed repair in the mouse Cdh23 gene, indicating that the CRISPR editing system has a potential for treating Usher syndrome (Mianne et al., 2016). Tauroursodeoxycholic acid, a taurine-conjugated bile acid that has been used in experimental research and for clinical applications in the liver, for diabetes, and neurodegenerative diseases, might prevent hearing loss and hair cell death in the study of Cdh23erl/erl mutant mice (Hu et al., 2016b). Salubrinal (Sal), the ER stress inhibitor, also delayed the progression of hearing loss and preserved hair cells in the Cdh23erl/erl mutant mice (Hu et al., 2016a). Therefore, the findings of this study assist in both understanding the molecular pathogenesis of USH1D and develop strategies for diagnosis, therapy, and genetic counseling for USH1D disease.
Data Availability Statement
The datasets for this article are not publicly available due to participant/patient anonymity concerns. Requests to access the datasets should be directed to JF, ZnVqdW5qaWFuZ0Bob3RtYWlsLmNvbQ==.
Ethics Statement
The protocol and procedures employed for mouse were ethically reviewed and approved by the Ethics Committee of Southwest Medical University/the Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University. The study has been approved by the Ethics Committee of Southwest Medical University/the Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University, and with the Helsinki Declaration of 1975, as revised in 2013. All three patients gave written informed consent agreeing to participate in the study and for the publication of their clinical cases.
Author Contributions
JuF, HL, and SS oversaw the project design and concept of the study. LZ, JiF, and JC performed DNA extraction, PCR amplification, sequencing, and data analysis. HL, QZ, and CD recruited the clinical patients and oversaw the clinical assessments. MK revised the manuscript. JuF wrote and revised the manuscript.
Funding
This project was funded by the National Natural Science Foundation of China (31701087, 30371493, and 81672887) and the Joint Research Foundation of Luzhou City and Southwest Medical University (2018LZXNYD-YL01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Rui Chen and Dr. Hui Wang at the Baylor College of Medicine for technique assistance and Shangyi Fu at the University of Houston/Baylor College of Medicine for assisting with this project. We would like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00422/full#supplementary-material
Footnotes
References
Adams, D. R., and Eng, C. M. (2018). Next-generation sequencing to diagnose suspected genetic disorders. New Eng. J. Med. 379, 1353–1362. doi: 10.1056/nejmra1711801
An, J., Yang, J., Wang, Y., Wang, Y., Xu, B., Xie, G., et al. (2019). Targeted next generation sequencing revealed a novel homozygous loss-of-function mutation in ILDR1 gene causes autosomal recessive nonsyndromic sensorineural hearing loss in a Chinese family. Front. Genet. 10:1. doi: 10.3389/fgene.2019.00001
Ben-Rebeh, I., Grati, M., Bonnet, C., Bouassida, W., Hadjamor, I., Ayadi, H., et al. (2016). Genetic analysis of Tunisian families with Usher syndrome type 1: toward improving early molecular diagnosis. Mol. Vis. 22, 827–835.
Bolz, H., von Brederlow, B., Ramirez, A., Bryda, E. C., Kutsche, K., Nothwang, H. G., et al. (2001). Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 27, 108–112. doi: 10.1038/83667
Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. doi: 10.1086/316954
Boughman, J. A., Vernon, M., and Shaver, K. A. (1983). Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 36, 595–603. doi: 10.1016/0021-9681(83)90147-9
Cheng, J., Fu, J., Zhou, Q., Xiang, X., Wei, C., Yang, L., et al. (2019). A novel splicing mutation in the PRPH2 gene causes autosomal dominant retinitis pigmentosa in a Chinese pedigree. J. Cell. Mol. Med. 23, 3776–3780. doi: 10.1111/jcmm.14278
Cheng, J., Peng, J., Fu, J., Khan, M. A., Tan, P., Wei, C., et al. (2020). Identification of a novel germline BRCA2 variant in a Chinese breast cancer family. J. Cell. Mol. Med. 24, 1676–1683. doi: 10.1111/jcmm.14861
Fu, J., Cheng, J., Zhou, Q., Wei, C., Chen, H., Lv, H., et al. (2019). A novel missense variant c.G644A (p.G215E) of the RPGR gene in a Chinese family causes X-linked retinitis pigmentosa. Biosci Rep. 39:BSR20192235.
Fu, J., Li, L., and Lu, G. (2002). Relationship between microdeletion on Y chromosome and patients with idiopathic azoospermia and severe oligozoospermia in the Chinese. Chin. Med. J. 115, 72–75.
Fu, J., Ma, L., Cheng, J., Yang, L., Wei, C., Fu, S., et al. (2018). A novel, homozygous nonsense variant of the CDHR1 gene in a Chinese family causes autosomal recessive retinal dystrophy by NGS-based genetic diagnosis. J. Cell. Mol. Med. 22, 5662–5669. doi: 10.1111/jcmm.13841
Fu, Q., Xu, M., Chen, X., Sheng, X., Yuan, Z., Liu, Y., et al. (2017). CEP78 is mutated in a distinct type of Usher syndrome. J. Med. Genet. 54, 190–195. doi: 10.1136/jmedgenet-2016-104166
Hu, J., Li, B., Apisa, L., Yu, H., Entenman, S., Xu, M., et al. (2016a). ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23(erl/erl) mutant mice. Cell Death Dis. 24:e2485. doi: 10.1038/cddis.2016.386
Hu, J., Xu, M., Yuan, J., Li, B., Entenman, S., Yu, H., et al. (2016b). Tauroursodeoxycholic acid prevents hearing loss and hair cell death in Cdh23(erl/erl) mice. Neuroscience. 316, 311–320. doi: 10.1016/j.neuroscience.2015.12.050
Huang, J., Fu, J., Fu, S., Yang, L., Nie, K., Duan, C., et al. (2019). Diagnostic value of a combination of next-generation sequencing, chorioretinal imaging and metabolic analysis: lessons from a consanguineous Chinese family with gyrate atrophy of the choroid and retina stemming from a novel OAT variant. Br. J. Ophthalmol. Mar. 103, 428–435. doi: 10.1136/bjophthalmol-2018-312347
Imani, S., Cheng, J., Fu, J., Mobasher-Jannat, A., Wei, C., Mohazzab-Torabi, S., et al. (2019). Novel splicing variant c. 208+2T>C in BBS5 segregates with Bardet-Biedl syndrome in an Iranian family by targeted exome sequencing. Biosci. Rep. 39:BSR20.
Imani, S., Ijaz, I., Shasaltaneh, M. D., Fu, S., Cheng, J., and Fu, J. (2018). Molecular genetics characterization and homology modeling of the CHM gene mutation: a study on its association with choroideremia. Mut. Res. 775, 39–50. doi: 10.1016/j.mrrev.2018.02.001
Ivanova, M. E., Trubilin, V. N., Atarshchikov, D. S., Demchinsky, A. M., Strelnikov, V. V., Tanas, A. S., et al. (2018). Genetic screening of Russian Usher syndrome patients toward selection for gene therapy. Ophthalmic Genet. 39, 706–713. doi: 10.1080/13816810.2018.1532527
Jiang, L., Liang, X., Li, Y., Wang, J., Zaneveld, J. E., Wang, H., et al. (2015). Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J. Rare Dis. 10:110. doi: 10.1186/s13023-015-0329-3
Jouret, G., Poirsier, C., Spodenkiewicz, M., Jaquin, C., Gouy, E., Arndt, C., et al. (2019). Genetics of usher syndrome: new insights from a meta-analysis. Otol. Neurotol. 40, 121–129. doi: 10.1097/mao.0000000000002054
Keats, B. J., and Corey, D. P. (1999). The usher syndromes. Am. J. Med. Genet. 89, 158–166. doi: 10.1002/(sici)1096-8628(19990924)89:3<158::aid-ajmg6>3.0.co;2-#
Koenekoop, R. K., Wang, H., Majewski, J., Wang, X., Lopez, I., Ren, H., et al. (2012). Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 44, 1035–1039. doi: 10.1038/ng.2356
Maddalena, A., Tornabene, P., Tiberi, P., Minopoli, R., Manfredi, A., Mutarelli, M., et al. (2018). Triple vectors expand AAV transfer capacity in the retina. Mol. TherJ. Am. Soc. Gene Ther. 26, 524–541. doi: 10.1016/j.ymthe.2017.11.019
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. doi: 10.1093/nar/gkw1129
Mathur, P., and Yang, J. (2015). Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420. doi: 10.1016/j.bbadis.2014.11.020
Mianne, J., Chessum, L., Kumar, S., Aguilar, C., Codner, G., Hutchison, M., et al. (2016). Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med. 8:16. doi: 10.1186/s13073-016-0273-4
Okano, S., Makita, Y., Katada, A., Harabuchi, Y., Kohmoto, T., Naruto, T., et al. (2019). Novel compound heterozygous CDH23 variants in a patient with Usher syndrome type I. Hum. Genome Variation. 6:8. doi: 10.1038/s41439-019-0037-y
Rahbaran, M., Hassani Doabsari, M., Salavitabar, S., Mokhberian, N., Morovvati, Z., and Morovvati, S. (2019). A novel frameshift mutation in the EDA gene in an Iranian patient affected by X-linked hypohidrotic ectodermal dysplasia. Cell. Mol. Biol. Lett. 24:54. doi: 10.1186/s11658-019-0174-9
Santana, E. E., Fuster-Garcia, C., Aller, E., Jaijo, T., Garcia-Bohorquez, B., Garcia-Garcia, G., et al. (2019). Genetic Screening of the Usher Syndrome in Cuba. Front. Genet. 10:501. doi: 10.3389/fgene.2019.00501
Soens, Z. T., Branch, J., Wu, S., Yuan, Z., Li, Y., Li, H., et al. (2017). Leveraging splice-affecting variant predictors and a minigene validation system to identify Mendelian disease-causing variants among exon-captured variants of uncertain significance. Hum. Mutat. 38, 1521–1533. doi: 10.1002/humu.23294
Soens, Z. T., Li, Y., Zhao, L., Eblimit, A., Dharmat, R., Li, Y., et al. (2016). Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet Med. 18, 1044–1051. doi: 10.1038/gim.2015.205
Sun, T., Xu, K., Ren, Y., Xie, Y., Zhang, X., Tian, L., et al. (2018). Comprehensive molecular screening in chinese usher syndrome patients. Invest. Ophthalmol. Vis. Sci. 59, 1229–1237.
Wei, C., Yang, L., Cheng, J., Imani, S., Fu, S., Lv, H., et al. (2018). A novel homozygous variant of GPR98 causes usher syndrome type IIC in a consanguineous Chinese family by next generation sequencing. BMC Med. Genet. 19:99. doi: 10.1186/s12881-018-0602-0
Zhang, Q., Peng, C., Song, J., Zhang, Y., Chen, J., Song, Z., et al. (2017). Germline mutations in CDH23, encoding cadherin-related 23, are associated with both familial and sporadic pituitary adenomas. Am. J. Hum. Genet. 100, 817–823.
Keywords: Usher syndrome type ID, CDH23 gene, missense mutation, targeted next-generation sequencing, genotype/phenotype correlation
Citation: Zhang L, Cheng J, Zhou Q, Khan MA, Fu J, Duan C, Sun S, Lv H and Fu J (2020) Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the CDH23 Gene Causing Usher Syndrome Type ID in a Chinese Patient. Front. Genet. 11:422. doi: 10.3389/fgene.2020.00422
Received: 19 October 2019; Accepted: 03 April 2020;
Published: 30 April 2020.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Xianjun Zhu, University of Electronic Science and Technology of China, ChinaAsifullah Khan, Abdul Wali Khan University Mardan, Pakistan
Copyright © 2020 Zhang, Cheng, Zhou, Khan, Fu, Duan, Sun, Lv and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suan Sun, aGF5eXNzYUAxNjMuY29t; Hongbin Lv, b2N1bGlzdGx2aG9uZ2JpbkAxNjMuY29t; Junjiang Fu, ZnVqdW5qaWFuZ0Bzd211LmVkdS5jbg==; ZnVqdW5qaWFuZ0Bob3RtYWlsLmNvbQ==