 Yue Xing
Yue Xing Xiao Li
Xiao Li Xiang Gao
Xiang Gao Qunfeng Dong
Qunfeng Dong- 1Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, TX, United States
- 2Department of Molecular and Cellular Medicine, Texas A&M University, College Station, TX, United States
- 3Department of Medicine, Stritch School of Medicine, Loyola University Chicago, Maywood, IL, United States
- 4Center for Biomedical Informatics, Stritch School of Medicine, Loyola University Chicago, Maywood, IL, United States
The furin cleavage site in the spike glycoprotein of the SARS-CoV-2 coronavirus is considered important for the virus to enter the host cells. By analyzing 45828 SARS-CoV-2 genome sequences, we identified 103 strains of SARS-CoV-2 with various DNA mutations including 18 unique non-synonymous point mutations, one deletion, and six gains of premature stop codon that may affect the furin cleavage site. Our results revealed that the furin cleavage site might not be required for SARS-CoV-2 to enter human cells in vivo. The identified mutants may represent a new subgroup of SARS-CoV-2 coronavirus with reduced tropism and transmissibility as potential live-attenuated vaccine candidates.
Introduction
A notable feature of the SARS-CoV-2 coronavirus is that its spike glycoprotein contains a polybasic furin cleavage site at the S1-S2 boundary (Andersen et al., 2020; Walls et al., 2020). Furin is a protease ubiquitously expressed in multiple organs and tissues in humans, such as the brain, lung, gastrointestinal tract, liver, pancreas, and reproductive tissues (Wang et al., 2020). Cleavage of the spike protein by the furin protease is considered to facilitate the entrance of SARS-CoV-2 into host cells. Due to the wide expression of furin in multiple tissues, the existence of the furin cleavage site in the spike glycoprotein may expand tropism and enhance the transmissibility of SARS-CoV-2 (Walls et al., 2020).
When the discovery of the furin cleavage site was published on April 16, 2020 (Walls et al., 2020), there only existed 144 SARS-CoV-2 genome sequences in the GISAID database (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) and the furin cleavage site was strictly conserved (Walls et al., 2020). As of June 13, 2020, the number of SARS-CoV-2 genome sequences in the GISAID database has significantly increased to 45828. Therefore, we sought to answer a straightforward yet important question: are there any natural polymorphisms in the furin cleavage site of the SARS-CoV-2 spike glycoprotein? The existence of natural polymorphisms in the furin cleavage site may represent a new subgroup of SARS-CoV-2 coronavirus with different tropism and transmissibility.
Methods
In total, 45828 SARS-CoV-2 genome sequences were downloaded from the GISAID database on June 13, 2020. The microbial genomics mutation tracker (MicroGMT) software, recently published by our group (Xing et al., 2020), was applied with default parameters to identify DNA mutations between each downloaded database sequence and the reference genome sequence of SARS-CoV-2 (i.e., SARA-CoV-2 isolate Wuhan-Hu-1 complete genome sequence with GenBank accession number NC_045512) (Wu et al., 2020). In brief, MicoGMT invokes (1) minimap2 (Li, 2018) to perform genome-wide pairwise alignments and (2) snpEff (Cingolani et al., 2012) to identify point mutations (synonymous and non-synonymous), insertions or deletions, and gains of stop codons from the genome alignments. The computation was performed for about 40 h in the high-performance research computer Ada at Texas A&M University. The NCBI Structure program1 was used to characterize the changes of the biochemical properties of non-synonymous mutations.
Results
From 45828 SARS-CoV-2 genome sequences available in the GISAID database as of June 13, 2020, 103 strains of SARS-CoV-2 carried various DNA mutations including 25 unique ones that may affect the furin cleavage site located at the amino acid residual positions 680–689 (S1/S2 region) (Coutard et al., 2020; Wang et al., 2020; Zhang et al., 2020) of the SARS-CoV-2 spike protein (Figure 1, Table 1, and Supplementary Table 1). Specifically, 96 SARS-CoV-2 strains were identified to carry a total of 23 unique point mutations in the furin cleavage site (each mutant strain carried only one non-synonymous point mutation in the furin cleavage site). Out of those 96 strains, 74 carried non-synonymous point mutations; out of the 23 unique point mutations, 18 were non-synonymous. Of those 18 non-synonymous changes, one changed from a non-polar amino acid residue (Ala) to a negatively charged residue (Glu); four changed from non-polar to neutral polar (Pro to Ser, Ala to Thr, and Ala to Ser at two different sites); one changed from non-polar (Pro) to positively charged (His); four changed from neutral polar to non-polar (Ser to Phe, Pro, Gly or Ile); two changed from positively charged to non-polar (Arg to Trp or Pro); two changed from positively charged to neutral polar (Arg to Gln at two different sites). Out of all the amino acid residues in the furin cleavage site, only Arg685 had no point mutations (neither synonymous nor non-synonymous). Besides point mutations, one strain (HongKong/XM-PII-S4/2020) contained a deletion in the furin cleavage site. The deletion spanned from Asn679 to Ala688, which is almost the entire length of the furin cleavage site (except for the last amino acid residue). In addition, we found six SARS-CoV-2 strains with the gains of stop codons in the spike protein between the position 258 and 516, which would abolish the downstream furin cleavage site located at the positions 680-689.
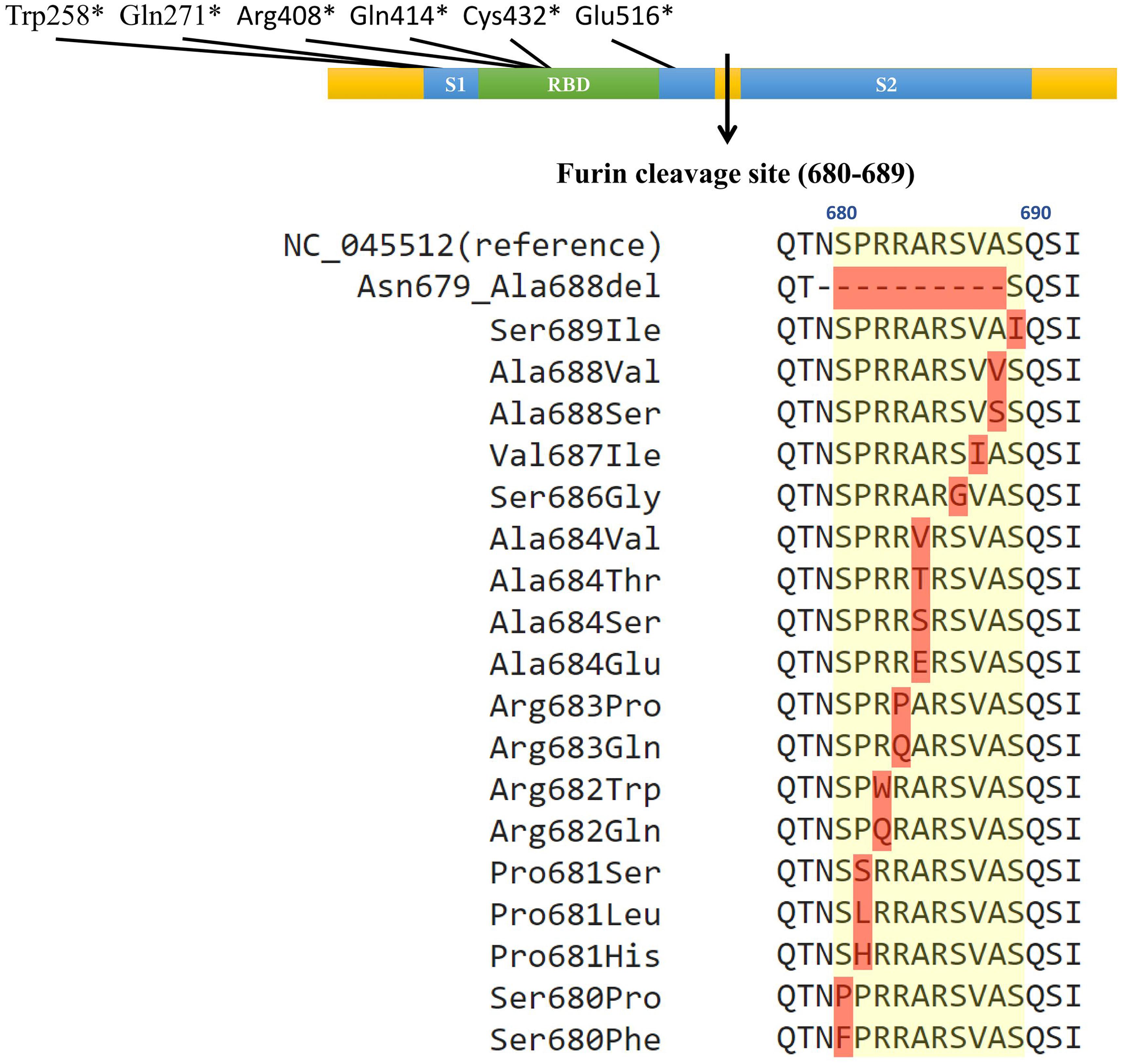
Figure 1. Naturally occurring polymorphisms in the furin cleavage site of the SARS-CoV-2 spike glycoprotein. The colored bar represents the spike glycoprotein, with the blue boxes indicating the S1 and S2 subunits, green box indicating the receptor binding domain, and the arrow indicating the furin cleavage site. The positions of the six gains of premature stop codons were illustrated on the top of the color bar (∗ indicates the stop codon). The yellow shade on the peptide sequences indicate the furin cleavage site, and red shades indicate mutations. The multiple sequence alignment shows each identified non-synonymous mutation with the yellow background indicating the furin cleavage site from the position 680 to 689.
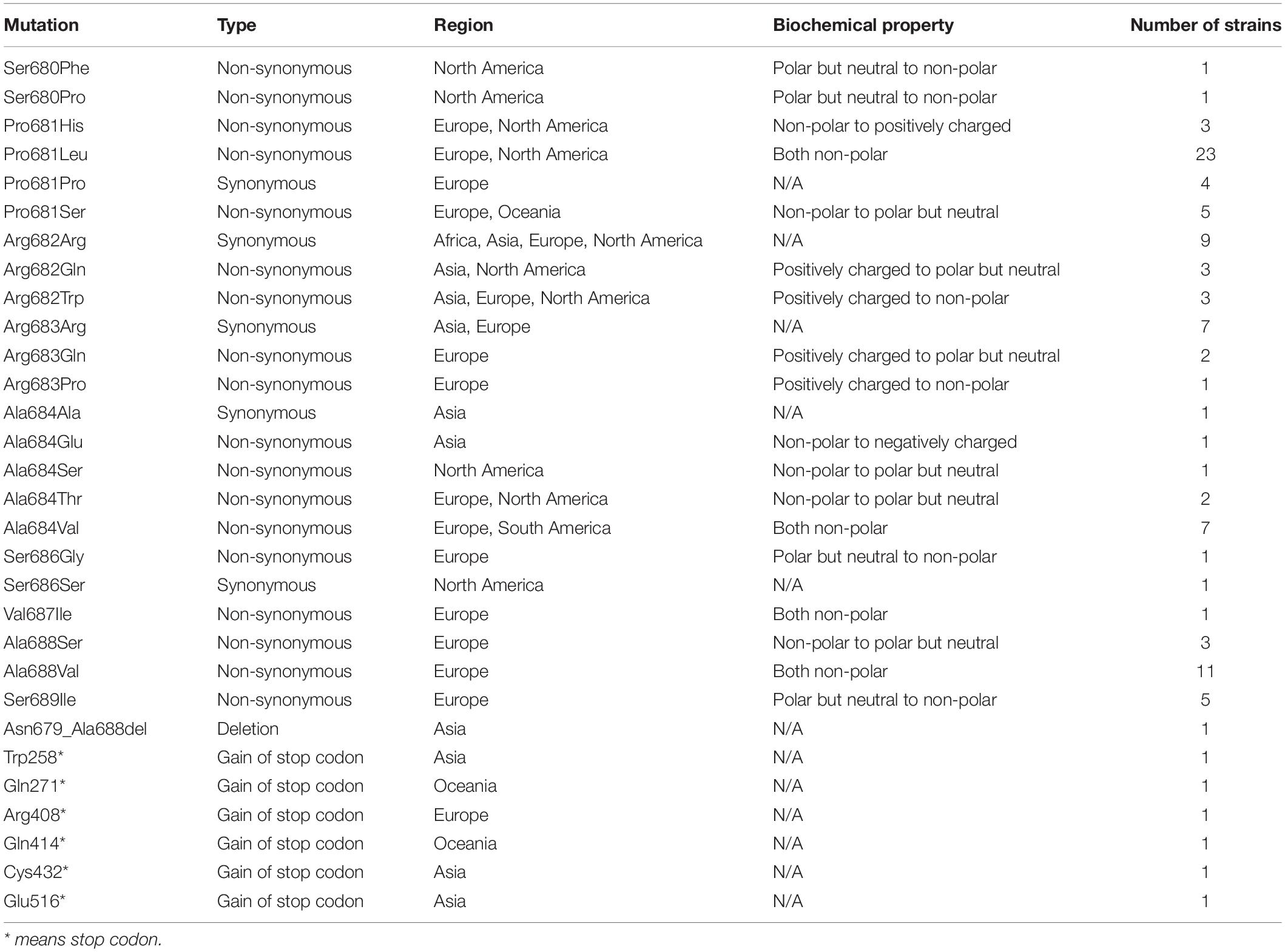
Table 1. Summary of mutations identified in the furin cleavage site.
The identified mutations in the furin cleavage site appeared in multiple geographic regions (Asia, Europe, North America, and Oceania) through January to May 2020. Most of them appeared in one or two geographic regions, but Arg682Trp appeared in three regions. Europe and North America had the most point mutations in the furin cleavage site. The only deletion was observed in Asia, and the gain of stop codons were observed in Asia, Europe, and Oceania.
Discussion
We uncovered 103 SARS-CoV-2 strains from multiple geographic regions, 81 of which carried 25 unique mutations that may affect the furin cleavage site in the spike glycoprotein. Out of the total 10 amino acid residues in the furin cleavage site, nine experienced non-synonymous changes. It is worth noting that the non-synonymous point mutations occurred at seven out of eight amino acid residues of the highly conserved region of 682 RRARSVAS689 (Anand et al., 2020). This conserved region included three of the four amino acid residues of 681PRRA684 that are unique to SARS-CoV-2 (Zhang et al., 2020) (non-synonymous point mutations also occurred at Pro681), containing the furin cleavage point between Arg685 and Ser686 (Coutard et al., 2020). Although no mutations were identified at Arg685, mutations existed at Ser686 (e.g., Gly) disabling furin-type cleavages. In addition, mutations around Arg685 and Ser686 may also affect the recognition of the cleavage site. Point mutations and deletions were also found upstream and downstream of positions 680-689 including two deletions from position 675–679 (data not shown). Finally, we also observed one deletion and six gains of premature stop codons, all of which completely abrogated the furin cleavage site. Interestingly, Davidson et al. (2020) also detected one deletion in the furin cleavage site based on RNA-Seq sequencing.
Since all the mutations were identified from live viral strains in COVID-19 patients, our results revealed that the furin cleavage site may not be required for SARS-CoV-2 to enter human cells in vivo, which agrees with the in vitro experimental results showing that SARS-CoV-2, with deletion of the furin cleavage site, could still enter the cell lines of humans, African green monkeys, and bay hamsters (Walls et al., 2020). Therefore, we speculate that our observed mutants may represent a new subgroup of SARS-CoV-2 coronavirus with reduced tropism and transmissibility, which requires further experimental validations. Analyzing clinical symptoms and infectiousness of the COVID-19 patients with those mutant strains may be also important in future studies. If tropism and transmissibility of those mutant strains were indeed reduced, they might serve as potential live-attenuated vaccine candidates (Turell et al., 2003; Lauring et al., 2010; Toth et al., 2011).
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Author Contributions
XL and YX performed the data analysis and drafted the manuscript. XG and QD designed the project and revised the manuscript. All authors approved the submitted version.
Funding
XG and QD were partially supported by NIH grant 5R01AI116706.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The high-performance research computer Ada at Texas A&M University was used for our data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00783/full#supplementary-material
Footnotes
References
Anand, P., Puranik, A., Aravamudan, M., Venkatakrishnan, A., and Soundararajan, V. (2020). SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. eLife 9:e58603.
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., and Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452. doi: 10.1038/s41591-020-0820-9
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Coutard, B., Valle, C., de Lamballerie, X., Canard, B., Seidah, N., and Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 176:104742. doi: 10.1016/j.antiviral.2020.104742
Davidson, A. D., Williamson, M. K., Lewis, S., Shoemark, D., Carroll, M. W., Heesom, K., et al. (2020). Characterisation of the transcriptome and proteome of SARS-CoV-2 using direct RNA sequencing and tandem mass spectrometry reveals evidence for a cell passage induced in-frame deletion in the spike glycoprotein that removes the furin-like cleavage site. bioRxiv [Preprint] doi: 10.1101/2020.03.22.002204
Elbe, S., and Buckland-Merrett, G. (2017). Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Challenges 1, 33–46. doi: 10.1002/gch2.1018
Lauring, A. S., Jones, J. O., and Andino, R. (2010). Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 28, 573–579. doi: 10.1038/nbt.1635
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Shu, Y., and McCauley, J. (2017). GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 22:30494.
Toth, A. M., Geisler, C., Aumiller, J. J., and Jarvis, D. L. (2011). Factors affecting recombinant Western equine encephalitis virus glycoprotein production in the baculovirus system. Protein Exp. Purif. 80, 274–282. doi: 10.1016/j.pep.2011.08.002
Turell, M. J., O’guinn, M. L., and Parker, M. D. (2003). Limited potential for mosquito transmission of genetically engineered, live-attenuated western equine encephalitis virus vaccine candidates. Am. J. Trop. Med. Hyg. 68, 218–221. doi: 10.4269/ajtmh.2003.68.218
Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281-292.e6.
Wang, Q., Qiu, Y., Li, J.-Y., Zhou, Z.-J., Liao, C.-H., and Ge, X.-Y. (2020). A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol. Sin. doi: 10.1007/s12250-020-00212-7 [Online ahead of print]
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Hu, Y., et al. (2020). Data from: Direct Submission of Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome to RefSeq. Shanghai: Fudan University.
Xing, Y., Li, X., Gao, X., and Dong, Q. (2020). MicroGMT: A mutation tracker for SARS-CoV-2 and other microbial genome sequences. Front. Microbiol. 11:1502. doi: 10.3389/fmicb.2020.01502
Keywords: COVID-19, SARS-CoV-2, spike glycoprotein, S protein, furin, mutation, live-attenuated vaccine
Citation: Xing Y, Li X, Gao X and Dong Q (2020) Natural Polymorphisms Are Present in the Furin Cleavage Site of the SARS-CoV-2 Spike Glycoprotein. Front. Genet. 11:783. doi: 10.3389/fgene.2020.00783
Received: 15 June 2020; Accepted: 01 July 2020;
Published: 17 July 2020.
Edited by:
Xian-Tao Zeng, Wuhan University, ChinaReviewed by:
Jun Sun, University of Illinois at Chicago, United StatesHong Cai, The University of Texas at San Antonio, United States
Copyright © 2020 Xing, Li, Gao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qunfeng Dong, cWRvbmdAbHVjLmVkdQ==