 Gayatri R. Iyer1,2
Gayatri R. Iyer1,2 Sayani Samajder1
Sayani Samajder1 Syeda Zubeda1
Syeda Zubeda1 Devi Soorya Narayana S1
Devi Soorya Narayana S1 Vishakha Mali1
Vishakha Mali1 Sharath Krishnan PV1
Sharath Krishnan PV1 Anuradha Sharma1
Anuradha Sharma1 Neyha Zainab Abbas3
Neyha Zainab Abbas3 Nandini Shyamali Bora1
Nandini Shyamali Bora1 Amulya Narravula1Qurratulain Hasan1*
Amulya Narravula1Qurratulain Hasan1*- 1Department of Genetics and Molecular Medicine, Kamineni Hospitals, Hyderabad, India
- 2Department of Genetics, Osmania University, Hyderabad, India
- 3Department of Genetics, Kamineni Hospitals, Hyderabad, India
Introduction: Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) has spread around the globe. Susceptibility has been associated with age, biological sex, and other prior existing health conditions. However, host genes are involved in viral infectivity and pathogenicity, and polymorphisms in these could be responsible for the interethnic/interindividual variability observed in infection and progression of COVID-19.
Materials and Methods: Clinical exome data of 103 individuals was analyzed to identify sequence variants in five selected candidate genes: ACE2, TMPRSS2, CD209, IFITM3, and MUC5B to assess their prevalence and role to understand the COVID-19 infectivity and progression in our population.
Results: A total of 497 polymorphisms were identified in the five selected genes in the exomes analyzed. Thirty-eight polymorphisms identified in our cohort have been reported earlier in literature and have functional significance or association with health conditions. These variants were classified into three groups: protective, susceptible, and responsible for comorbidities.
Discussion and Conclusion: The two polymorphisms described in literature as risk inducing are rs35705950 in MUC5B gene and TMPRSS2 haplotype (rs463727, rs34624090, rs55964536, rs734056, rs4290734, rs34783969, rs11702475, rs35899679, and rs35041537) were absent in our cohort explaining the slower infectivity of the disease in this part of India. The 38 functional variants identified can be used as a predisposition panel for the COVID-19 infectivity and progression and stratify individuals as “high or low risk,” which would help in planning appropriate surveillance and management protocols. A larger study from different regions of India is warranted to validate these results.
Introduction
Severe Acute Respiratory Syndrome Corona virus-2 (SARS-CoV-2) is a new virus responsible for an outbreak of respiratory illness, since December 2019, named as COVID-19, which has spread to several countries around the globe. Susceptibility initially was associated with age, biological sex, and other prior existing health conditions, which is true for all infectious diseases (Yi et al., 2020). The World Health Organization has declared it as a Public Health Emergency of International Concern. Although it is still early to predict susceptible populations, this needs to be addressed urgently to triage and safeguard individuals with high risk of infectivity and/or the potential to get this progressive disease with adverse prognosis.
SARS-CoV-2 belongs to the family of RNA viruses known as coronaviruses. Sequencing results from patient isolates have indicated that SARS-CoV-2 is similar to the beta (β) coronaviruses identified in bats. Two subtypes of coronaviruses have earlier been responsible for large-scale pandemics, Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS). However, COVID-19 has been found to have higher levels of transmissibility than the earlier two viruses (Adhikari et al., 2020).
It has been observed that susceptibility to COVID-19 shows geographical variation, and it has also been noted that individuals have different disease severities, indicating that host genomic variations might be playing an important role. These need to be determined to predict disease risk and outcome, as well as help in plan new specific interventions and vaccine delivery.
A single patient follow-up study from Australia indicated that robust multi-factorial immune responses can be elicited to SARS-CoV-2, which is similar to the avian H7N9 disease, suggesting that early adaptive immune responses might correlate with better clinical outcomes (Thevarajan et al., 2020).
Recent studies have found that the SARS-CoV-2 and SARS-CoV genomes share around 80% of homology and use the same cell entry receptor, angiotensin converting enzyme 2 (ACE2), for infectivity (Gralinski and Menachery, 2020). Although there is no direct evidence that supports that variants of ACE2 exhibit differential binding of SARS-CoV-2 with S-protein, 11 common variants and one rare variant (rs143695310) associated with high expression of ACE2 in tissues was reported by (Cao et al., 2020). There is only one report from the Indian population about polymorphisms and their in-silico functional significance (Sharma et al., 2020).
After analyzing data from > 200,000 exomes, homozygous/heterozygous/hemizygous loss of function mutations of ACE2 appear to be extremely deleterious (Cirulli et al., 2020). Although rare, some of these variants affect the regions that interact wit h SARS-CoV-2, and further research may identify some of these variants as conferring resistance or heightened susceptibility to the virus.
Binding of SARS-CoV S protein to ACE2 triggers subtle conformational rearrangements, which are believed to increase the sensitivity of the S protein to proteolytic digestion at the S1 and S2 subunits (Haga et al., 2008). Cleavage of the S protein by host cell proteases is essential for viral infectivity and the responsible enzyme is the type II transmembrane serine proteases (TMPRSS2) (Matsuyama et al., 2010).
TMPRSS2 is expressed in ACE2-positive cells, including the human lung, and results obtained with surrogate cell culture systems suggest that TMPRSS2 might play a significant role in coronavirus spread in the human respiratory tract (Bertram et al., 2013). TMPRSS2 facilitates infection via two independent mechanisms, cleavage of ACE2, which might promote viral uptake, and cleavage of SARS-S, which activates the S protein for membrane fusion (Heurich et al., 2014).
Viruses use multiple alternative receptors to enter host cells and a study by Cai (2020) reported that apart from ACE2, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) known as CD209 may be important for microbial infectivity (Cai, 2020). DC-SIGN acts as an adhesion molecule and also initiates innate immunity, although the exact mechanism is not clear, but it is known to be involved in microbial clearance through capture, destruction, and presentation of antigens. High expression of DC-SIGN in older individuals and higher gene expression of L-SIGN in Caucasians when compared to Asians was reported. It was also shown that significantly higher DC-SIGN gene expression occurs in the lungs of smokers, especially former smokers, indicating that this may affect SARS-CoV-2 infection (Cai, 2020). It is commonly believed that RNA virus entry into cells by endocytosis is regulated with interferon (IFN)-induced transmembrane (IFITM) proteins, which detect and eliminate viral invaders before the establishment of infection (Weidner et al., 2010). Therefore, mutation in the genes coding for these proteins can lead to variability in establishing infections in the host. IFITM3 is a cellular restriction factor that inhibits infection of influenza virus and many other pathogenic viruses, and sequence variants of this gene exhibits ethnic differences (Yount et al., 2010; Zhang et al., 2013) This highlights the significant role of IFITM3 genetic variants on the epidemiology of influenza. The IFITM3 rs12252 polymorphism has different allele frequencies among different ethnicities ranging from 0% among Japanese to 44% among Chinese populations. Zhang et al. (2013) found a significant association between rs12252 and susceptibility to severe, but not mild flu, among Asians and evidence of an association between rs12252 C allele homozygotes and susceptibility to mild flu in patients with Caucasian ancestry (Zhang et al., 2013).
Respiratory surfaces are exposed to pathogens continuously and a protective mucus barrier traps and eliminates them via mucociliary clearance. MUC5B is an evolutionarily conserved gene that encodes the principal macromolecules in airway mucus (Roy et al., 2014). Genetic variants of MUC genes are linked to diverse lung diseases. MUC5B deficiency causes organisms to accumulate in upper and lower airways and is responsible for development of idiopathic pulmonary fibrosis (Kaur et al., 2017). MUCB5 variants may also be linked to COVID-19 progression. A gain-of-function promoter polymorphism, rs35705950 of MUC5B has been indicated to be responsible for increased severity of lung disease. This variant has been considered to confer the largest risk, genetically for pulmonary fibrosis (Roy et al., 2014; Helling et al., 2017).
The aim of the present study was to identify variants in these five selected candidate genes from the clinical exome data available with us for more than 100 individuals and make an attempt to classify them as relevant to the present COVID-19 aetiopathology, especially for the Indian population.
Methods
We performed a literature survey to study the global trends of COVID-19 infection and its severity in different populations to understand and predict the impact of the pandemic in the Indian population. Scientific reports, publications, and Genome Wide Association Studies (GWAS) results were studied with respect to severe acute respiratory conditions both viral and non-viral. In view of limited data on COVID-19 susceptibility studies, we included studies on other pandemics like H1N1, H7N9, and MERS, and we identified five candidate genes and their polymorphisms, which may be responsible for infectivity, disease progression, and disease outcome. The selected genes are ACE2, TMPRSS2, IFITM3, CD209 (DC-SIGN), and MUC5B.
The Department of Genetics & Molecular Medicine, Kamineni Hospitals has a genetic counseling and diagnostic facility where patients from different specialties are referred for genetic evaluation. All the patients included in the study were provided with pre- and post-test genetic counseling. Written informed consent to utilize genetic information for research/academic purposes was obtained prior to collecting blood samples.
The variant calling format (vcf) and annotated file data generated from clinical exome studies from individuals/families were stored in a data bank. The selected candidate gene variants were assessed in our internal cohort of 103 individuals, who had earlier provided consent, to perform a pilot study on the susceptibility and disease severity of Indians for COVID-19.
The variant mining flow carried out is briefly described below:
1. Files in vcf format were uploaded on Base Space Variant Interpreter, an Illumina freeware platform. The variants were identified using inbuilt filters.
2. Frequency of the variants was tabulated and compared to global frequencies using TOPMED, EXAC, 1000G databases.
Results
Data from a total of 103 exomes were analyzed of which 53 were females and 50 were males, and 497 polymorphisms were identified in the five selected candidate genes (Table 1).
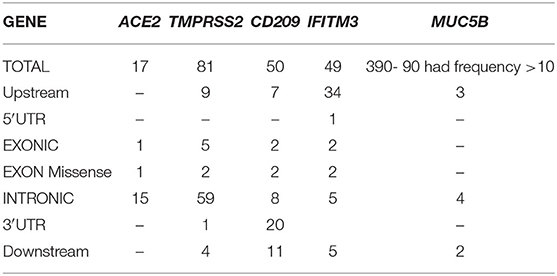
Table 1. Polymorphisms identified in five genes from preliminary exome data analysis of individuals from India (Note−390 variants in MUC5B gene were identified in our cohort, functionally relevant nine are mentioned here, full data can be accessed from Supplementary Data).
In the ACE2 gene, 17 polymorphisms were identified of which rs2285666 was the most frequent (9/103), followed by rs113691336, rs971249, and rs4646174, which were observed in 4/103 individuals (Table 2). One missense variant rs4646116 was identified in one individual. This variant causes a protein change lysine to arginine at 26th position, which lies in the extracellular membrane and inhibits interaction with Sar-COV2 Spike protein.
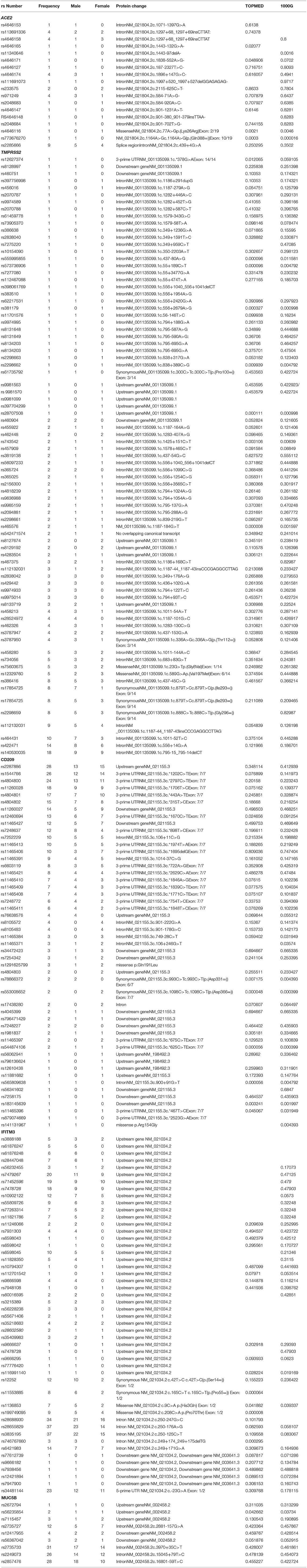
Table 2. Showing genewise frequencies of polymorphisms identified with gender stratification (Note−390 variants in MUC2B gene were identified in our cohort, functionally relevant nine are mentioned here, full data can be accessed from Supplementary Data).
In TMPRSS2 gene 81 variants were identified and the most common polymorphism identified was rs140530035 (18/103), followed by rs422471 (14/103) and rs464431 (10/103). Both missense variants were in the non-catalytic domain, which does not interact directly with SAR-COV2 S protein (Table 2).
In the CD209 gene, 50 variants were identified, and the most frequent ones were rs2287886 (28/103), rs1544766 (26/103), and rs4804800 (24/103) followed by rs11260028 (18/103), rs4804801 (17/103), rs4804802 (15/103), rs11260027 (14/103), and rs12460694 (13/103) (Table 2). Both missense variants rs1291625799 (3/103) causing p.Gln191Leu and rs141131967 (1/103) causing p.Arg154Gly alter disulfide formation in the C-type lectin domain, which functions as a calcium-dependent carbohydrate-recognition domain, were identified in our cohort.
A total of 49 variants were identified in the IFITM3 gene. Most frequent variants identified were rs28655829 (37/103), rs3835195(37/103), rs28688930 (34/103), rs34481144 (23/103), rs7479267 (20/103), rs71452596 (19/103), rs7478728 (18/103), rs6421983 (14/103), rs10902122 (12/103), rs12252 (12/103), and rs12252 (10/103).
MUC5B gene exhibited the maximum number of variants. Of the total 390 variants identified, 93 of them had frequency of above 10, the most frequent variants observed were rs2735733 (31/103), rs2249073 (26/103), and rs2857476 (28/103). Polymorphisms rs2735727 (12/103) and rs12417955 (4/103) that lead to alternative splicing of MUC5B and rs56367042 (3/103) are speculated to be involved in the pathogenesis of idiopathic pulmonary fibrosis.
Discussion
A wide range of inter individual variation is being observed in the infectivity, disease symptoms, progression of disease, and mortality of COVID-19 between different populations. Host genetic factors have frequently been implicated in respiratory infectious diseases, single nucleotide polymorphisms (SNPs), or commonly known gene polymorphisms have been considered responsible for both ethnic and inter individual variation (Patarčić et al., 2015). A recent twin study indicated that there is 50% genetic heritability in response to the SARS-CoV2 infection justifying the need to evaluate sequence variants in candidate genes (Williams et al., 2020). Until exome and genome data of individuals with specific SARS-CoV2 infection, unaffected contacts and individuals with range of disease symptoms, as well as, those who succumbed to disease is available, analysis of existing data from random population is the only option to identify variants, which may help to develop a polymorphism panel to identify individuals who are susceptible to infection and also those at risk of developing severe COVID-19 disease.
Since host genetic polymorphisms have been demonstrated to be associated with vulnerability to human infection, in this study five candidate genes—ACE2, TMPRSS2, CD209, IFITM3, and MUC5B—were selected based on their relevance to the current pandemic. All variants reported from each gene were identified from the sequence data available with us as vcf files. The data belonged to 103 individuals of Indian origin (who had consented for use of their data for research) and was selected randomly without any prior bias. Maximum number of variants (n = 390) were identified in MUC5B gene and the least (n = 17) in ACE2 gene.
ACE2 is a human homolog for ACE, which is well-known for its role in the Renin-Angiotensin pathway. ACE2 was identified in the year 2000, it consists of 805 amino acids, and it is a type I transmembrane glycoprotein with a single extracellular catalytic motif (Kuba et al., 2010). It is a carboxypeptidase that catalyzes liberation of vasodilator peptide, angiotensin, from angiotensin II, thus is responsible for counterbalancing the potent vasoconstrictor effects of angiotensin II (Schindler et al., 2007). It also has various other physiological functions, like being a key regulator of dietary amino acid homeostasis in colitis (Hashimoto et al., 2012). But in the context of COVID-19, ACE2 is the major viral receptor and is important for SARS-CoV-2 entry into the cell, making it extremely relevant for infectivity.
Several polymorphisms in ACE2 were reported from a large commercial dataset (Cai, 2020). A recent study from Italian population states that ACE2 variants underlie interindividual variability and susceptibility to COVID-19 (Hashimoto et al., 2012). In our analysis, 17 variants were identified, and the earlier reported missense variant rs4646116, which is responsible for Lys26Arg change in exon 2, was seen with a frequency of 0.0048 in our preliminary data. The frequency of rs 4646116 is reported as 0.0021 in TOPMED and 0.0046 in 1000 Genomes, it has been reported from most populations with Europeans having the highest frequency (Asselta et al., 2020). Another intronic polymorphism rs4646171 was identified with a frequency of 0.0048 and was reported with a higher frequency of 0.048 in TOPMED and 0.070 in 1000 Genome data. The most common polymorphism in our cohort was rs2285666, which exhibited a frequency of 0.0436, which was higher than what was observed in the two databases (Table 2). The A/A genotype of rs2285666 has a 50% lower expression level of ACE2 compared to G/G genotype and may be protective (Asselta et al., 2020).
Earlier studies reported that the A allele of ACE2 rs4646127 intronic SNP is responsible for decreased tissue expression of ACE2 and was considerably more common in people of European (44.1%) descent and less common in people of East Asian (1%) descent. Another polymorphism rs4646174 has been associated with central pulse pressure, Brain Natriuretic Peptide, and NYHA classification of patients with chronic heart failure (Malard et al., 2013). In several studies, different ACE2 polymorphisms with altered ACE2 expression have been associated with systolic blood pressure, diabetes, cardiovascular disorders, stroke, etc. These could be the likely reason behind the individuals with comorbidities succumbing severely to the COVID-19 infection than others.
TMPRSS2 is considered to play a role in SARS-CoV-2 virus entry in human cells along with ACE2. The host cell protease TMPRSS2 is involved in the fusion of the virus with cell membrane, thereby it is important to understand the role of its variants in COVID-19 infection and disease progression (Bertram et al., 2013; Heurich et al., 2014). TMPRSS2 is known to cleave the influenza A virus and knock out TMPRSS2−/− mice are resistant to infection, indicating the importance of this gene in the spread and pathogenesis of viral infection (Lambertz et al., 2020).
The role of TMPRSS2 gene in prostate cancer is well-known; however, there are very few reports about the association of TMPRSS2 polymorphism with respiratory distress. An SNP of the TMPRSS2 gene (rs12329760 C>T; Met160Val) present in an exonic splicing enhancer srp40 site, which is highly conserved across mammals, has been found to be associated positively with TMPRSS2–ERG fusion by translocation due to an increased chance of exon skipping in prostate cancer (Bhanushali et al., 2018). This rs12329760 polymorphism was seen in 4.85% of individuals from our study.
However, TMPRSS2 gene eQTLs in lungs appear to cluster at the 3′ end and are potentially associated with expression of alternative transcripts in lungs. Amongst these common eQTLs, HaploReg annotations indicated rs 4818239 as an important SNP (Sharma et al., 2020). This was identified in about 2% of the individuals in our population. A recent Indian paper assessed the in-silico functional analysis of various variants some of which were identified in our cohort, like rs4818239 (2/103), rs734056 (5/103), and rs62217531 (1/103) (Sharma et al., 2020). It has been demonstrated that genetic variants with higher TMPRSS2 expression confer greater risk to severe influenza A(H1N1). Notably, rs2070788 and rs383510 had high expression of TMPRSS2 and were significantly associated with the susceptibility to Influenza A(H7N9) (Cheng et al., 2015). The rs2298662 showed high LD with rs2070788 associated with respiratory disorders (Cheng et al., 2015). All these three variants—rs2070788, rs2298662 (1/103), and rs383510 (1/103)—were found in our study at a low frequency. The same variants also increase susceptibility to human Influenza A(H7N9) and may be relevant for COVID-19 infectivity. A haplotype, predicted to be associated with higher TMPRSS2 expression, is characterized by three SNPs (rs2070788, rs9974589, rs7364083), whose Minor Allele Frequency (MAF) is significantly increased in Europeans and was 9% higher in Italians with respect to East Asians (Asselta et al., 2020). The polymorphism rs9974995 is nominally associated with phenotype related to respiratory function or respiratory medication (Salmeterol or fluticasone propionate) (Wang et al., 2020).
Higher expression of ACE2 and TMPRSS2 in males, African Americans, and patients with diabetes mellitus provides rationale for monitoring these subgroups for high infectivity and poor COVID-19 outcomes. The lower expression of ACE2 and TMPRSS2 with inhaled corticosteroid use warrants prospective study of inhaled corticosteroid use as a predictor of decreased susceptibility to SARS-CoV-2 infection and decreased COVID-19 morbidity (Peters et al., 2020). The two polymorphisms rs112132031 and rs75603675 identified in 10.67 and 4.85% of our cohort, respectively, may be relevant for COVID-19 Cytokine Release syndrome and conjunctival infection according to earlier reports (Peters et al., 2020).
The CD209 gene, which encodes the DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), a key effector of the innate immunity and antiviral defense, is a receptor expressed in the dendritic cells involved in recognition of oligosaccharides present in several pathogens (Granelli-Piperno et al., 2005). Hence, polymorphisms in this gene may explain susceptibility/resistance to infection as well as severity of symptoms in several infectious diseases. In our cohort, 50 variants have been identified in this gene. The GG genotype of rs2287886 present in the promoter region of CD209 was identified in 27.18% individuals in our cohort. It is reported to be associated with development of dengue fever requiring hospitalization, cytomegalovirus disease after allogeneic stem-cell transplantation, predisposition to developing tick-borne encephalitis, invasive pulmonary Aspergillosis infection, Kawazaki infection, and colorectal cancer (Mezger et al., 2008; Barkhash et al., 2012; Sainz et al., 2012; Alagarasu et al., 2013; Lu et al., 2013; Portman et al., 2013; Czupryna et al., 2017). Individuals with genotype AA at rs2287886, whose frequency in Central Asian Mongoloids is high, have shown to express higher levels of CD209, thereby having a higher rate of infection by cytomegalovirus than DCs carrying the GG genotype creating an analogous situation for the existence of Epstein-Barr virus (EBV), since glycoproteins on the viral surface, which are conserved are similar with those on cytomegalovirus (Barkhash et al., 2012). CD209 is also known to transport viruses via immature DCs from the periphery to lymph nodes, where CD4 cells get activated and infected, elucidating a link between CD209 genetic variation and CD4 count. In our dataset, the rs8105483 was observed in 3.88% subjects and rs2287886 in 27.18%, which together form a protective haplotype, which has a higher CD4 count (Geijtenbeek et al., 2003; Tailleux et al., 2003; Tassaneetrithep et al., 2003; Barreiro et al., 2006; Hennig et al., 2011). These polymorphisms may similarly be protective for COVID-19.
In addition, significant associations were found between high risk of Kawasaki disease with CD209 polymorphisms rs4804800 and rs2287886 (Kuo et al., 2014). A study by Ovsyannikova et al. (2011) has shown associations between promoter SNP rs11881682 and intronic SNPs: rs8105572 and rs7252229 of the CD209 gene and measles-specific IFN-γ Elispot responses in Caucasian subjects (Ovsyannikova et al., 2011). SNPs in CD209 rs4804800, rs11465384, rs7248637, and rs7252229 have shown association with an increased risk to develop invasive pulmonary aspergillosis infection (Sainz et al., 2012). The polymorphism rs7248637 in CD209, which showed association with dengue in the Colombian population (Avendaño-Tamayo et al., 2019), was observed in 11.65% subjects in our dataset, and 9.7% subjects in our dataset showed rs11465413 associated with atopic sensitization (Penders et al., 2010).
Interferon-induced membrane protein that inhibits the entry of viruses into the host cell cytoplasm by preventing viral fusion with cholesterol depleted endosomes is encoded by the IFITM3 gene (Zhao et al., 2019). It has a capacity to inactivate new enveloped viruses, which bud out of the infected cell. It has been shown to be active against multiple viruses, including influenza A virus, SARS coronavirus (SARS-CoV), Ebola virus (EBOV), Dengue virus (DNV), human immunodeficiency virus type 1 (HIV-1), etc. (Brass et al., 2009; Lu et al., 2011). Pathways through which IFITM3 functions are: Innate Immune System and Interferon gamma signaling. Studies have shown that the first 21 amino acids of the N-terminus of IFITM3 gene are required for attenuation of vesicular stomatitis virus replication, and that truncated IFITM3 protein fails to restrict the replication of various strains of influenza virus, as well as HIV-1 (Jia et al., 2012; Bailey et al., 2014). Williams et al. (2014) and Kim et al. (2019) showed that even full-length IFITM3 restricts entry and replication of H1N1. Polymorphisms of this gene have been studied in several infections. The polymorphism rs6598045 c.−188T > C (4.85% in our cohort) induces a difference in the binding capacity of the transcription factor causing a difference in the transcription efficiency of the IFITM3 gene, which was reported to exhibit a strong association with influenza H1N1 2009 pandemic virus infection (Shen et al., 2013). Another functional polymorphism rs3888188, showed that peripheral-blood mononuclear cells carrying GG genotype had reduced IFITM3 mRNA level compared to those with TT or GT genotype, which predisposes toward pulmonary tuberculosis in Iranian and Han Chinese populations (Shen et al., 2013).
Previous studies predicted that rs12252 C allele might produce an alternate spliced transcript that encodes an aberrant truncated protein Δ21 of IFITM3, which reduces the cellular resistance to influenza viruses by blocking early stage of viral replication (Everitt et al., 2012; Compton et al., 2016). Association of this polymorphism has been observed in Chinese population with pandemic influenza (H1N1 09pdm), seasonal influenza (H3N2 and influenza B), and avian influenza (H7N9) (Carter et al., 2018). In our cohort, 11.65% of the individuals carried this allele. Seasonal influenza hospital admissions were associated with rs7948108, which was observed with a low (0.97%) percentage in our cohort.
The rs34481144 is considered to directly affect IFITM3 promoter function. The risk allele A is linked to diminished promoter activity by increasing the binding of the methylation-sensitive CTCF transcriptional repressor. This ablation of methylation site controls IFITM3-promoter methylation in memory Cytotoxic T Lymphocytes (CTLs) reducing CTCF binding to increase IFITM3 expression, which leads to increased memory CTL survival and more efficient viral clearance from infected airways (Eisfeld and Kawaoka, 2017). This was observed in 22% of individuals of our cohort and may be a relevant variant for SARS-CoV-2 clearance and reduced progression of disease.
MUC5B gene encodes a member of the mucin family of proteins, which are highly glycosylated macromolecular components of mucus secretions. This family member is the major gel-forming mucin in mucus (Ridley and Thornton, 2018). It is a major contributor to the lubricating and viscoelastic properties of whole saliva, normal lung mucus, and cervical mucus. This gene has been found to be upregulated in some human diseases, while pathogenic variants have been reported to cause pulmonary fibrosis—a lung disease characterized by shortness of breath and varying degrees of inflammation and fibrosis, which is rapidly progressive and acute lung injury with subsequent scarring and end-stage lung disease (Zhang et al., 2019). Many of these symptoms are similar to that reported in COVID-19 disease.
Airway lining mucus serves as the first line of defense during upper respiratory infection. Pathogens trapped in the mucus layer are first removed by the mucociliary clearance mechanism of the underlying airway epithelium as well as macrophages and then by neutrophils recruited into the airways in response to inflammatory mediators released by epithelial cells and macrophages (Kim, 2012). Adult MUC5B-deficient mice displayed bronchial hyperplasia and metaplasia, interstitial thickening, alveolar collapse, immune cell infiltrates, fragmented and disorganized elastin fibers, and collagen deposits that were, for approximately one-fifth of the mice, associated with altered pulmonary function leading to respiratory failure demonstrating that the mouse MUC5B is essential for maintaining normal lung function (Valque et al., 2019).
MUC5B gene had the maximum number (n = 390) of variants in our cohort. The SNP rs2672794 is associated significantly with increased susceptibility to coal workers' pneumoconiosis in a Chinese population (Ji et al., 2014). While the rs56235854 polymorphism is associated with severe asthma (Johnson, 2020), this was identified in 1.94% of individuals analyzed in our dataset. The MUC5B gene rs2735733, rs2249073, and rs2857476 were associated with dental caries; all the three variants were present in 30.09, 25.24, and 27.18%, respectively, of individuals from our cohort, indicating that Indians are highly susceptible to dental caries.
Polymorphisms rs2735727 (12/103) and rs12417955 (4/103) that lead to alternative splicing of MUC5B and rs56367042 (3/103) are speculated to be involved in the pathogenesis of idiopathic pulmonary fibrosis (Nance et al., 2014). A promoter variant rs7115457 is associated with diffuse pan bronchiolitis (Kamio et al., 2005), while another MUC5B promoter polymorphism, rs35705950, is the strongest risk factor, genetic or otherwise, accounting for 30–35% risk of developing Idiopathic pulmonary fibrosis (IPF), a disease that was previously considered idiopathic. This MUC5B variant can potentially be used to identify individuals with preclinical pulmonary fibrosis and is predictive of radiologic progression of disease. The excessive production of MUC5B either enhances injury due to reduced mucociliary clearance or impedes repair consequent to disruption of normal regenerative mechanisms in the distal lung (Evans et al., 2016). This variant rs35705950 (1000G frequency T = 0.0467), was not identified in our cohort and maybe responsible for protecting us from COVID-19 full-blown symptoms.
A total of 497 polymorphisms were identified in five genes in 103 exomes analyzed; 38 polymorphisms identified in our cohort have been reported earlier in literature and have functional significance. Two polymorphic variants rs35705950 of MUC5B that increase susceptibility to pulmonary fibrosis and the common “European” haplotype of TMPRSS2 gene (composed of SNPs rs463727, rs34624090, rs55964536, rs734056, rs4290734, rs34783969, rs11702475, rs35899679, and rs35041537) are totally absent in the Asian population. This may be one of the plausible factors for reduced severity of COVID-19 in Asians compared to Europeans.
Based on the function and expression, we categorized the 38 polymorphisms into three groups: polymorphisms increasing susceptibility to the infection (respiratory illnesses), polymorphisms that confer enhanced immunity, and the polymorphisms that are involved in other pathologies, which can induce comorbidities and make an individual fall under high risk category. The six protective polymorphisms, three in ACE2, two in CD209, and one in IFITM3 genes (Table 3) were identified in our cohort, at least one was present in 38.83% of the individuals analyzed. There were 27 risk susceptibility polymorphisms identified in four genes (Table 3) and cumulative count of individuals with at least one risk polymorphism was 47.57%. There were five polymorphisms, three in MUC5B and one each in ACE2 and TMPRSS2 genes, which are associated with comorbidities and the cumulative count of individuals with at least one risk polymorphism in our cohort was 40.77% (Table 3).
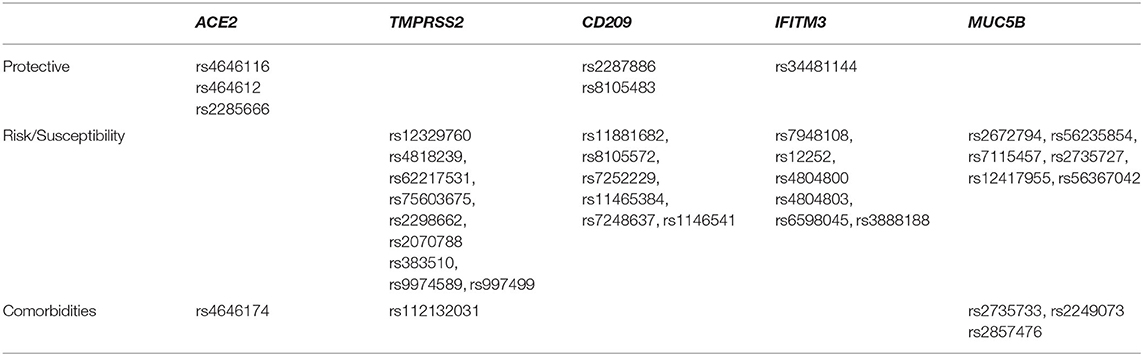
Table 3. Showing stratification of polymorphisms into protective, risk inducing, and associated with comorbidities.
A larger study for validating our results from Indian population is required, and the sequence data for this maybe already available with the Council of Scientific and Industrial Research—Institute of Genomics and Integrative Biology (CSIR-IGIB) consortium and commercial companies doing testing for Indian patients since 2014. These will also include individuals from different regions of India, unlike the present study where the majority of individuals were from South India. Preliminary sequence data analysis results from five selected candidate genes presented in this paper highlights the importance of identifying polymorphisms from COVID-19 infected asymptomatic and symptomatic patients to give more meaningful results, which will help in managing this pandemic.
Conclusion
This is the first study from an Indian population presenting polymorphisms from five selected candidate genes, which may be important for understanding the infectivity and progression of COVID-19 in our population. A larger dataset needs to be analyzed to validate the results and develop a panel of polymorphisms useful for identifying individuals at risk, as well as, those likely to have severe disease symptoms.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
GI co-conceptualized the study, performed data analysis, and wrote the first draft of the manuscript. SS compiled and analyzed the data. SZ co-conceptualized the study and assisted in scientific editing of the manuscript. DS, VM, SP, AS, NA, and NB co-compiled the data. AN co-conceptualized the study. QH conceptualized, supervised the study, and performed scientific editing of the manuscript. All authors approved the final draft of the manuscript and agree to be accountable for the content of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00861/full#supplementary-material
References
Adhikari, S. P., Meng, S., Wu, Y. J., Mao, Y. P., Ye, R. X., Wang, Q. Z., et al. (2020). Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: a scoping review. Infect. Dis Poverty 9, 1–2. doi: 10.1186/s40249-020-00646-x
Alagarasu, K., Damle, I. M., Bachal, R. V., Mulay, A. P., Shah, P. S., and Dayaraj, C. (2013). Association of promoter region polymorphisms of CD209 gene with clinical outcomes of dengue virus infection in Western India. Infect. Genet. Evol. 17, 239–242. doi: 10.1016/j.meegid.2013.04.024
Asselta, R., Paraboschi, E. M., Mantovani, A., and Duga, S. (2020). ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 12, 10087–10098. doi: 10.1101/2020.03.30.20047878
Avendaño-Tamayo, E., Rúa, A., Parra-Marín, M. V., Rojas, W., Campo, O., Chacón-Duque, J., et al. (2019). Evaluation of variants in IL6R, TLR3, and DC-SIGN genes associated with dengue in a sampled Colombian population. Biomédica 39, 88–101. doi: 10.7705/biomedica.v39i1.4029
Bailey, C. C., Zhong, G., Huang, I. C., and Farzan, M. (2014). IFITM-family proteins: the cell's first line of antiviral defense. Annu Rev Virol. 1, 261–283. doi: 10.1146/annurev-virology-031413-085537
Barkhash, A. V., Perelygin, A. A., Babenko, V. N., Brinton, M. A., and Voevoda, M. I. (2012). Single nucleotide polymorphism in the promoter region of the CD209 gene is associated with human predisposition to severe forms of tick-borne encephalitis. Antiviral Res. 93, 64–68. doi: 10.1016/j.antiviral.2011.10.017
Barreiro, L. B., Neyrolles, O., Babb, C. L., Tailleux, L., Quach, H., McElreavey, K., et al. (2006). Promoter variation in the DC-SIGN–encoding gene CD209 is associated with tuberculosis. PLoS Med. 3:e20. doi: 10.1371/journal.pmed.0030020
Bertram, S., Dijkman, R., Habjan, M., Heurich, A., Gierer, S., Glowacka, I., et al. (2013). TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 87, 6150–6160. doi: 10.1128/JVI.03372-12
Bhanushali, A., Rao, P., Raman, V., Kokate, P., Ambekar, A., Mandva, S., et al. (2018). Status of TMPRSS2–ERG fusion in prostate cancer patients from India: correlation with clinico-pathological details and TMPRSS2 Met160Val polymorphism. Prostate Int. 6, 145–150. doi: 10.1016/j.prnil.2018.03.004
Brass, A. L., Huang, I. C., Benita, Y., John, S. P., Krishnan, M. N., Feeley, E. M., et al. (2009). The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254. doi: 10.1016/j.cell.2009.12.017
Cai, G. (2020). Bulk and single-cell transcriptomics identify tobacco-use disparity in lung gene expression of ACE2, the receptor of 2019-nCov. MedRxiv [Preprint]. doi: 10.1101/2020.02.05.20020107
Cao, Y., Li, L., Feng, Z., Wan, S., Huang, P., Sun, X., et al. (2020). Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 6, 1–4. doi: 10.1038/s41421-020-0147-1
Carter, T. C., Hebbring, S. J., Liu, J., Mosley, J. D., Shaffer, C. M., Ivacic, L. C., et al. (2018). Pilot screening study of targeted genetic polymorphisms for association with seasonal influenza hospital admission. J. Med. Virol. 90, 436–446. doi: 10.1002/jmv.24975
Cheng, Z., Zhou, J., To, K. K., Chu, H., Li, C., Wang, D., et al. (2015). Identification of TMPRSS2 as a susceptibility gene for severe 2009 pandemic A (H1N1) influenza and A (H7N9) influenza. J. Infect. Dis. 212, 1214–1221 doi: 10.1093/infdis/jiv246
Cirulli, E. T., Riffle, S., Bolze, A., and Washington, N. L. (2020). Revealing variants in SARS-CoV-2 interaction domain of ACE2 and loss of function intolerance through analysis of> 200,000 exomes. bioRxiv [Preprint]. doi: 10.1101/2020.04.07.030544
Compton, A. A., Roy, N., Porrot, F., Billet, A., Casartelli, N., Yount, J. S., et al. (2016). Natural mutations in IFITM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep. 17, 1657–1671. doi: 10.15252/embr.201642771
Czupryna, P., Parczewski, M., Grygorczuk, S., Pancewicz, S., Zajkowska, J., Dunaj, J., et al. (2017). Analysis of the relationship between single nucleotide polymorphism of the CD209, IL-10, IL-28 and CCR5 D32 genes with the human predisposition to developing tick-borne encephalitis. Adv. Hyg. Exp. Med. 71, 788–796. doi: 10.5604/01.3001.0010.3856
Eisfeld, A. J., and Kawaoka, Y. (2017). Calculated risk: a new single-nucleotide polymorphism linked to severe influenza disease. Nat. Med. 23:911. doi: 10.1038/nm.4383
Evans, C. M., Fingerlin, T. E., Schwarz, M. I., Lynch, D., Kurche, J., Warg, L., et al. (2016). Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol. Rev. 96, 1567–1591. doi: 10.1152/physrev.00004.2016
Everitt, A. R., Clare, S., Pertel, T., John, S. P., Wash, R. S., Smith, S. E., et al. (2012). IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523. doi: 10.1038/nature10921
Geijtenbeek, T. B., Van Vliet, S. J., Koppel, E. A., Sanchez-Hernandez, M., Vandenbroucke-Grauls, C. M., Appelmelk, B., et al. (2003). Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17. doi: 10.1084/jem.20021229
Gralinski, L. E., and Menachery, V. D. (2020). Return of the Coronavirus: 2019-nCoV. Viruses 12:135. doi: 10.3390/v12020135
Granelli-Piperno, A., Pritsker, A., Pack, M., Shimeliovich, I., Arrighi, J. F., Park, C. G., et al. (2005). Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J. Immunol. 175, 4265–4273. doi: 10.4049/jimmunol.175.7.4265
Haga, S., Yamamoto, N., Nakai-Murakami, C., Osawa, Y., Tokunaga, K., Sata, T., et al. (2008). Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. U.S.A. 105, 7809–7814. doi: 10.1073/pnas.0711241105
Hashimoto, T., Perlot, T., Rehman, A., Trichereau, J., Ishiguro, H., Paolino, M., et al. (2012). ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487, 477–481. doi: 10.1038/nature11228
Helling, B. A., Gerber, A. N., Kadiyala, V., Sasse, S. K., Pedersen, B. S., Sparks, L., et al. (2017). Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 57, 91–99. doi: 10.1165/rcmb.2017-0046OC
Hennig, B. J., Velez-Edwards, D. R., Van Der Loeff, M. F., Bisseye, C., Edwards, T. L., Tacconelli, A., et al. (2011). CD4 intragenic SNPs associate with HIV-2 plasma viral load and CD4 count in a community-based study from Guinea-Bissau, West Africa. J. Acquired Immune Def. Syndr. 56, 1–8. doi: 10.1097/QAI.0b013e3181f638ed
Heurich, A., Hofmann-Winkler, H., Gierer, S., Liepold, T., Jahn, O., and Pöhlmann, S. (2014). TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 88, 1293–1307. doi: 10.1128/JVI.02202-13
Ji, X., Wu, B., Jin, K., Luo, C., Han, R., Chen, M., et al. (2014). MUC5B promoter polymorphisms and risk of coal workers' pneumoconiosis in a Chinese population. Mol. Biol. Rep. 41, 4171–4176. doi: 10.1007/s11033-014-3100-2
Jia, R., Pan, Q., Ding, S., Rong, L., Liu, S. L., Geng, Y., et al. (2012). The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J. Virol. 86, 13697–13707. doi: 10.1128/JVI.01828-12
Johnson, L. (2020). Genetically determined variation of respiratory mucins: disease and demography (Doctoral dissertation), UCL, University College London.
Kamio, K., Matsushita, I., Hijikata, M., Kobashi, Y., Tanaka, G., Nakata, K., et al. (2005). Promoter analysis and aberrant expression of the MUC5B gene in diffuse panbronchiolitis. Am. J. Respir. Crit. Care Med. 171, 949–957. doi: 10.1164/rccm.200409-1168OC
Kaur, A., Mathai, S. K., and Schwartz, D. A. (2017). Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front. Med. 4:154. doi: 10.3389/fmed.2017.00154
Kim, K. C. (2012). Role of epithelial mucins during airway infection. Pulm. Pharmacol. Ther. 25, 415–419. doi: 10.1016/j.pupt.2011.12.003
Kim, Y. C., Jeong, M. J., and Jeong, B. H. (2019). Strong association of regulatory single nucleotide polymorphisms (SNPs) of the IFITM3 gene with influenza H1N1 2009 pandemic virus infection. Cell. Mol. Immunol. 17, 662–664. doi: 10.1038/s41423-019-0322-1
Kuba, K., Imai, Y., Ohto-Nakanishi, T., and Penninger, J. M. (2010). Trilogy of ACE2: a peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 128, 119–128. doi: 10.1016/j.pharmthera.2010.06.003
Kuo, H. C., Huang, Y. H., Chien, S. C., Yu, H. R., Hsieh, K. S., Hsu, Y. W., et al. (2014). Genetic variants of CD209 associated with Kawasaki disease susceptibility. PLoS ONE 9:e105236. doi: 10.1371/journal.pone.0105236
Lambertz, R. L., Gerhauser, I., Nehlmeier, I., Gärtner, S., Winkler, M., Leist, S. R., et al. (2020). H2 influenza A virus is not pathogenic in TMPRSS2 knock-out mice. Virol. J. 17, 1–7. doi: 10.1186/s12985-020-01323-z
Lu, J., Pan, Q., Rong, L., Liu, S. L., and Liang, C. (2011). The IFITM proteins inhibit HIV-1 infection. J. Virol. 85, 2126–2137. doi: 10.1128/JVI.01531-10
Lu, S., Bevier, M., Huhn, S., Sainz, J., Lascorz, J., Pardini, B., et al. (2013). Genetic variants in C-type lectin genes are associated with colorectal cancer susceptibility and clinical outcome. Int. J. Cancer 133, 2325–2333. doi: 10.1002/ijc.28251
Malard, L., Kakinami, L., O'Loughlin, J., Roy-Gagnon, M. H., Labbe, A., Pilote, L., et al. (2013). The association between the angiotensin-converting enzyme-2 gene and blood pressure in a cohort study of adolescents. BMC Med. Genet. 14:117. doi: 10.1186/1471-2350-14-117
Matsuyama, S., Nagata, N., Shirato, K., Kawase, M., Takeda, M., and Taguchi, F. (2010). Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 84, 12658–12664. doi: 10.1128/JVI.01542-10
Mezger, M., Steffens, M., Semmler, C., Arlt, E. M., Zimmer, M., Kristjanson, G. I., et al. (2008). Investigation of promoter variations in dendritic cell-specific ICAM3-grabbing non-integrin (DC-SIGN)(CD209) and their relevance for human cytomegalovirus reactivation and disease after allogeneic stem-cell transplantation. Clin. Microbiol Infect. 14, 228–234. doi: 10.1111/j.1469-0691.2007.01902.x
Nance, T., Smith, K. S., Anaya, V., Richardson, R., Ho, L., Pala, M., et al. (2014). Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS ONE 9:e97550. doi: 10.1371/journal.pone.0092111
Ovsyannikova, I. G., Haralambieva, I. H., Vierkant, R. A., O'Byrne, M. M., Jacobson, R. M., and Poland, G. A. (2011). The association of CD46, SLAM and CD209 cellular receptor gene SNPs with variations in measles vaccine-induced immune responses: a replication study and examination of novel polymorphisms. Hum. Hered. 72, 206–223. doi: 10.1159/000331585
Patarčić, I., Gelemanović, A., Kirin, M., Kolčić, I., Theodoratou, E., Baillie, K. J., et al. (2015). The role of host genetic factors in respiratory tract infectious diseases: systematic review, meta-analyses and field synopsis. Sci. Rep. 5:16119. doi: 10.1038/srep16119
Penders, J., Thijs, C., Mommers, M., Stobberingh, E., Dompeling, E., Reijmerink, N., et al. (2010). Intestinal lactobacilli and the DC-SIGN gene for their recognition by dendritic cells play a role in the aetiology of allergic manifestations. Microbiology 156, 3298–3305. doi: 10.1099/mic.0.042069-0
Peters, M. C., Sajuthi, S., Deford, P., Christenson, S., Rios, C. L., Montgomery, M. T., et al. (2020). COVID-19 related genes in sputum cells in asthma: relationship to demographic features and corticosteroids. Am. J. Respir. Crit. Care Med. 202, 83–90. doi: 10.1164/rccm.202003-0821OC
Portman, M. A., Wiener, H. W., Silva, M., Shendre, A., and Shrestha, S. (2013). DC-SIGN gene promoter variants and IVIG treatment response in Kawasaki disease. Pediatr. Rheumatol. 11:32. doi: 10.1186/1546-0096-11-32
Ridley, C., and Thornton, D. J. (2018). Mucins: the frontline defence of the lung. Biochem. Soc. Trans. 46, 1099–1106. doi: 10.1042/BST20170402
Roy, M. G., Livraghi-Butrico, A., Fletcher, A. A., McElwee, M. M., Evans, S. E., Boerner, R. M., et al. (2014). MUC5B is required for airway defence. Nature 505, 412–416. doi: 10.1038/nature12807
Sainz, J., Lupiáñez, C. B., Segura-Catena, J., Vazquez, L., Ríos, R., Oyonarte, S., et al. (2012). Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS ONE 7:e32273. doi: 10.1371/journal.pone.0032273
Schindler, C., Bramlage, P., Kirch, W., and Ferrario, C. M. (2007). Role of the vasodilator peptide angiotensin-(1–7) in cardiovascular drug therapy. Vasc. Health Risk Manage. 3, 125–137.
Sharma, S., Singh, I., Haider, S., Malik, M. Z., Ponnusamy, K., and Rai, E. (2020). ACE2 homo-dimerization, human genomic variants and interaction of host proteins explain high population specific differences in outcomes of COVID19. bioRxiv [Preprint]. doi: 10.1101/2020.04.24.050534
Shen, C., Wu, X. R., Jiao, W. W., Sun, L., Feng, W. X., Xiao, J., et al. (2013). A functional promoter polymorphism of IFITM3 is associated with susceptibility to pediatric tuberculosis in Han Chinese population. PLoS ONE 8:e67816. doi: 10.1371/journal.pone.0067816
Tailleux, L., Schwartz, O., Herrmann, J. L., Pivert, E., Jackson, M., Amara, A., et al. (2003). DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 197, 121–127. doi: 10.1084/jem.20021468
Tassaneetrithep, B., Burgess, T. H., Granelli-Piperno, A., Trumpfheller, C., Finke, J., Sun, W., et al. (2003). DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197, 823–829. doi: 10.1084/jem.20021840
Thevarajan, I., Nguyen, T. H., Koutsakos, M., Druce, J., Caly, L., van de Sandt, C. E., et al. (2020). Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. 26, 453–455. doi: 10.1038/s41591-020-0819-2
Valque, H., Gouyer, V., Duez, C., Leboeuf, C., Marquillies, P., Le Bert, M., et al. (2019). MUC5B-deficient mice develop early histological lung abnormalities. Biol. Open 8:bio046359. doi: 10.1101/705962
Wang, A., Chiou, J. A., Poirion, O. B., Buchanan, J., Valdez, M. J., Verheyden, J. M., et al. (2020). Single nucleus multiomic profiling reveals age-dynamic regulation of host genes associated with SARS-CoV-2 infection. bioRxiv [Preprint]. doi: 10.1101/2020.04.12.037580
Weidner, J. M., Jiang, D., Pan, X. B., Chang, J., Block, T. M., and Guo, J. T. (2010). Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84, 12646–12657. doi: 10.1128/JVI.01328-10
Williams, D. E., Wu, W. L., Grotefend, C. R., Radic, V., Chung, C., Chung, Y. H., et al. (2014). IFITM3 polymorphism rs12252-C restricts influenza A viruses. PLoS ONE 9:e110096. doi: 10.1371/journal.pone.0110096
Williams, F. M., Freydin, M., Mangino, M., Couvreur, S., Visconti, A., Bowyer, R. C., et al. (2020). Self-reported symptoms of covid-19 including symptoms most predictive of SARS-CoV-2 infection, are heritable. medRxiv [Preprint]. doi: 10.1101/2020.04.22.20072124
Yi, Y., Lagniton, P. N., Ye, S., Li, E., and Xu, R. H. (2020). COVID-19: what has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 16:1753. doi: 10.7150/ijbs.45134
Yount, J. S., Moltedo, B., Yang, Y. Y., Charron, G., Moran, T. M., López, C. B., et al. (2010). Palmitoylome profiling reveals S-palmitoylation–dependent antiviral activity of IFITM3. Nat. Chem. Biol. 6, 610–614. doi: 10.1038/nchembio.405
Zhang, Q., Wang, Y., Qu, D., Yu, J., and Yang, J. (2019). The possible pathogenesis of idiopathic pulmonary fibrosis considering MUC5B. Biomed. Res. Int. 2019:9712464. doi: 10.1155/2019/9712464
Zhang, Y. H., Zhao, Y., Li, N., Peng, Y. C., Giannoulatou, E., Jin, R. H., et al. (2013). Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat. Commun. 4, 1–6. doi: 10.1038/ncomms2433
Keywords: COVID-19, candidate genes, variants, polymorphisms, infectivity
Citation: Iyer GR, Samajder S, Zubeda S, S DSN, Mali V, PV SK, Sharma A, Abbas NZ, Bora NS, Narravula A and Hasan Q (2020) Infectivity and Progression of COVID-19 Based on Selected Host Candidate Gene Variants. Front. Genet. 11:861. doi: 10.3389/fgene.2020.00861
Received: 28 May 2020; Accepted: 15 July 2020;
Published: 04 September 2020.
Edited by:
Tarunveer Singh Ahluwalia, Steno Diabetes Center Copenhagen (SDCC), DenmarkReviewed by:
Sandhya Kiran Pemmasani, Mapmygenome, IndiaMala Ganesan, Centre for Cellular & Molecular Biology (CCMB), India
Copyright © 2020 Iyer, Samajder, Zubeda, S, Mali, PV, Sharma, Abbas, Bora, Narravula and Hasan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qurratulain Hasan, cWhhc2FuMjAwMEB5YWhvby5jb20=