 Manman Shen1,2,3
Manman Shen1,2,3 Tingting Li1Fuxiang Chen1Pengfeng Wu1Ying Wang1Lan Chen1Kaizhou Xie1Jinyu Wang1*Genxi Zhang1*
Tingting Li1Fuxiang Chen1Pengfeng Wu1Ying Wang1Lan Chen1Kaizhou Xie1Jinyu Wang1*Genxi Zhang1*- 1College of Animal Science and Technology, Yangzhou University, Yangzhou, China
- 2Jiangsu Key Laboratory of Sericultural Biology and Biotechnology, College of Biotechnology, Jiangsu University of Science and Technology, Zhenjiang, China
- 3Jiangsu Institute of Poultry Science, Yangzhou, China
Follicular development plays a key role in poultry reproduction, affecting clutch traits and thus egg production. Follicular growth is determined by granulosa cells (GCs), theca cells (TCs), and oocyte at the transcription, translation, and secretion levels. With the development of bioinformatic and experimental techniques, non-coding RNAs have been shown to participate in many life events. In this study, we investigated the transcriptomes of GCs and TCs in three different physiological stages: small yellow follicle (SYF), smallest hierarchical follicle (F6), and largest hierarchical follicle (F1) stages. A differential expression (DE) analysis, weighted gene co-expression network analysis (WGCNA), and bioinformatic analyses were performed. A total of 18,016 novel circular RNAs (circRNAs) were detected in GCs and TCs, 8127 of which were abundantly expressed in both cell types. and more circRNAs were differentially expressed between GCs and TCs than mRNAs. Enrichment analysis showed that the DE transcripts were mainly involved in cell growth, proliferation, differentiation, and apoptosis. In the WGCNA analysis, we identified six specific modules that were related to the different cell types in different stages of development. A series of central hub genes, including MAPK1, CITED4, SOD2, STC1, MOS, GDF9, MDH1, CAPN2, and novel_circ0004730, were incorporated into a Cytoscape network. Notably, using both DE analysis and WGCNA, ESR1 was identified as a key gene during follicular development. Our results provide valuable information on the circRNAs involved in follicle development and identify potential genes for further research to determine their roles in the regulation of different biological processes during follicle growth.
Introduction
Follicle development is a complex biological process involving a series of events, such as follicle recruitment, follicle selection, and oviposition. The process of follicle development extends from the primordial follicles to the establishment of a well-organized follicular hierarchy and is an important factor in the maintenance of high reproductive efficiency in hens. An intact hen follicle consists of one oocyte and two main types of somatic cells, theca cells (TCs) and granulosa cells (GCs). Abundant blood vessels surround or penetrate the basement membrane and the granulosa layer (Yoshimura and Koga, 1982; Wulff et al., 2001). GCs have been extensively studied because they are easily obtained and cultured in vitro (Johnson, 2015). Research has shown that GCs in the granulosa layer are responsive to follicle-stimulating hormone (FSH) during follicle selection (Hocking, 2009), and are the primary sites of progesterone secretion. In preovulatory follicles, GCs are involved in yolk deposition (Kim et al., 2016). Several genes, such as BMP15 (Elis et al., 2007), GDF9 (Paradis et al., 2009), and STAR (Zhu et al., 2015), have been shown experimentally to play important roles in the proliferation and differentiation of follicles, and the regulation of the genes that encode them involves multiple signaling pathways, including the steroidogenic pathway (Lee et al., 1998), transforming growth factor β (TGF-β) signaling pathway (Zhu et al., 2014), and oocyte meiosis (Peng et al., 2018).
With the development of molecular biological techniques, TCs have been widely studied in vivo and vitro (Kang et al., 2017; Wang and Gong, 2017). Previous reports have shown that the main function of TCs in birds is the production of androgens and estrogens (Bahr, 1991; Lee et al., 1998), which differs from the situation in mammals. Molecules such as CYP19A1 (Wang and Gong, 2017) and the hormone prostaglandin (Jia et al., 2010) promote the proliferation of TCs. The pathways involved in TC growth include the GPCR/cAMP signaling pathway and lipid/amino acid metabolism pathways in the horse (Donadeu et al., 2014) and the angiogenesis pathway in primates (Wulff et al., 2001).
Although GCs and TCs play different roles in follicle development, their involvement is interactive. Follicular atresia, the main process by which follicles are lost, is not only associated with GCs (Kim and Johnson, 2018) but also with TCs, albeit less strongly (Lebedev et al., 2006), and with the inadequate penetration of blood vessels among TCs (Kim et al., 2016).
An increased number of studies have demonstrated that transcriptional regulation is modulated by both protein coding RNAs and non-coding RNAs. Protein-coding RNAs play key roles in life processes, whereas non-coding RNAs can act in mediation roles during the expression of protein-coding RNAs. Non-coding RNAs have recently emerged as integral components of folliculogenesis (Cheng et al., 2017; Kang et al., 2017; Peng et al., 2018). Circular RNA (circRNA), single-stranded RNA which unlike the better known linear RNA, forms a covalently closed continuous loop, has become a research hotspot since Salzman et al. (2012) identified numerous circRNAs in 2012. Structure feature of circRNA display stability (Li et al., 2017) that are not easily degraded by RNase R (Alvaro Mercadal et al., 2015; Lu et al., 2015). They are also conserved across species (Jeck et al., 2013). The expression levels of circRNAs are tissue- and developmental-stage specific (Xu et al., 2017), and they are potential biomarkers of cancers, with diagnostic implications (Shang et al., 2019). With technical advances, thousands of circRNAs have been identified in the liver and embryonic muscle of chickens (Ouyang et al., 2017; Zhang et al., 2017; Chen et al., 2019), but there is only limited information on the circRNAs involved in follicle development, especially in chickens. However, the transcriptomic profiles of circRNAs in the follicles and ovary have been reported in mice (Jia et al., 2018), humans (Cheng et al., 2017; Cai et al., 2018), goats (Tao et al., 2017), and pigs (Guo et al., 2019). Research on mice has shown that estrogen signaling is upregulated in adult ovaries relative to that in neonatal ovaries (Jia et al., 2018). A study of ovarian senescence in humans showed that the differentially expressed circRNAs in aging ovaries compared with young ovaries were enriched in the steroid hormone biosynthesis and insulin secretion pathways, and that circDDX10 may be a sponge for microRNA miR-1301-3p or miR-4660 in modulating the expression of the SIRT3 gene, which may be associated with ovarian function (Cai et al., 2018). Another study of human GCs reported that circRNAs expressed during maternal aging may participate in glucose metabolism, ovarian steroidogenesis, etc. (Cheng et al., 2017). Quan and Li (2018) hypothesized that ciRS-7 influences oocyte maturation by regulating epidermal growth factor receptor (EGFR) expression. A study in goats showed that the target genes of circRNAs derived from the follicle may be involved in the GnRH signaling pathway, progesterone-mediated oocyte maturation, and the FoxO signaling pathway (Tao et al., 2017). The results of these studies suggest that circRNAs play functional roles in the ovary. However, there is only limited information on the roles of circRNAs during follicle development in chickens, which is a useful model in which to investigate reproductive diseases and follicular development (Bahr, 1991; Mellouk et al., 2018).
Although follicle development is a complex process involving multiples genes and interactions between GCs and TCs, no follicle-derived circRNAs that control its growth have been identified. In this study, we investigated the functions of circRNAs and mRNAs and their interactions in folliculogenesis.
Materials and Methods
Ethics Statement
All procedures involving birds were approved by the Animal Care Committee of Yangzhou University, Yangzhou, China (no. SYXK [Su] 2012-0029) and were conducted in accordance with the guidelines of the Chinese Ministry of Science and Technology. The birds were humanely killed as necessary and all animal suffering was minimized.
Birds and Sample Collection
Jinghai Yellow chickens used in present study were sixteen parental generation population from Jiangsu Jinghai Industry Poultry Group Co, Ltd (Nantong, Jiangsu, China). The birds in this Company had been purified of Salmonella and Avian leukosis for more than 5 years. The positive rate was none in the present populations. Routine immunization procedures were used throughout the whole life of all the chickens. All hens were reared under natural light and were transferred to single cages from 16 weeks old, with 10% restriction of food and free access to water. Light was increased by 1 h per week until 16 h of light was provided. The laying nutrient levels content 11.3–11.92 MJ/kg metabolizable energy, 15.0–16.0% crude protein, 3.35% Ca and 0.45% P. Egg reproduction and body weight were recorded during the egg-laying period. The mean egg laying rate and the body weight of the birds used was 82.3% and 1983 ± 125 g, respectively. At 27 weeks of age, the birds sharing half-sib relationships were used for follicle collection. The hens with laying sequences of 5 were killed with 60–70% carbon dioxide, and the ovaries of each bird were immediately removed. The GCs and TCs were separated from three birds with eggs in their oviducts which were in three different physiological stages: (small yellow follicle [SYF], smallest hierarchical follicle [F6], and largest hierarchical follicle [F1] stages), according to the methods described by Gilbert and Eresheim (Gilbert et al., 1977; Eresheim et al., 2014). SYF which has a diameter about 4–8 mm is a stage of yolk accumulation beginning, with a yellowish appearance. The hierarchical follicles are numbered from the largest (F1, with a diameter about 40 mm) to the smallest (F6, with a diameter 9–12 mm), where F1 will be the next to ovulate (Johnson, 1990). F6 is a result of follicle selection from SYF, and F1 is a result of rapid proliferation from F6 after about 6 days. The tissues were placed immediately in liquid nitrogen and stored at −70°C.
RNA Library Preparation
Total RNA was extracted with TRIzol Reagent (Invitrogen, Carlsbad, CA, United States), according to the manufacturer’s instructions. RNA contamination was checked on 1% agarose gels. The purity and integrity of the RNA were measured with a NanoPhotometer (Implen, United States) and an Agilent 6000 Nano Kit (Agilent Technologies, United States). The RNA concentrations were determined with a Qubit RNA Assay Kit in a Qubit 2.0 Fluorometer (Life Technologies, United States).
The total RNA from each tissue, with an RNA integrity number (RIN) > 8.5, was used as the input material for the RNA sample preparation. All total RNA samples were treated with the Epicentre Ribo-ZeroTM rRNA Removal Kit (Epicentre, Madison, WI, United States) to remove rRNA. Moreover, libraries of previously identified circRNAs were also digested with 3 U of RNase R (Epicentre). A total of 36 RNA libraries were constructed with the NEBNext® UltraTM Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, United States), according to the manufacturer’s instructions. The index-coded samples were then clustered with the cBot cluster generation system using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, United States). Finally, the libraries were sequenced on an Illumina HiSeq 4000 platform and 150-bp paired-end reads were produced. RNA-seq raw sequence reads from the follicle samples in current study can be accessed from PRJNA481176 and PRJNA511712 BioProject, available at the NCBI SRA repository (Supplementary Table S3E).
Identification of Transcripts
We filtered the raw reads in the fastq format to remove those reads containing adapter, poly-N, or low-quality sequences. The Q30 scores and G + C contents of the clean data were calculated. For the genome-wide identification of circRNA transcripts, we aligned the clean paired reads to the reference genome Galgal 5.0 using Bowtie (Langmead and Salzberg, 2012). Two methods, find_circ (Memczak et al., 2013) and CIRI2 (Gao et al., 2017), were used to detect and identify circRNAs. Based on the features of circRNAs, we have summarized the characteristics of circRNAs in the chicken follicle. The features of circRNAs were determined based on their sequences.
To annotate the protein-coding transcripts, the clean reads were aligned to Galgal 5.0 with HISAT2 (Kim et al., 2019) using the parameter “—rna-strandness RF” and other default settings. The mapped reads were assembled with StringTie (v1.3.1) using a reference-based method (Pertea et al., 2016).
Differential Expression Analysis
The raw counts of circRNAs were normalized as transcripts per million (TPM) with the following criteria (Zhou et al., 2010). To identify circRNAs differentially expressed in TCs and GCs, an analysis based on the negative binomial distribution was performed with the DESeq R package (1.10.1) (Love et al., 2014). The expression of protein-coding transcripts was calculated as the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) with Cuffdiff (v2.1.1) (Trapnell et al., 2010). The differentially expressed circRNAs were detected with TPM. All transcripts with fold changes of >2 and P-adjusted < 0.05 were deemed to be differentially expressed. To study the relationships between the circRNAs and the corresponding host protein-coding genes, we analyzed this fold change in expression in GCs relative to TCs in different follicles.
Co-expression of Transcripts
A weighted gene co-expression network analysis (WGCNA) (Langfelder and Horvath, 2008) was used to assess the potential functions of transcripts expressed in the granulosa layer or thecal layer during follicle development. Briefly, genes with the top 25% variance of expression values among samples were included (which included 7217 protein coding RNAs and 4504 circRNAs) as the input matrix for the WGCNA analysis. The best power was set to 12 to balance the scale-free property of the co-expression network (Supplementary Table S1). The cutreeDynamamic function was used with the parameters minModuleSize = 30, deepSplit = 2, pamRespectsDendro = F, and cutHeight = 0.3 to merge these modules, whose expression profiles were very similar. Gene significance (GS) was defined as the absolute value of the correlation between the gene and the differentiation stage. Module membership (MM) was defined as the correlation between the module eigengene and the gene expression profile. Hub genes were filtered with GS > 0.4 and MM > 0.8, and a bioinformatic analysis was performed in KOBAS (Xie et al., 2011). The top 200 connects based on the hub genes ranked by weighted values were visualized with Cytoscape (v3.6.1) (Shannon et al., 2003).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment
A GO enrichment analysis of the differentially expressed genes, the host genes of the circRNAs, and the hub genes was performed at the KOBAS. The GO terms or KEGG pathways with P-values < 0.05 or the top 20 terms are shown in the figures and tables.
Validation of Transcripts
The splice sites of the circRNAs were first confirmed with divergent primers (Jeck et al., 2013) (Supplementary Table S2), which were designed with the “out-facing” strategy to exclude linear mRNAs by RNase R (Epicentre, NC, United States). The reverse transcription reaction was carried out with PrimeScriptTM RT Master Mix (Takara, Dalian, China), and the PCR products were tested with agarose gel electrophoresis. The PCR products were sequenced with Sanger sequencing at Sango Biotech Co. Ltd (Shanghai, China). Divergent primers and exon–exon-spanning primers were then used to confirm the expression levels of the circRNAs and their corresponding host genes on an ABI 7500 thermocycler (Life Technologies, United States). The expression levels of the transcripts were quantified with AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, China), in a final volume of 20 μl, according to the user manual. Each reaction was performed in triplicate under the same experimental conditions: 95°C for 30 s and 40 cycles of 95°C for 5 s and 60°C for 34 s. The 2–ΔΔCt method was used to calculate the gene expression levels, with β-actin as the reference gene.
Results
Summary of Transcriptome Profiles
The quality control parameters for the RNA-seq data are shown in Supplementary Table S3. A total of 864,689,266 and 828,121,626 raw reads of circRNA were obtained from the GCs and TCs, respectively. A total of 972,646,432 and 831,515,782 raw reads of linear RNA were obtained from the GCs and TCs, respectively. The percentages of reads that mapped to the chicken reference genome (Gallus-gallus-5.0/galGal5) were all >92%; the error rates were all <0.03%; and the Q30 scores were all >90%. Interestingly, the G + C contents of the circRNAs identified in the GCs or TCs were all >60%, whereas those of the linear RNAs were around 47%. The G + C contents in the TCs were higher than those in the GCs for both the circRNAs and linear RNAs.
A total of 18,016 novel circRNAs were detected in the GCs and TCs, 8127 (with red font in Supplementary Table S4) of which were abundant in both cell types (Figure 1A and Supplementary Table S4). A total of 25,781 mRNAs were identified in the GCs and TCs, 23,769 of which were detected in both cell types (Figure 1B). The distribution of circRNAs on chromosomes 1–28 and the Z and W chromosomes were analyzed, and showed that the longer the chromosome, the greater the percentage of circRNAs. Interestingly, the percentage distribution was lowest on chromosome 16 (Figure 1C).
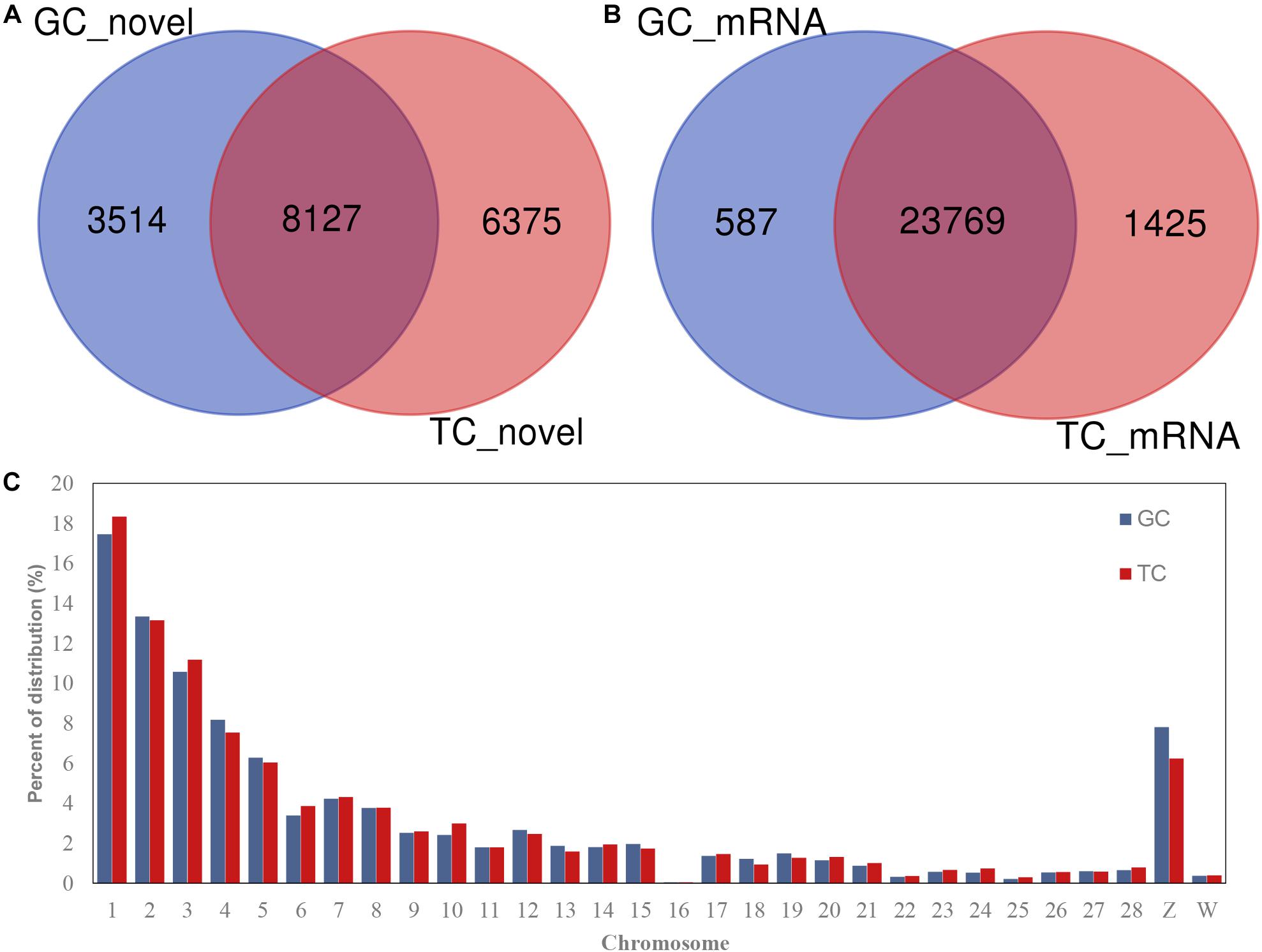
Figure 1. circRNAs and mRNAs detected in GCs or TCs. (A) Novel circRNAs detected in GCs and TCs; (B) mRNA identified in GCs and TCs; (C) circRNA distribution on chromosome. GCs, granulosa cells; TCs, theca cells.
Of the linear reads, ∼88.3% of the paired reads mapped to unique sequences in the chicken genome. Both read1 and read2 mapped to 44% of the reference genome. The numbers of reads that mapped to the “+” strand or “−” strand were nearly the same. The number of reads that mapped to splice sites was higher in the F1 follicles than in the F6 or SYF follicles (Supplementary Table S5a).
The features of the circRNAs were analyzed to clarify their roles in the follicle production process. The features of the circRNAs derived from GCs and TCs were almost the same in Supplementary Table S5. The source of the circRNAs, either coding sequences or intronic regions, was close to 40% (Supplementary Table S5b). The lengths of the circRNAs were predominantly 100–500 nt, with most in the range of 200–400 nt (Supplementary Table S5c). Almost a third of exonic-type circRNAs (38.44 and 40.39% in GCs and TCs, respectively) involved two exons, and about 27% of exonic-type circRNAs involved three exons. In brief, most exonic type circRNAs involved 1–5 exons (Supplementary Table S5d), with host gene lengths of >8000 kb (Supplementary Table S5e) and were separated by genomic distance of <10 kb (Supplementary Table S5f). The circularization of circRNA is affected by the length of the flanking intron, and our data show that the most frequent flank length (more than 40%) was 104 kb (Supplementary Tables S5g–i).
Differentially Expressed Transcripts
The numbers of differentially expressed (DE) transcripts are listed in Table 1, which shows that the number of DE circRNAs exceeded the number of DE mRNAs, and that DE mRNAs were only detected in the SYFG vs. SYFT comparison. A total of 625 DE circRNAs were identified in SYFG relative to SYFT, 271 of which were upregulated and 354 downregulated, whereas 145 DE mRNAs were detected in this comparison, with 37 regulated and 108 downregulated. In the F6 development stage, a total of 806 circRNAs were differentially expressed in GCs and TCs, but only five mRNAs were differentially expressed. Most DE circRNAs were detected in F1G vs. F1T comparison, 614 of which were upregulated and 828 downregulated.
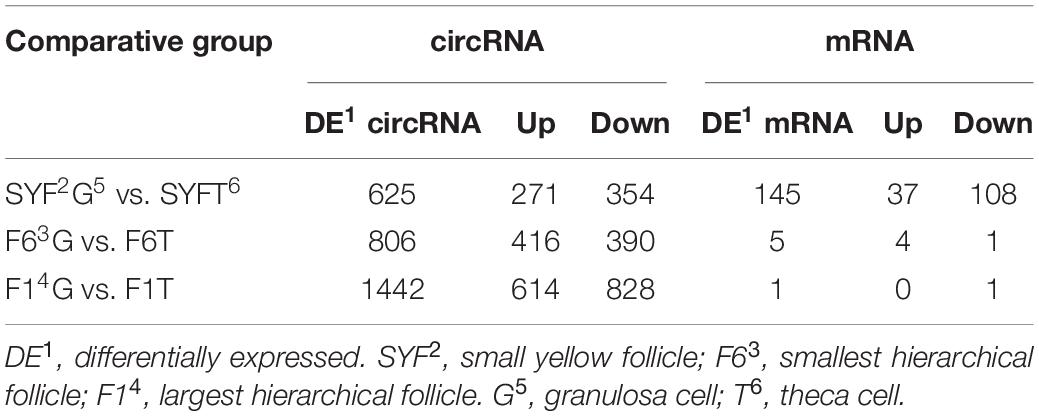
Table 1. Numbers of differentially expressed transcripts in different comparisons of RNA-Seq experiments.
The top DE circRNAs in the GCs and TCs are shown in Tables 2, 3, respectively. About 8/10 and 7/10 circRNAs were derived from protein-coding genes in the GCs and TCs, respectively. Host genes of DE circRNAs in the GCs included SSPBP2, IGF1R, TOX2, and USP22, whereas in the TCs, they included IL1R1, MINDY4, FGFR2, and CRIIM1. Notably, novel_circ_0002934 produced from the RAB11A gene was expressed at high levels in both GCs and TCs. A total of 2027 DE circRNAs were identified, 207 of which were abundantly expressed in the GCs and TCs at all three follicular stages (Figure 2A). However, their expression levels showed opposite trends in GCs and TCs (Figure 2B): in GCs, the expression levels were first clustered in two groups, SYFG and F6G, and then in F1G, whereas in TCs, the circRNAs were first clustered in F6T and F1T.
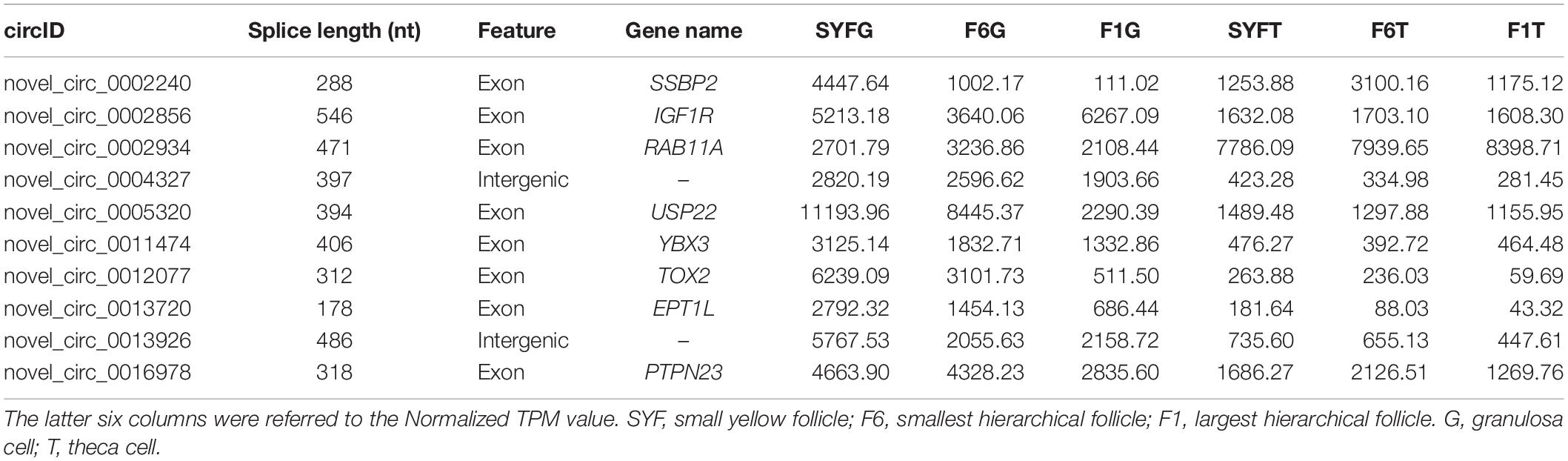
Table 2. Top 10 differentially expressed circRNAs in granulosa cells.
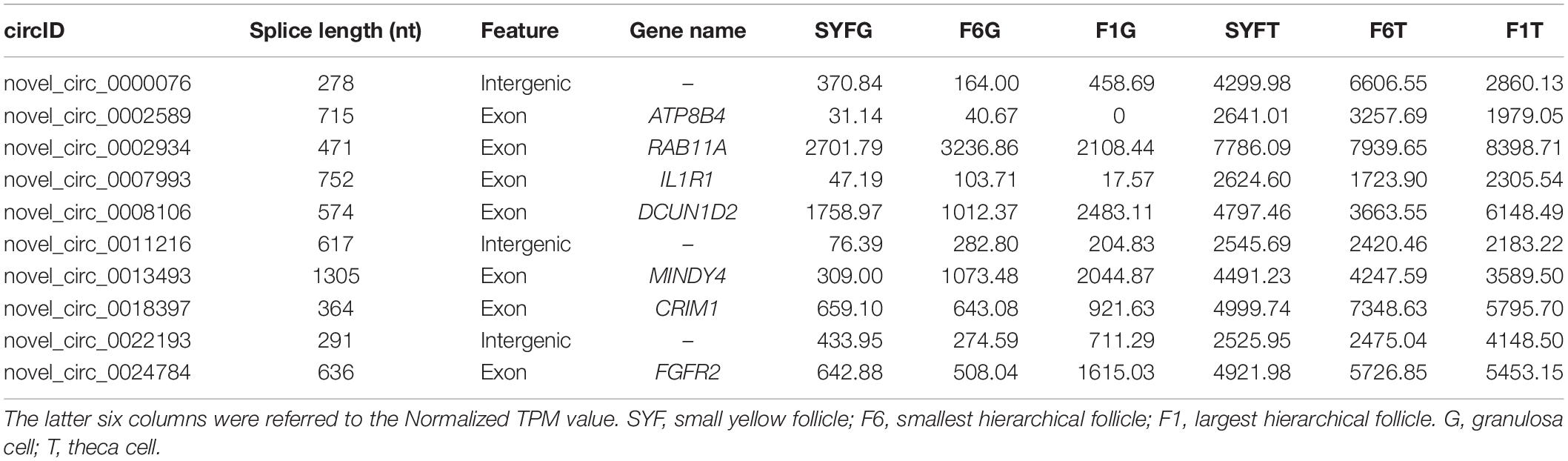
Table 3. Top 10 differentially expressed circRNAs in theca cells.
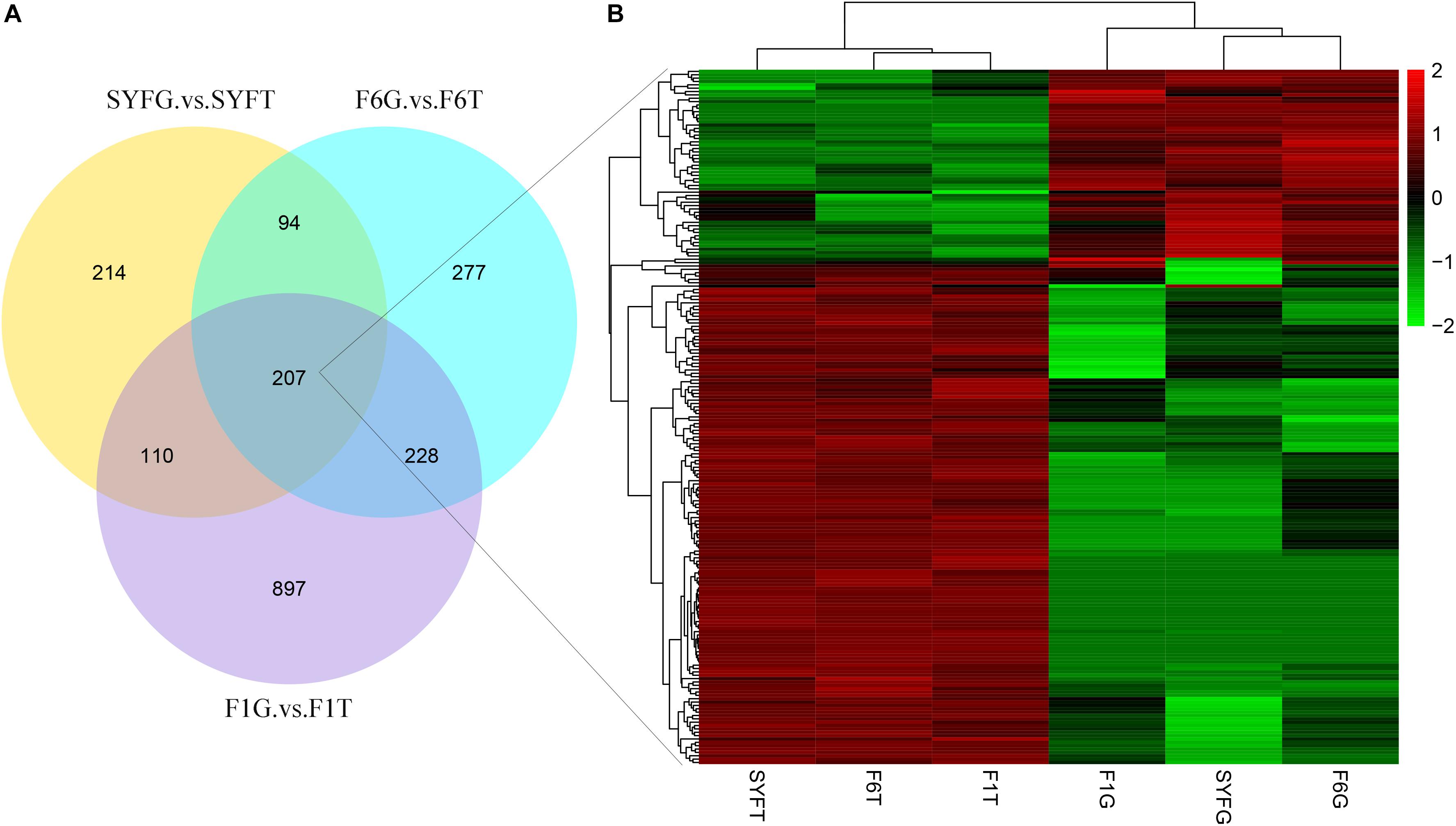
Figure 2. Differentially expressed circRNAs in venn diagram and heatmap. (A) Venn diagram of differentially expressed circRNAs; (B) heatmap of 207 pervasive circRNAs in three follicular development stage. SYF, small yellow follicle; F6, the smallest hierarchical follicle; F1, the largest hierarchical follicle. GCs, granulosa cells; TCs, theca cells.
Many mRNAs were differentially expressed in the SYF stage, so the relationship between circRNA and mRNA expression was investigated in terms of the fold changes and the overlap with host gene expression (Figure 3). The results showed that the fold changes in circRNA and mRNA expression were irregular, with the fold changes in the circRNAs greater, smaller, or the same as those in the mRNAs (Figure 3A). The expression of only nine genes overlapped with the expression of the host genes of the DE circRNAs and mRNAs in the SYFG vs. SYFT comparison (Figure 3B). Detailed information on the overlapping genes is given in Supplementary Table S6.
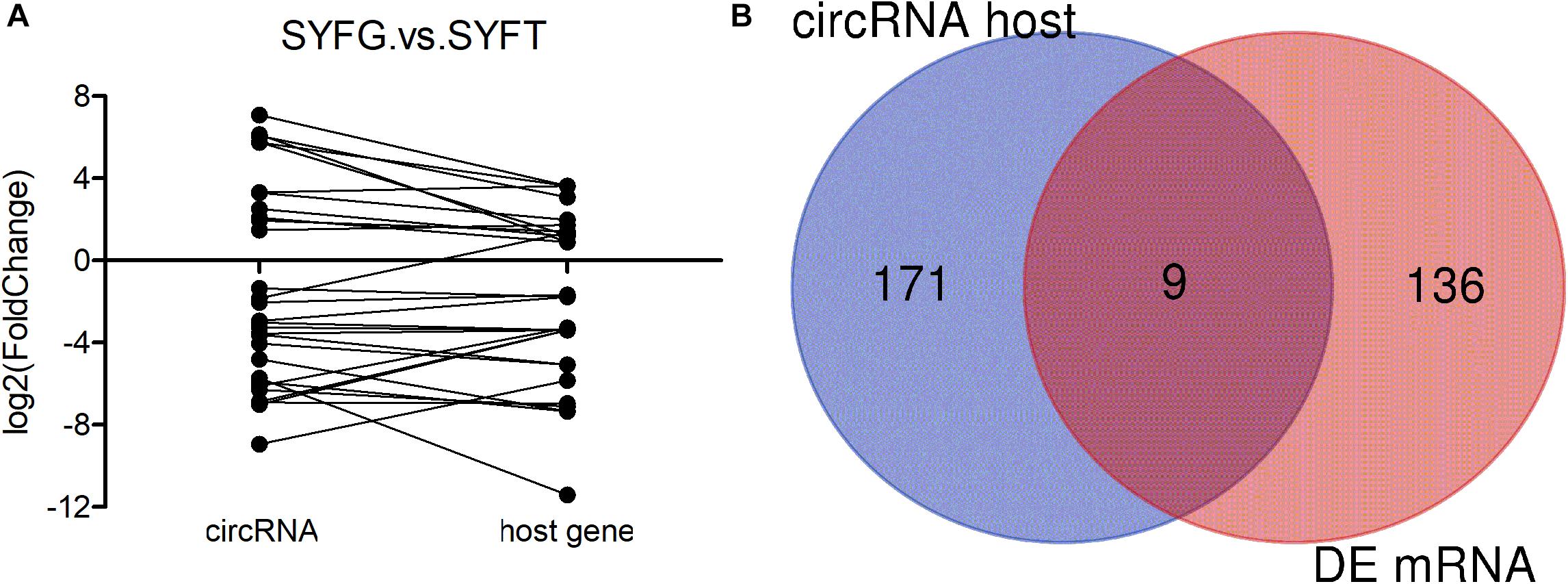
Figure 3. Expression level of relationship between circRNA and its host genes. (A) Fold change of circRNA and its host genes; (B) Venn diagram of DE circRNA host genes and DE mRNA in SYFG vs. SYFT comparison group. SYFG, granulosa cells of small yellow follicle; SYFT, theca cells of small yellow follicle.
Validation of circRNA and RNA-seq Data
We selected four circRNAs from the nine circRNAs mentioned above to test their splice sites, the accuracy of the RNA-seq of the circRNAs, and their host genes. Divergent primers were designed to confirm the back-splice sites of the predicted circRNAs (Supplementary Table S2) and the products were detected with agarose gel electrophoresis (Figure 4A). The products of the expected sizes were sequenced with Sanger sequencing (Figure 4B), which showed that the splice sites were those predicted by the software.
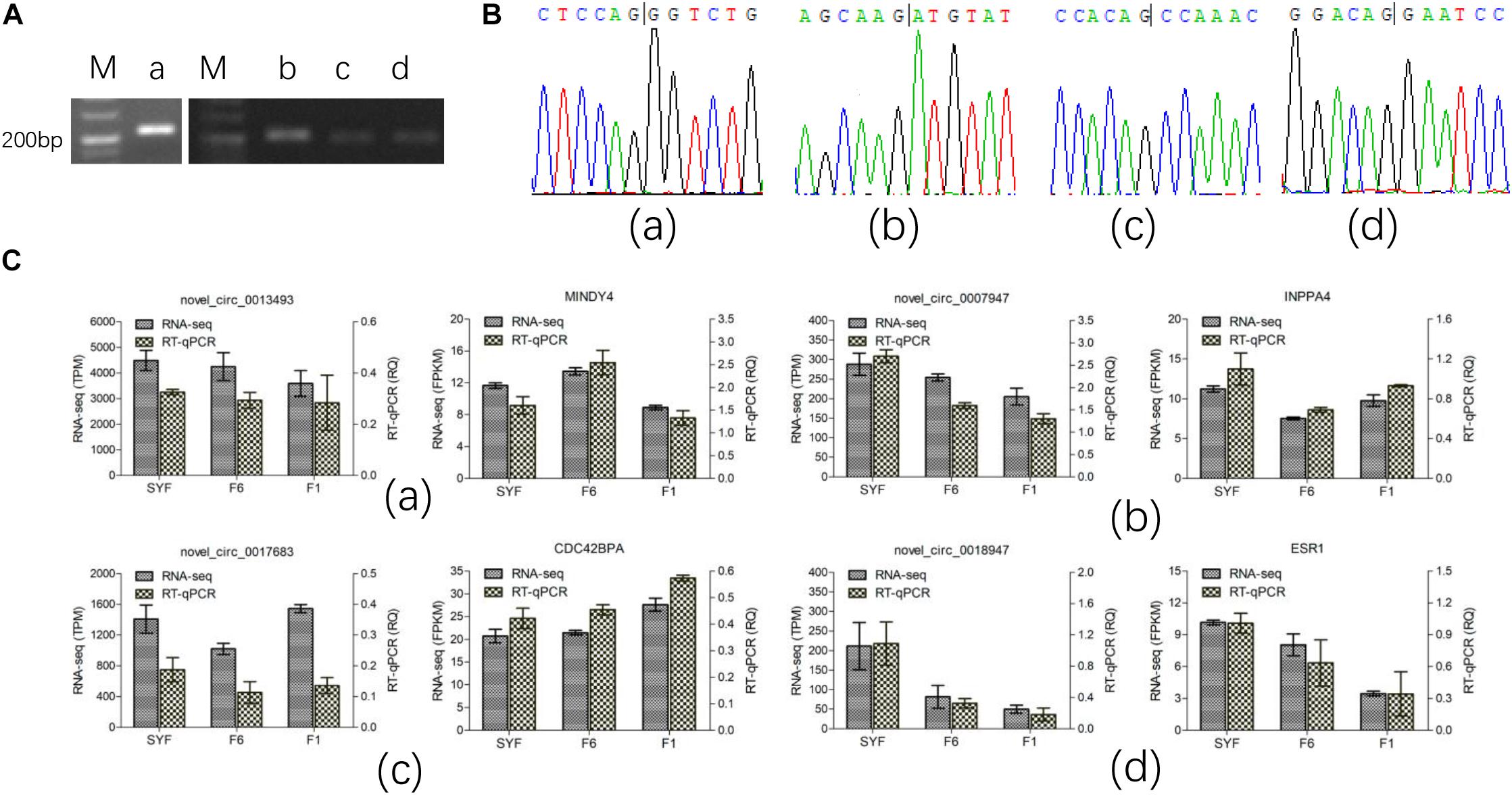
Figure 4. Experimental validation of circRNA. (A) PCR products were detected in agarose gel electrophoresis; (B) Sanger sequencing for the back-splice site of circRNA circulation; (C) RT-qPCR test for the expression level. Figures of small letter a, b, c and d represents 4 circRNAs and its host genes as following order: novel_circ_0013493(MINDY4), novel_circ_0007947(INPP4), novel_circ_0017683(CDC42BPA) and novel_circ_0018947(ESR1), respectively.
Only the transcripts in TCs were validated with RT–qPCR because the volume of GCs available was limited. The results are shown in Figure 4C, and are consistent with the RNA-seq data, confirming that the results of RNA-seq were accurate.
Bioinformatic Analysis of DE Transcripts
We performed an enrichment analysis of the host genes of the DE circRNAs to predict the functions of the circRNAs with KOBAS, and the results are shown in Figures 5, 6. Figure 5 shows that most of the same GO terms were enriched in the SYFG vs. SYFT and F1G vs. F1T comparisons, and included regulation of biological process, cellular development, cell differentiation, and cellular metabolic process. However, in the F6G vs. F6T comparison, the GO terms enriched were cellular response to chemical stimulus/organic substance, biological adhesion, biological proliferation, and endothelial cell proliferation.
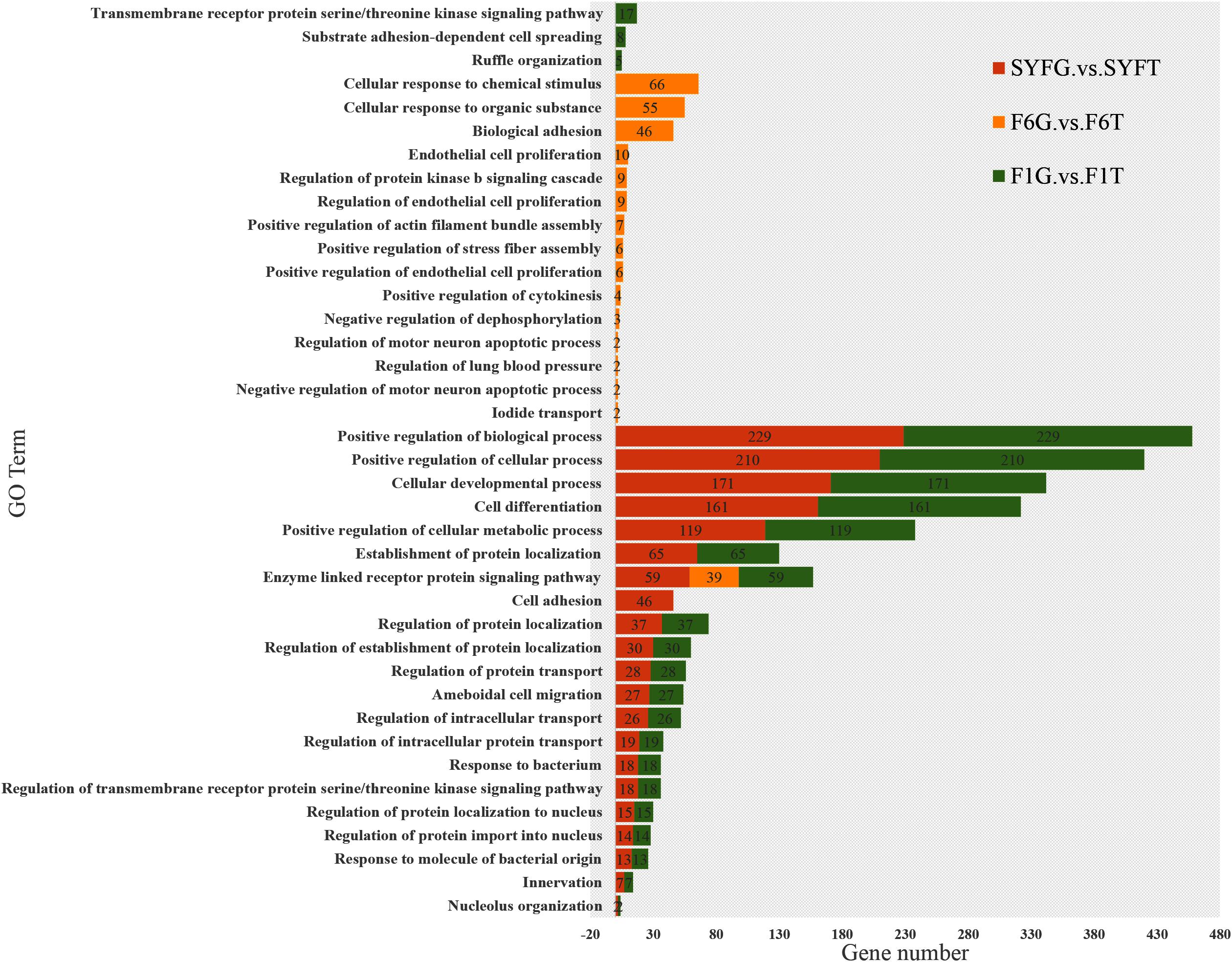
Figure 5. Biological process of host genes of DE circRNAs in different comparison group. DE, differentially expressed. SYF, small yellow follicle; F6, the smallest hierarchical follicle; F1, the largest hierarchical follicle. G, granulosa cell; T, theca cell.
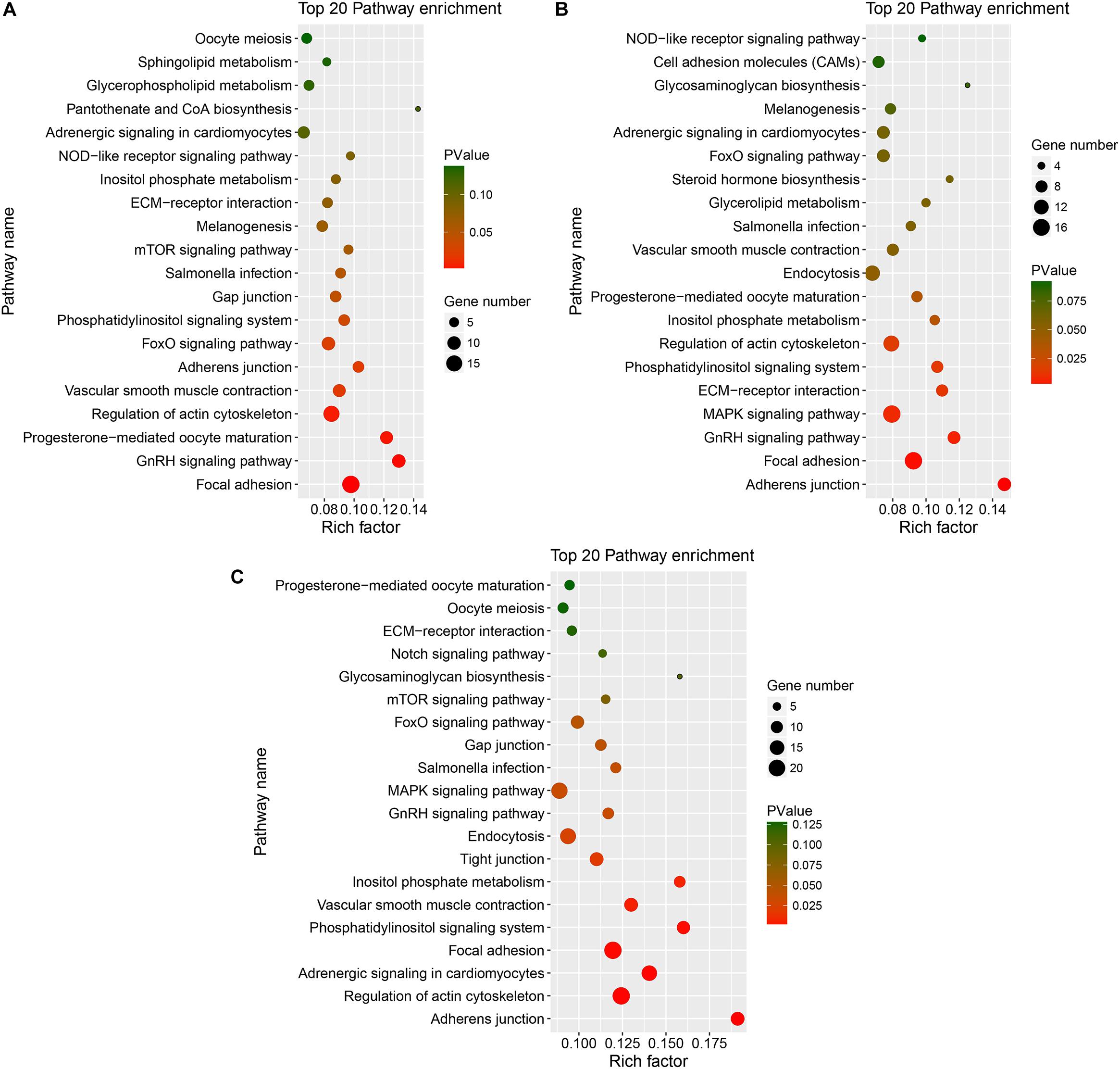
Figure 6. KEGG of host genes of DE circRNAs in different comparison group. (A–C) represents Top 20 pathways for host genes of circRNAs for SYFG vs. SYFT, F6G vs. F6T, and F1G vs. F1T comparison group, respectively. DE, differentially expressed; SYF, small yellow follicle; F6, the smallest hierarchical follicle; F1, the largest hierarchical follicle. G, granulosa cell; T, theca cell.
Figure 6 shows the major KEGG pathways enriched in DE circRNAs in the three comparison groups. The same 12 pathways were enriched in all three groups, including the FoxO signaling pathway, GnRH signaling pathway, ECM–receptor interaction, and progesterone-mediated oocyte maturation. The NOD-like receptor signaling pathway and melanogenesis were also enriched in the SYFG vs SYFT comparison. Cell adhesion molecules and steroid hormone biosynthesis were enriched in the F6G vs. F6T comparison. The MAPK signaling pathway was enriched in the F1G vs. F1T comparison.
Because DE mRNAs were only detected in the SYFG vs. SYFT comparison, GO and KEGG analyses were only performed for these stages, and the results are shown in Figure 7. The significant GO terms included cytokine receptor activity, cell periphery, membrane, and some fatty acid biosynthetic processes. The KEGG pathways included the FoxO signaling pathway, calcium signaling pathway, and apoptosis and some immunity-associated pathways, such as the PPAR signaling pathway and p53 signaling pathway were also enriched.
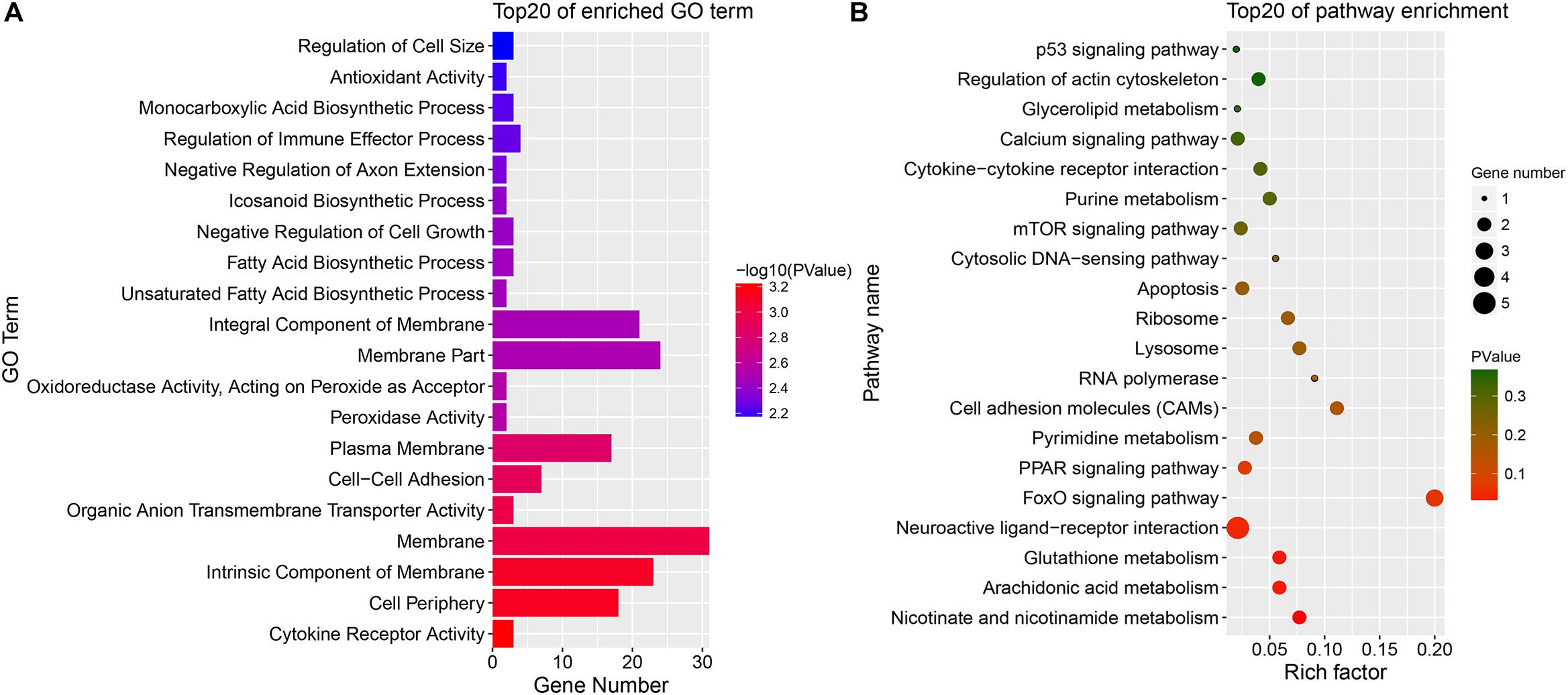
Figure 7. Enrichment DE mRNAs in different comparison group. (A) GO enrichment for DE mRNA; (B) KEGG enrichment for DE mRNA. SYF, small yellow follicle; F6, the smallest hierarchical follicle; F1, the largest hierarchical follicle.
Co-expression of Transcripts
To evaluate the potential functions of the circRNAs and their interactions with mRNAs, the WGCNA method was used to systematically identify the gene sets related to specific biological processes. After a series of calculations, a total of 18 modules were identified, with an average of 651.17 transcripts per module (Supplementary Table S7). The module visualization and module–trait correlations are show in Figure 8. Sixteen modules were positively associated with different follicle cells (correlation ≥ 0.5, p < 0.05; Figure 8B). Notably, five modules were strongly related to five follicle cells, except in the SYFT stage (correlation ≥ 0.8, p < 0.05; Figure 8B).
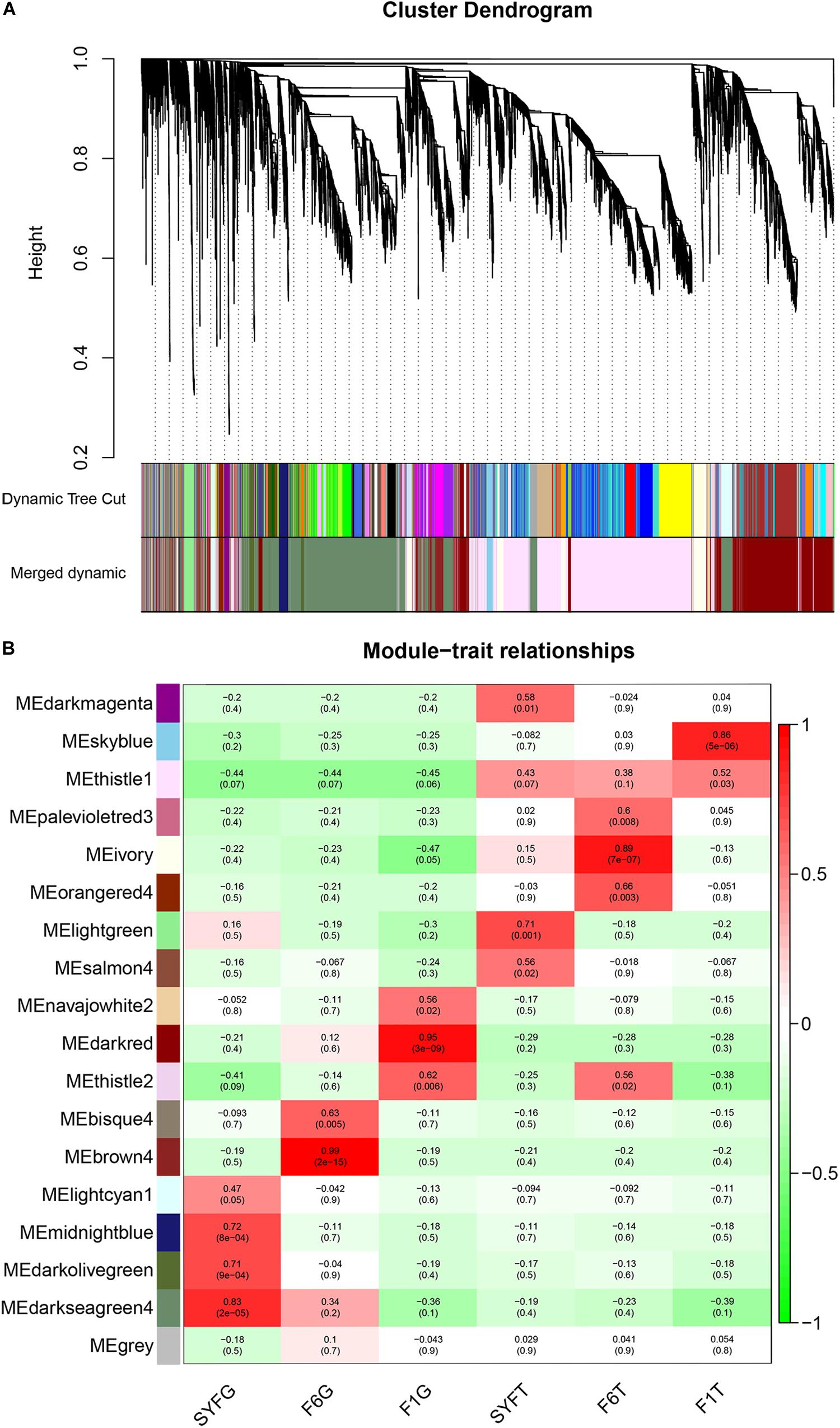
Figure 8. Visualization of transcripts co-expression modules and module-trait relationships. (A) Visualization of transcripts co-expression modules, clustering dendrogram of transcripts, with dissimilarity based on topological overlap; (B) Module-stage/tissue relationships and corresponding p-values, on the left, different colors represent different modules. Each cell contains the correlation and p-value given in the parentheses. Red, positive correlation; green, negative correlation; white, none correlation. SYF, small yellow follicle; F6, the smallest hierarchical follicle; F1, the largest hierarchical follicle. G, granulosa cells; T, theca cells.
GS and MM were calculated for all transcripts within each module, especially those with high module–trait correlations (correlation > 0.5, p < 0.01), to identify the modules specific to the different stages of follicle development (Supplementary Figure S1). The correlations between GS and MM usually showed that the transcripts were strongly associated with a specific stage or tissue (correlation > 0.5, p < 0.01), except the thistle1 module (correlation = 0.39). The module eigengene (ME) was calculated to construct a heatmap and bar plot of the co-expression of the mRNAs and circRNAs in the different modules, and six stage-specific modules were identified (Figure 9). The transcripts in the modules darkseagreen4, darkred, lightgreen, ivory, and skyblue were highly upregulated in the SYFG, F1G, SYFT, F6T, and F1T stages, respectively, whereas the transcripts in brown4 were up-regulated in the F6G stage and down-regulated in the other modules. To simplify the downstream analysis, the six modules darkseagreen4, brown4, darkred, lightgreen, ivory, and skyblue are referred to as SYFGm, F6Gm, F1Gm, SYFTm, F6Tm, and F1Tm, respectively.
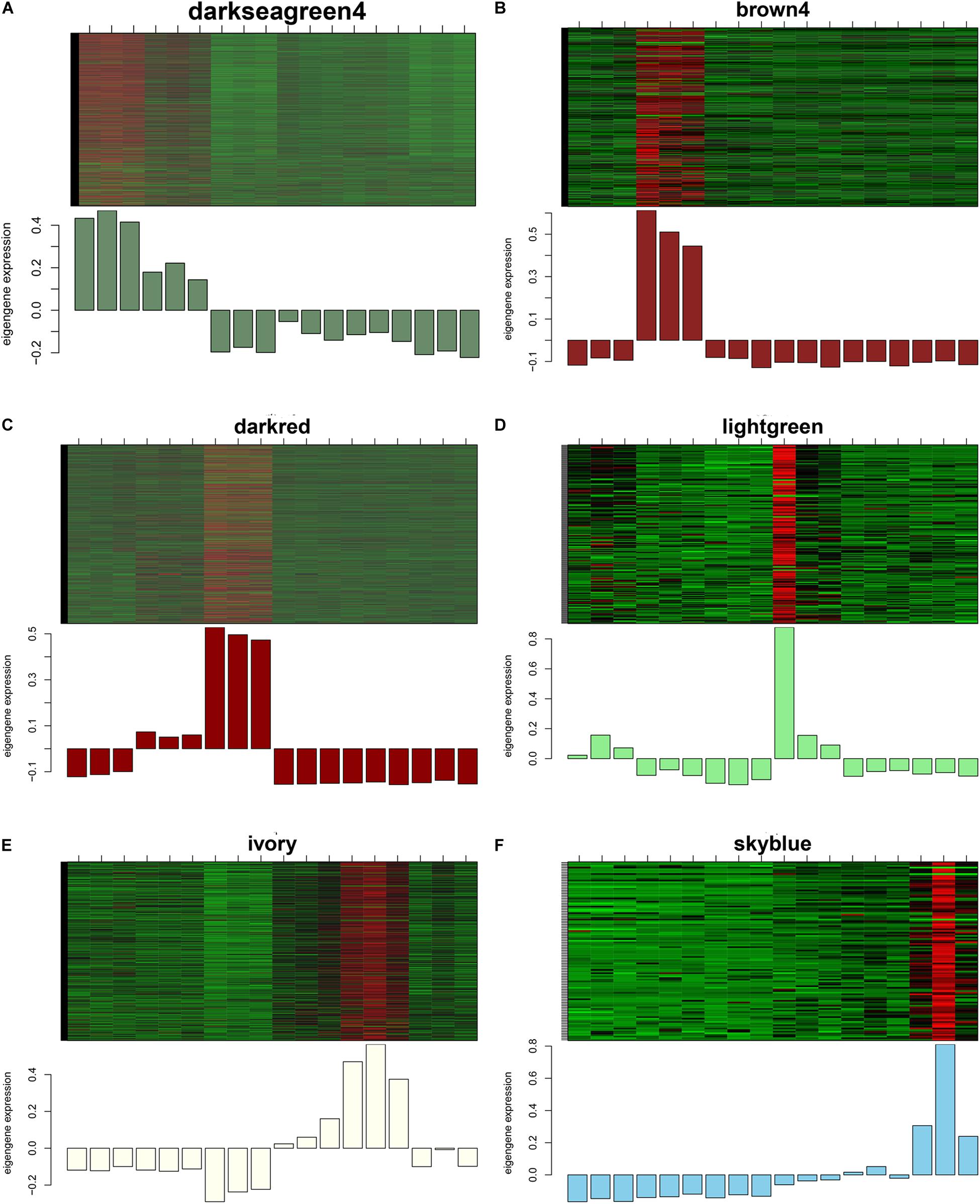
Figure 9. Heatmap and bar plot of expression level in six stage-specific modules. Six modules represent (A–F) darksegreen4, brown4, darkred, lightgreen, ivory, and skyblue with specific expression in SYFG, F6G, F1T, SYFT, F6T, and F1T, respectively. Samples were sorted in the order of: SYFG1-3, F6G1-3, F1G1-3, SYFT1-3, F6T1-3 and F1T1-3.
Bioinformatic Analysis of Hub Genes
Recently, the analysis of hub genes has been recognized as a promising approach to identifying key biological process genes (Nibbe et al., 2010). In the abovementioned six modules, we considered those transcripts with GS > 0.4 and MM > 0.8 to be putative hub genes (Supplementary Table S8), and 1469, 405, 1088, 78, 378, and 81 hub transcripts were identified in the SYFGm, F6Gm, F1Gm, SYFTm, F6Tm, and F1Tm modules, respectively. We performed a GO enrichment analysis on the mRNAs and host genes of the circRNAs in each module (Table 4). The hub genes in SYFGm were mainly significantly enriched in intracellular and organelle activity including intracellular part, intracellular organelle part, and intracellular organelle. Examples of these genes include RPL23, ORC5, HMGB3, and PIK3CA. The GO terms of the hub genes in F6Gm were mainly enriched in cellular process and protein regulation process, and included PAK1, HDAC2, SENP5, and TMED5. The hub genes in F1Gm, such as ATF2, SUPT3H, and TUBGCP2, may be involved in mitosis and cell development. The hub genes in SYFTm, such as SYPL1, SGMS2, and SOD1, may be involved in germ cell development. The hub genes in F6Tm, such as C1D, SOD1, and IPO13, may participate in intracellular processes. KEGG pathways analyses were performed for the specific stages (Table 5). The pathways identified in different specific modules differed. The hub genes in SYFGm may be involved in the cell cycle, RNA transport, and polymerase. The hub genes in F6Gm were enriched in oxocarboxylic acid metabolism, oocyte meiosis, and the MAPK signaling pathway. The hub-gene-enriched pathways in F1Gm included endocytosis, steroid biosynthesis, and the FoxO signaling pathway. The hub-gene-enriched pathways in SYFTm included cell cycle, drug metabolism cytochrome P450, and the calcium signaling pathway; in F6Tm, they included the biosynthesis of unsaturated fatty acids, fatty acid metabolism and protein export; and in F1Tm, they included focal adhesion, vascular smooth muscle contraction, and apoptosis.
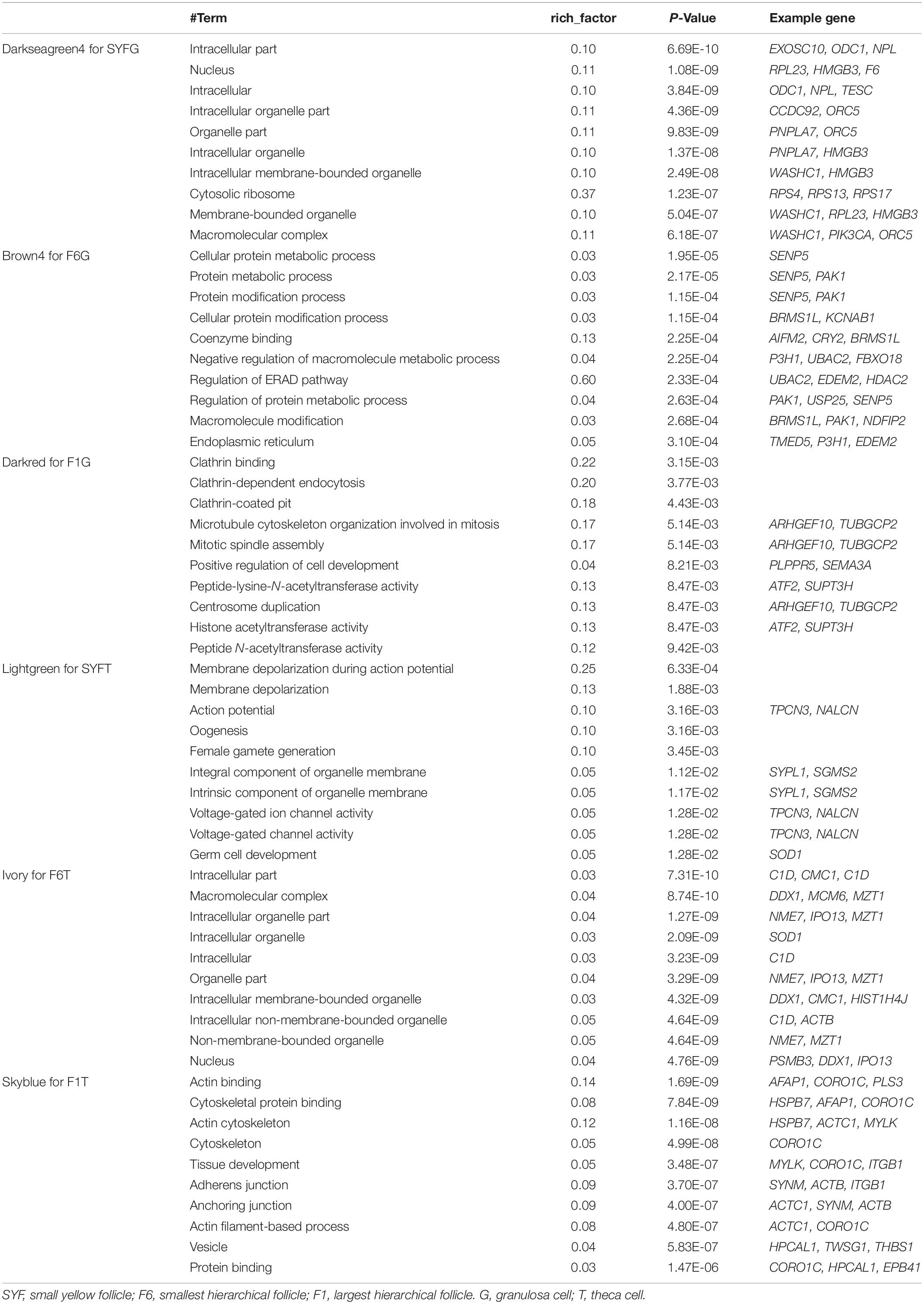
Table 4. Top 10 GO terms of hub genes in different specific modules.
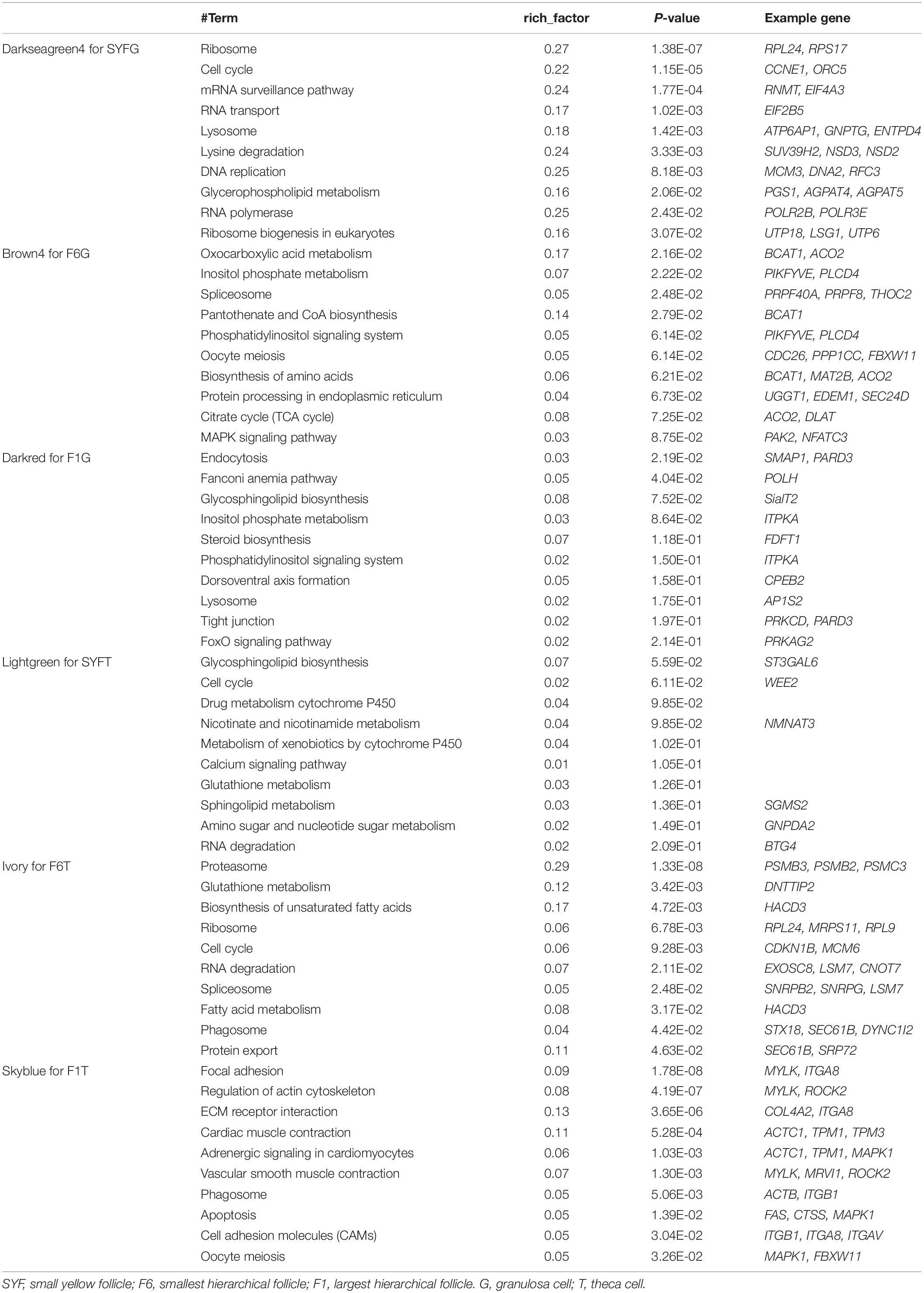
Table 5. Top 10 KEGG pathways of hub genes in different specific modules.
The larger the weighted value, the stronger the proof that a gene is a trait-associated hub gene. Therefore, networks of the top 200 edges ranked with weighted values were visualized with Cytoscape (Figure 10). In the network, the central hub genes with the edge numbers ≥ 15 were considered to be highly connected intramodular hub genes (designated ‘central hub genes’). In SYFGm, six central hub genes were identified: UHMK1, FZR1, MAPK1, INTS4, LMNB2, and CITED4. In F6Gm, five central hub genes were identified: ENSGALG00000028247, P3H1, SLC30A6, DST, and SOD2. In F1Gm, eight central hub genes were identified: SCT1, TAF12, SEPN1, RNF122, RAB8B, CAPZB, STRA6, and MAT2B. In SYFTm, more than 10 central hub genes were identified, including GDF9, BTG4, TCTE3, BIRC7, and PLS1. In F6Tm, seven central hub genes were identified: ATP5F1, NUDT21, PSMC1, SCFD1, MDH1, ARL3, and ENSGALG00000007127. In F1Tm, one circRNA (novel_circ_0004730) and nine protein-coding genes (CAPN2, PALLD, MYL4, ACTC1, MRVI1, etc.) were identified.
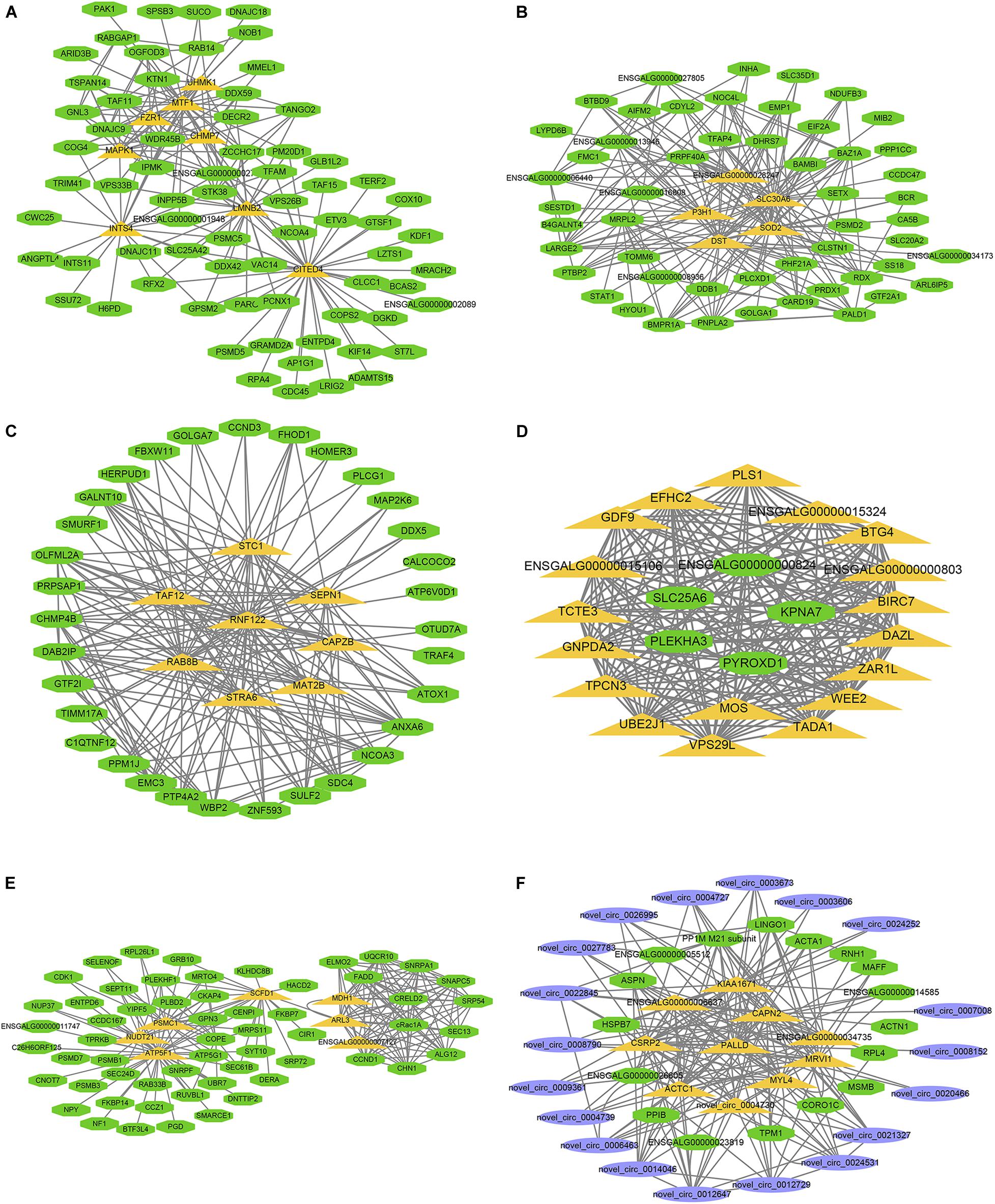
Figure 10. Visualization of hub genes in specific module. Network of (A–F) represent top hub genes in module darksegreen4, brown4, darkred, lightgreen, ivory, and skyblue, respectively. Green nodes represent protein-coding gene, blue nodes represent circRNA, orange nodes represent highly connected intramodular hub genes with edge number ≥ 15.
Discussion
Follicle development is a correctly ordered series of biological processes, including follicle recruitment, follicle selection, and ovulation, and plays a key role in the laying performance of hens, which is a major research hotspot for poultry breeders (Stephens and Johnson, 2016, 2017; Peng et al., 2018). Follicle cells mainly include three cell types, GCs in the granulosa layer, TCs in the theca layer, and oocyte, and each cell type has specific functions in follicle development. To provide a comprehensive view of the transcriptomes during GC and TC growth in the follicles of chickens, including both circRNAs and protein-coding genes, whole-transcriptome analyses were performed to identify the genes involved and their regulatory roles.
circRNAs are a novel type of non-coding RNA that plays various roles in life events. To our knowledge, this is the first study of the circRNAs involved in chicken follicle development in terms of GCs and TCs, although systematic searches for chicken circRNAs have been conducted in the liver (Zhang et al., 2017) and embryonic muscle (Ouyang et al., 2017). In this study, we identified over ten thousand circRNAs in GCs and TCs, 8127 of which were abundant in both cell types, and found that they share similar features in GCs and TCs. In total, 23,769 mRNAs were detected in GCs and TCs. Notably, more circRNAs and mRNAs were detected in TCs than in GCs, consistent with a previous study (Jing et al., 2018). This suggests that transcripts are preferentially expressed in TCs because of the complexity of the thecal layer, which contains aromatase cells, fibroblast, and interstitial cells (Bahr, 1991), whereas the components of the granulosa layer are much simpler (Johnson, 2015). A DE analysis of these circRNAs and mRNAs showed that circRNAs are preferentially expressed in a tissue-specific manner, suggesting that the regulatory roles played by non-protein-coding genes at the transcription level differ from those played by protein-coding genes.
Previous reports have shown that circRNA biogenesis sometimes competes with pre-mRNA splicing (Ashwal-Fluss et al., 2014), and that some circRNAs do not colocalize with their host genes (You et al., 2015). In this study, we found that circRNA expression levels were independent of the abundance of the linear RNA isoform and that only nine genes that overlapped at the circRNA and mRNA levels were differentially expressed in the SYFG vs. SYFT comparison. A GO analysis of DE circRNAs showed that most genes differentially expressed in the SYFG vs. SYFT and F1G vs. F1T comparisons were associated with cell differentiation, cell development, and metabolic process, suggesting that the same biological events occur in the SYF and F1 follicles. The GO terms enriched in the F6G vs. F1T comparison differed from those for the other two comparisons, and were enriched in cellular response, cellular response, and endothelial cell proliferation. A possible explanation is that the processes in each stage of follicular cell development in the ovary differ, and that the SYF and F1 follicles undergo selection and ovulation, whereas in the F6 stage, GCs and TCs proliferate.
Most KEGG pathways identified in the three comparisons were related to some reproduction trait, such as the FoxO signaling pathway, GnRH signaling pathway, and progesterone-mediated oocyte maturation, which have been reported to play key roles in follicular development (Peng et al., 2018). In the enrichment analysis of DE mRNAs, only the mRNAs differentially expressed in the SYFG vs. SYFT comparison were enhanced in some fatty acid biosynthetic process and reproduction pathways. SYF is the main stage in which estrogen is produced (Bahr, 1991), and fatty acid biosynthetic processes provide the components of estrogen. The differences detected in the enrichment analyses of circRNAs and mRNAs may imply their different regulatory roles at the transcription level, insofar as circRNAs may function in cell development whereas mRNA may play a key role in physiological functions.
Remarkably, most of the highly expressed circRNAs were derived from exons in both GCs and TCs, as shown in Tables 2, 3 but only about 40% of the circRNAs identified in the RNA-seq data were produced from exons (Supplementary Table S4). The host genes of the DE circRNAs that were most strongly differentially expressed in the GCs and TCs in chickens in this study also produce a series of isoforms as circRNAs in humans (Salzman et al., 2013; Rybak-Wolf et al., 2015), which have been published in circBase (Glazar et al., 2014) (except YBX3, EPT1L, and MINDY4). To date, no research has examined the functions of these circRNAs in vitro or in vivo, but isoforms of the mRNAs transcribed from the host genes of these circRNAs play a regulatory role in mammalian reproduction. Insulin-like growth factor receptor (IGF1R) is a functional receptor for IGF1 and IGF2, which has been shown to promote the nuclear maturation of oocytes and to stimulate cell proliferation and inhibit apoptosis (Sirotkin, 2011). ESR1 encodes one of the estrogen receptors that mediate the regulation of mammalian reproduction by the hormone estradiol (Edson et al., 2009). Interleukin 1 receptor (IL1R1) increases the expression of the FSH receptor (FSHR) in mouse primary GCs (Uri-Belapolsky et al., 2017). However, there is little information on the functions of these genes in the chicken follicle, in which GCs and TCs have different functions from those in mammalian ovaries (Johnson, 2015). Interestingly, novel_circ_0002934 produced from the RAB11A gene was highly expressed in both cell types. A previous study reported that RAB11A may act as a major regulator of membrane delivery during cytokinesis (Chu et al., 2009) and plays a supporting role in autophagy (Huttenhower et al., 2009). The regression of the chicken postovulatory follicle is mediated by autophagy (Lin et al., 2018), and autophagy signaling pathways are critical in controlling broodiness in the goose (Yu et al., 2016). During the follicular growth process, most SYF follicles undergo atresia (Hocking, 2009), which involves a complex array of mechanisms that modulate GC and TC apoptosis (Johnson, 2003; Lebedev et al., 2006; Kim et al., 2016). However, the effect of autophagy on follicle atresia before follicle selection has received little attention. Therefore, research in autophagy during follicle development requires further investigation with reference to the RAB11A gene and its circular RNA.
Functional studies of circRNAs usually focus on their microRNA (miRNA) sponges (Fu and Jiang, 2018), and research on the expression networks of circRNAs and protein-coding genes has been limited. In this study, we used WGCNA (Langfelder and Horvath, 2008) to construct specific modules and networks of interactions during follicle development. According to the expression profiles of these modules, we finally identified six specific functional modules and thousands of hub genes that specific positively correlate with the six follicular tissues. Expression level of the darkseagreen4, brown4, darkred, lightgreen, ivory, and skyblue modules peaked at SYFG, F6G, F1G, SYFT, F6T, and F1T, respectively. Consequently, we explored enrichment analysis in the six specific modules. The genes present in a single module have a common function and affect tissue- or stage-specific biological processes. Enrichment analyses of the hub genes in the different modules showed that different terms and pathways were enriched in the different modules. SYFGs are undifferentiated and steroidogenically inactive (Tilly et al., 1991a), while SYFTs is the stage at which the differentiation of the internal and external thecal layers occurs. Apoptotic DNA cleavage was detected in GCs and TCs (Tilly et al., 1991b), and the majority of SYFs undergo atresia, with only one SYF growing to the hierarchical follicle each day in a clutch, which suggests that SYF is a complex tissue, involving multiple biological processes, including growth and apoptosis (Tilly et al., 1991b). To understand the fine mechanisms operating in this special stage, more studies are required of differently sized follicles in the SYF pool, using new technologies, such as single-cell sequencing, a powerful new technology for studying rare cells (Picelli, 2017).
Another module enrichment analysis may identify the main functions of different tissues, e.g., the regulation of the endoplasmic-reticulum-associated protein degradation (ERAD) pathway (which had a high enrichment factor of 0.6 in F6Gm) and oxocarboxylic acid metabolism (with an enrichment factor of 0.17). The ERAD and oxocarboxylic acid pathways have been shown to act in cell proliferation (McKeehan and McKeehan, 1979; Diaferia et al., 2013). The proliferation of GCs and TCs in F6 follicles is the beginning of a rapidly growing hierarchy (Dunn et al., 2003). The genes in the F6 module may participate in the proliferation of the follicle. The hub genes in F1Gm were enriched in clathrin binding, endocytosis, steroid biosynthesis, and so on, and the hub genes in F1Tm were enriched in actin binding, focal adhesion, vascular smooth muscle contraction, and so on. The F1 follicle is the main site of progesterone production (Lee et al., 1998). The reduction in tight junctions in the follicular theca is the key factor affecting ovulation (Lebedev et al., 2006). From our WGCNA analysis and the different functions of GCs and TCs, we draw the conclusion that in the different stages of follicle development, the cells undertake different biological functions, for which different genes are required.
As hub genes are principle regulators in modules, we designated the genes with ≥15 edge numbers connectivity as central hubs in each network. Clearly, most hub components in specific modules are different from each other. The central hub genes in specific modules, such as MAPK1 in SYFGm (Woods et al., 2009), SOD2 in F6Gm (Jia et al., 2016), and GDF9 in SYFTm (Johnson, 2012), have been described in previous reports, but most of them were first reported in our study. CITED4 was one of the very central hub genes in SYFGm, and CITED4 targets luteinizing hormone (LH) and ERK1/2, triggering ovulation in mice (Zhang et al., 2014). Before follicle selection, the LH receptor (LHR) is present at low levels in prehierarchical follicles (Johnson et al., 2004), and GCs preferentially express LHR compared with TCs (Yao and Bahr, 2001). Therefore, we hypothesized that CITED4 is involved in the regulation of LHR expression in SYFGs, thus mediating follicle selection. GDF9, MOS, BTG4, and another 15 genes were the central hub genes in SYFTm. GDF9 increased the number of human ovary TCs in culture, but inhibited thecal steroidogenesis (Yamamoto et al., 2002). MOS is differentially expressed in the ovarian follicles of Bashang long tail chickens and Hy-line brown layers, and its expression may be regulated by several of long non-coding RNAs (lncRNAs) (Peng et al., 2018). BTG4 has antiproliferative properties and is a novel tumor suppressor (Toyota et al., 2008). It is speculated that these genes and other genes or transcripts in SYFTm are involved in a large network that regulates thecal growth and its functions.
SOD2, a central hub gene in F6Gm, is antioxidant activity-related genes playing an important role in inhibiting ROS biosynthesis. It was previously reported that SOD2 involved in pharmacological protection of the mouse ovaries (Qin et al., 2018) and had a role in goat granulosa cells proliferation (Yao et al., 2017). Given its roles in antioxidation and proliferation, SOD2 is likely a potential key gene modulating granulosa cell proliferation and protection in F6G stage. STC1, a central hub gene in F1Gm, may be involved in human ovarian tumorigenesis via promoting proliferation and inhibiting apoptosis of tumor cells (Liu et al., 2010). In a granulosa cells research, SCT1 had been proved to dampen the gonadotropin stimulation of granulosa cell differentiation by paracrine regulation and inhibit progesterone secretion (Luo et al., 2004). It is tempting to speculate that STC1 may have a pivotal role in the F1G stage. MDH1, a central hub gene in F6Tm, is a glucose metabolism-related enzyme gene may have an important role of the activation and maintenance of the mouse ovarian follicular fool (Sutherland et al., 2012). A study from Seol et al. (2006) showed that genes involved in glucose utilization ensure chickens have a rapid follicle development from F6 to F1 follicle, even though glycolysis and β-oxidation are not modulated by follicle growth. F6 stage is the begining of hierarchal follile phase which suggested that MDH1 may be involved in energy utilization to participate in rapid follicle growth. CAPN2, a central hub gene in F1Tm, was implicated to important cellular process including apoptosis and survival (Storr et al., 2012), and had been reported to be upregulated by hCG in bovin granulosa cells of ovulatory follicle and may contribute to ovulation processes (Lussier et al., 2017). Thus we speculated that CAPN2 may have a similar function on the F1T cells in the ovulation. Despite these candidate hub genes findings, questions remain. Further functional characterization of these hub genes in the future will shed light on how they interact with each other to modulate follicle development.
It is noteworthy that we only detected one very top hub circRNA, novel_circ_0004730, in F1Tm. This may be because only a few circRNAs function at the transcription level, and that most of them are involved in crosstalk (Tay et al., 2014). novel_circ_0004730 is produced from the SAR1B gene (Supplementary Table S4), which regulates cholesterol homeostasis (Sane et al., 2015). Previous studies have mainly addressed the function of SAR1B in lipid secretion and cholesterol biosynthesis (Fryer et al., 2014), and there is little information on the role of this gene in reproduction, especially in follicle development.
An intriguing observation is that ESR1 was differentially expressed as circRNA and mRNA isoforms in the SYFG vs. SYFT comparison, and was one of the hub genes in F1Gm, with GS = 0.96 and MM = 0.89. The ESR1 gene is mainly expressed in the theca in the mouse ovary (Britt and Findlay, 2003), and is one of the estrogen receptors that mediate the regulation of mammalian reproduction by estradiol through autocrine and paracrine mechanisms (Edson et al., 2009). It has also been suggested that estrogen is required to maintain the integrity of the follicle wall (Bahr, 1991). A previous study showed that the expression of CYP19 was strongly associated with ESR1 expression in ewe large follicles (Foroughinia et al., 2017), and CYP19A1 was a hub gene in F6Tm in this study (Supplementary Table S8). Estrogen is involved in both positive and negative feedback in the hypothalamic–pituitary–gonadal axis (Hocking, 2009). ESR1 localized in SYF tissues may increase their estrogen secretion by positive feedback to estrogen, because about 50% of estrogen is produced from small follicles (Bahr, 1991), and the estrogen in F1 follicle tissues is reduced by negative feedback to estrogen (Johnson et al., 1996). The mechanism of follicular development is more complex than a simple molecular interaction. Our results indicate that ESR1 may be a key regulator of the development of the granulosa and thecal layers and play a role in the interactions between them. Further investigation of the ESR1 gene as the source of circRNAs and mRNAs involved in GC and TC development in chickens is required.
Conclusion
Ovarian follicular development requires a continuous supply of endocrine, paracrine, and autocrine factors and these factors affect cell differentiation, proliferation, and apoptosis. This study extends our understanding of the circRNAs and mRNAs that affect follicle development. The numbers of circRNAs and mRNAs and the levels of their differential expression in GCs and TCs differ at different stages, suggesting that these transcripts or genes have specific functions in cells or tissues. In a WGCNA analysis, we identified six specific modules that are associated with different follicular cells or stages of follicle development. Based on the WGCNA regulatory network, we conclude that the MAPK1 and CITED4 in the SYFGm, SOD2 in the F6Gm, STC1 in the F1Gm, GDF9 and MOS in the SYFTm, MDH1 in the F6Tm, CAPN2 and novel_circ_0004730 in the F1Tm may participate the specific function of follicle development. Moreover, another interesting gene ESR1, detected by both DE analysis and WGCNA method, may be an important gene involved in follicle development, in both GCs and TCs, and is expressed as both circRNA and mRNA isoforms. This study lays a preliminary foundation for research into circRNAs and mRNAs in the development of follicles. However, the mechanisms of their actions remain to be clarified.
Data Availability Statement
The raw data were submitted to the National Center for Biotechnology Information (NCBI) under BioProject accession numbers PRJNA481176 and PRJNA511712.
Ethics Statement
The animal study was reviewed and approved by Animal Care Committee of Yangzhou University.
Author Contributions
MS, JW, and GZ designed the work and wrote the manuscript. MS, TL, and FC performed the transcriptome data and prepared the figures. PW, YW, and KX collected the samples. All authors read and approved the final manuscript.
Funding
This work was supported by Priority Academic Program Development of Jiangsu Higher Education Institutions, Natural Science Foundation of Jiangsu Province (Grant No. BK20181217), and China’s Agricultural Research Systems (Grant No. CARS-41).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the help of data analysis from colleague of Jiangsu Institute of Poultry Science.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00503/full#supplementary-material
FIGURE S1 | Visualization of GS vs. MM. The scatter plots display the distribution of GS and MM of transcripts in stage-specific modules related to individual sample.
TABLE S1 | Soft power estimated according to TOM.
TABLE S2 | Primers for the validation of circRNAs or RT-qPCR.
TABLE S3 | Quality control results for circRNAs and mRNAs.
TABLE S4 | Novel circRNAs detected in follicles. The overlapped circRNAs in GCs and TCs were marked with red font.
TABLE S5 | Feature of transcripts detected in current study.
TABLE S6 | Detail information of nine overlapped genes.
TABLE S7 | Modules and genes number identified in WGCNA analysis.
TABLE S8 | Transcripts information in different specific modules.
References
Alvaro Mercadal, B., Imbert, R., Demeestere, I., Gervy, C., De Leener, A., Englert, Y., et al. (2015). AMH mutations with reduced in vitro bioactivity are related to premature ovarian insufficiency. Hum. Reprod. 30, 1196–1202. doi: 10.1093/humrep/dev042
Ashwal-Fluss, R., Meyer, M., Pamudurti, N. R., Ivanov, A., Bartok, O., Hanan, M., et al. (2014). circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 56, 55–66. doi: 10.1016/j.molcel.2014.08.019
Bahr, J. M. (1991). The chicken ovary as a model of follicular development. Semin. Reprod. Med. 9, 352–359.
Britt, K. L., and Findlay, J. K. (2003). Regulation of the phenotype of ovarian somatic cells by estrogen. Mol. Cell. Endocrinol. 202, 11–17. doi: 10.1016/s0303-7207(03)00055-8
Cai, H., Li, Y., Li, H., Niringiyumukiza, J. D., Zhang, M., Chen, L., et al. (2018). Identification and characterization of human ovary-derived circular RNAs and their potential roles in ovarian aging. Aging 10, 2511–2534. doi: 10.18632/aging.101565
Chen, B., Yu, J., Guo, L., Byers, M. S., Wang, Z., Chen, X., et al. (2019). Circular RNA circHIPK3 promotes the proliferation and differentiation of chicken myoblast cells by sponging miR-30a-3p. Cells 8:177. doi: 10.3390/cells8020177
Cheng, J., Huang, J., Yuan, S., Zhou, S., Yan, W., Shen, W., et al. (2017). Circular RNA expression profiling of human granulosa cells during maternal aging reveals novel transcripts associated with assisted reproductive technology outcomes. PLoS One 12:e0177888. doi: 10.1371/journal.pone.0177888
Chu, B. B., Ge, L., Xie, C., Zhao, Y., Miao, H. H., Wang, J., et al. (2009). Requirement of myosin Vb.Rab11a.Rab11-FIP2 complex in cholesterol-regulated translocation of NPC1L1 to the cell surface. J. Biol. Chem. 284, 22481–22490. doi: 10.1074/jbc.M109.034355
Diaferia, G. R., Cirulli, V., and Biunno, I. (2013). SEL1L regulates adhesion, proliferation and secretion of insulin by affecting integrin signaling. PLoS One 8:e79458. doi: 10.1371/journal.pone.0079458
Donadeu, F. X., Fahiminiya, S., Esteves, C. L., Nadaf, J., Miedzinska, K., McNeilly, A. S., et al. (2014). Transcriptome profiling of granulosa and theca cells during dominant follicle development in the horse. Biol. Reprod. 91:111. doi: 10.1095/biolreprod.114.118943
Dunn, I., Miao, Y., Morris, A., Romanov, M., Wilson, P., and Waddington, D. (2003). A study of association between genetic markers in candidate genes and reproductive traits in one generation of a commercial broiler breeder hen population. Heredity 92, 128–134. doi: 10.1038/sj.hdy.6800396
Edson, M. A., Nagaraja, A. K., and Matzuk, M. M. (2009). The mammalian ovary from genesis to revelation. Endocr. Rev. 30, 624–712. doi: 10.1210/er.2009-0012
Elis, S., Dupont, J., Couty, I., Persani, L., Govoroun, M., Blesbois, E., et al. (2007). Expression and biological effects of bone morphogenetic protein-15 in the hen ovary. J. Endocrinol. 194, 485–497. doi: 10.1677/JOE-07-0143
Eresheim, C., Leeb, C., Buchegger, P., and Nimpf, J. (2014). Signaling by the extracellular matrix protein Reelin promotes granulosa cell proliferation in the chicken follicle. J. Biol. Chem. 289, 10182–10191. doi: 10.1074/jbc.M113.533489
Foroughinia, G., Fazileh, A., and Eghbalsaied, S. (2017). Expression of genes involved in BMP and estrogen signaling and AMPK production can be important factors affecting total number of antral follicles in ewes. Theriogenology 91, 36–43. doi: 10.1016/j.theriogenology.2016.12.023
Fryer, L. G., Jones, B., Duncan, E. J., Hutchison, C. E., Ozkan, T., Williams, P. A., et al. (2014). The endoplasmic reticulum coat protein II transport machinery coordinates cellular lipid secretion and cholesterol biosynthesis. J. Biol. Chem. 289, 4244–4261. doi: 10.1074/jbc.M113.479980
Fu, Y., and Jiang, H. (2018). Genome-wide analysis of circular RNAs in bovine cumulus cells treated with BMP15 and GDF9. Sci. Rep. 8:7944. doi: 10.1038/s41598-018-26157-2
Gao, Y., Zhang, J., and Zhao, F. (2017). Circular RNA identification based on multiple seed matching. Brief Bioinform. 19, 803–810. doi: 10.1093/bib/bbx014
Gilbert, A. B., Evans, A. J., Perry, M. M., and Davidson, M. H. (1977). A method for separating the granulosa cells, the basal lamina and the theca of the preovulatory ovarian follicle of the domestic fowl (Gallus domesticus). J. Reprod. Fertil. 50, 179–181. doi: 10.1530/jrf.0.0500179
Glazar, P., Papavasileiou, P., and Rajewsky, N. (2014). circBase: a database for circular RNAs. RNA 20, 1666–1670. doi: 10.1261/rna.043687.113
Guo, T., Huang, L., Yao, W., Du, X., Li, Q., Ma, M., et al. (2019). The potential biological functions of circRNAs during the initiation of atresia in pig follicles. Domestic Anim. Endocrinol. 72:106401. doi: 10.1016/j.domaniend.2019.106401
Huttenhower, C., Haley, E. M., Hibbs, M. A., Dumeaux, V., Barrett, D. R., Coller, H. A., et al. (2009). Exploring the human genome with functional maps. Genome Res. 19, 1093–1106. doi: 10.1101/gr.082214.108
Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., et al. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157. doi: 10.1261/rna.035667.112
Jia, W., Xu, B., and Wu, J. (2018). Circular RNA expression profiles of mouse ovaries during postnatal development and the function of circular RNA epidermal growth factor receptor in granulosa cells. Metabolism 85, 192–204. doi: 10.1016/j.metabol.2018.04.002
Jia, Y., Lin, J., Zeng, W., and Zhang, C. (2010). Effect of prostaglandin on luteinizing hormone-stimulated proliferation of theca externa cells from chicken prehierarchical follicles. Prostaglandins Other Lipid Mediat. 92, 77–84. doi: 10.1016/j.prostaglandins.2010.03.005
Jia, Y., Yang, M., Zhu, K., Wang, L., Song, Y., Wang, J., et al. (2016). Melatonin implantation improved the egg-laying rate and quality in hens past their peak egg-laying age. Sci. Rep. 6:39799. doi: 10.1038/srep39799
Jing, R., Gu, L., Li, J., and Gong, Y. (2018). A transcriptomic comparison of theca and granulosa cells in chicken and cattle follicles reveals ESR2 as a potential regulator of CYP19A1 expression in the theca cells of chicken follicles. Comp. Biochem. Physiol. D Genomics Proteomics 27, 40–53. doi: 10.1016/j.cbd.2018.04.002
Johnson, A. L. (1990). Steroidogenesis and action of steroids in the hen ovary. Crit. Rev. Poult. Biol. 2, 319–346.
Johnson, A. L. (2003). Intracellular mechanisms regulating cell survival in ovarian follicles. Anim. Reprod. Sci. 78, 185–201. doi: 10.1016/s0378-4320(03)00090-3
Johnson, A. L. (2015). Ovarian follicle selection and granulosa cell differentiation. Poult. Sci. 94, 781–785. doi: 10.3382/ps/peu008
Johnson, A. L., Bridgham, J. T., and Wagner, B. (1996). Characterization of a chicken luteinizing hormone receptor (cLH-R) complementary deoxyribonucleic acid, and expression of cLH-R messenger ribonucleic acid in the ovary. Biol. Reprod. 55, 304–309. doi: 10.1095/biolreprod55.2.304
Johnson, A. L., Bridgham, J. T., and Woods, D. C. (2004). Cellular mechanisms and modulation of activin A- and transforming growth factor beta-mediated differentiation in cultured hen granulosa cells. Biol. Reprod. 71, 1844–1851. doi: 10.1095/biolreprod.104.032573
Johnson, P. A. (2012). Follicle selection in the avian ovary. Reprod. Domest. Anim. 47(Suppl. 4), 283–287. doi: 10.1111/j.1439-0531.2012.02087.x
Kang, L., Yang, C., Wu, H., Chen, Q., Huang, L., Li, X., et al. (2017). miR-26a-5p Regulates TNRC6A expression and facilitates theca cell proliferation in chicken ovarian follicles. DNA Cell Biol. 36, 922–929. doi: 10.1089/dna.2017.3863
Kim, D., and Johnson, A. L. (2018). Differentiation of the granulosa layer from hen prehierarchal follicles associated with follicle-stimulating hormone receptor signaling. Mol. Reprod. Dev. 85, 729–737. doi: 10.1002/mrd.23042
Kim, D., Lee, J., and Johnson, A. L. (2016). Vascular endothelial growth factor and angiopoietins during hen ovarian follicle development. Gen. Comp. Endocrinol. 232, 25–31. doi: 10.1016/j.ygcen.2015.11.017
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lebedev, V. A., Lebedeva, I. Y., Grossmann, R., Kuzmina, T. I., and Parvizi, N. (2006). Ovulatory cycle-related alterations in the thecal growth and membrane protein content of thecal tissue of hen preovulatory follicles. Theriogenology 66, 217–223. doi: 10.1016/j.theriogenology.2005.11.004
Lee, K. A., Volentine, K. K., and Bahr, J. M. (1998). Two steroidogenic pathways present in the chicken ovary: theca layer prefers delta 5 pathway and granulosa layer prefers delta 4 pathway. Domest. Anim. Endocrinol. 15, 1–8. doi: 10.1016/s0739-7240(97)00057-x
Li, L., Guo, J., Chen, Y., Chang, C., and Xu, C. (2017). Comprehensive CircRNA expression profile and selection of key CircRNAs during priming phase of rat liver regeneration. BMC Genomics 18:80. doi: 10.1186/s12864-016-3476-6
Lin, X., Liu, X., Ma, Y., Mi, Y., Zeng, W., Li, J., et al. (2018). Coherent apoptotic and autophagic activities involved in regression of chicken postovulatory follicles. Aging 10, 819–832. doi: 10.18632/aging.101436
Liu, G., Yang, G., Chang, B., Mercado-Uribe, I., Huang, M., Zheng, J., et al. (2010). Stanniocalcin 1 and ovarian tumorigenesis. J. Natl. Cancer Inst. 102, 812–827. doi: 10.1093/jnci/djq127
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15;550. doi: 10.1186/s13059-014-0550-8
Lu, T., Cui, L., Zhou, Y., Zhu, C., Fan, D., Gong, H., et al. (2015). Transcriptome-wide investigation of circular RNAs in rice. RNA 21, 2076–2087. doi: 10.1261/rna.052282.115
Luo, C.-W., Kawamura, K., Klein, C., and Hsueh, A. J. W. (2004). Paracrine regulation of ovarian granulosa cell differentiation by stanniocalcin (STC) 1: mediation through specific STC1 receptors. Mol. Endocrinol. 18, 2085–2096. doi: 10.1210/me.2004-0066
Lussier, J. G., Diouf, M. N., Lévesque, V., Sirois, J., and Ndiaye, K. (2017). Gene expression profiling of upregulated mRNAs in granulosa cells of bovine ovulatory follicles following stimulation with hCG. Reprod. Biol. Endocrinol. 15:88. doi: 10.1186/s12958-017-0306-x
McKeehan, W. L., and McKeehan, K. A. (1979). Oxocarboxylic acids, pyridine nucleotide-linked oxidoreductases and serum factors in regulation of cell proliferation. J. Cell Physiol. 101, 9–16. doi: 10.1002/jcp.1041010103
Mellouk, N., Rame, C., Barbe, A., Grandhaye, J., Froment, P., and Dupont, J. (2018). Chicken is a useful model to investigate the role of adipokines in metabolic and reproductive diseases. Int. J. Endocrinol. 2018;4579734. doi: 10.1155/2018/4579734
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. doi: 10.1038/nature11928
Nibbe, R. K., Koyuturk, M., and Chance, M. R. (2010). An integrative -omics approach to identify functional sub-networks in human colorectal cancer. PLoS Comput. Biol. 6:e1000639. doi: 10.1371/journal.pcbi.1000639
Ouyang, H., Chen, X., Wang, Z., Yu, J., Jia, X., Li, Z., et al. (2017). Circular RNAs are abundant and dynamically expressed during embryonic muscle development in chickens. DNA Res. 25, 71–86. doi: 10.1093/dnares/dsx039
Paradis, F., Novak, S., Murdoch, G. K., Dyck, M. K., Dixon, W. T., and Foxcroft, G. R. (2009). Temporal regulation of BMP2, BMP6, BMP15, GDF9, BMPR1A, BMPR1B, BMPR2 and TGFBR1 mRNA expression in the oocyte, granulosa and theca cells of developing preovulatory follicles in the pig. Reproduction 138, 115–129. doi: 10.1530/rep-08-0538
Peng, Y., Chang, L., Wang, Y., Wang, R., Hu, L., Zhao, Z., et al. (2018). Genome-wide differential expression of long noncoding RNAs and mRNAs in ovarian follicles of two different chicken breeds. Genomics 111, 1395–1403. doi: 10.1016/j.ygeno.2018.09.012
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Picelli, S. (2017). Single-cell RNA-sequencing: the future of genome biology is now. RNA Biol. 14, 637–650. doi: 10.1080/15476286.2016.1201618
Qin, Y., Iwase, A., Murase, T., Bayasula, Ishida, C., Kato, N., et al. (2018). Protective effects of mangafodipir against chemotherapy-induced ovarian damage in mice. Reprod. Biol. Endocrinol. 16:106. doi: 10.1186/s12958-018-0426-y
Quan, G., and Li, J. (2018). Circular RNAs: biogenesis, expression and their potential roles in reproduction. J. Ovarian Res. 11:9. doi: 10.1186/s13048-018-0381-4
Rybak-Wolf, A., Stottmeister, C., Glazar, P., Jens, M., Pino, N., Giusti, S., et al. (2015). Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 58, 870–885. doi: 10.1016/j.molcel.2015.03.027
Salzman, J., Chen, R. E., Olsen, M. N., Wang, P. L., and Brown, P. O. (2013). Cell-type specific features of circular RNA expression. PLoS Genet. 9:e1003777. doi: 10.1371/journal.pgen.1003777
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N., and Brown, P. O. (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7:e30733. doi: 10.1371/journal.pone.0030733
Sane, A., Seidman, E., Spahis, S., Lamantia, V., Garofalo, C., Montoudis, A., et al. (2015). New insights in intestinal Sar1B GTPase regulation and role in cholesterol homeostasis. J. Cell Biochem. 116, 2270–2282. doi: 10.1002/jcb.25177
Seol, H. S., Sato, K., Murakami, H., Toyomizu, M., and Akiba, Y. (2006). Changes in gene expression involved in energy utilization during chicken follicle development. Anim. Reprod. Sci. 95, 283–294. doi: 10.1016/j.anireprosci.2005.09.016
Shang, Q., Yang, Z., Jia, R., and Ge, S. (2019). The novel roles of circRNAs in human cancer. Mol. Cancer 18:6. doi: 10.1186/s12943-018-0934-6
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sirotkin, A. V. (2011). Growth factors controlling ovarian functions. J. Cell Physiol. 226, 2222–2225. doi: 10.1002/jcp.22588
Stephens, C. S., and Johnson, P. A. (2016). Bone morphogenetic protein 15 may promote follicle selection in the hen. Gen. Comp. Endocrinol. 235, 170–176. doi: 10.1016/j.ygcen.2016.06.027
Stephens, C. S., and Johnson, P. A. (2017). Occludin expression and regulation in small follicles of the layer and broiler breeder hen. Gen. Comp. Endocrinol. 248, 106–113. doi: 10.1016/j.ygcen.2017.02.010
Storr, S. J., Lee, K. W., Woolston, C. M., Safuan, S., Green, A. R., Macmillan, R. D., et al. (2012). Calpain system protein expression in basal-like and triple-negative invasive breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 23, 2289–2296. doi: 10.1093/annonc/mds176
Sutherland, J. M., Keightley, R. A., Nixon, B., Roman, S. D., Robker, R. L., Russell, D. L., et al. (2012). Suppressor of cytokine signaling 4 (SOCS4): moderator of ovarian primordial follicle activation. J. Cell. Physiol. 227, 1188–1198. doi: 10.1002/jcp.22837
Tao, H., Xiong, Q., Zhang, F., Zhang, N., Liu, Y., Suo, X., et al. (2017). Circular RNA profiling reveals chi_circ_0008219 function as microRNA sponges in pre-ovulatory ovarian follicles of goats (Capra hircus). Genomics [Epub ahead of print]. doi: 10.1016/j.ygeno.2017.10.005
Tay, Y., Rinn, J., and Pandolfi, P. P. (2014). The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–352. doi: 10.1038/nature12986
Tilly, J. L., Kowalski, K. I., and Johnson, A. L. (1991a). Stage of ovarian follicular development associated with the initiation of steroidogenic competence in avian granulosa cells. Biol. Reprod. 44, 305–314. doi: 10.1095/biolreprod44.2.305
Tilly, J. L., Kowalski, K. I., Johnson, A. L., and Hsueh, A. J. (1991b). Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology 129, 2799–2801. doi: 10.1210/endo-129-5-2799
Toyota, M., Suzuki, H., Sasaki, Y., Maruyama, R., Imai, K., Shinomura, Y., et al. (2008). Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 68, 4123–4132. doi: 10.1158/0008-5472.can-08-0325
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Uri-Belapolsky, S., Miller, I., Shaish, A., Levi, M., Harats, D., Ninio-Many, L., et al. (2017). Interleukin 1-alpha deficiency increases the expression of Follicle-stimulating hormone receptors in granulosa cells. Mol. Reprod. Dev. 84, 460–467. doi: 10.1002/mrd.22799
Wang, J., and Gong, Y. (2017). Transcription of CYP19A1 is directly regulated by SF-1 in the theca cells of ovary follicles in chicken. Gen. Comp. Endocrinol. 247, 1–7. doi: 10.1016/j.ygcen.2017.03.013
Woods, D. C., Schorey, J. S., and Johnson, A. L. (2009). Toll-like receptor signaling in hen ovarian granulosa cells is dependent on stage of follicle maturation. Reproduction 137, 987–996. doi: 10.1530/rep-08-0320
Wulff, C., Wiegand, S. J., Saunders, P. T., Scobie, G. A., and Fraser, H. M. (2001). Angiogenesis during follicular development in the primate and its inhibition by treatment with truncated Flt-1-Fc (vascular endothelial growth factor Trap(A40)). Endocrinology 142, 3244–3254. doi: 10.1210/endo.142.7.8258
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xu, T., Wu, J., Han, P., Zhao, Z., and Song, X. (2017). Circular RNA expression profiles and features in human tissues: a study using RNA-seq data. BMC Genomics 18(Suppl. 6):680. doi: 10.1186/s12864-017-4029-3
Yamamoto, N., Christenson, L. K., McAllister, J. M., and Strauss, J. F. III (2002). Growth differentiation factor-9 inhibits 3’5’-adenosine monophosphate-stimulated steroidogenesis in human granulosa and theca cells. J. Clin. Endocrinol. Metab. 87, 2849–2856. doi: 10.1210/jcem.87.6.8551
Yao, H. H., and Bahr, J. M. (2001). Chicken granulosa cells show differential expression of epidermal growth factor (EGF) and luteinizing hormone (LH) receptor messenger RNA and differential responsiveness to EGF and LH dependent upon location of granulosa cells to the germinal disc. Biol. Reprod. 64, 1790–1796. doi: 10.1095/biolreprod64.6.1790
Yao, X., Zhang, G., Guo, Y., Ei-Samahy, M., Wang, S., Wan, Y., et al. (2017). Vitamin D receptor expression and potential role of vitamin D on cell proliferation and steroidogenesis in goat ovarian granulosa cells. Theriogenology 102, 162–173. doi: 10.1016/j.theriogenology.2017.08.002
Yoshimura, Y., and Koga, O. (1982). Ultrastructural changes of the stigma of the follicle during the process of ovulation in the hen. Cell Tissue Res. 224, 349–359. doi: 10.1007/bf00216878
You, X., Vlatkovic, I., Babic, A., Will, T., Epstein, I., Tushev, G., et al. (2015). Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 18, 603–610. doi: 10.1038/nn.3975
Yu, J., Lou, Y., and Zhao, A. (2016). Transcriptome analysis of follicles reveals the importance of autophagy and hormones in regulating broodiness of Zhedong white goose. Sci. Rep. 6:36877. doi: 10.1038/srep36877
Zhang, X., Yan, Y., Lei, X., Li, A., Zhang, H., Dai, Z., et al. (2017). Circular RNA alterations are involved in resistance to avian leukosis virus subgroup-J-induced tumor formation in chickens. Oncotarget 8, 34961–34970. doi: 10.18632/oncotarget.16442
Zhang, Y. L., Xia, Y., Yu, C., Richards, J. S., Liu, J., and Fan, H. Y. (2014). CBP-CITED4 is required for luteinizing hormone-triggered target gene expression during ovulation. Mol. Hum. Reprod. 20, 850–860. doi: 10.1093/molehr/gau040
Zhou, L., Chen, J., Li, Z., Li, X., Hu, X., Huang, Y., et al. (2010). Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One 5:e15224. doi: 10.1371/journal.pone.0015224
Zhu, G., Kang, L., Wei, Q., Cui, X., Wang, S., Chen, Y., et al. (2014). Expression and regulation of MMP1, MMP3, and MMP9 in the chicken ovary in response to gonadotropins, sex hormones, and TGFB1. Biol. Reprod. 90:57. doi: 10.1095/biolreprod.113.114249
Keywords: follicle, transcriptome, circRNA, WGCNA, hub genes, ESR1
Citation: Shen M, Li T, Chen F, Wu P, Wang Y, Chen L, Xie K, Wang J and Zhang G (2020) Transcriptomic Analysis of circRNAs and mRNAs Reveals a Complex Regulatory Network That Participate in Follicular Development in Chickens. Front. Genet. 11:503. doi: 10.3389/fgene.2020.00503
Received: 03 December 2019; Accepted: 23 April 2020;
Published: 15 May 2020.
Edited by:
Enrique Medina-Acosta, State University of the North Fluminense Darcy Ribeiro, BrazilReviewed by:
Xiquan Zhang, South China Agricultural University, ChinaTruong Anh Duc, National Institute of Veterinary Research (NIVR), Vietnam
Copyright © 2020 Shen, Li, Chen, Wu, Wang, Chen, Xie, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinyu Wang, anl3YW5nQHl6dS5lZHUuY24=; Genxi Zhang, emd4MTU4OEAxMjYuY29t