 Wei-Bo Niu1,2,3†
Wei-Bo Niu1,2,3† Mei-Rong Bai1,2,3†Huan-Lei Song1,2,3Yan-Jiao Lu1,2,3Wen-Jie Wu1,2,3Yi-Ming Gong1,2,3
Mei-Rong Bai1,2,3†Huan-Lei Song1,2,3Yan-Jiao Lu1,2,3Wen-Jie Wu1,2,3Yi-Ming Gong1,2,3 Xian-Xian Yu1,2,3Zhi-Liang Wei1,2,3Wen-Wen Yu1,2,3Bei-Lin Gu1,2,3Wei Cai1,2,3*
Xian-Xian Yu1,2,3Zhi-Liang Wei1,2,3Wen-Wen Yu1,2,3Bei-Lin Gu1,2,3Wei Cai1,2,3* Xun Chu1,2,3*
Xun Chu1,2,3*- 1Department of Pediatric Surgery, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 2Shanghai Key Laboratory of Pediatric Gastroenterology and Nutrition, Shanghai, China
- 3Shanghai Institute of Pediatric Research, Shanghai, China
Background and Aims: Hirschsprung’s disease (HSCR) is a rare genetically heterogeneous congenital disorder. A recent study based on whole genome sequencing demonstrated that common variants at four novel loci, which contained two intronic variants on CASQ2 and PLD1, and intergenic variants located between SLC4A7 and EOMES at 3p24.1, and between LINC01518 and LOC283028 at 10q11.21, were associated with HSCR susceptibility. To validate these associations with HSCR susceptibility, we performed a case–control study in a Han Chinese sample set.
Methods: We selected four previously identified single nucleotide polymorphisms (SNPs) for replication, along with tag SNPs to cover the four associated regions. In total, 61 SNPs were genotyped in 420 HSCR patients and 1,665 healthy controls from the Han Chinese population.
Results: None of the 14 tag SNPs in the CASQ2 gene region, including the previously associated rs9428225, showed an association with HSCR. Among the 24 tag SNPs from the SLC4A7-EOMES region at 3p24.1, rs2642925 [odds ratio (OR) = 1.41, 95% confidence interval (95% CI) = 1.10–1.79; PAdditive = 0.007] and the previously associated SNP rs9851320 showed a suggestive association (OR = 1.22, 95% CI = 1.01–1.47; PAdditive = 0.042). A non-synonymous SNP, rs2287579, in PLD1 showed a suggestive association with HSCR susceptibility (OR = 1.71, 95% CI = 1.18–2.46; PAdditive = 0.004). Additionally, the previously associated PLD1 SNP rs12632766 showed a suggestive significance (OR = 1.20, 95% CI = 1.01–1.42, PAdditive = 0.038). In the LINC01518-LOC283028 region at 10q11.21, three SNPs meet the study-wide significance threshold. Rs17153309 was the most associated SNP (OR = 1.60, 95% CI = 1.34–1.90; PAdditive = 1.13 × 10–7). The previously associated SNP rs1414027 also showed significant association (OR = 1.43, 95% CI = 1.20–1.70, PAdditive = 3.92 × 10–5). Two associated SNPs at 10q11.21 (rs1414027 and rs624804) were expression quantitative trait loci in digestive tract tissues from GTEx databases.
Conclusions: Our results confirmed that variants of the LINC01518-LOC283028 region were associated with HSCR in the Han Chinese population. Additionally, the susceptibility of SNPs in the LINC01518-LOC283028 region were associated with the expression levels of nearby genes. These results provide new insight into the pathogenesis of HSCR.
Introduction
Hirschsprung’s Disease (HSCR) is a polygenic congenital disease (Wallace, 2011; Tilghman et al., 2019) characterized by the absence of enteric neurons in the variable length of the distal gastrointestinal tract (Heuckeroth, 2018). Children with HSCR manifest a functional intestinal obstruction. Constipation, chronic abdominal distension, vomiting, repeated enterocolitis, loss of appetite, and delayed growth and development caused by HSCR result in heavy burdens for individuals, families, and society (Amiel et al., 2008; Langer et al., 2017). The worldwide incidence rate of HSCR is 1/5,000 live births, and 2.8/10,000 live births in the Asian population (Amiel et al., 2008). Hirschsprung’s Disease patients can be divided into three subtypes according to the extent of aganglionosis: short-segment HSCR (S-HSCR), long-segment HSCR (L-HSCR), and total colonic aganglionosis (TCA; Tang et al., 2016).
It is well known that genetic factors play an essential role in the development of HSCR, with RET as the major risk gene, as genome-wide association studies (GWASs) using single nucleotide polymorphism (SNP) chip revealed that RET, SEMA3, and NRG1 were associated with HSCR, which was also confirmed in subsequent independent studies (Garcia-Barcelo et al., 2009; Kim et al., 2014; Jiang et al., 2015; Fadista et al., 2018). Earlier studies adapting whole genome or exome sequencing strategies also revealed new risk variants with high penetrance. Using genome-wide copy number analysis and exome sequencing, NRG3 was found to contribute to HSCR susceptibility (Tang et al., 2012; Yang et al., 2013). FAT3 gene variants were seen to be significantly enriched in five HSCR families (Luzón-Toro et al., 2015). Whole exome sequencing coupled with functional analysis found that rare variants of DENND3, NCLN, NUP98, and TBATA were enriched in HSCR patients (Gui et al., 2017). Exome sequencing of samples from 190 patients of European ancestry revealed that seven genes, including ACSS2, ADAMTS17, ENO3, FAM213A, SH3PXD2A, SLC27A4, and UBR4, harbored rare HSCR risk variants with high penetrance (Tilghman et al., 2019). Notably, Tang et al. (2018) performed whole-genome sequence analyses of 464 patients with sporadic S-HSCR and 498 control individuals, and performed association analysis for both rare and common variants with HSCR susceptibility. Except for BACE2 that harbors an excess of rare protein-altering variants, they found that a common variation in four novel loci was associated with HSCR, which contains two intronic variants on calsequestrin 2 (CASQ2) and phospholipase D1 (PLD1), and two intergenic regions (one between SLC4A7 and EOMES on 3p24.1, another between LINC01518 and LOC283028 on 10q11.21).
We conducted a case–control study to further investigate the association of the common variations in CASQ2, intergenic region of SLC4A7-EOMES, PLD1, and intergenic region of LINC01518-LOC283028 with HSCR susceptibility. We selected previously identified SNPs from the study of Tang et al. (2018) and tag SNPs of the four associated regions. In total, 61 SNPs were genotyped in 420 patients with HSCR and 1,665 healthy controls within the Han Chinese population.
Materials and Methods
Subjects
Study design and protocol conformed to the ethical guidelines of the Declaration of Helsinki and were approved by the Ethics Committee of the Xin Hua Hospital affiliated to Shanghai Jiao Tong University School of Medicine. Each individual, or the legal guardians of each child, received a detailed description of the purpose of this study and signed a written informed consent form. Sporadic HSCR patients were recruited from people who had received treatments in Xinhua hospital, affiliated to Shanghai Jiao Tong University school of Medicine, between 2008 and 2018. Diagnosis of HSCR was determined by histological examination of biopsy specimens for the absence of the enteric ganglia. We recruited 420 sporadic patients (322 males and 98 females, the male: female ratio of 3.29:1) with HSCR (323/58/39 for S-HSCR/L-HSCR/TCA), and the mean age of HSCR patients was 1.16 ± 1.71 years. A total of 1,665 gender-matched healthy controls, who visited Xinhua hospital for routine health check-ups, were randomly selected as controls, including 1281 males and 384 females (the male: female ratio of 3.34:1) with a mean age of 36.14 ± 7.54 years. Each control subject was in good health and without a history of HSCR or any other neurological disorders. All the cases and controls were unrelated individuals of Han Chinese origin. Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit, according to the manufacturer’s protocol (Qiagen, Hilden, Germany).
SNP Selection
Four new loci were identified in a previous study by Tang et al. (2018) showing a moderate association (P < 1 × 10–6) with HSCR, which includes 2 intergenic (rs1414027 between LINC01518 and LOC283028 on 10q11.21, and rs9851320 between SLC4A7 and EOMES on 3p24.1) and 2 intronic variants (rs12632766 on PLD1 and rs9428225 on CASQ2) (Tang et al., 2018). We genotyped these four SNPs for replication and selected tag SNPs to cover the common variation of these associated regions. The Genome Variation Server1 with MAF (minor allele frequency) ≥ 0.01 and r2 ≥ 0.8 based on the HapMap HCB (Han Chinese in Beijing) data was used to select. The four reported SNPs were forced to be tag SNPs in the tag SNP selection procedure. We selected 14 tag SNPs, including rs9428225 to cover the CASQ2 gene region and a 10 kb region flanking the 5′ and 3′ end of the gene. Twenty-four tag SNPs, including rs9851320, were selected to cover the intergenic region between SLC4A7 and EOMES on 3p24.1. Additionally, we selected 13 tag SNPs, including rs12632766, to cover the PLD1 gene region and a 10 kb region flanking the 5′ and 3′ end of the gene. Ten SNPs, including rs1414027, were selected to cover the intergenic region between LINC01518 and LOC283028. In total we genotyped 61 SNPs to investigate the associations of the four regions with HSCR susceptibility.
Genotyping
Sixty-one SNPs were genotyped using a Fluidigm 192.24 Dynamic Array with EP1 (Fluidigm Corp., CA). Allele-specific fluorescent (FAM or VIC) primers and common reverse primers were employed for genotyping. We used EP1 SNP Genotyping Analysis software to analyze the data. We placed one duplicate sample on each 96-well sample plate to assess genotyping accuracy. The PCR was conducted on a total volume of 4 μl per inlet. The primer sequences and PCR reaction conditions are shown in Supplementary Tables S1,S2.
In silico Functional Annotation
Functional annotation of associated SNPs was explored by HaploReg v4.1. The expression quantitative trait locus (eQTL) evidence of disease-associated SNPs was investigated using GTEx database Release V8 (dbGap Accession phs000424.v8.p2) (GTExConsortium, 2015).
To quantify the probability that the HSCR association and eQTL signals at a given locus were attributable to a shared causal variant, we conducted colocalization analysis by coloc using our association summary statistics and eQTLs data from GTEx (Guo et al., 2015). eQTLs originating from all digestive tract regions were extracted from the GTEx database, including tissues of esophagus muscularis (n = 465), esophagus mucosa (n = 497), esophagus – gastroesophageal junction (n = 330), stomach (n = 324), small intestine – terminal ileum (n = 174), colon transverse (n = 368), and colon – sigmoid (n = 318). We restricted analyses to genes within 1 Mb of the associated SNPs of interest [eQTL P < 0.01 and m-value (Posterior Probability from METASOFT) > 0.9] and ran coloc.abf with default priors. We considered coloc tests with a Posterior probability of hypothesis 4 (PPH4) ≥ 0.9 or PPH4.ABF > 0.5 as having strong or moderate evidence for colocalization.
Statistical Analysis
Hardy–Weinberg equilibrium tests and quality control were performed using PLINK1.9 (Purcell et al., 2007). Linkage disequilibrium of all pairs of SNPs was estimated using D’ and r2 according to the standard measurement. Haplotype block was determined using Haploview 4.2 (Barrett et al., 2005). Association analysis was performed using PLINK1.9 under the additive genetic model. SNP–SNP interaction was assessed using a logistic regression approach with additive encoding as implemented in PLINK1.9 (Purcell et al., 2007). Conditional logistic analysis was performed to find SNPs with an independent effect by adding the top associated SNPs as covariates in the model. We computed statistical power using the Genetic Power Calculator2 (Purcell et al., 2003). A study-wide level P value < 0.00086 (0.05/58) was considered statistically significant.
Results
Case-Control Association Study
Among the 61 SNPs genotyped, three were excluded due to poor genotyping success rate (<5%). The remaining 58 SNPs with genotyping success rate >95% conformed to Hardy–Weinberg equilibrium expectations (P > 0.01). The distributions of the allele and genotype frequencies of these SNPs in controls and HSCR patients are shown in Table 1. The LD pattern of SNPs and haplotype block was plotted in Figure 1.
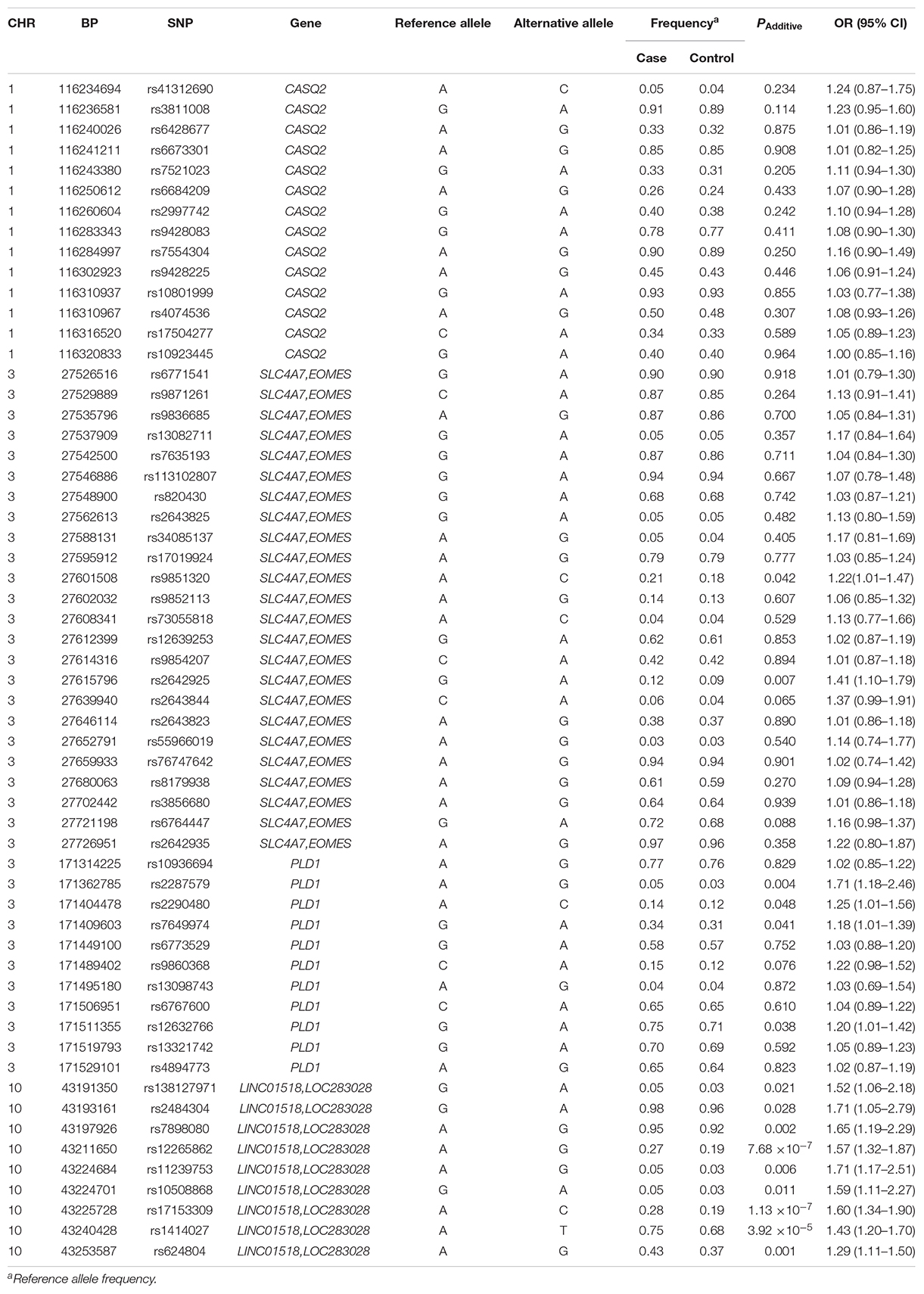
Table 1. Allele distribution and association analysis for 58 SNPs in HSCR patients and healthy controls.
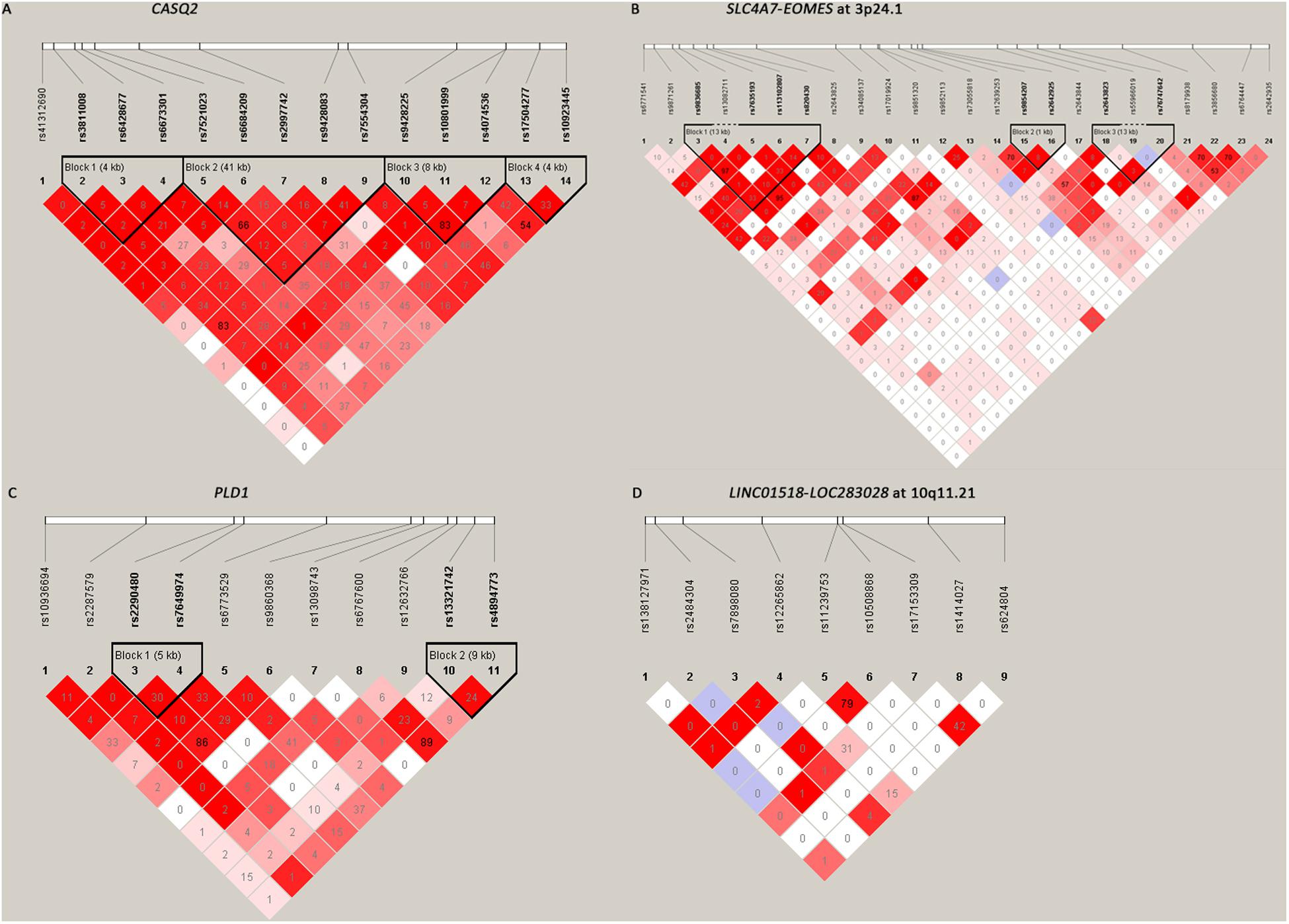
Figure 1. The linkage disequilibrium (LD) patterns of the four loci. (A) 14 SNPs in CASQ2 regions; (B) 24 SNPs between SLC4A7 and EOMES at 3p24.1; (C) 11 SNPs in the PLD1 region; (D) 9 SNPs between LINC01518 and LOC283028 at 10q11.21. Blocks were defined according to the default method of Haploview. The numbers in the boxes are the pairwise LD coefficient r2 between SNPs. r2 values of 1 represent complete LD; values greater than 0.8 represent strong evidence of LD; values of 0.2–0.8 represent modest LD; and values less than 0.2 represent negligible evidence of LD.
None of the 14 SNPs in the CASQ2 gene region showed significant association with HSCR susceptibility. Two tag SNPs located between SLC4A7 and EOMES on 3p24.1 showed a suggestive significant association (Table 1): rs2642925 [odds ratio (OR) = 1.41, 95% confidence interval (95% CI) = 1.10–1.79; PAdditive = 0.007; Table 1] and the previously associated SNP rs9851320 (OR = 1.22, 95% CI = 1.01–1.47; PAdditive = 0.042; Table 1). A coding SNP, rs2287579, in PLD1 showed a suggestive association with HSCR susceptibility (OR = 1.71, 95% CI = 1.18–2.46; PAdditive = 0.004; Table 1). This SNP resulted in an amino acid substitution from a valine to methionine (p.Val > Met) at position 820 of PLD1 protein isoform a. The previously associated PLD1 SNP rs12632766 showed a suggestive significance (OR = 1.20, 95% CI = 1.01–1.42, PAdditive = 0.038; Table 1). Two new SNPs, rs2290480 and rs7649974, also showed suggestive association with HSCR susceptibility, PAdditive = 0.048 and 0.041, respectively. However, none of the P values of SNPs in 3p24.1 and the PLD1 region could reach the study-wide significance (P > 0.00086).
All the nine tag SNPs located between LINC01518 and LOC283028 on 10q11.21 demonstrated a suggestive association (P < 0.05), and three of them met the Bonferroni multiple testing corrected P value threshold (P < 0.00086). The top SNP was rs17153309 (OR = 1.60, 95% CI = 1.34–1.90; PAdditive = 1.13 × 10–7; Table 1). Rs1414027 was reported to exhibit an association with HSCR risk in the previous study (Tang et al., 2018), and it was the third most associated SNP in the current data, with a PAdditive value of 3.92 × 10–5 (OR = 1.43, 95% CI = 1.20–1.70). Nine SNPs genotyped in this region were in low to moderate LD (Figure 1D). No significant SNP-SNP interaction was detected among the SNPs genotyped.
Furthermore, we applied stepwise conditional regression analysis to determine which SNPs in the LINC01518-LOC283028 region confer independent risks on HSCR. When conditioning on the top SNP, rs17153309, rs1414027 showed the most significant independent evidence of association (conditional P = 4.73 × 10–5; OR = 1.44, 95% CI = 1.21–1.72). Similarly, rs17153309 was significantly associated with HSCR when conditioning on rs1414027 (P = 1.70 × 10–7; OR = 1.60, 95% CI = 1.34–1.90). Therefore, it was obvious these two SNPs represent independent associations. When conditioning on rs17153309 and rs1414027, no SNPs showed significant independent associations (conditional P > 0.00086). These results suggest that independent association signals existed in the LINC01518-LOC283028 region and the combination of the above two SNPs could explain the majority of risk for this region.
In addition, considering that the clinical manifestations, surgical options, and post-operation complications differ significantly between S-HSCR, L-HSCR, and TCA patients, we performed association analysis stratified by clinical subtype and did not find SNPs associated with a specific subtype (Supplementary Table S3).
Functional Annotation
Putative function of associated SNPs was annotated by HaploReg (Table 2). At the 10q11.21 locus, the six associated SNPs altered the sequences of regulatory motifs (Table 2). Additionally, rs624804 maps to a DNase I hypersensitivity site (Table 2). Two associated SNPs (rs1414027 and rs624804) at 10q11.21 were eQTLs of nearby genes (Table 3). Rs1414027 was an eQTL of ZNF33B in the small intestine (P = 4.60 × 10–3; PP.H4 = 0.853), transverse colon (P = 3.40 × 10–9; PP.H4 = 0.999), and sigmoid colon (P = 9.30 × 10–5; PP.H4 = 0.682). Rs624804 was an eQTL of LINC00839 in the esophagus (Pmuscularis = 6.50 × 10–12, PP.H4 = 0.777; Pmucosa = 2.80 × 10–9, PP.H4 = 0.992; PGastroesophageal Junction = 9.30 × 10–10, PP.H4 = 0.995), while rs624804 was also an eQTL of RP11-313J2.1 in the stomach (P = 1.20 × 10–11; PP.H4 = 0.999).

Table 2. Functional annotation of positive SNPs associated with HSCR using data from Haploreg and ENCODE.

Table 3. The expression quantitative trait loci (eQTL) evidence of the positive SNPs.
Discussion
Four common SNPs at different chromosome regions were found to be associated with HSCR susceptibility in an earlier study (Tang et al., 2018). In the current study, we genotyped 61 SNPs to validate the association and fine-map the susceptibility loci in the Han Chinese population. Our results indicated a suggestive association of HSCR susceptibility with variants in SLC4A7-EOMES and PLD1, and significant association with variants in the LINC01518-LOC283028 regions.
None of the 14 tag SNPs in the CASQ2 region showed any association with HSCR susceptibility. The previously associated CASQ2 SNP, rs9428225, also displayed no significance in our sample-set. The risk allele A of rs9428225 was 0.472 in cases and 0.356 in controls in the previous study on Chinese and Vietnamese populations, as shown in Supplementary Table S4 (Tang et al., 2018). In the current sample-set of the Han Chinese population, the frequencies of rs9428225 A allele were 0.448 in cases and 0.434 in controls. Thus, the rs9428225 A allele had the same effect direction in the current sample as in the previous data, though the difference of allele frequency between cases and controls was not significant. Owing to the modest effect of this locus, further studies are needed to validate this association.
In the intergenic region between SLC4A7 and EOMES on 3p24.1, the previously associated SNP rs9851320 showed a suggestive association in our samples. The frequencies of rs9851320 minor allele A were 0.213 in cases and 0.181 in controls in the current Han Chinese population, which followed the same trend as that of as previous data on Chinese and Vietnamese populations, with 0.248 in cases and 0.164 in controls (Tang et al., 2018). The frequency of rs9851320 minor allele A is 0.186 in the South Han Chinese data in the 1000 genomes project, which is close to that observed in the current sample set (0.181) (Supplementary Table S4). SLC4A7 encodes a sodium bicarbonate cotransporter. The highest expression level of SLC4A7 was detected in the duodenum [reads per kilobase per million mapped reads (RPKM), 27.5] and the small intestine (RPKM 8.8) from quantitative transcriptomics analysis (Fagerberg et al., 2014). EOMES is also known as T-brain gene 2 (TBR2) and in mammals, the transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone (Arnold et al., 2008). In addition, Eomes was indispensable for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons (Hodge et al., 2012). Without Tbr2, intermediate neuronal progenitors were depleted in spite of augmented neural stem cell proliferation, and neurogenesis ceased due to the failure of neuronal differentiation (Hodge et al., 2012). Hirschsprung’s disease is caused by the developmental defect of the enteric neuron system (ENS; Heuckeroth, 2018). Therefore, SLC4A7 and EOMES may play a role in the development of ENS and this could provide a link to the pathogenesis of HSCR. However, further studies are required to validate the association at these loci with HSCR.
We identified a non-synonymous SNP, rs2287579, (p.Val 820 Met) in PLD1 associated with HSCR susceptibility with suggestive evidence. The minor allele A frequency of previously associated rs12632766 was 0.251 in cases and 0.286 in controls in the current Han Chinese population. The frequencies were 0.196 and 0.293 in cases and controls, respectively, in the previous study of Chinese and Vietnamese populations, as shown in Supplementary Table S4 (Tang et al., 2018). The effect direction of current data was consistent with previous data that rs12632766 minor allele A exhibited a protective effect in HSCR susceptibility, although the association could not reach the significance threshold. PLD1 encodes a phosphatidylcholine-specific phospholipase, which could catalyze the hydrolysis of phosphatidylcholine to produce phosphatidic acid and choline. This enzyme may play a crucial role in signal transduction. PLD1 signaling pathway genes might account for disease severity of HSCR (Tang et al., 2018). Notably, the BACE1-APP-BACE2 pathway may be an important pathway underlying the etiology of HSCR (Tang et al., 2018). PLD1 was found to be involved in Abeta production via regulating amyloid precursor protein (APP) metabolism and trafficking (Cai et al., 2006a, b). All this evidence suggests that PLD1 may play a role in the pathogenesis of HSCR. However, the association of PLD1 with HSCR needs to be confirmed in additional samples.
Variants located between LINC01518 and LOC283028 on 10q11.21 showed significant associations in the current study. The previously associated SNP, rs1414027, was successfully replicated in our samples. Two SNPs represented independent association signals and accounted for the association signal of the locus. Both LINC01518 and LOC283028 are long non-coding RNAs (lncRNAs). Recent studies have revealed that lncRNAs played an important role in the pathogenesis of HSCR. Dysregulated lncRNAs were found in HSCR tissues, which suggested their probable use as novel diagnostic biomarkers for HSCR (Shen et al., 2016). Notably, the associated SNP, rs1414027, was an eQTL of ZNF33B in multiple tissues, and rs624804 was an eQTL of LINC00839 and RP11-313J2.1. However, no colocalized signals were observed convincingly across all digestive tract tissues, emphasizing the tissue specific effects of disease causal variants. ZNF33B encodes a member of the zinc finger family of proteins and was expressed in the colon and small intestine tissues, implicating its potential role in the development of gut tissues (Fagerberg et al., 2014). LINC00839 was highly expressed in the colon, esophagus, small intestine, and stomach (Fagerberg et al., 2014). Since all the above-mentioned genes may be potential susceptibility genes, further functional studies are needed to clarify the causal gene in this region.
The relatively small sample size limits the current study. Under the assumption of 0.0002 disease prevalence, the study-wide significance level, and odds ratios of 1.3/1.6 for heterozygotes/rare homozygotes, 420 cases and 1,665 controls could achieve 38.8, 24.7, 39.1, and 37.5% statistical power for rs9428225 (MAF = 0.43), rs9851320 (MAF = 0.18), rs12632766 (MAF = 0.39), and rs1414027 (MAF = 0.32). However, the statistical power would be much lower with the threshold of P-value, calculated using a strict Bonferroni correction for multiple comparisons. Much larger sample sizes are required to achieve an adequate statistical power.
In conclusion, we replicated the association of variants of LINC01518-LOC283028 at 10q11.21 with HSCR susceptibility. Our fine-mapping also revealed new associated variants, and two independent SNPs accounting for the association of the LINC01518-LOC283028 region with HSCR. Additionally, eQTL analysis indicated that these disease-associated SNPs influenced the expression levels of nearby genes. These findings provide new insights toward understanding the pathogenesis of HSCR.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA597838.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of the Xin Hua Hospital affiliated to Shanghai Jiao Tong University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
WC and XC conceived the study. M-RB, Y-JL, X-XY, Z-LW, W-JW, H-LS, Y-MG, W-WY, and B-LG conducted the experiment. W-BN and XC participated in data analysis and figure preparation and drafted the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31671317, 31471190, and Key Program 81630039), Foundation of Shanghai Municipal Health Commission (201840014), Foundation for Shanghai Key Laboratory of Pediatric Gastroenterology and Nutrition (17DZ2272000), Foundation of Science and Technology Commission of Shanghai Municipality (19495810500), Shanghai Sailing Program (19YF1440700), and the Interdisciplinary Program of Shanghai Jiao Tong University (ZH2018QNA57).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the contribution of the members participating in this study, as well as the clinicians who helped us with diagnosis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00738/full#supplementary-material
Footnotes
References
Amiel, J., Sproat-Emison, E., Garcia-Barcelo, M., Lantieri, F., Burzynski, G., Borrego, S., et al. (2008). Hirschsprung disease, associated syndromes and genetics: a review. J. Med. Genet. 45, 1–14. doi: 10.1136/jmg.2007.053959
Arnold, S. J., Huang, G. J., Cheung, A. F. P., Era, T., Nishikawa, S. I., Bikoff, E. K., et al. (2008). The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 22, 2479–2484. doi: 10.1101/gad.475408
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Cai, D., Netzer, W. J., Zhong, M., Lin, Y., Du, G., Frohman, M., et al. (2006a). Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 103, 1941–1946. doi: 10.1073/pnas.0510708103
Cai, D., Zhong, M., Wang, R., Netzer, W. J., Shields, D., Zheng, H., et al. (2006b). Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 1936–1940. doi: 10.1073/pnas.0510710103
Fadista, J., Lund, M., Skotte, L., Geller, F., Nandakumar, P., Chatterjee, S., et al. (2018). Genome-wide association study of Hirschsprung disease detects a novel low-frequency variant at the RET locus. Eur. J. Hum. Genet. 26, 561–569. doi: 10.1038/s41431-017-0053-7
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Garcia-Barcelo, M. M., Tang, C. S., Ngan, E. S., Lui, V. C., Chen, Y., So, M. T., et al. (2009). Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 2694–2699. doi: 10.1073/pnas.0809630105
GTExConsortium. (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. doi: 10.1126/science.1262110
Gui, H., Schriemer, D., Cheng, W. W., Chauhan, R. K., Antiòolo, G., Berrios, C., et al. (2017). Whole exome sequencing coupled with unbiased functional analysis reveals new Hirschsprung disease genes. Genome Biol. 18:48. doi: 10.1186/s13059-017-1174-6
Guo, H., Fortune, M. D., Burren, O. S., Schofield, E., Todd, J. A., and Wallace, C. (2015). Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum. Mol. Genet. 24, 3305–3313. doi: 10.1093/hmg/ddv077
Heuckeroth, R. O. (2018). Hirschsprung disease — integrating basic science and clinical medicine to improve outcomes. Nat. Rev. Gastroenterol. Hepatol. 15, 152–167. doi: 10.1038/nrgastro.2017.149
Hodge, R. D., Nelson, B. R., Kahoud, R. J., Yang, R., Mussar, K. E., Reiner, S. L., et al. (2012). Tbr2 is essential for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons. J. Neurosci. 32, 6275–6287. doi: 10.1523/JNEUROSCI.0532-12.2012
Jiang, Q., Arnold, S., Heanue, T., Kilambi, K. P., Doan, B., Kapoor, A., et al. (2015). Functional Loss of Semaphorin 3C and/or Semaphorin 3D and their epistatic interaction with ret are critical to hirschsprung disease liability. Am. J Hum. Genet. 96, 581–596. doi: 10.1016/j.ajhg.2015.02.014
Kim, J. H., Cheong, H. S., Sul, J. H., Seo, J. M., Kim, D. Y., Oh, J. T., et al. (2014). A genome-wide association study identifies potential susceptibility loci for Hirschsprung disease. PLoS One 9:e110292. doi: 10.1371/journal.pone.0110292
Langer, J. C., Rollins, M. D., Levitt, M., Gosain, A., Torre, L., Kapur, R. P., et al. (2017). Guidelines for the management of postoperative obstructive symptoms in children with Hirschsprung disease. Pediatr. Surg. Int. 33, 523–526. doi: 10.1007/s00383-017-4066-7
Luzón-Toro, B., Gui, H., Ruiz-Ferrer, M., Sze-Man Tang, C., Fernández, R. M., Sham, P., et al. (2015). Exome sequencing reveals a high genetic heterogeneity on familial Hirschsprung disease. Sci. Rep. 5:16473. doi: 10.1038/srep16473
Purcell, S., Cherny, S. S., and Sham, P. C. (2003). Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150. doi: 10.1093/bioinformatics/19.1.149
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Shen, Z., Du, C., Zang, R., Xie, H., Lv, W., Li, H., et al. (2016). Microarray expression profiling of dysregulated long non-coding RNAs in Hirschsprung’s disease reveals their potential role in molecular diagnosis. Neurogastroenterol. Motil. 28, 266–273. doi: 10.1111/nmo.12722
Tang, C. S., Cheng, G., So, M. T., Yip, B. H., Miao, X. P., Wong, E. H., et al. (2012). Genome-wide copy number analysis uncovers a new HSCR gene: NRG3. PLoS Genet. 8:e1002687. doi: 10.1371/journal.pgen.1002687
Tang, C. S., Gui, H., Kapoor, A., Kim, J., Luzón-Toro, B., Pelet, A., et al. (2016). Trans -ethnic meta-analysis of genome-wide association studies for Hirschsprung disease. Hum. Mol. Genet. 25, 5265–5275. doi: 10.1093/hmg/ddw333
Tang, C. S., Li, P., Lai, F. P., Fu, A. X., Lau, S. T., So, M. T., et al. (2018). Identification of genes associated with hirschsprung disease, based on whole-genome sequence analysis, and potential effects on enteric nervous system development. Gastroenterology 155, 1908–1922. doi: 10.1053/j.gastro.2018.09.012
Tilghman, J. M., Ling, A. Y., Turner, T. N., Sosa, M. X., Krumm, N., Chatterjee, S., et al. (2019). Molecular genetic anatomy and risk profile of Hirschsprung’s disease. N. Engl. J. Med. 380, 1421–1432. doi: 10.1056/NEJMoa1706594
Wallace, A. S. (2011). Genetic interactions and modifier genes in Hirschsprung’s disease. World J. Gastroenterol. 17:4937. doi: 10.3748/wjg.v17.i45.4937
Keywords: association, Hirschsprung’s disease, case-control study, single nucleotide polymorphisms, Han Chinese population
Citation: Niu W-B, Bai M-R, Song H-L, Lu Y-J, Wu W-J, Gong Y-M, Yu X-X, Wei Z-L, Yu W-W, Gu B-L, Cai W and Chu X (2020) Association of Variants in PLD1, 3p24.1, and 10q11.21 Regions With Hirschsprung’s Disease in Han Chinese Population. Front. Genet. 11:738. doi: 10.3389/fgene.2020.00738
Received: 02 December 2019; Accepted: 19 June 2020;
Published: 10 July 2020.
Edited by:
Inês Barroso, University of Exeter, United KingdomCopyright © 2020 Niu, Bai, Song, Lu, Wu, Gong, Yu, Wei, Yu, Gu, Cai and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Cai, Y2FpdzIwNEBzanR1LmVkdS5jbg==; Xun Chu, Y2h1eHVuQHhpbmh1YW1lZC5jb20uY24=
†These authors have contributed equally to this work