 Guang-Can Zhou
Guang-Can Zhou Wen Li1
Wen Li1 Yan-Mei Zhang
Yan-Mei Zhang- 1College of Agricultural and Biological Engineering (College of Tree Peony), Heze University, Heze, China
- 2Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China
- 3State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, China
Nucleotide-binding site (NBS)-type disease resistance genes (R genes) play key roles in plant immune responses and have co-evolved with pathogens over the course of plant lifecycles. Comparative genomic studies tracing the dynamic evolution of NBS-encoding genes have been conducted using many important plant lineages. However, studies on Sapindaceae species have not been performed. In this study, a discrepant number of NBS-encoding genes were identified in the genomes of Xanthoceras sorbifolium (180), Dinnocarpus longan (568), and Acer yangbiense (252). These genes were unevenly distributed and usually clustered as tandem arrays on chromosomes, with few existed as singletons. The phylogenetic analysis revealed that NBS-encoding genes formed three monophyletic clades, RPW8-NBS-LRR (RNL), TIR-NBS-LRR (TNL), and CC-NBS-LRR (CNL), which were distinguished by amino acid motifs. The NBS-encoding genes of the X. sorbifolium, D. longan, and A. yangbiense genomes were derived from 181 ancestral genes (three RNL, 23 TNL, and 155 CNL), which exhibited dynamic and distinct evolutionary patterns due to independent gene duplication/loss events. Specifically, X. sorbifolium exhibited a “first expansion and then contraction” evolutionary pattern, while A. yangbiense and D. longan exhibited a “first expansion followed by contraction and further expansion” evolutionary pattern. However, further expansion in D. longan was stronger than in A. yangbiense after divergence, suggesting that D. longan gained more genes in response to various pathogens. Additionally, the ancient and recent expansion of CNL genes generated the dominance of this subclass in terms of gene numbers, while the low copy number status of RNL genes was attributed to their conserved functions.
Introduction
Nucleotide-binding site (NBS)-encoding genes are the largest type (∼80%) of disease resistance genes (R genes) found in plants and are responsible for the protection against various pathogens (Liu et al., 2007; Dangl et al., 2013). A typical NBS-encoding gene consists of a variable domain at the N-terminal, a highly conserved NBS domain in the middle, and a diverse leucine-rich repeat (LRR) domain at the C-terminal. Therefore, according to the structure of N-terminal domains that possess a coiled-coil (CC), Drosophila toll and mammalian interleukin-1 receptor-like (TIR), or resistance to powdery mildew8 (RPW8) domain, NBS-encoding genes can be classified into three subclasses: CC-NBS-LRR (CNL), TIR-NBS-LRR (TNL), and RPW8-NBS-LRR (RNL) (Meyers et al., 2003; Shao et al., 2014, 2016, 2019; Zhang et al., 2016, 2020; Qian et al., 2017; Xue et al., 2020). As an example of function, the CNL gene, RPP8, in Arabidopsis thaliana provides resistance against downy mildew after Peronospora parasiticia infection (Mcdowell et al., 1998). Another CNL gene, Pik, confers resistance to rice blast caused by Magnaporthe grisea infection (Zhai et al., 2011). The tobacco TNL gene, N, prevents tobacco mosaic virus invasion (Whitham et al., 1994). Functionally, CNL and TNL genes act as detectors that recognize specific pathogen effectors encoded by avirulence genes and initiate downstream hypersensitive reactions in the resistance pathway (Dangl and Jones, 2001; Mchale et al., 2006), while RNL genes appear to function downstream and transduce signals from CNL or TNL genes through interactions with corresponding partners (Bonardi et al., 2011; Collier et al., 2011; Tamborski and Krasileva, 2020). Therefore, NBS-encoding R genes are of great importance to plant growth and would tangibly benefit humankind if properly used in disease resistance breeding.
NBS-encoding genes constitute a large gene family found in plant genomes. With the accumulation of more plant whole-genome sequences, genome-wide evolutionary analyses of NBS-encoding genes have been performed in many plants since the first comprehensive study on NBS-encoding genes in A. thaliana (Meyers et al., 2003). Furthermore, comparative genomic studies on the NBS-encoding gene family have been performed among a few closely related plant species, which exhibited different evolutionary patterns. For example, frequent gene losses and limited gene duplications resulted in small number of NBS-encoding genes in three Cucurbitaceae species (Lin et al., 2013). Both Li et al. (2010) and Luo et al. (2012) investigated the NBS-encoding genes in four Poaceae species, including rice, maize, sorghum, and brachypodium, which exhibited a “contraction” evolutionary pattern and may have been caused by gene losses or frequent gene deletions and translocations. Similar analyses were performed on Fabaceae, Rosaceae, and Brassicaceae species, of which Fabaceae and Rosaceae species exhibited a “consistent expansion” evolutionary pattern (Shao et al., 2014; Jia et al., 2015), while five Brassicaceae species exhibited a “first expansion and then contraction” evolutionary pattern (Zhang et al., 2016). Moreover, although plants belonged to the same family, the evolutionary patterns of NBS-encoding genes were also diverse. For example, in three Solanaceae crop species, pepper exhibited a “contraction” pattern, tomato showed a “first expansion and then contraction” pattern, and potato presented a “consistent expansion” pattern (Qian et al., 2017). In four orchid species, Phalaenopsis equestris and Dendrobium catenatum exhibited an “early contraction to recent expansion” evolutionary pattern, while Gastrodia elata and Apostasia shenzhenica showed a “contraction” evolutionary pattern (Xue et al., 2020). Recently, a large scale analysis of the NBS-encoding genes in 22 representative angiosperms demonstrated that CNL genes exhibited a “gradual expansion” evolutionary pattern during the first 100 million years of angiosperm evolution, then underwent intense expansion along with TNL genes, which corresponded with the explosion of fungal diversity (Shao et al., 2016).
The soapberry family (Sapindaceae) consists of 135 genera and ∼1500 species, which is comprised mostly of trees or shrubs and some herbaceous climbers, which are widely distributed throughout the tropics and subtropics, especially in Southeastern Asia (Flora of China1). Sapindaceae species possess many great economic uses. For example, the seed kernels of Yellowhorn (Xanthoceras sorbifolium), a major woody oil plant species, contains as much as 67% oil (Venegas-Calerón et al., 2017), and extractions from its husks improve learning and memory that could be used to treat Alzheimer’s disease (Ji et al., 2017; Zhang et al., 2018). Longan (Dinnocarpus longan), an important evergreen fruit tree, is mainly grown in Southern China and serves as a source of traditional medicine and timber (Chung et al., 2010; Mei et al., 2014). Acer yangbiense is a charismatic landscape plant that possesses a colorful foliage is a newly described critically endangered endemic maple tree found in Southwestern China (Chen et al., 2003), and it possesses many bioactive compounds (Bi et al., 2016; Yang et al., 2019).
Recently, the high-quality genome sequences of X. sorbifolium, D. longan, and A. yangbiense were made available and all recovered >94% complete BUSCO genes by BUSCO analysis (Lin et al., 2017; Bi et al., 2019; Yang et al., 2019). An analysis of the NBS-encoding genes in D. longan was performed, and researchers found high number and recent expansions/contractions of these genes that may be attributed to the genomic basis for insect, fungus, and bacteria resistance (Lin et al., 2017). However, these findings require further elucidation of this phenomenon caused by gene duplication/loss events. Additionally, the systematic evaluation and comparison of NBS-encoding genes at the genome level in more Sapindaceae species is needed to obtain a better understanding of this family’s molecular evolutionary history. In this study, X. sorbifolium, D. longan, and A. yangbiense genome sequence data were utilized to perform comparative genomic analyses in order to uncover the evolutionary features and patterns of NBS-encoding genes in the Sapindaceae family, as well as further investigate the mechanisms of these evolutionary changes.
Materials and Methods
Identification and Classification of the NBS-Encoding Genes
The whole genomes of the X. sorbifolium, D. longan, and A. yangbiense were used in this study (Supplementary Figure S1). Genomic sequences and annotation files were obtained from the GigaScience database (X. sorbifolium2, Bi et al., 2019; D. longan3, Lin et al., 2017; A. yangbiense4, Yang et al., 2019). The method for NBS-encoding gene identification was described in a previous study (Shao et al., 2016). Briefly, BLAST and hidden Markov model (HMM) searches were simultaneously conducted using the NB-ARC domain (Pfam accession No.: PF00931) as the query sequence to identify candidate NBS-encoding genes in the three genomes. The threshold expectation value was set to 1.0 for the BLAST search. For the HMM search5, default settings were used. Then, the identified candidate sequences were merged and the redundant hits were removed. In order to confirm the presence of the NBS domain, the remaining sequence hits were subjected to online Pfam analysis6 with an E-value of 10–4. All of the identified NBS-encoding genes were subjected to NCBI’s conserved domain database7 using the default settings to determine whether they encoded CC, TIR, RPW8, or LRR domains.
Chromosomal Distribution and Cluster Arrangement of Identified NBS-Encoding Genes
The chromosomal locations of all identified NBS-encoding genes in Sapindaceae genomes were determined by retrieving relevant information from the downloaded gff files. Gene cluster was determined according to the criterion used for Medicago truncatula (Ameline-Torregrosa et al., 2008): if two neighboring NBS-encoding genes were located within 250 kb on a chromosome, these two genes were regarded as members of the same gene cluster. Based on this criterion, the NBS-encoding genes in Sapindaceae genomes were assigned to clustered loci and singleton loci, which were mapped along the chromosomes.
Sequence Alignment and Conserved Motif Analysis of the NBS Domain
Amino acid sequences of the NBS domain were extracted from the identified NBS-encoding genes and used for multiple alignments using ClustalW integrated in MEGA 7.0 using the default settings (Kumar et al., 2016). Sequences that were too short (<190 amino acids, less than two-thirds of a regular NBS domain) or too divergent were removed to prevent interference with the alignments and subsequent phylogenetic analysis. The resulting alignments were manually corrected and improved using MEGA 7.0. The conserved protein motifs within the NBS domain of the three subclasses of NBS-encoding genes were analyzed by Multiple Expectation Maximization for Motif Elicitation (MEME) and WebLogo using the default settings (Crooks et al., 2004; Bailey et al., 2006). Additionally, structural motif annotation was performed using the Pfam analysis8 and SMART tools9.
Phylogenetic and Gene Loss/Duplication Analysis of the NBS-Encoding Genes
To explore the relationships of NBS-encoding genes in the three Sapindaceae genomes, a phylogenetic tree was constructed based on the amino acid sequences of the conserved NBS domain using the NBS-encoding genes of A. thaliana as a reference. The NBS-encoding genes of A. thaliana were identified using the same method, and these NBS-encoding genes were also used in a previous study (Zhang et al., 2011). Amino acid sequences were aligned as described above. The phylogenetic tree was constructed using IQ-TREE and the maximum likelihood method based on the best-fit model estimated by ModelFinder (Nguyen et al., 2015; Kalyaanamoorthy et al., 2017); branch support values were assessed using UFBoot2 tests (Minh et al., 2013). The short or divergent sequences that were removed from the phylogenetic analyses were BLASTp searched against all of the identified NBS genes to identify their potential phylogenetic positions by identifying their closest relatives. Additionally, in order to identify the gene duplication/loss events during the speciation of the three Sapindaceae species, the NBS-encoding gene phylogenetic tree was reconciled with the real species tree using Notung software (Stolzer et al., 2012). The NBS-encoding genes on the tree that formed a monophyletic branch and originated from one ancestral gene inherited from the common ancestor of the three Sapindaceae species was defined as a Sapindaceae lineage gene. One or more Sapindaceae lineage genes that formed a monophyletic branch with the A. thaliana NBS-encoding genes were inherited from a common ancestor of the three Sapindaceae species and A. thaliana, and were defined as Malvids lineage genes.
Synteny Analyses Across/Within Sapindaceae Genomes and Gene Duplication Type Determination
The MCScanX package (Lee et al., 2012; Wang et al., 2012) was used to identify syntenic blocks within a genome or between different genomes through pair-wise all-against-all blast of protein sequences. The purposes of synteny analysis were to explore the pattern of conservation of NBS-encoding gene loci among the Sapindaceae genomes and determine the types of NBS-encoding gene duplication. Synteny relationship of NBS-encoding genes was displayed by TBtools10.
Results
Identification and Classification of the NBS-Encoding Genes
A total of 180, 252, and 568 non-redundant NBS-encoding genes were identified from the genomes of X. sorbifolium, A. yangbiense, and D. longan, respectively (Table 1 and Supplementary Table S1), accounting for 0.73, 0.89, and 1.83% of the 24,672, 28,320, and 31,007 annotated protein-coding genes in X. sorbifolium, A. yangbiense, and D. longan genomes, respectively (Lin et al., 2017; Bi et al., 2019; Yang et al., 2019). The average lengths of NBS-encoding genes in X. sorbifolium, D. longan, and A. yangbiense were 6081, 5569, and 6199 bp, while the average lengths of the coding region of NBS-encoding genes in X. sorbifolium, D. longan, and A. yangbiense were 3082, 3132, and 3142 bp, respectively. D. longan possessed the largest number of NBS-encoding genes and was 3.16- and 2.25-times greater than X. sorbifolium and A. yangbiense, respectively. The identified NBS-encoding genes from the three Sapindaceae species were divided into the CNL, TNL, and RNL subclasses based on their domain compositions and primary phylogenies. Among the three subclassses, CNL genes overwhelmingly outnumbered TNL and RNL genes with proportions of 81.11, 89.26, and 89.68% in X. sorbifolium, D. longan, and A. yangbiense, respectively. The proportion of TNL genes was 17.78, 9.15, and 7.94% in X. sorbifolium, D. longan, and A. yangbiense, respectively, while the number of RNL genes was the smallest with only two, nine, and six genes, respectively. Not all of the identified NBS-encoding genes had intact structures possessing all three domains (CC/TIR/RNL) with many either lacking the CC/TIR/RPW8 domain at the N-terminus, the LRR domain at the C-terminus, or domains at both termini (Table 1). Additionally, some identified NBS-encoding genes were classified as “other” in TNL and CNL due to their atypical structural domain compositions (Table 1). For example, the D. longan genome encoded two “other” genes in TNL, including LNTIRL and TLTN, and 13 “other” genes in CNL, including CNLNL(2), NCCLNCCLNCCL(1), NCCNCCLNCCL(1), NLCNL(1), CNLCNL(4), CNLN(1), NCCNCCL(2), and CNN(1).
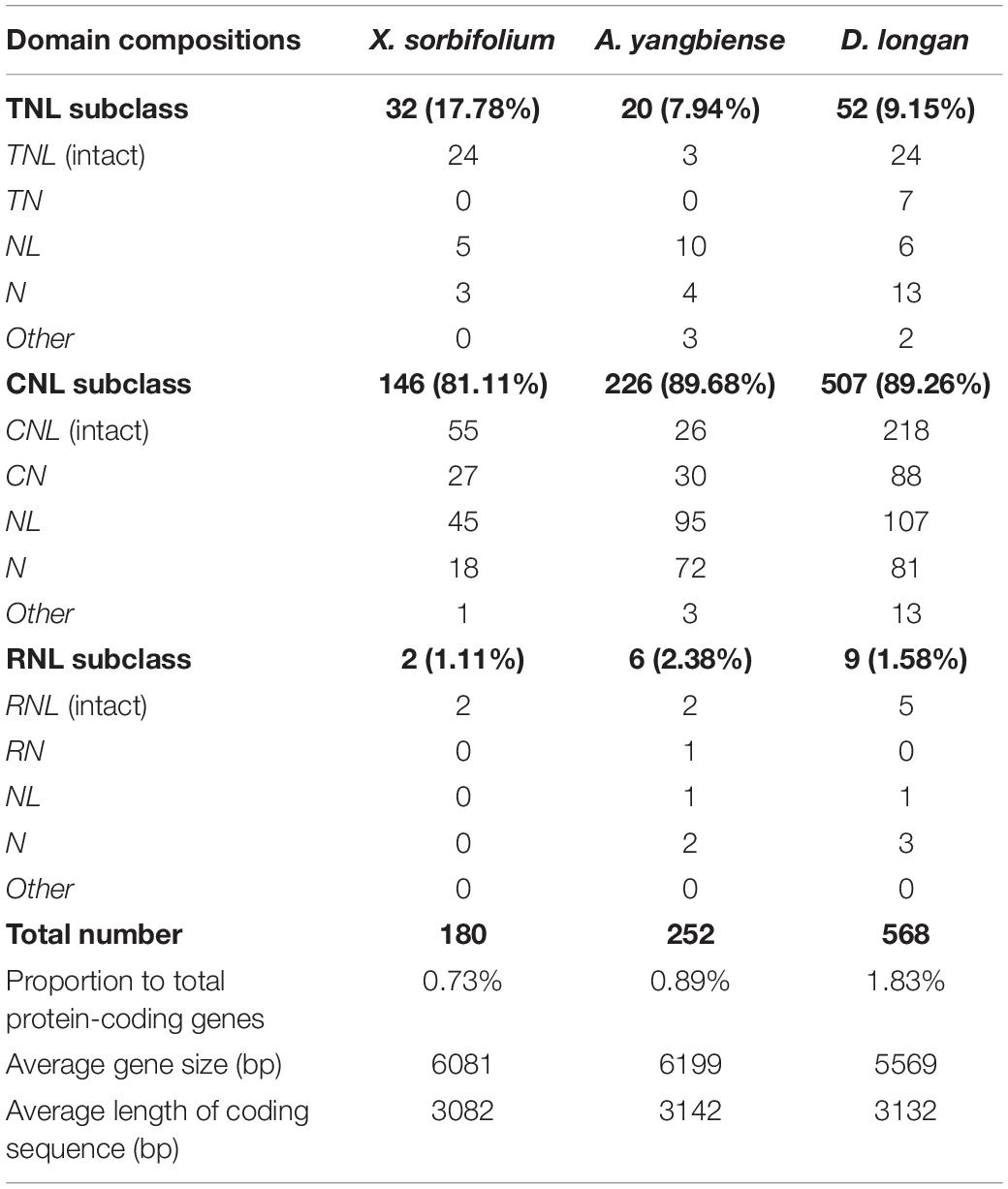
Table 1. The number of identified NBS-encoding genes in the three Sapindaceae genomes.
Distribution and Organization of NBS-Encoding Genes in Sapindaceae Genomes
The NBS-encoding genes were unevenly distributed among different chromosomes. For example, Chrom (chromosome) 9 of X. sorbifolium genome contains the most genes (42 genes), whereas the Chrom 1 and 11 contain the fewest (only two genes in each Chrom) (Supplementary Figure S3). In A. yangbiense, Chrom 3 contains the most genes (89 genes), whereas no NBS-encoding gene was detected on Chrom 11 (Supplementary Figure S4). Uneven distributions were also observed among the three subclasses of NBS-encoding genes (Supplementary Figures S3, S4). Chrom 9 (41 genes) of X. sorbifolium and Chrom 3 (86 genes) of A. yangbiense contained the most CNL genes, whereas Chrom 4 (20 genes) of X. sorbifolium and Chrom 2 (five genes) and 8 (five genes) of A. yangbiense contained the most TNL genes. All chromosomes of the two species contained CNL genes except Chrom 10 and 11 of A. yangbiense, whereas only seven chromosomes of X. sorbifolium and six chromosomes of A. yangbiense contain TNL genes. There were too few RNL genes for this analysis. The majority of NBS-encoding genes were organized into clusters rather than singletons in X. sorbifolium and A. yangbiense genomes, and their ratios were 2.98 and 5.31, respectively (Table 2). A. yangbiense contained more clustered genes than X. sorbifolium (207 genes vs 131 genes), and the largest gene cluster of X. sorbifolium and A. yangbiense were on Chrom 14 (14 genes) and Chrom 3 (21 genes), respectively. Since the NBS-encoding genes in D. longan genome were not anchored to chromosomes yet, the chromosomal locations and cluster assignments of these genes were not examined.

Table 2. Organization of NBS-encoding genes in the three Sapindaceae genomes.
Motif Analysis of the NBS Domain
The NBS domain consists of several functional motifs that were conserved and strictly ordered among the NBS-encoding genes (Yue et al., 2012). A total of six conserved motifs were identified in each subclass of the NBS domains in the three Sapindaceae species using MEME and WebLogo (Crooks et al., 2004; Bailey et al., 2006). From the N-terminus to the C-terminus, these motifs included the P-loop, Kinase-2, Kinase-3, RNBS-C, GLPL, and RNBS-D motifs. Comparisons of the amino acid sequences of these motifs are presented in Figure 1. The P-loop, Kinase-2, Kinase-3, and GLPL motifs exhibited high similarity among the three subclasses of NBS-encoding genes, suggesting that the NBS domains with critical functions regulating the immune responses were homologous. The other two motifs, especially RNBS-D, were poorly conserved among the three subclasses of NBS-encoding genes. The variation of these motifs may be responsible for further functional divergences within these subclasses. The subclass-specific signatures within the six motifs were further analyzed, which could be used as preliminary labels to identify CNL, TNL, or RNL genes. For example, Tryptophan (W) at the seventh position of the RNBS-C and Cysteine (C) at the seventh position of RNBS-D in CNL genes, phenylalanine (F) at the 11th position of the RNBS-D in TNL genes, and serine (S) at the fourth position of the P-loop and glutamic acid (E) at the second position of the RNBS-D in RNL genes. Additionally, the amino acids at the final position of Kinase-2 could also be used to distinguish TNL genes from CNL and RNL genes (Figure 1). Therefore, the subclass of a given NBS-encoding gene could be classified based on the amino acid signatures of the motif sequences.
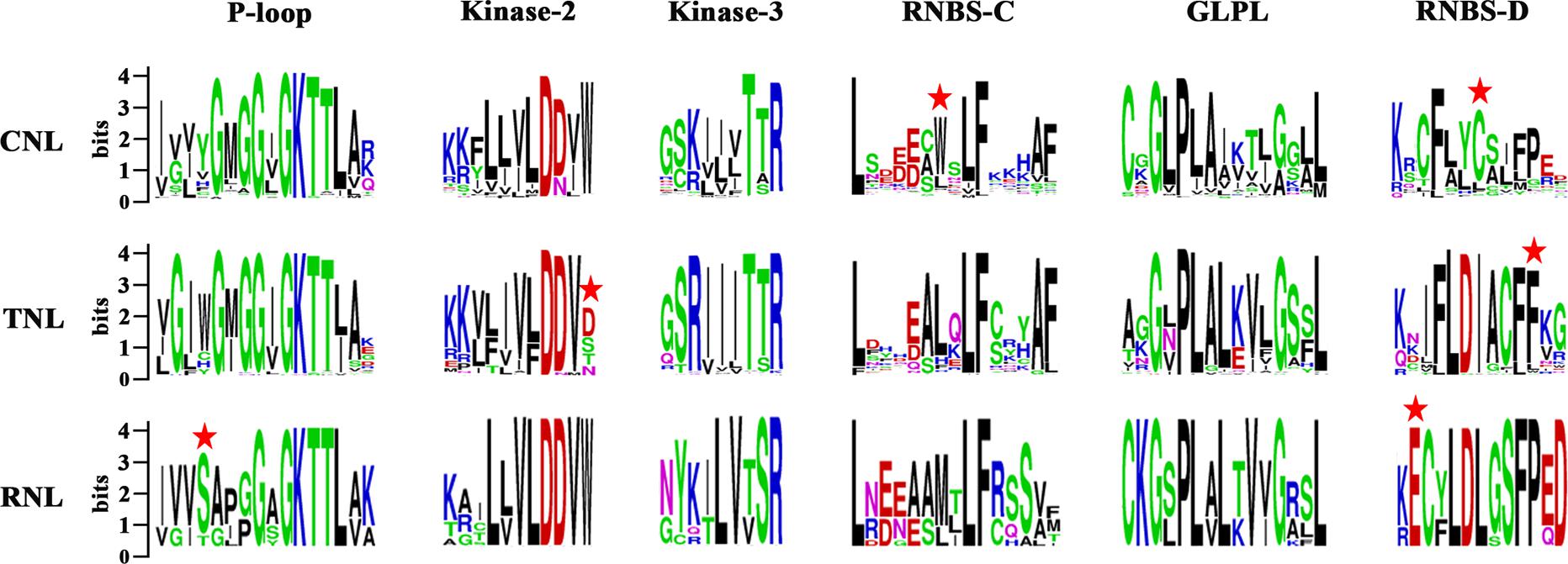
Figure 1. Six conserved motifs of NBS domains in the three Sapindaceae species. Amino acids of the six conserved motifs were extracted. Different conserved amino acids among CNL, TNL, and RNL subclass genes are labeled with a red star. Details of the amino acids of the whole NBS domain are presented in Supplementary Figure S2.
Phylogenetic Analysis of the NBS-Encoding Genes
To reconstruct the phylogenetic relationship of the NBS-encoding genes in Sapindaceae, a phylogenetic tree was constructed based on the amino acid sequences of NBS domain alignments using the NBS-encoding genes of A. thaliana as a reference. To attain better phylogeny, too short or extremely divergent NBS domains were removed from the data matrix. A total of 957 genes (X. sorbifolium, 159; D. longan, 468; A. yangbiense, 173; A. thaliana, 157) were obtained and used to reconstruct the evolutionary history of the NBS-encoding genes. The phylogenetic tree was composed of three monophyletic clades, RNL, TNL, and CNL, with support values > 99%; many internal nodes had high (>70%) support values (Figure 2 and Supplementary Figure S5). The three clades represented the divergence of RNL, TNL, and CNL genes. Compared to TNL and CNL, the branch lengths of RNL genes were short (Figure 2), suggesting that they had a low evolutionary rate.
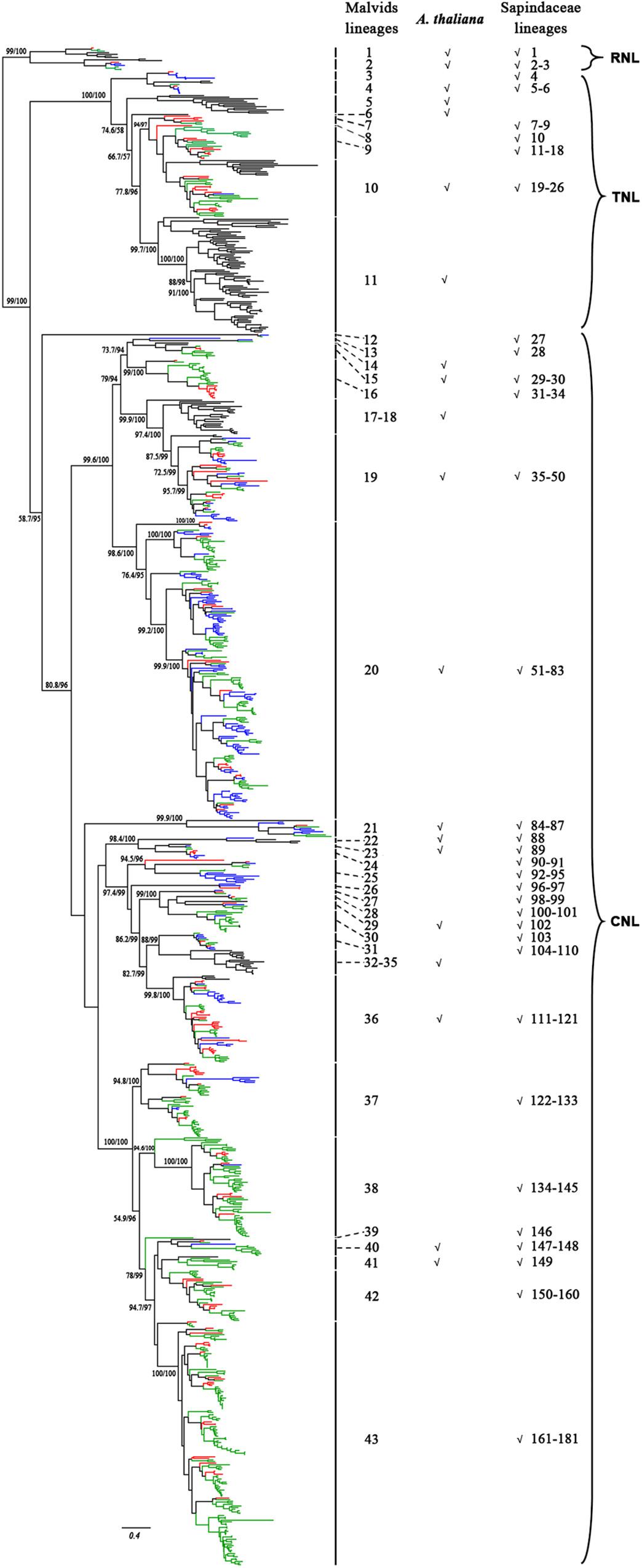
Figure 2. Phylogenetic relationships of the NBS-encoding genes in X. sorbifolium, A. yangbiense, and D. longan based on the amino acids of conserved NBS domains. Red, blue, green, and black lines represent the NBS-encoding genes in X. sorbifolium, A. yangbiense, D. longan, and A. thaliana, respectively. Support values > 70% for basal nodes are shown. The reconstructed phylogeny was divided into 43 Malvids and 181 Sapindaceae lineage NBS-encoding genes. The first column number 1–43 represents the Malvids lineage NBS-encoding genes, and the last column number 1–181 represents the Sapindaceae lineage NBS-encoding genes. The presence of Malvids lineage NBS-encoding genes in A. thaliana and the common ancestor of the three Sapindaceae species is indicated by “√.” The scale bar indicated amino acid substitutions/sites. The detailed phylogenetic tree of the NBS-encoding genes, including gene names, evolutionary relationships among genes, and supporting values of all of the nodes, is presented in Supplementary Figure S5.
To gain insight on the evolution of the NBS-encoding genes before and after divergence of A. thaliana and the three Sapindaceae species, Notung software was used to reconcile gene duplication/loss events of the NBS-encoding genes at each node of the phylogenetic tree (Stolzer et al., 2012). Based on the definitions (refer to section “Materials and Methods” for details), 43 Malvids lineage genes were retrospected (Figure 2). The ratio of RNL:TNL:CNL genes was 2:9:32. Among them, only 13 genes (two RNL, two TNL, and nine CNL) were reserved by both A. thaliana and the common ancestor of the three Sapindaceae species, while 19 genes (four TNL, 15 CNL) were lost by A. thaliana and 10 genes (three TNL, seven CNL) were lost by the common ancestor.
A total of 33 Malvids lineage genes (two RNL, six TNL, 25 CNL) were inherited by the common ancestor of the three Sapindaceae species and these genes intensively expanded to 181 Sapindaceae lineage genes (three RNL, 23 TNL, 155 CNL) (Figure 2). CNL genes exhibited a more active expansion rate than RNL or TNL genes, especially the CNL Malvids lineage 19, 20, and 43 expanded 16, 33, and 21 Sapindaceae lineages, respectively (Figure 2). Further analysis revealed that 93, 122, and 58 of 181 Sapindaceae lineages were inherited by X. sorbifolium, D. longan, and A. yangbiense, respectively (Figure 3), while only eight lineages were reserved in all three genomes. A total of 76 Sapindaceae lineages (X. sorbifolium and D. longan, 42; D. longan and A. yangbiense, 16; X. sorbifolium and A. yangbiense, 18) were maintained in only two genomes and 97 lineages were species-specific (X. sorbifolium, 25; D. longan, 56; A. yangbiense, 16). The distribution patterns suggest that these ancestral gene lineages have experienced differential gene duplication/loss events when the three Sapindaceae species diverged.
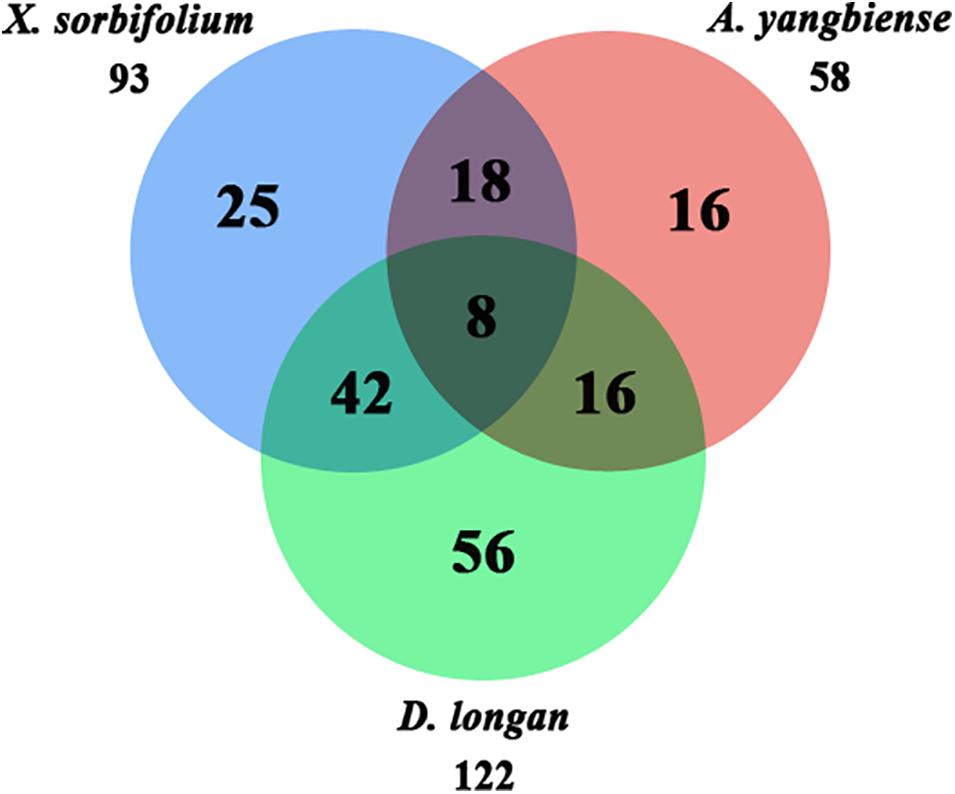
Figure 3. Ancestral Sapindaceae linage genes in the X. sorbifolium, A. yangbiense, and D. longan genomes.
Syntenic Analysis of NBS-Encoding Genes in Sapindaceae Genomes
The synteny analysis was performed between and within the three Sapindaceae genomes (the D. longan genome was presented as scaffold form). A total of 33 syntenic NBS-encoding genes were detected between X. sorbifolium and A. yangbiense genome (Figure 4A and Supplementary Table S2). Among them, the number of syntenic CNL and RNL subclass genes was 32 and one, respectively, whereas no syntenic TNL subclass genes were detected. The CNL, TNL, and RNL subclass syntenic genes were all detected between X. sorbifolium and D. longan genome (Figure 4B and Supplementary Table S2), and the gene number was 26, 2, and 1, respectively. However, there were only 16 CNL subclass syntenic genes were detected between A. yangbiense and D.longan (Figure 4C and Supplementary Table S2). Moreover, there were only seven syntenic NBS-encoding genes preserved by the three genomes and they all belong to the CNL subclass (Supplementary Table S2). The syntenic NBS-encoding genes revealed three different forms: singleton to singleton, singleton to cluster, and cluster to cluster. These forms were resulted from different extents of gene duplication/loss of the ancestor genes. The large number of NBS-encoding genes showing presence/absent (P/A) polymorphism and asymmetric gene numbers among co-linear loci indicate that the NBS-encoding genes have experienced dramatic gain and loss during the evolution of Sapindaceae species.
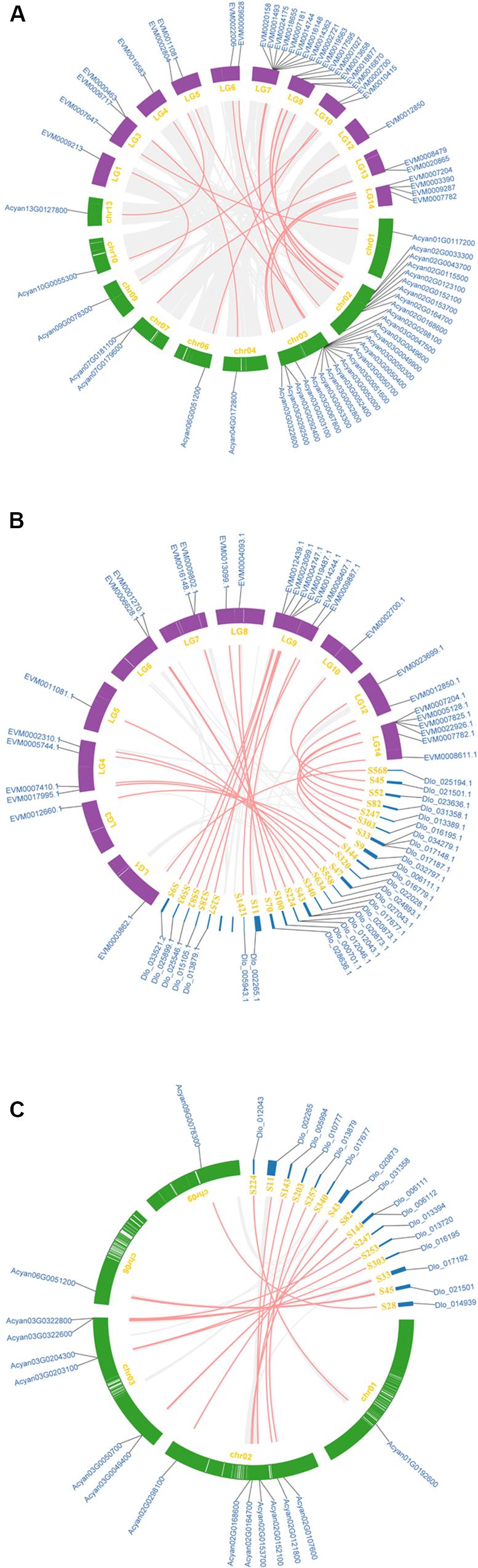
Figure 4. Synteny of the three Sapindaceae species NBS-encoding genes. (A) Synteny of X. sorbifolium and A. yangbiense NBS-encoding genes. (B) Synteny of X. sorbifolium and D. longan NBS-encoding genes. (C) Synteny of A. yangbiense and D. longan NBS-encoding genes. LG1-15, X. sorbifolium chromosomes; chr01-13, A. yangbiense chromosomes; S, D. longan scaffolds. Red and gray lines represent synteny NBS-encoding genes and non-NBS-encoding genes, respectively.
There were three types of NBS-encoding gene duplications: local tandem duplication, dispersed (or ectopic) duplication, and whole genome duplications (WGDs) or segmental duplication (Leister, 2004). MCScanX software was adopted to determine the type of gene duplications in producing NBS-encoding genes during the evolution of Sapindaceae species. As shown in Table 3, tandem and dispersed duplication were the main contributors to gene expansion events in X. sorbifolium and A. yangbiense genomes. However, the proportion of segmental duplicated genes might have been underestimated because of the syntenic relationship of NBS-encoding genes would be disrupted during long-term evolution. The gene duplication types of the NBS-encoding genes in D. longan genome were not examined, since these genes were not anchored to chromosomes yet.
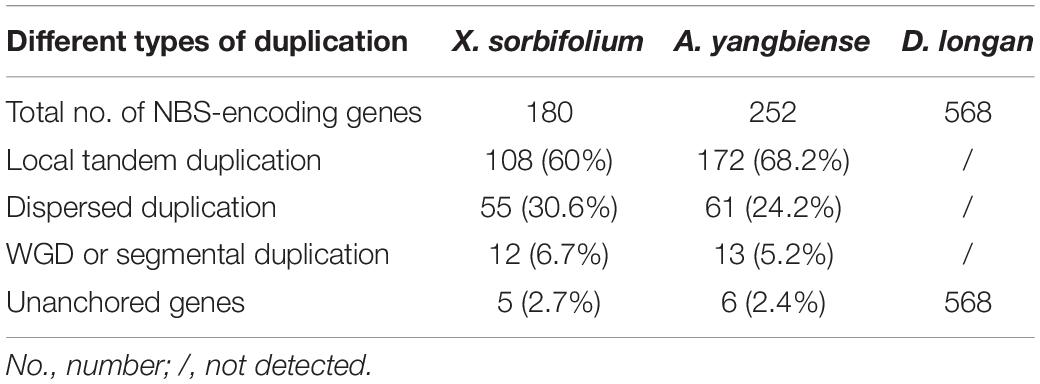
Table 3. Contributions of three duplication types in producing NBS-encoding genes during the evolution of Sapindaceae species.
Gene Duplications/Losses Resulting in Dynamic Evolutionary Patterns of the NBS-Encoding Genes
To assess the evolutionary patterns of the NBS-encoding genes during the speciation of the three Sapindaceae species, the gene duplication/loss events of the NBS-encoding genes were restored in the phylogenetic tree based on the conserved NBS domain sequences. The retrospective analysis revealed that 43 ancient NBS-encoding genes were shared by the common ancestor of A. thaliana and the three Sapindaceae species (Figure 2). Among these ancient genes, 33 were inherited and duplicated into 181 genes in the common ancestor of the three Sapindaceae species. These 181 ancestral Sapindaceae genes experienced different evolutionary patterns, which were reflected by species-specific gene duplication/loss events during the speciation of X. sorbifolium, D. longan, and A. yangbiense. X. sorbifolium duplicated 66 genes (57 CNL, nine TNL) and lost 88 genes (80 CNL, seven TNL, one RNL) during speciation (Figures 5A,B and Supplementary Figure S6), resulting in a slight decrease in the number of the NBS-encoding genes in its genome. Similarly, the common ancestor of D. longan and A. yangbiense after diverging from X. sorbifolium lost 25 genes (18 CNL, seven TNL) and duplicated 12 genes (11 CNL, one TNL), resulting in a decrease in the total number of genes (168). However, D. longan and A. yangbiense underwent considerably different evolutionary processes after divergence (Figures 5A,B and Supplementary Figure S6). Specifically, D. longan duplicated 340 genes (321 CNL, 16 TNL, three RNL), but lost 40 genes (37 CNL, two TNL, one RNL), thus, the gene number clearly increased in its genome. A. yangbiense duplicated 106 genes (97 CNL, eight TNL, one RNL) and lost 101 genes (85 CNL, 14 TNL, two RNL), resulting in a slight increase in the number of NBS-encoding genes in its genome. Therefore, the NBS-encoding genes in the three Sapindaceae species exhibited dynamic and distinct evolutionary patterns.
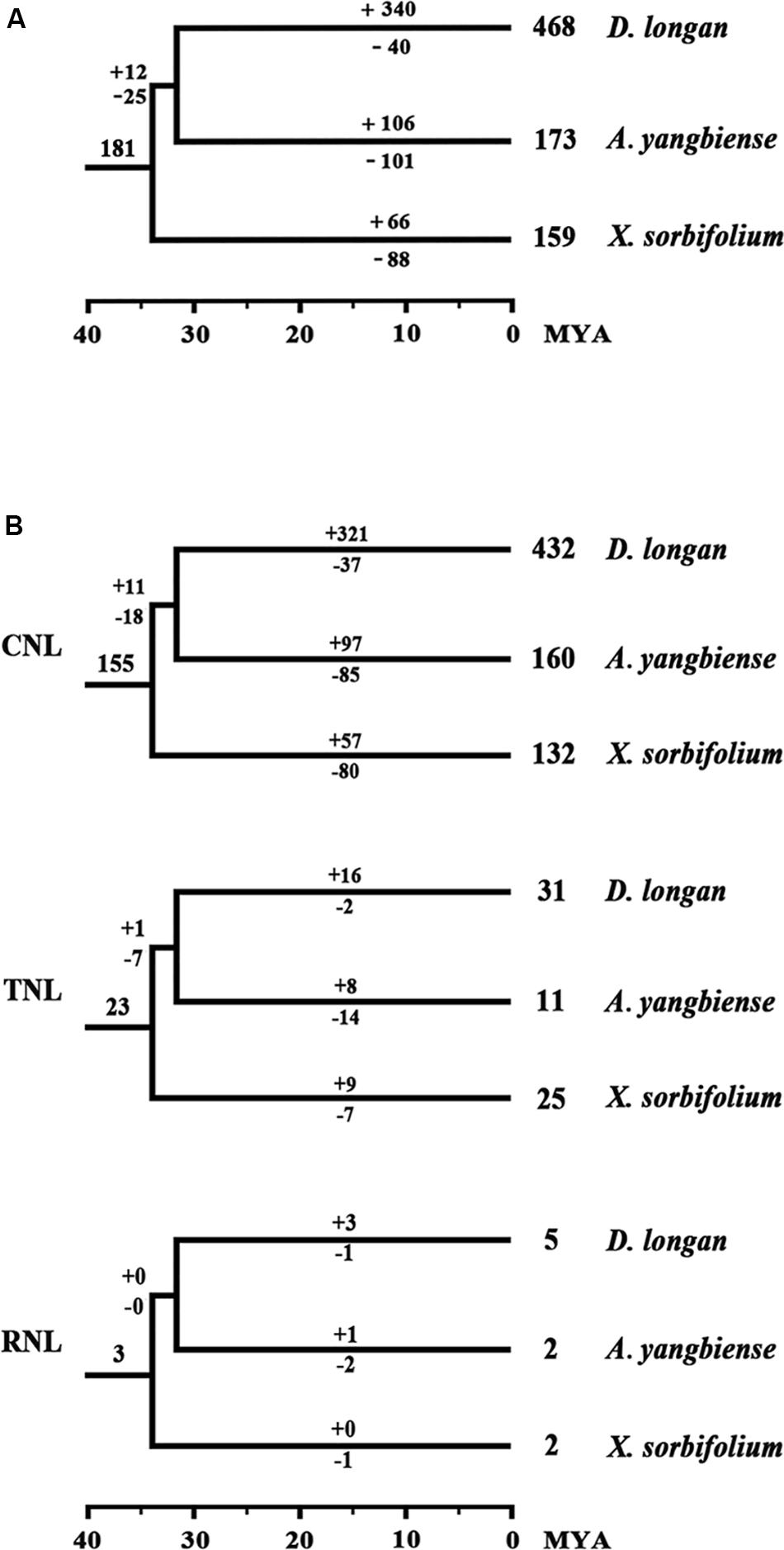
Figure 5. Duplication/loss events of the NBS-encoding genes during the speciation of X. sorbifolium, A. yangbiense, and D. longan. (A) Differential gene duplication and loss events of NBS-encoding genes. (B) Duplication/loss events of CNL, TNL, and RNL genes. Gene duplication/loss events are indicated by numbers with “+” or “−” on each branch, respectively. Detailed information for the duplication/loss events of the NBS-encoding genes are presented in Supplementary Figure S6.
Discussion
Copy Number Variation of the NBS-Encoding Genes Among Different Species
The NBS-encoding gene family is one of the most divergent plant genome families with high copy number discrepancies among plant lineages/species (Jacob et al., 2013). For example, several NBS-encoding gene sequences were identified in some orchid species genomes (Xue et al., 2020), which possess the smallest number of genes observed thus far. Dozens of NBS-encoding genes have been identified in papaya, melon, cucumber, and watermelon (Lin et al., 2013; Zhang et al., 2016). Hundreds of gene copies in Arabidopsis, rice, potato, tomato, pepper, cotton, soybean, grape, poplar, barley, and sunflower have also been identified (Yang et al., 2008; Li et al., 2010; Guo et al., 2011; Wei et al., 2013; Shao et al., 2014; Wu et al., 2014; Andersen et al., 2016; Qian et al., 2017; Neupane et al., 2018). Moreover, 1219 and 1303 copies were identified in wheat and apple genomes, respectively (Jia et al., 2013, 2015). In this study, the gene number variation among the three Sapindaceae species was also quite large, with 180, 568, and 252 genes identified in X. sorbifolium, D. longan, and A. yangbiense, respectively. The proportion of NBS-encoding genes to all predicted genes in the three Sapindaceae genomes (0.73–1.83%) was higher than the proportions reported for Cucurbitaceae species (0.19–0.27%), similar to Rosaceae (0.78–2.05%) and Solanaceae (0.73–1.15%) species (Jia et al., 2015; Qian et al., 2017). The discrepancy of NBS-encoding genes among the three species should not be the result of ploidy variation, because all of the three analyzed species are diploids and have similar chromosomal numbers (Lin et al., 2017; Bi et al., 2019; Yang et al., 2019). Actually, a previous study has shown that no correlation exists between the NBS-encoding genes and species phylogeny or genome size (Jacob et al., 2013). For example, in Fabaceae, soybean (465) possessed fewer NBS-encoding genes than M. truncatula (571), although the genomes size of the former was twice that of the latter (Shao et al., 2014). In Brassicaceae, the numbers of NBS-encoding genes were similar across some species even though these species exhibited variations in genome size (Zhang et al., 2016).
The A. yangbiense genome was sequenced using the combined Pacific Biosciences Single-molecule Real-time, Illumina HiSeq X, and Hi-C technologies, and BUSCO analysis recovered 95.5% complete BUSCO genes (Yang et al., 2019); the X. sorbifolium genome was sequenced using the combined Illumina HiSeq, Pacific Biosciences Sequel, and Hi-C technologies, and BUSCO analysis recovered 94.7% complete BUSCO genes (Bi et al., 2019); for D. longan genome sequencing: using the standard Illumina library preparation protocols, and BUSCO analysis recovered 94% complete BUSCO genes (Lin et al., 2017). Additionally, the assembly quality of D. longan genome sequence was assessed by aligning the scaffolds to a D. longan transcriptome assembly from the NCBI Sequence Read Archive (SRA; SRA050205). Of the 96,251 D. longan transcriptome sequences reported previously (Lai and Lin, 2013), 97.55% were identified in the genome assembly (Lin et al., 2017). Although all of the three analyzed genomes are of high quality (all recovered > 94% complete BUSCO genes) according to the BUSCO analysis of the genome papers (Lin et al., 2017; Bi et al., 2019; Yang et al., 2019), the D. longan genome assembly has not reached a chromosomal level. A previous study revealed that genome sequence and annotation quality may affect the NBS-encoding gene identification (Bayer et al., 2018). This raises a possibility that the fragmented assembled genome of D. longan may have contributed to its high NBS-encoding gene number by splitting of one NBS-encoding gene into multiple genes during annotation. However, this possibility was largely rejected by coding sequence length analysis, which revealed that the average coding sequence length of NBS-encoding genes in D. longan is larger than that of X. sorbifolium. Furthermore, a close look at NBS-encoding gene profiles in the three species revealed that the number of intact NBS-encoding genes in D. longan (247) is even larger than the total NBS-encoding gene number in X. sorbifolium (180) and close to the total NBS-encoding gene number in A. yangbiense. Therefore, the relatively larger number of NBS-encoding genes in D. longan is more likely a consequence of evolution rather than other artificial reasons.
The rapid evolutionary feature of the NBS-encoding gene family caused by frequent gene duplications/loss events could elicit copy number discrepancies. The phylogenetic analysis and ancestral gene classification results of the NBS-encoding genes indicated that species-specific gene duplication/loss events occurred after species diverged from the common ancestor (Figures 2, 5A and Supplementary Figure S6). Lots of D. longan-specific lineage genes were found in D. longan and these specific lineage genes experienced recent sharp expansions by more frequent gene duplications and less gene loss events (+340 vs. -40) after the divergence of D. longan from A. yangbiense (Figure 4). The differences between gene duplication/loss events in X. sorbifolium (+66 vs. -88) or A. yangbiense (+106 vs. -101) were not as large (Figure 5A). Therefore, a copy number discrepancy of NBS-encoding genes was observed among the three Sapindaceae species. The NBS-encoding gene number variations caused by independent gene duplication/loss events were common in Cucurbitaceae, Fabaceae, Rosaceae, Poaceae, Brasssicaceae, Solanaceae, and orchid species (Li et al., 2010; Lin et al., 2013; Shao et al., 2014; Jia et al., 2015; Zhang et al., 2016; Qian et al., 2017; Xue et al., 2020). The high copy number of NBS-encoding genes that resulted from recent expansions in D. longan could represent candidate resistance genes that responded to various pathogens, such as witches broom disease (Lai et al., 2000; Guo, 2002). From another perspective, high copy number of NBS-encoding genes might be a disadvantage for plants in the absence of corresponding pathogens due to the fitness cost of resistance genes (Tian et al., 2003). There should be some balancing mechanisms between resistance and fitness cost provided by the large number of NBS-encoding genes in D. longan genome.
Chromosomal distribution analysis of NBS-encoding genes suggested an uneven distribution pattern among different chromosomes (Supplementary Figure S3, S4). The uneven distributing pattern was commonly observed in other lineages, like legume, Brassicaceae, Solanaceae species, Vitis vinifera, and Populus trichocarpa (Yang et al., 2008; Shao et al., 2014; Zhang et al., 2016; Qian et al., 2017). Tandem duplication and dispersed duplication played major roles in the NBS-encoding gene expansion in X. sorbifolium and A. yangbiense genomes (Table 3). Random dispersed gene duplications and gene losses likely brought out the uneven distributions of these genes on different chromosomes, and this difference was made more apparent through local tandem duplications. This strategy made the NBS-encoding gene form clusters to overcome the limitations of R gene diversity during the coevolution of plants and pathogens (Michelmore and Meyers, 1998; Le Roux et al., 2015).
The dynamic evolution of NBS-encoding genes in the three Sapindaceae species and A. thaliana was traced back to different time periods by reconciling ancient genes in the common ancestor of the three Sapindaceae species and the common ancestor of Sapindaceae and A. thaliana. In total, 43 ancient Malvids lineage genes were restored in the common ancestor of Sapindaceae and A. thaliana (Figure 2). Among these ancient genes, 10 were lost by the Sapindaceae common ancestor before the three Sapindaceae species diverged. The remaining 33 Malvids lineage genes were inherited and duplicated into 181 genes in the common ancestor of the three Sapindaceae species. These genes experienced further species-specific evolution during speciation and resulted in the currently observed NBS-encoding genes of the three genomes. Over time, the ancient NBS-encoding genes in the common ancestor of Sapindaceae and A. thaliana experienced different evolutionary patterns during the speciation of the three Sapindaceae species (Figure 6). X. sorbifolium exhibited a “first expansion and then contraction” evolutionary pattern (Figure 6I); more genes were duplicated in the common ancestor of Sapindaceae, but subsequently lost more genes during speciation. Although A. yangbiense and D. longan exhibited similar evolutionary patterns (Figures 6II,III), expansion followed by contraction and further expansion, subsequent expansion in D. longan was stronger and gained more genes than A. yangbiense after their divergence (Figure 5A). Furthermore, the evolutionary patterns of these three subclasses of NBS-encoding genes were also diverse among the three species. For example, the CNL and RNL genes exhibited similar patterns as the NBS-encoding genes of corresponding species (Figure 5B), while the patterns of TNL genes were altered in A. yangbiense (recent further contraction) and X. sorbifolium (recent further expansion) (Figure 5B). Such small changes in TNL genes had little effect on the overall trends of the NBS-encoding genes in A. yangbiense and X. sorbifolium due to the dominance of CNL genes (see discussion below). Therefore, distinct and dynamic evolutionary patterns of the NBS-encoding genes caused by independent gene duplication/loss events appeared during the speciation of the three Sapindaceae species.
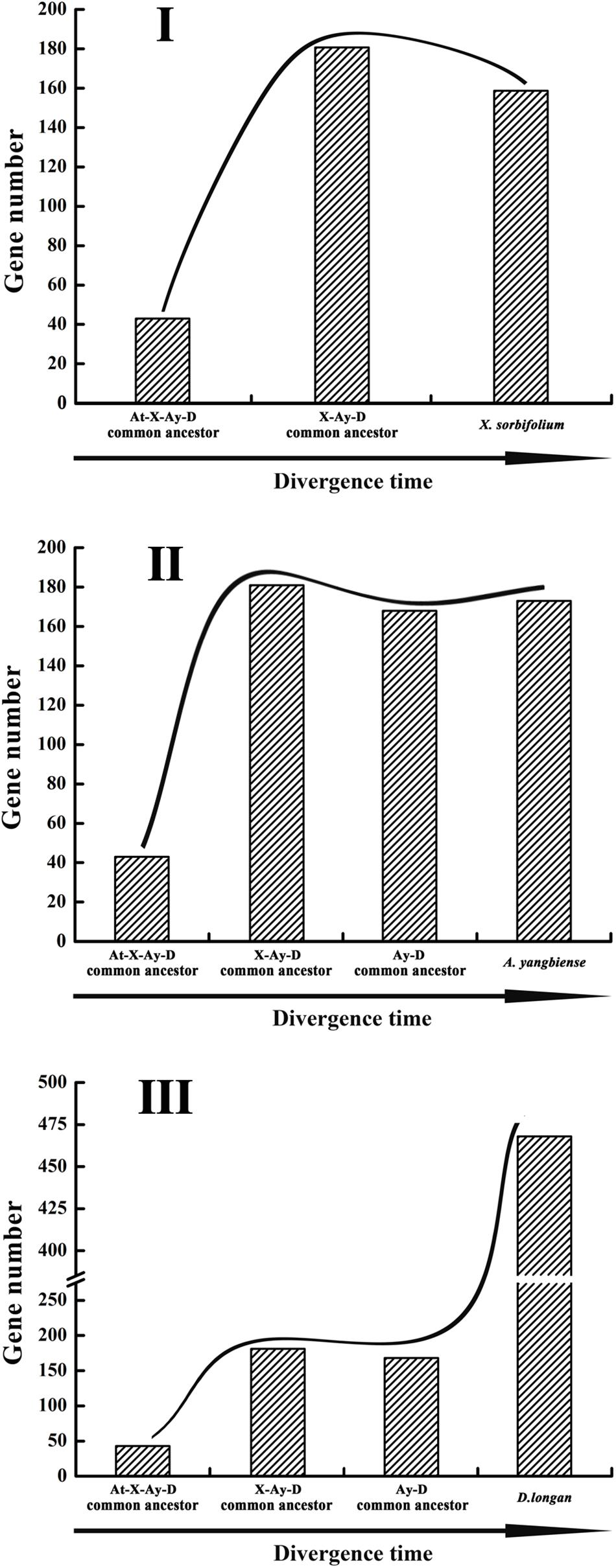
Figure 6. Evolutionary patterns of the NBS-encoding genes in the three Sapindaceae genomes. Gene number variation from the A. thaliana, X. sorbifolium, A. yangbiense, and D. longan (At-X-Ay-D) ancestor to X. sorbifolium (I), A. yangbiense (II), and D. longan (III).
Numbers of Subclass Genes and the Causes
The number of CNL subclass genes in X. sorbifolium, D. longan, and A. yangbiense was considerably higher than TNL and RNL genes, accounting for 81.11, 89.26, and 89.68% of the NBS-encoding genes in each species, respectively. The greater number of CNL subclass genes is a common phenomenon observed among plant species, except for the Brassicaceae (Zhang et al., 2016). For example, CNL subclass genes are the majority in the genome of Glycine max (57.8%), Phaseolus vulgaris (66.5%), V. vinifera (82.8%), Capsicum annuum (94.1%), Solanum lycopersicum (87.0%), Solanum tuberosum (83.7%), and Amborella trichopoda (84.8%) (Yang et al., 2008; Shao et al., 2014, 2016; Jia et al., 2015; Qian et al., 2017). Extremely, monocots have only CNL genes due to the loss of TNL genes near dicot/monocot differentiation (Li et al., 2010; Shao et al., 2016). Previously, it was reported that CNL genes underwent twice gene expansions and expanded earlier than TNL genes during angiosperm evolution (Shao et al., 2016). A total of 155 CNL, 23 TNL, and three RNL ancestral genes were reconciled in the common ancestor of the three Sapindaceae species. Therefore, when the common ancestor of the three Sapindaceae species emerged, its genome possessed more CNL genes than TNL or RNL genes. Although these ancestral genes have experienced frequent gene duplication/loss events during the divergence of the three Sapindaceae species, gene expansion in their common ancestor maintained the large number of CNL genes. Additionally, lots of recent expansions of CNL genes in the three Sapindaceae species may have given rise to the dominance of this subclass’s genes (Figures 2, 5 and Supplementary Figure S6). An extremely small number of RNL genes were previously reported in angiosperms (Shao et al., 2016; Zhang et al., 2016; Qian et al., 2017; Xue et al., 2020). In this study, two, six, and nine RNL genes were identified in X. sorbifolium, A. yangbiense, and D. longan, respectively. It was speculated that CNL and TNL genes were involved in recognizing specific pathogens, while RNL genes helped transduce signals in the downstream pathways of plant immunity (Bonardi et al., 2011; Collier et al., 2011; Tamborski and Krasileva, 2020). Therefore, RNL genes clearly participated in basic defense responses and without the divergent selection of pathogens, these genes did not necessarily expand in large numbers. Moreover, pairwise synteny analysis detected more syntenic CNL (74 genes) subclass genes than TNL (two genes) and RNL (one gene) subclass genes among the three Sapindaceae genomes (Figure 4 and Supplementary Table S2). These differences might be caused by the different evolutionary feature of the three subclass genes during angiosperm evolution: long-term contraction and fast evolution rate of TNL genes, gradual expansion of CNL genes, and conservative evolution of RNL genes (Zhang et al., 2011; Shao et al., 2016).
Notably, not all of the identified NBS-encoding genes in X. sorbifolium, A. yangbiense, and D. longan genomes had intact structures possessing all three domains. There were only 81 (24 TNL, 55 CNL, two RNL), 31 (three TNL, 26 CNL, two RNL), and 247 (24 TNL, 218 CNL, five RNL) NBS-encoding genes with intact structures in X. sorbifolium, A. yangbiense, and D. longan genomes (Table 1), respectively, accounting for 45.0, 12.3, and 43.5% of all identified NBS-encoding genes in these three species, respectively. Similarly, the relative small proportion of NBS-encoding genes with intact structures were also reported in C. annuum (23.2%), S. lycopersicum (42.7%), S. tuberosum (28.2%), P. trichocarpa (46.2%), M. truncatula (39.1%), Lotus japonicus (31.0%), and Oryza sativa (30.6%) genomes (Yang et al., 2008; Zhang et al., 2011; Shao et al., 2014; Qian et al., 2017). In plants, the highly conserved NBS domain has been demonstrated to be able to bind and hydrolyze ATP or GTP and act as molecular switches in immune signaling, while the LRR and N-terminal domains (TIR and CC) are typically involved in the recognition of, and the activation of, corresponding partners, respectively (Meyers et al., 1999; Dangl and Jones, 2001; Lukasik and Takken, 2009). Theoretically, in order to trigger immune responses and transfer defense signals, an NBS-encoding gene should possess an intact structure. However, transient overexpression results showed that those NBS-encoding genes without intact structures also function in plant immunity (Nandety et al., 2013; Kato et al., 2014).
Data Availability Statement
The datasets generated for this study can be found in the X. sorbifolium: http://dx.doi.org/10.5524/100606, D. longan: http://dx.doi.org/10.5524/100276, and A. yangbiense: http://dx.doi.org/10.5524/100610.
Author Contributions
G-CZ, WL, and Y-LW conceived and directed the project. G-CZ, WL, Y-MZ, and YL obtained and analyzed the data. Y-MZ, ML, MZ, YL, and G-QM conducted the phylogenetic analysis and constructed the discussion. G-CZ wrote the manuscript. All of the authors contributed to the discussion of the results, reviewed the manuscript, and approved of the final article.
Funding
This work was funded by the Doctoral Fund (No. XY19BS15) and the First Training Program of Institute of Peony of Heze University (provided to G-CZ), and Qingchuang Science and Technology Support Program of Shandong Provincial College. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00737/full#supplementary-material
FIGURE S1 | Phylogenetic relationship of X. sorbifolium, A. yangbiense, and D. longan. Time of divergence: million years ago (MYA) (Koenen et al., 2015; Liu et al., 2015; Lin et al., 2017; Bi et al., 2019; Yang et al., 2019).
FIGURE S2 | Details of the amino acid frequencies of the whole NBS domain of CNL, TNL, and RNL genes in the three Sapindaceae species (WebLogo).
FIGURE S3 | The chromosomal distribution of identified NBS-encoding genes in the X. sorbifolium genome.
FIGURE S4 | The chromosomal distribution of identified NBS-encoding genes in the A. yangbiense genome.
FIGURE S5 | The detailed ML tree of the NBS-encoding genes. Sequence names and supporting values are presented on all of the nodes. The tree was reconstructed based on the amino acids of the NBS domain of the NBS-encoding genes in X. sorbifolium, A. yangbiense, and D. longan using NBS-encoding genes in the A. thaliana genome were used as a reference.
FIGURE S6 | The reconciled NBS-encoding gene tree with real species phylogeny and restored various duplication/loss events. “n4” indicates a loss event that occurred in the common ancestor of X. sorbifolium, A. yangbiense, and D. longan. “n2” indicates a loss event that occurred in the common ancestor of A. yangbiense and D. longan.
TABLE S1 | A list of NBS-encoding genes identified from the three Sapindaceae genomes.
TABLE S2 | Detailed information of the synteny NBS-encoding genes in three Sapindaceae genomes.
Footnotes
- ^ www.iplant.cn/foc/
- ^ http://dx.doi.org/10.5524/100606
- ^ http://dx.doi.org/10.5524/100276
- ^ http://dx.doi.org/10.5524/100610
- ^ http://hmmer.org
- ^ http://pfam.sanger.ac.uk/
- ^ https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^ http://pfam.janelia.org/
- ^ http://smart.embl-heidelberg.de/
- ^ https://github.com/CJ-Chen/TBtools
References
Ameline-Torregrosa, C., Wang, B. B., Obleness, M., Deshpande, S., Zhu, H., Roe, B. A., et al. (2008). Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21. doi: 10.1104/pp.107.104588
Andersen, E. J., Ali, S., Reese, R. N., Yen, Y., Neupane, S., and Nepal, M. P. (2016). Diversity and evolution of disease resistance genes in barley (Hordeum vulgare L.). Evolut. Bioinform. 12:S38085.
Bailey, T. L., Williams, N., Misleh, C., and Li, W. W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373.
Bayer, P. E., Edwards, D., and Batley, J. (2018). Bias in resistance gene prediction due to repeat masking. Nat. Plants 4, 762–765. doi: 10.1038/s41477-018-0264-0
Bi, Q., Zhao, Y., Du, W., Lu, Y., Gui, L., Zheng, Z., et al. (2019). Pseudomolecule-level assembly of the Chinese oil tree yellowhorn (Xanthoceras sorbifolium) genome. GigaScience 8:giz070.
Bi, W., Gao, Y., Shen, J., He, C., Liu, H., Peng, Y., et al. (2016). Traditional uses, phytochemistry, and pharmacology of the genus Acer (maple): a review. J. Ethnopharmacol. 189, 31–60. doi: 10.1016/j.jep.2016.04.021
Bonardi, V., Tang, S., Stallmann, A., Roberts, M., Cherkis, K., and Dangl, J. L. (2011). Expanded functions for a family of plant intracellular immune receptors beyond specific recognition of pathogen effectors. Proc. Natl. Acad. Sci. U.S.A. 108, 16463–16468. doi: 10.1073/pnas.1113726108
Chen, Y., Yang, Q., and Zhu, G. (2003). Acer yangbiense (Aceraceae), a new species from Yunnan, China. Novon 13, 296–299.
Chung, Y. C., Lin, C. C., Chou, C. C., and Hsu, C. P. (2010). The effect of Longan seed polyphenols on colorectal carcinoma cells. Eur. J. Clin. Investigat. 40, 713–721. doi: 10.1111/j.1365-2362.2010.02322.x
Collier, S. M., Hamel, L.-P., and Moffett, P. (2011). Cell death mediated by the N-terminal domains of a unique and highly conserved class of NB-LRR protein. Mol. Plant Microbe Interact. 24, 918–931. doi: 10.1094/mpmi-03-11-0050
Crooks, G. E., Hon, G., Chandonia, J.-M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Dangl, J. L., Horvath, D. M., and Staskawicz, B. J. (2013). Pivoting the plant immune system from dissection to deployment. Science 341, 746–751. doi: 10.1126/science.1236011
Dangl, J. L., and Jones, J. D. (2001). Plant pathogens and integrated defence responses to infection. Nature 411, 826–833. doi: 10.1038/35081161
Guo, J.-H. (2002). A list of diseases on litchi and longan in China mainland. Subtrop. Plant Sci. 31, 48–50.
Guo, Y.-L., Fitz, J., Schneeberger, K., Ossowski, S., Cao, J., and Weigel, D. (2011). Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes in Arabidopsis. Plant Physiol. 157, 757–769. doi: 10.1104/pp.111.181990
Jacob, F., Vernaldi, S., and Maekawa, T. (2013). Evolution and conservation of plant NLR functions. Front. Immunol. 4:297.
Ji, X.-F., Chi, T.-Y., Liu, P., Li, L.-Y., Xu, J.-K., Xu, Q., et al. (2017). The total triterpenoid saponins of Xanthoceras sorbifolia improve learning and memory impairments through against oxidative stress and synaptic damage. Phytomedicine 25, 15–24. doi: 10.1016/j.phymed.2016.12.009
Jia, J., Zhao, S., Kong, X., Li, Y., Zhao, G., He, W., et al. (2013). Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95.
Jia, Y., Yuan, Y., Zhang, Y., Yang, S., and Zhang, X. (2015). Extreme expansion of NBS-encoding genes in Rosaceae. BMC Genet. 16:48.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14:587. doi: 10.1038/nmeth.4285
Kato, H., Saito, T., Ito, H., Komeda, Y., and Kato, A. (2014). Overexpression of the TIR-X gene results in a dwarf phenotype and activation of defense-related gene expression in Arabidopsis thaliana. J. Plant Physiol. 171, 382–388. doi: 10.1016/j.jplph.2013.12.002
Koenen, E. J., Clarkson, J. J., Pennington, T. D., and Chatrou, L. W. (2015). Recently evolved diversity and convergent radiations of Rainforest mahoganies (Meliaceae) shed new light on the origins of rainforest hyperdiversity. New Phytol. 207, 327–339. doi: 10.1111/nph.13490
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lai, Z., Chen, C., Zeng, L., and Chen, Z. (2000). “Somatic embryogenesis in longan [Dimocarpus longan Lour.],” in Somatic Embryogenesis in Woody Plants. Forestry Sciences, Vol. 67, eds S. M. Jain, P. K. Gupta, and R. J. Newton (Dordrecht: Springer), 415–431. doi: 10.1007/978-94-017-3030-3_13
Lai, Z., and Lin, Y. (2013). Analysis of the global transcriptome of longan (Dimocarpus longan Lour.) embryogenic callus using Illumina paired-end sequencing. BMC Genomics 14:561. doi: 10.1186/1471-2164-14-561
Le Roux, C., Huet, G., Jauneau, A., Camborde, L., Tremousaygue, D., Kraut, A., et al. (2015). A receptor pair with an integrated decoy converts pathogen disabling of transcription factors to immunity. Cell 161, 1074–1088. doi: 10.1016/j.cell.2015.04.025
Lee, T., Tang, H., Wang, X., and Paterson, A. H. (2012). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41, 1152–1158.
Leister, D. (2004). Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance genes. Trends Genet. 20, 116–122. doi: 10.1016/j.tig.2004.01.007
Li, J., Ding, J., Zhang, W., Zhang, Y., Tang, P., Chen, J.-Q., et al. (2010). Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol. Genet. Genom. 283, 427–438. doi: 10.1007/s00438-010-0527-6
Lin, X., Zhang, Y., Kuang, H., and Chen, J. (2013). Frequent loss of lineages and deficient duplications accounted for low copy number of disease resistance genes in Cucurbitaceae. BMC Genom. 14:335. doi: 10.1186/1471-2164-14-335
Lin, Y., Min, J., Lai, R., Wu, Z., Chen, Y., Yu, L., et al. (2017). Genome-wide sequencing of longan (Dimocarpus longan Lour.) provides insights into molecular basis of its polyphenol-rich characteristics. Gigascience 6:gix023.
Liu, B., Ye, J., Liu, S., Wang, Y., Yang, Y., Lai, Y., et al. (2015). Families and genera of Chinese angiosperms: a synoptic classification based on APG III. Biodiv. Sci. 23, 225–231. doi: 10.17520/biods.2015052
Liu, J., Liu, X., Dai, L., and Wang, G. (2007). Recent progress in elucidating the structure, function and evolution of disease resistance genes in plants. J. Genet. Genom. 34, 765–776. doi: 10.1016/s1673-8527(07)60087-3
Lukasik, E., and Takken, F. L. W. (2009). STANDing strong, resistance proteins instigators of plant defence. Curr. Opin. Plant Biol. 12, 427–436. doi: 10.1016/j.pbi.2009.03.001
Luo, S., Zhang, Y., Hu, Q., Chen, J., Li, K., Lu, C., et al. (2012). Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol. 159, 197–210. doi: 10.1104/pp.111.192062
Mcdowell, J. M., Dhandaydham, M., Long, T. A., Aarts, M. G., Goff, S., Holub, E. B., et al. (1998). Intragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of Arabidopsis. Plant Cell 10, 1861–1874. doi: 10.1105/tpc.10.11.1861
Mchale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7:212.
Mei, Z., Fu, S., Yu, H., Yang, L., Duan, C., Liu, X., et al. (2014). Genetic characterization and authentication of Dimocarpus longan Lour. using an improved RAPD technique. Genet. Mol. Res. 13, 1447–1455. doi: 10.4238/2014.march.6.3
Meyers, B. C., Dickerman, A. W., Michelmore, R. W., Sivaramakrishnan, S., Sobral, B. W. S., and Young, N. D. (1999). Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J. 20, 317–332. doi: 10.1046/j.1365-313x.1999.t01-1-00606.x
Meyers, B. C., Kozik, A., Griego, A., Kuang, H., and Michelmore, R. W. (2003). Genome-wide analysis of NBS-LRR–encoding genes in Arabidopsis. Plant Cell 15, 809–834. doi: 10.1105/tpc.009308
Michelmore, R. W., and Meyers, B. C. (1998). Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 8, 1113–1130. doi: 10.1101/gr.8.11.1113
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evolut. 30, 1188–1195. doi: 10.1093/molbev/mst024
Nandety, R. S., Caplan, J. L., Cavanaugh, K. A., Perroud, B., Wroblewski, T., Michelmore, R. W., et al. (2013). The role of TIR-NBS and TIR-X proteins in plant basal defense responses. Plant Physiol. 162, 1459–1472. doi: 10.1104/pp.113.219162
Neupane, S., Andersen, E. J., Neupane, A., and Nepal, M. P. (2018). Genome-wide identification of NBS-encoding resistance genes in sunflower (Helianthus annuus L.). Genes 9:384. doi: 10.3390/genes9080384
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evolut. 32, 268–274. doi: 10.1093/molbev/msu300
Qian, L.-H., Zhou, G.-C., Sun, X.-Q., Lei, Z., Zhang, Y.-M., Xue, J.-Y., et al. (2017). Distinct patterns of gene gain and loss: diverse evolutionary modes of NBS-encoding genes in three Solanaceae crop species. G3 Genes Genomes Genetics 7, 1577–1585. doi: 10.1534/g3.117.040485
Shao, Z.-Q., Xue, J.-Y., Wang, Q., Wang, B., and Chen, J.-Q. (2019). Revisiting the origin of plant NBS-LRR genes. Trends Plant Sci. 24, 9–12. doi: 10.1016/j.tplants.2018.10.015
Shao, Z.-Q., Xue, J.-Y., Wu, P., Zhang, Y.-M., Wu, Y., Hang, Y.-Y., et al. (2016). Large-scale analyses of angiosperm nucleotide-binding site-leucine-rich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns. Plant Physiol. 170, 2095–2109. doi: 10.1104/pp.15.01487
Shao, Z.-Q., Zhang, Y.-M., Hang, Y.-Y., Xue, J.-Y., Zhou, G.-C., Wu, P., et al. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol. 166, 217–234. doi: 10.1104/pp.114.243626
Stolzer, M., Lai, H., Xu, M., Sathaye, D., Vernot, B., and Durand, D. (2012). Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28, i409–i415. doi: 10.1093/bioinformatics/bts386
Tamborski, J., and Krasileva, K. V. (2020). Evolution of plant NLRs: from natural history to precise modifications. Ann. Rev. Plant Biol. 71, 355–378. doi: 10.1146/annurev-arplant-081519-035901
Tian, D., Traw, M. B., Chen, J., Kreitman, M., and Bergelson, J. (2003). Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423, 74–77. doi: 10.1038/nature01588
Venegas-Calerón, M., Ruíz-Méndez, M. V., Martínez-Force, E., Garcés, R., and Salas, J. J. (2017). Characterization of Xanthoceras sorbifolium Bunge seeds: lipids, proteins and saponins content. Industr. Crops Product. 109, 192–198. doi: 10.1016/j.indcrop.2017.08.022
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:E49.
Wei, H., Li, W., Sun, X., Zhu, S., and Zhu, J. (2013). Systematic analysis and comparison of nucleotide-binding site disease resistance genes in a diploid cotton Gossypium raimondii. PLoS One 8:e68435. doi: 10.1371/journal.pone.0068435
Whitham, S., Dinesh-Kumar, S. P., Choi, D., Hehl, R., Corr, C., and Baker, B. (1994). The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell 78, 1101–1115. doi: 10.1016/0092-8674(94)90283-6
Wu, P., Shao, Z.-Q., Wu, X.-Z., Wang, Q., Wang, B., Chen, J.-Q., et al. (2014). Loss/retention and evolution of NBS-encoding genes upon whole genome triplication of Brassica rapa. Gene 540, 54–61. doi: 10.1016/j.gene.2014.01.082
Xue, J.-Y., Zhao, T., Liu, Y., Liu, Y., Zhang, Y.-X., Zhang, G.-Q., et al. (2020). Genome-wide analysis of the nucleotide binding site leucine-rich repeat genes of four orchids revealed extremely low numbers of disease resistance genes. Front. Genet. 10:1286.
Yang, J., Wariss, H. M., Tao, L., Zhang, R., Yun, Q., Hollingsworth, P., et al. (2019). De novo genome assembly of the endangered Acer yangbiense, a plant species with extremely small populations endemic to Yunnan Province, China. GigaScience 8:giz085.
Yang, S., Zhang, X., Yue, J.-X., Tian, D., and Chen, J.-Q. (2008). Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genom. 280, 187–198. doi: 10.1007/s00438-008-0355-0
Yue, J. X., Meyers, B. C., Chen, J. Q., Tian, D., and Yang, S. (2012). Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS−LRR) genes. New Phytol. 193, 1049–1063. doi: 10.1111/j.1469-8137.2011.04006.x
Zhai, C., Lin, F., Dong, Z., He, X., Yuan, B., Zeng, X., et al. (2011). The isolation and characterization of Pik, a rice blast resistance gene which emerged after rice domestication. New Phytol. 189, 321–334. doi: 10.1111/j.1469-8137.2010.03462.x
Zhang, X., Feng, Y., Cheng, H., Tian, D., Yang, S., and Chen, J. (2011). Relative evolutionary rates of NBS-encoding genes revealed by soybean segmental duplication. Mol. Genet. Genom. 285, 79–90. doi: 10.1007/s00438-010-0587-7
Zhang, Y., Xiao, L., Xiao, B., Yin, M., Gu, M., Zhong, R., et al. (2018). Research progress and application prospect of Xanthoceras sorbifolia for treating Alzheimer’s disease. Drug Eval. Res. 25, 912–917.
Zhang, Y.-M., Chen, M., Wang, Y., Yin, J., Liu, J., Sun, X.-Q., et al. (2020). Genome-wide identification and evolutionary analysis of NBS-LRR genes from Dioscorea rotundata. Front. Genet. 11:484.
Keywords: Sapindaceae, NBS-encoding gene, phylogeny, gene duplication/loss, evolutionary pattern
Citation: Zhou G-C, Li W, Zhang Y-M, Liu Y, Zhang M, Meng G-Q, Li M and Wang Y-L (2020) Distinct Evolutionary Patterns of NBS-Encoding Genes in Three Soapberry Family (Sapindaceae) Species. Front. Genet. 11:737. doi: 10.3389/fgene.2020.00737
Received: 12 March 2020; Accepted: 19 June 2020;
Published: 10 July 2020.
Edited by:
Madhav P. Nepal, South Dakota State University, United StatesReviewed by:
Anna Maria Mastrangelo, Council for Agricultural and Economics Research (CREA), ItalyJacqueline Batley, The University of Western Australia, Australia
Copyright © 2020 Zhou, Li, Zhang, Liu, Zhang, Meng, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Can Zhou, emdjYW4yMDA5QDE2My5jb20=; Yi-Lei Wang, d2FuZ3lpbGVpMDAxQDE2My5jb20=