 Ronghua Li1,2,3
Ronghua Li1,2,3 Weijia Zhang1Junkai Lu1,2Zhouyi Zhang1
Weijia Zhang1Junkai Lu1,2Zhouyi Zhang1 Changkao Mu1,2
Changkao Mu1,2 Weiwei Song1,2Herve Migaud1,2,3
Weiwei Song1,2Herve Migaud1,2,3 Chunlin Wang1,2*
Chunlin Wang1,2* Michaël Bekaert3*
Michaël Bekaert3*- 1Key Laboratory of Applied Marine Biotechnology, Ministry of Education, Ningbo University, Ningbo, China
- 2Collaborative Innovation Center for Zhejiang Marine High-Efficiency and Healthy Aquaculture, Ningbo University, Ningbo, China
- 3Faculty of Natural Sciences, Institute of Aquaculture, University of Stirling, Stirling, United Kingdom
The hard-shelled mussel (Mytilus coruscus) is an economically important shellfish that has been cultivated for the last decade. Due to over-exploitation, most mussel stocks have dramatically declined. Efforts to study this species' natural distribution, genetics, breeding, and cultivation have been hindered by the lack of a high-quality reference genome. To address this, we produced a hybrid high-quality reference genome of M. coruscus using a long-read platform to assemble the genome and short-read, high-quality technology to accurately correct for sequence errors. The genome was assembled into 10,484 scaffolds, a total length of 1.90 Gb, and a scaffold N50 of 898 kb. Ab initio annotation of the M. coruscus genome assembly identified a total of 42,684 genes. This accurate reference genome of M. coruscus provides an essential resource with the advantage of enabling the genome-scale selective breeding of M. coruscus. More importantly, it will also help in deciphering the speciation and local adaptation of the Mytilus species.
1. Introduction
The marine mussel Mytilus is among the foremost cosmopolitan marine genera and is present in estuarine and oceanic habitats, in both the subtidal and intertidal zones (Koehn, 1991). The global distribution of Mytilus species combined with certain features, such as partial reproductive isolation, which produces natural hybrids in areas of sympatry (Hilbish et al., 2000), doubly uniparental inheritance (DUI) of mitochondria (Zouros et al., 1994), very high bio-accumulation, and a low bio-transformation potential for both organic and inorganic contaminants (Smolders et al., 2003), make them attractive models for genetic, evolution, and ecological research. Besides, mussels are commercially important molluscs: the global production of farmed mussels reached 2,164,000 tons in 2017 (Food and Agricultural Organization, 2019).
Mytilus galloprovincialis was the first sequenced marine mussel, and, its genome provided valuable information for the research and sustainable management of this species (Murgarella et al., 2016). Mytilus coruscus Gould, 1861 (also recognized as Mytilus unguiculatus Valenciennes, 1858) is another important mussel species distributed along the coast of China (Ye et al., 2012), Korea (An and Lee, 2012), and Japan (Okutani, 2000). In addition to its ecological importance in the intertidal and subtidal communities, it is also a popular edible shellfish in many Asian countries. As a large-bodied mussel species, M. coruscus is valued for its high nutritional value and good commercial price (Zhang et al., 2020). It has also been reported that some of its lipids have anti-inflammatory properties (Fu et al., 2015). The mariculture of M. coruscus has been carried out in several regions of China in the past; the Shengsi Islands (Zhejiang province) are one of the oldest and largest culture areas in the Eastern China Sea and have an annual production in excess of 500,000 tons (Guo et al., 2017). The cultured juveniles primarily originate from natural populations. In the last few decades, natural juvenile stocks have decreased, while mussel farms have increased (Shen et al., 2009). Recently, breeding programs of M. coruscus have been initiated, mainly aiming to improve growth rates and disease resistance under aquaculture conditions. Despite its ecological and economic importance, genome information on this endemic mussel species is still lacking.
This report presents the first draft genome assembly for M. coruscus, performed using a hybrid assembly strategy. An Oxford Nanopore Technologies PromethION long-read platform was used to assemble the genome and Illumina HiSeq X Ten short-read, high-quality technologies were used to accurately correct for sequence errors. The resulting assembled genome sequence has 10,484 scaffolds, a total length of 1.90 Gb, GC content of 32.22%, and a scaffold N50 of 898 kb. Furthermore, we identified 1.01 Gb (52.83% of the assembly) of repeat content, 42,684 protein-coding genes, 278 rRNAs, and a high heterozygosity of 1.64%. This high-quality reference genome will serve as a substantial resource for future studies of basic genetics as well as genome-scale selective breeding programs for M. coruscus.
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
An adult female specimen of M. coruscus (133.73 g) was collected in November 2018 from the Shengsi Islands in Zhejiang province, China. Gills were dissected and stored in liquid nitrogen until DNA extraction. Genomic DNA was extracted as previously reported (Venier and Canova, 1996) with small modifications. Briefly, gill samples were digested with RNase and proteinase K, adjusted to 2% SDS, and heated at 60°C for 10 min. Sodium perchlorate was added, and DNA was extracted once by gently shaking with 24:1 chloroform:isoamyl alcohol for 30 min at room temperature. DNA was then precipitated with cold 75% ethanol and suspended in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 7.4). The quality and concentration of the extracted genomic DNA were checked using 0.3% agarose gel electrophoresis and a Qubit fluorimeter (Invitrogen, Carlsbad, CA, USA).
2.2. Library Construction and Sequencing
High-quality DNA was used for subsequent library preparation and sequencing using PromethION and Illumina platforms (Biomarker Technologies Corporation, Beijing, China). To obtain long non-fragmented sequence reads, ~15 μg of genomic DNA was sheared and size-selected (30–80 kb) with a BluePippin (Sage Science, Beverly, MA, USA). The selected fragments were processed using the Ligation Sequencing 1D Kit (Oxford Nanopore, Oxford, UK) according to the manufacturer's instructions and sequenced using the PromethION DNA sequencer (Oxford Nanopore, Oxford, UK) for 48 h. For the estimation and correction of genome assembly, an Illumina DNA paired-end (PE) library with an insert size of 350 bp was constructed in accordance with the manufacturer's protocol and sequenced with an Illumina HiSeq X Ten platform (Illumina, Inc., San Diego, CA, USA) with paired-end 150 read layout.
2.3. De novo Assembly
Reads from the two types of sequencing platforms were used at different assembly stages (Figure 1A). Long reads were filtered for length (>15,000 nt) and complexity (entropy over 15), while all short reads were filtered for quality (QC > 25), length (150 nt), absence of primers/adaptors, and complexity (entropy over 15) by using fastp (Chen et al., 2018). Using Jellyfish (Marçais and Kingsford, 2011), the frequency of 31-mers in the Illumina filtered data was calculated with a 1 bp sliding window (Vurture et al., 2017). Long reads were then assembled using wtdbg2 (Ruan and Li, 2020), which uses a fuzzy Bruijn graph. As it assembles raw reads without error correction and then builds the consensus from intermediate assembly output, multiple stages of error correction, gap closing, and polishing were applied. Original output was realigned against the long reads and polished using Minimap2 (Li, 2018) and Racon (Vaser et al., 2017), first with filtered reads to bridge potential gaps, and then with the filtered reads to correct for error. Finally, Pilon (Walker et al., 2014) was used to polish and correct for sequencing error using the short reads.
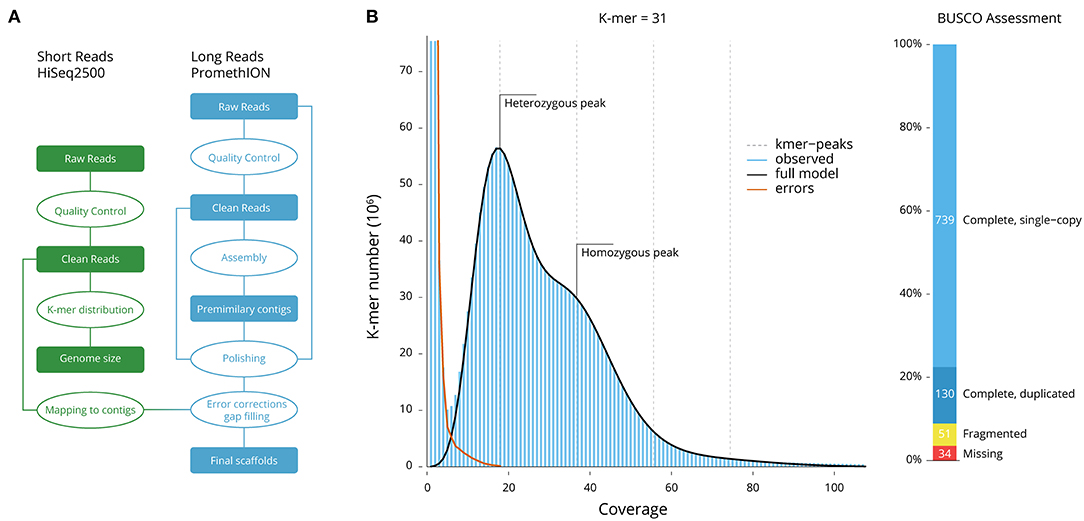
Figure 1. Genome size and quality estimations. (A) Genome assembly workflow. (B) The 31-mer distribution used for the estimation of genome size and heterozygosity. The heterozygous and homozygous peaks of k-mer depth are clearly markers, suggesting a high-complexity genome and high heterozygosity of 1.64%. (C) BUSCO assessment (Metazoa database; number of framework genes 954); 96.44% of the genes were recovered.
2.4. Gene Models
We used Braker (Hoff et al., 2019) to perform ab initio gene prediction, combining methods that integrate ab initio gene prediction and RNA-seq-based prediction to annotate the protein-coding genes in the M. coruscus genome. These raw RNA-seq reads were downloaded from the EBI for three independent transcriptomic projects, covering multiple tissues and multiple conditions: PRJNA301064 (Xu et al., 2016), PRJNA269003 & PRJNA269004. The resulting predictions were then filtered for the presence of at least one InterPro (Jones et al., 2014) pattern using InterProScan (Mitchell et al., 2019).
2.5. Repeat Sequences
The transposable elements were annotated using a de novo prediction by using RepeatModeler (Smit and Hubley, 2017) and LTR-Finder (Stanke et al., 2008). The repetitive sequences returned from these two algorithms were combined to compile a non-redundant repeat sequence library. With this library, we scanned the representative sequences in the M. coruscus genome using RepeatMasker (Smit et al., 2019).
2.6. Completeness
The completeness of gene regions was further assessed using BUSCO (Simão et al., 2015), using a Metazoa (release 10) benchmark of 954 conserved Metazoa genes.
2.7. Synteny With M. galloprovincialis
To assess the macro-synteny between M. coruscus and M. galloprovincialis (Murgarella et al., 2016) genomes, we reciprocally mapped all M. coruscus and M. galloprovincialis scaffolds with a minimum overlap of 100 nt.
2.8. Code Availability
The versions, settings, and parameters of the software used in this work are as follows:
Genome assembly: (1) fastp: version 0.20.0, short-read parameters: -q 25 -y -Y 15 -l 150 -detect_adapter_for_pe; (2) fastp: version 0.20.0, long-read parameters: -Q -y -Y 15 -l 15000; (3) wtdbg2: version 2.4, parameters: -x rs -k 23 -p 0 -AS 6 -R -g 1567m -rescue-low-cov-edges; (4) wtpoa-cns: version 2.4, default parameters; (5) minimap2: version 2.17, parameters: -x map-ont -r2k; (6) racon: version 1.4.3, default parameters; (7) bwa: version 0.7.17, mode mem, default parameters; (8) pilon: version 1.23, parameters: -diploid -fix all -changes; (9) BUSCO: version 4.0.2, parameters: -l metazoa_odb10; (10) RepeatModeler: version 1.0.11, parameters: -database mussel; (11) LTR_Finder: version 1.07, default parameters; (12) RepteatMasker: version 4.0.9, parameters: -lib mussel-families.fa; (13) Braker: version 2.1.4, parameters: -gff3 -softmasking; (14) InterProScan: version 5.42-78.0, parameters: -f tsv -dp -iprlookup -goterms.
K-mer analysis: (1) jellyfish: version 2.3.0, parameters: -m 31 -C -s 10G; (2) GenomeScope: version 2.0, default parameters.
Mitochondria annotation: (1) MITOS: revision 999, online version, parameters: “Genetic code 5”.
Synteny blocks: (1) nucmer: version 3.23, parameters: -simplify -maxgap = 500 -mincluster = 100; (2) circos: version 0.69-9, default parameters (tutorial 5.9).
3. Results and Discussion
3.1. Sequencing Results
After sequencing with the PromethION platform, a total of 11.31 million (161.04 Gb) long reads were generated, and these were used for the subsequent genome assembly. The N50 of the sequences produced was 21,771 nt. The Illumina HiSeq X Ten platform produces 288.22 million (86.47 Gb) paired-ended short reads (150 nt). The genome size of M. coruscus was estimated to have 2n = 28 chromosomes (Pérez-García et al., 2014) and a C-value of 1.90 pg (Ieyama et al., 1994) or 1.85 Gb; therefore, the average sequencing coverage was 87× and 46×, respectively (Table 1).

Table 1. Sequencing data statistics.
3.2. De novo Assembly of the M. coruscus Genome
The frequency of 31-mers in the Illumina filtered data was calculated using Jellyfish and followed a Poisson distribution (Figure 1B). The proportion of heterozygosity in the M. coruscus genome was evaluated to be 1.64%, and the genome size was estimated as 1.57 Gb, with a repeat content of 36.35% (Table 2). Long-read assembly using wtdbg2 (Ruan and Li, 2020), polished using Racon and sequence-corrected using short reads and Pilon, produced an assembled genome of M. coruscus containing 10,484 contigs with a total length and contig N50 of 1.90 Gb and 898 kb, respectively (Table 2).
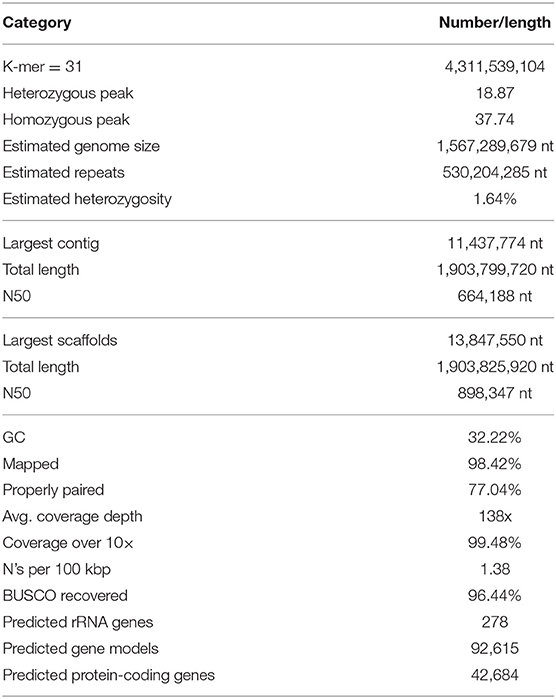
Table 2. Statistics of the genome assembly of Mytilus coruscus.
3.3. Repeat Sequences and Gene Models
The transposable elements and repetitive sequences were annotated using RepeatMasker and LTR-Finder. In total, 1.01 Gb (52.83%) of the genome was identified as repetitive sequences (Table 3).
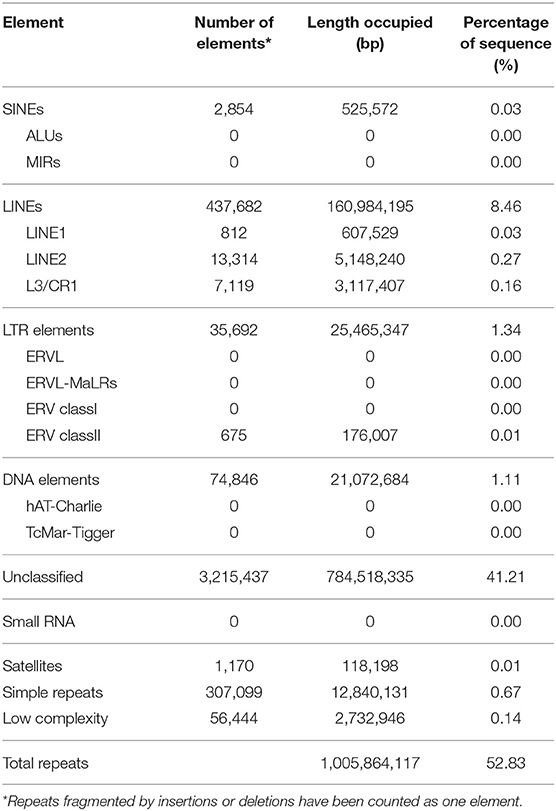
Table 3. RepeatMasker statistics.
We used a combined method that integrates ab initio gene prediction and RNA-seq-based prediction to annotate the protein-coding genes in the M. coruscus genome. In total, 42,684 distinct gene models were annotated.
3.4. Evaluating the Completeness of the Genome Assembly
To estimate the quality of the hybrid genome assembly, short reads were aligned to the consensus genome, and 98.42% did align overall, suggesting that our assembly results contained comprehensive genomic information.
The completeness of the gene models was also assessed using BUSCO (Simão et al., 2015) using a Metazoa (release 10) benchmark of 954 conserved Metazoa genes: 91.09% had complete gene coverage (including 13.63% duplicated ones), 5.35% were fragmented, and only 3.56% were missing (Figure 1C). This largely supports a high-quality M. coruscus genome assembly and gene models.
3.5. Synteny With M. galloprovincialis
To uncover the macro-synteny between the M. coruscus and M. galloprovincialis (Murgarella et al., 2016) genomes, we reciprocally mapped all M. coruscus and M. galloprovincialis scaffolds with a minimum overlap of 100 nt (Table 4). A total of 28.12% of M. galloprovincialis scaffolds mapped to a M. coruscus scaffold while 70.25% M. coruscus scaffolds mapped to a M. galloprovincialis scaffold. Figure 2A reports the synteny blocks of over 3,000 nt; the map shows a good synteny between the two genomes despite M. galloprovincialis genome fragmentation.

Table 4. Comparison between Mytilus spp. assemblies.
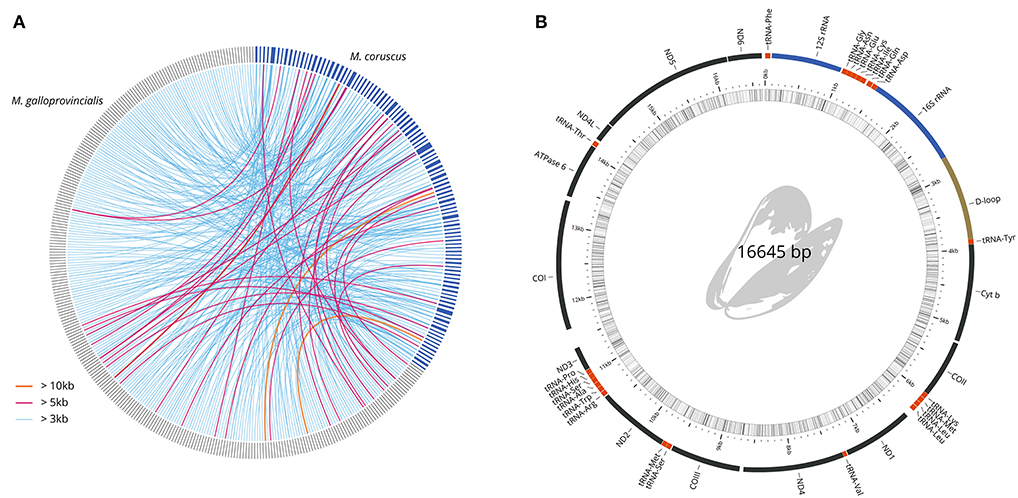
Figure 2. Genome comparisons. (A) Circos Krzywinski et al. (2009) mapping of longest synteny blocks between M. galloprovincialis (287 dark gray scaffolds) and M. coruscus (104 blue scaffolds). (B) M. coruscus annotated mitochondrial genome.
3.6. Mitochondrial Genome
The mitochondrial genome was manually recovered from the genome assembly. The contig was validated for continuity and circularity and annotated using MITOS (Bernt et al., 2013). The complete mitochondrial genome (Figure 2B) was compared to the reference M. coruscus genome (Lee and Lee, 2016). Only one haplotype was recovered, which differed from the reference by only 6 SNPs and one 2-nucleotide insertion.
4. Conclusion
This study is the first to present a high-quality genome sequence assembly of the hard-shelled mussel M. coruscus. We generated a hybrid genome assembly of 1.90 Gb with an N50 of 898 kb. The assembled genome was predicted to contain 42,684 protein-coding genes, 278 rRNAs, 1.01 Gb (52.83%) of repetitive elements, and high heterozygosity of 1.64%. We also recovered and annotated the whole circular mitochondrial genome of 16.65 kb lacking atp8 gene. This high-quality reference genome will serve as a substantial resource for future studies of basic genetics as well as genome-scale selective breeding programs for M. coruscus.
Data Availability Statement
The raw sequencing reads of all libraries are available from EBI/ENA via the accession numbers ERR3415816 and ERR3431204. The assembled genomes are available in EBI with the accession numbers ERZ1292486 (nuclear genome) and ERZ1195933 (mitochondrial genome), Project PRJEB33342.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use committee at the School of Marine Sciences, Ningbo University.
Author Contributions
RL, MB, and CW conceived, initialized, and guided the entire project. JL, ZZ, CM, and WS collected the sample. WZ, JL, and ZZ prepared the sample and performed the genome sequencing. MB performed the data processing and genome, and gene model analysis. RL and MB drafted the manuscript. HM helped with the manuscript preparation. All authors read and approved the final manuscript.
Funding
This work was supported by the grants from the Science and Technology correspondent of Zhejiang Province, the United Kingdom Biotechnology and Biological Sciences Research Council China—UK Partnering Award (BB/S020357/1), CSC Scholarship (201708330421), and the K. C. Wong Magana Fund at Ningbo University. The funders had no role in study design, data collection, and analyses, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
An, H. S., and Lee, J. W. (2012). Development of microsatellite markers for the korean mussel, Mytilus coruscus (mytilidae) using next-generation sequencing. Int. J. Mol. Sci. 13, 10583–10593. doi: 10.3390/ijms130810583
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). Mitos: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one fastq preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Food and Agricultural Organization (FAO) (2019). FAO Yearbook. Fishery and Aquaculture Statistics 2017. Aquaculture Production. Rome: FAO.
Fu, Y., Li, G., Zhang, X., Xing, G., Hu, X., Yang, L., et al. (2015). Lipid extract from hard-shelled mussel (Mytilus coruscus) improves clinical conditions of patients with rheumatoid arthritis: a randomized controlled trial. Nutrients 7, 625–645. doi: 10.3390/nu7010625
Guo, Y., Zhao, W., Gao, H., Wang, S., Yu, P., Yu, H., et al. (2017). China Fishery Statistical Yearbook. Beijing: Bureau of Fisheries and Fishery Management, China Agriculture Press.
Hilbish, T. J., Mullinax, A., Dolven, S. I., Meyer, A., Koehn, R. K., and Rawson, P. D. (2000). Origin of the antitropical distribution pattern in marine mussels (Mytilus spp.): routes and timing of transequatorial migration. Mar. Biol. 136, 69–77. doi: 10.1007/s002270050010
Hoff, K. J., Lomsadze, A., Borodovsky, M., and Stanke, M. (2019). “Whole-genome annotation with braker,” in Gene Prediction: Methods and Protocols, ed M. Kollmar (New York, NY: Springer New York), 65–95. doi: 10.1007/978-1-4939-9173-0_5
Ieyama, H., Kameoka, O., Tan, T., and Yamasaki, J. (1994). Chromosomes and nuclear dna contents of some species in mytilidae. Venus 53, 327–331.
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). Interproscan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Koehn, R. K. (1991). The genetics and taxonomy of species in the genus >Mytilus. Aquaculture 94, 125–145. doi: 10.1016/0044-8486(91)90114-M
Krzywinski, M. I., Schein, J. E., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lee, Y.-C., and Lee, Y.-H. (2016). The f type mitochondrial genome of hard-shelled mussel: Mytilus coruscus (mytiloida, mytilidae). Mitochondr. DNA Part A 27, 624–625. doi: 10.3109/19401736.2014.908375
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2019). Interpro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360. doi: 10.1093/nar/gky1100
Murgarella, M., Puiu, D., Novoa, B., Figueras, A., Posada, D., and Canchaya, C. (2016). A first insight into the genome of the filter-feeder mussel Mytilus galloprovincialis. PLoS ONE 11:e0151561. doi: 10.1371/journal.pone.0151561
Pérez-García, C., Morán, P., and Pasantes, J. J. (2014). Karyotypic diversification in Mytilus mussels (bivalvia: Mytilidae) inferred from chromosomal mapping of rrna and histone gene clusters. BMC Genet. 15:84. doi: 10.1186/1471-2156-15-84
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1101/530972
Shen, Y.-B., Li, J.-L., and Feng, B.-B. (2009). Genetic analysis of cultured and wild populations of Mytilus coruscus based on mitochondrial dna. Zool. Res. 30, 240–246. doi: 10.3724/SP.J.1141.2009.03240
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). Busco: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smit, A. F. A., and Hubley, R. (2017). RepeatModeler. Available online at: http://www.repeatmasker.org/RepeatModeler/
Smit, A. F. A., Hubley, R., and Green, P. (2019). RepeatMasker. Available online at: http://www.repeatmasker.org
Smolders, R., Bervoets, L., Wepener, V., and Blust, R. (2003). A conceptual framework for using mussels as biomonitors in whole effluent toxicity. Hum. Ecol. Risk Assess. Int. J. 9, 741–760. doi: 10.1080/713609965
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cdna alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Venier, P., and Canova, S. (1996). Formation of dna adducts in the gill tissue of Mytilus galloprovincialis treated with benzo[a]pyrene. Aquat. Toxicol. 34, 119–133. doi: 10.1016/0166-445X(95)00035-3
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). Genomescope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Xu, M., Jiang, L., Shen, K.-N., Wu, C., He, G., and Hsiao, C.-D. (2016). Transcriptome response to copper heavy metal stress in hard-shelled mussel (Mytilus coruscus). Genom. Data 7, 152–154. doi: 10.1016/j.gdata.2015.12.010
Ye, Y.-Y., Li, J.-J., Wu, C.-W., Xu, M.-Y., and Guo, B.-Y. (2012). Genetic analysis of mussel (Mytilus coruscus) populations on the coast of east china sea revealed by issr-pcr markers. Biochem. Syst. Ecol. 45, 1–6. doi: 10.1016/j.bse.2012.07.022
Zhang, W., Li, R., Chen, X., Wang, C., Gu, Z., Mu, C., et al. (2020). Molecular identification reveals hybrids of Mytilus coruscus × Mytilus galloprovincialis in mussel hatcheries of china. Aquacult. Int. 28, 85–93. doi: 10.1007/s10499-019-00445-8
Keywords: Mytilus coruscus, hard-shelled mussel, sequencing, genome assembly and annotation, mitochondria, syntheny
Citation: Li R, Zhang W, Lu J, Zhang Z, Mu C, Song W, Migaud H, Wang C and Bekaert M (2020) The Whole-Genome Sequencing and Hybrid Assembly of Mytilus coruscus. Front. Genet. 11:440. doi: 10.3389/fgene.2020.00440
Received: 18 February 2020; Accepted: 09 April 2020;
Published: 08 May 2020.
Edited by:
Peng Xu, Xiamen University, ChinaReviewed by:
Chao Bian, Beijing Genomics Institute (BGI), ChinaJian Xu, Chinese Academy of Fishery Sciences (CAFS), China
Copyright © 2020 Li, Zhang, Lu, Zhang, Mu, Song, Migaud, Wang and Bekaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunlin Wang, d2FuZ2NodW5saW5AbmJ1LmVkdS5jbg==; Michaël Bekaert, bWljaGFlbC5iZWthZXJ0QHN0aXIuYWMudWs=