 Kevin C. Deitz
Kevin C. Deitz Willem Takken2
Willem Takken2 Michel A. Slotman
Michel A. Slotman- 1Department of Entomology, Texas A&M University, College Station, TX, United States
- 2Laboratory of Entomology, Wageningen University and Research, Wageningen, Netherlands
The Anopheles gambiae complex is comprised of eight morphologically indistinguishable species and has emerged as a model system for the study of speciation genetics due to the rapid radiation of its member species over the past two million years. Male hybrids between most An. gambiae complex species pairs are sterile, and some genotype combinations in hybrid males cause inviability. We investigated the genetic basis of hybrid male inviability and sterility between An. coluzzii and An. quadriannulatus by measuring segregation distortion and performing a QTL analysis of sterility in a backcross population. Hybrid males were inviable if they inherited the An. coluzzii X chromosome and were homozygous at one or more loci in 18.9 Mb region of chromosome 3. The An. coluzzii X chromosome has a disproportionately large effect on hybrid sterility when introgressed into an An. quadriannulatus genetic background. Additionally, an epistatic interaction between the An. coluzzii X and a 1.12 Mb, pericentric region of the An. quadriannulatus 3L chromosome arm has a statistically significant contribution to the hybrid sterility phenotype. This same epistatic interaction occurs when the An. coluzzii X is introgressed into the genetic background of An. arabiensis, the sister species of An. quadriannulatus, suggesting that this may represent one of the first Dobzhansky–Muller incompatibilities to evolve early in the radiation of the Anopheles gambiae species complex. We describe the additive effects of each sterility QTL, epistatic interactions between them, and genes within QTL with protein functions related to mating behavior, reproduction, spermatogenesis, and microtubule morphogenesis, whose divergence may contribute to post-zygotic reproductive isolation between An. coluzzii and An. quadriannulatus.
Introduction
The study of speciation genetics and genomics has largely focused on the evolution of hybrid dysfunction (Dobzhansky, 1937; Coyne and Orr, 2004) because it represents the first step in the formation of post-zygotic reproductive isolation between diverging populations. The evolution of post-zygotic isolation mechanisms is positively correlated with the time of divergence between two putative species, and is impacted by the number of mutations separating two allopatric lineages at any given time, the number of incompatibilities per mutation, and the fitness of each incompatibility (Orr and Turelli, 2001).
Post-zygotic isolation occurs as Dobzhansky-Müller incompatibilities (DMI) arise between populations evolving in allopatry (Dobzhansky, 1936; Müller, 1940). Upon secondary contact of genetically divergent populations, epistatic loci on parental chromosomes are unable to function effectively in hybrids. The resulting gene products in hybrids can have unpredictable, deleterious epistatic interactions (Maheshwari and Barbash, 2011). Orr (1995) and Orr and Turelli (2001) proposed the “snowball effect” of DMI evolution, which predicts that the number of DMIs accumulating between lineages “snowballs,” or increases faster than linearly (see Kalirad and Azevedo, 2017 for explicit testing of the underlying assumptions of this model). The snowball effect has been supported by empirical evidence of the rate of DMI evolution between Drosophila (Matute et al., 2010; Matute and Gavin-Smyth, 2014) and Solanum sibling species pairs (Moyle and Nakazato, 2010).
DMI can evolve as a result of various mechanisms in either an ecological speciation (divergent evolution due to selection pressures imposed by novel environments) or mutation-order speciation scenarios (fixation of different, incompatible mutations in allopatric populations experiencing the same environmental conditions) (Mani and Clarke, 1990; Nosil and Flaxman, 2011). These mechanisms include mutation-driven co-evolution (e.g., copper tolerance in Mimulus guttatus, MacNair, 1983; MacNair and Christie, 1983), gene duplication (e.g., the OdsH gene in Drosophila simulans × D. mauritiania backcross males, Sun et al., 2004; Ting et al., 2004), gene transposition (e.g., the JYAlpha gene in D. melanogaster × D. simulans backcross males, Masley et al., 2006), or a molecular arms race (e.g., divergent NB-LRR alleles in Arabidopsis thaliana strains, Bakker et al., 2006; Bomblies et al., 2007; Bomblies and Weigel, 2007). Furthermore, divergence in gene regulatory regions between parental species can result in in allelic imbalance in hybrids due to incompatible cis- and/or trans-acting factors (Graze et al., 2012), and segregation distortion of parental loci can skew sex ratios of hybrid progeny (Orr and Irving, 2005; Phadnis and Orr, 2009).
The Anopheles (An.) gambiae complex is comprised of eight morphologically indistinguishable species and has emerged as a model system for the study of speciation genetics due to the rapid radiation of its member species over the past two million years (Fontaine et al., 2015). All members of the An. gambiae complex are competent vectors of human malaria parasites, though they differ in their host specificity, ranging from highly anthropophilic (An. gambiae s.s. and An. coluzzii) (Takken and Knols, 1990; Dekker and Takken, 1998; Pates et al., 2001b) to almost entirely zoophilic (An. quadriannulatus A) (White, 1974; Gibson, 1996; Dekker and Takken, 1998; but see Pates et al., 2001a). From a vector control stand point, recent advances in the development of transgenic mosquito control methods have made understanding the permeability of species boundaries even more urgent (e.g., Kyrou et al., 2018) because it is important to know whether transgenic alleles have the potential to introgress between target and non-target species.
F1 hybrid female progeny of crosses between species of the Anopheles gambiae complex are fertile, but, with the exception of the An. gambiae – An. coluzzii species pair, males range from semi- to completely sterile (White, 1974; Hunt et al., 1998). A previous QTL analyses of sterility in bi-directional backcross hybrids identified regions in the An. coluzzii genome that cause male and female sterility when introgressed into an An. arabiensis genetic background, and vice versa (Slotman et al., 2004, 2005). One or more loci on the X chromosome contribute to the effect of the X chromosome on the hybrid sterility phenotype in both directions of the cross. Unfortunately, recombination on the X chromosomes is suppressed in hybrids by the Xag inversion, which is fixed within An. coluzzii but absent in An. arabiensis. The Xag inversion comprises ∼75% of the An. coluzzii X chromosome. This limits our ability to map genes to the X chromosome between species in the complex that harbor the Xag inversion (An. gambiae, An. coluzzii, and An. merus), and those that have the standard arrangement, including An. quadriannulatus.
Slotman et al. (2004) also identified both partially dominant and partially recessive male sterility QTL on the 2nd and 3rd chromosomes in both backcrosses. Consistent with the Dobzhansky-Muller model (Dobzhansky, 1936; Müller, 1940), and previous results from Drosophila (Wu and Beckenback, 1983; Orr and Coyne, 1989) the position of autosomal sterility QTL differs between each backcross. Complete sterility is achieved through the inheritance of at least three to four autosomal sterility loci (Slotman et al., 2004), including a pericentromeric QTL on the 3L chromosome arm. Hybrid male inviability is caused by an incompatibility between the An. coluzzii X chromosome and at least one locus on each autosome of An. arabiensis.
Female hybrid sterility QTL are present on the third chromosomes of both An. coluzzii and An. arabiensis (Slotman et al., 2005). A QTL on the second chromosome An. coluzzii causes female hybrid sterility when it is introgressed into an An. arabiensis genomic background. However, the female sterility phenotype is affected primarily by the interaction of a foreign X chromosome with the two autosomal QTL. Approximately 75% of observed phenotypic variance is due to the X chromosome, which only represents 8.8% of the genome (Holt et al., 2002). Due to the lack of recombination between X chromosomes in the cross, the number of loci on the X chromosome that effect the hybrid sterility phenotype is unknown. Therefore, the impact of the X on the hybrid sterility phenotype could result from one locus of major effect, or the effects could be spread across multiple loci. A comparison of the data between the male (Slotman et al., 2004) and female (Slotman et al., 2005) An. coluzzii × An. arabiensis hybrid sterility data shows that there are a greater number of male than female hybrid sterility QTL, supporting previous studies that found male sterility factors to evolve faster than female sterility factors (Charlesworth et al., 1987; Kirkpatrick and Hall, 2004). This is consistent with faster male evolution observed in Aedes mosquitoes (Presgraves and Orr, 1998), and the higher numbers of male vs. female sterility factors in Drosophila (e.g., Hollocher and Wu, 1996; True et al., 1996; Tao and Hartl, 2003).
In order to better understand how post-zygotic reproductive isolation evolved during the rapid radiation of the of the An. gambiae species complex we performed a QTL analysis of male hybrid sterility in a (An. coluzzii × An. quadriannulatus) F1 × An. quadriannulatus backcross, and identified regions of the An. coluzzii genome that contribute to hybrid male sterility and inviability when introgressed into an An. quadriannulatus genetic background. We discuss how these QTL relate to those identified in the (An. coluzzii × An. arabiensis) F1 × An. arabiensis backcross (Slotman et al., 2004), and the implications these findings have on our understanding of the evolution of post-zygotic reproductive isolation in the Anopheles gambiae species complex, and more broadly, in the context of rapid species radiations.
Materials and Methods
Backcross and Phenotype Scoring
We performed a backcross between the SUA2La strain of An. coluzzii and the SANGUA strain of An. quadriannulatus to identify regions of the An. coluzzii genome that exhibit DMI with the An. quadriannulatus genome, resulting in F1 male sterility. The An. coluzzii strain was established from collections in Suakoko, Libera and the An. quadriannulatus strain was established in from populations in Sangwe, Zimbabwe. Both strains have been lab reared for hundreds of generations. We used a standard backcross scheme that has been used in studies to investigate the genetic basis of hybrid sterility and inviability going back to Dobzhansky (1936). F1 An. coluzzii × An. quadriannulatus (females × males, respectively) hybrids were reared and backcrossed to An. quadriannulatus males en masse. Consistent with crosses between An. coluzzii and An. arabienesis, the Xag inversion was expected to suppress recombination between the X chromosomes of An. coluzzii and An. quadriannulatus.
The 2La inversion is also fixed between the mosquito strains used in this cross, limiting our mapping resolution in this region of the genome. While the 2La is polymorphic within An. coluzzii, and An. coluzzii strains with the standard arrangement (co-linear with An. quadriannulatus) are available, we used the same An. coluzzii strain as Slotman et al. (2004) to allow a comparison between them.
The testes of male hybrids were dissected and squashed between a microscope slide and a cover slip. Each hybrid male mosquito was assigned a sterility phenotype following Slotman et al. (2004). Sterility scores were primarily based upon the proportion of normal to abnormal sperm. If no sperm was present, testes were often atrophied as well. In some instances, testes were missing completely. Phenotype scores were as follows: (1) normal testes and sperm, (2) slightly abnormal sperm, (3) 50/50 normal and abnormal sperm, (4) mostly abnormal sperm, (5) entirely abnormal sperm, (6) no sperm present and/or severely atrophied testes, (7) no testes present. Phenotyping was conducted by a single observer, without repeat observations. If unambiguous scoring was not possible, or if the reproductive tissues were damaged during dissection, the sample was discarded. Mosquitoes were preserved in 100% ethanol after phenotyping for DNA extraction an genotyping.
DNA Sequencing
DNA extractions were performed on a Qiagen Biosprint DNA extraction machine (Qiagen Inc., Valencia, CA, United States). Extracted DNA was suspended in 200 μl Qiagen elution buffer and stored at −20°C. Each mosquito was genotyped using the restriction enzyme-based, genotype-by-sequencing approach according to the multiplexed shotgun genotyping (MSG) approach published by Andolfatto et al. (2011).
Briefly, 10 ng of genomic DNA of each mosquito was digested with the restriction enzyme MseI, which cuts the An. gambiae genome on average every 183.2 bp. Samples were digested for 3 h at 37°C, followed by a 20 min, 65°C enzyme deactivation step. Non-ligated bar-coded adapters were removed using an isopropanol precipitation, and bar-coded samples with unique adapters were pooled (96 per plate). Pooled samples were precipitated via centrifugation and re-suspended in TE buffer. Next, DNA was bead-purified using an Agencourt AMPure PCR purification kit. Bar-coded, purified DNA was size selected to a range of 250–400 bp using a Pippen Prep and then amplified using a Phusion High-Fidelity PCR kit. Samples were amplified using a common primer, and an indexed, library-specific primer (one per plate) that also incorporates the Illumina flow-cell adapter. Thus, individuals were identifiable to a library prep by their index and to the individual level by their barcoded adapter. Library amplification (PCR) was performed at a range of 12–18 cycles to identify the minimum number necessary to amplify the library to a target of 10–30 ng/μL (post clean up) in an effort to mitigate the incorporation of PCR errors. Amplified libraries were bead-purified using an Agencourt AMPure PCR purification kit. Library size distribution, integrity, and DNA concentration was confirmed by running each library on a NanoDrop and BioAnalyzer prior to sequencing. We phenotyped and sequenced a total of 841 male backcross hybrids. Hybrids were sequenced at the Texas A&M Genomics and Bioinformatics core facility on an Illumina HiSeq 2500 using 125 bp, single-end chemistry to 0.5–2.0x depth per individual.
We also performed pool-seq of ten An. coluzzii females and ten An. quadriannulatus females in order to identify fixed genetic difference between parental strains that would be diagnostic of the origin of genomic regions introgressed from An. coluzzii into An. quadriannulatus in the backcross. Libraries were sequenced on a single lane of Illumina HiSeq 2500 using 150 bp, paired-end chemistry to an average depth of 110.5x (An. coluzzii) and 66.9x (An. quadriannulatus). Additionally, we prepared and sequenced MSG libraries for 192 An. coluzzii and 192 An. quadriannulatus females to further validate ancestry informative markers and tune the MSG software error model.
Parental Pseudo-Reference Construction
An. coluzzii pool-seq reads were aligned to the An. gambiae AgamP4.4 reference genome using the BWA-MEM alignment algorithm (Li and Durbin, 2009) and SNPs were called using bcftools mpileup (v1.3) (Li et al., 2009). SNPs within three bases of indels were excluded. A pseudo-reference for An. coluzzii was created by updating the AgamP4.4 reference genome (vectorbase.org) with SNPs that had read depth between greater than 10 and less than 1000, map quality greater than 20, and a phred-scaled quality score for the alternative allele greater than 26. The An. quadriannulatus pseudo-reference was created in a similar fashion, but by aligning it to the An. coluzzii pseudo-reference and updating this with fixed SNPs (An. coluzzii – An. quadriannulatus divergent) that met the thresholds above.
Hybrid Genotyping
We used Trimmomatic version 0.30 (Bolger et al., 2014) to trim MSG sequencing reads by quality by simultaneously soft clipping the reads from both 5′ and 3′ ends to an average phred quality score of 20, with no single bp in a four bp window below a phred quality score of 20. Only reads ≥ 50 bp were retained for alignment and subsequent analyses.
We used the MSG software package (Andolfatto et al., 2011) to align hybrid MSG data to parental pseudo-references and estimate the posterior probabilities of ancestry at genome-wide SNPs that were informative of parental ancestry. We removed 33 hybrids from this analysis due to low sequencing coverage (<10K reads). Posterior probabilities of ancestry were thinned using pull_thin_tsv.py1. We considered a marker to be informative if its conditional probability of being homozygous differed by more than 0.10 from its neighboring marker. Missing genotypes were imputed using the hidden Markov model implemented in the sim.geno() function of R/qtl (Broman et al., 2003).
Sterility QTL Mapping
We used R/qtl (Broman et al., 2003) to identify redundant, adjacent markers and further thinned our marker set to include only informative markers. It is important to note that this experiment is not marker, but recombination limited. Therefore, many markers are redundant, and we removed these to reduce computational time. Next, we used R/qtl to estimate the genetic map of 808 hybrid males using the Kosambi map function (Kosambi, 1943). We performed quantitative trait locus mapping with the composite interval mapping (CIM) method using 15 markers as co-factors, and also assessed the significance of QTL models using multiple imputation mapping. This method drops one QTL at a time to calculate the probability of QTL locations and their interactions using an ANOVA (Sen and Churchhill, 2001). In all analyses, sterility was treated as a normal trait, and we used 1000 permutation replicates to assess the significance of log of odds (LOD) scores.
Ultimately, we took three approaches to identify sterility QTL in the dataset. The first was to scan for QTL using the composite interval mapping and calculating the logarithm of these odds (LOD) score every 1 cM. When calculating LOD scores we controlled for the genetic background of adjacent markers by using 15 marker covariates across the genome. Due to the skew of the phenotype scores toward “normal,” and severe segregation distortion of the X chromosome, we hypothesized that the QTL identified through CIM of the full dataset were in fact comprised of two types: (1) autosomal An. coluzzii loci that are incompatible with the An. quadriannulatus X, and (2) homozygous An. quadriannulatus autosomal loci that interact epistatically with the An. coluzzii X in individuals that are otherwise primarily heterozygous at autosomal loci (recovering viability despite the inheritance of a non-recombinant An. coluzzii X chromosome). Thus, we also split the dataset into individuals that had inherited the An. quadriannulatus X (N = 568) and An. coluzzii X (N = 211), respectively, and performed CIM (as above) on these datasets separately.
Next, we tested all peaks identified by CIM, regardless of significance or LOD score, in the full dataset, and any peaks that were unique to either the An. quadriannulatus X or An. coluzzii X datasets, respectively, by adding each peak independently to the multiple imputation QTL model that also contained the highly significant X chromosome QTL. After identifying all peaks that had a significant additive effect in the multiple imputation QTL model, we tested epistatic interactions between each, in a pairwise fashion, by adding each of these interactions to the model independently. This approach allowed us to identify peaks (and their interactions) which, despite having non-significant LOD scores in the CIM permutation test, were found to have a significant impact on sterility when incorporated into the multiple imputation QTL model.
We identified candidate sterility genes within QTL by determining the 1.5 LOD interval of all QTL peaks in base pairs, and used these coordinates to download the list of annotated genes within QTL from the AgamP4 genome using the VectorBase (vectorbase.org) genome browser using the “Export Data” function. Next we used the PANTHER classification system (Mi et al., 2019) gene list analysis search function (pantherdb.org) to search for the protein annotations and functions of these genes. Finally, we performed a protein network analysis on the entire gene list using STRING (Szklarczyk et al., 2019) in order to identify direct interactions between putative sterility genes.
Identification of Inviability Loci
We identified regions of the An. coluzzii genome that result in hybrid inviability when introgressed into an An. quadriannulatus genetic background by analyzing segregation distortion of autosomal loci in the An. quadriannulatus X chromosome and An. coluzzii X chromosome datasets. We used the mean, genome-wide proportion of An. quadriannulatus homozygosity in the full An. coluzzii × An. quadriannulatus backcross population as the expected homozygosity proportion, assuming Hardy-Weinberg equilibrium. We used a chi-square test to identify loci that deviate significantly (p < 6.01e-05 after Bonferroni correction) from expected homozygosity.
Results
Pseudo-Reference Construction, Genotyping, and Genetic Map
After whole genome alignment, SNP calling, and variant filtering of the An. coluzzii pool-seq data, we generated the An. coluzzii pseuo-reference by incorporating 4,391,200 filtered SNPs into the An. gambiae s.s. AgamP4 reference genome. To construct the An. quadriannulatus pseuo-reference, we updated the An. coluzzii pseudo-reference with 4,242,426 An. quadriannulatus SNPs that were found to be fixed between the An. quadriannulatus and An. coluzzii pool-seq samples. MSG data is inherently low coverage, so not all fixed differences between the pool-seq datasets were subsequently used for hybrid genotyping.
We calculated genotype probabilities at 239,420 ancestry informative SNPs for 808 An. coluzzii × An. quadriannulatus backcross hybrid males. Ultimately, we trimmed the dataset to 860 non-redundant markers (Table 1). The mean proportion of missing data per individual was 0.021 (SD = 0.060, range = 0.0–0.445). These markers were used to calculate a genetic map (Figure 1) with Kosambi distances of 206.1 cM for the second chromosome (110.9 Mb), 100.7 cM for the third (95.2 Mb), and 3.7 cM for the X (24.4 Mb) (Table 1 and Figure 1). The larger relative genetic map of the second chromosome is likely due to the presence of the 2La inversion, which is fixed between these species and can significantly increase recombination frequency on the 2R (i.e., Schultz-Redfield Effect, Schultz and Redfield, 1951; Lucchesi and Suzuki, 1968; Stevison et al., 2011), and segregation distortion resulting from male inviability for some genotype combinations (Hackett and Broadfoot, 2003; Zuo et al., 2019).

Table 1. The number of ancestry informative, thinned, and non-duplicate markers genotyped in hybrids, and the physical and map distances of each chromosome in the cross.
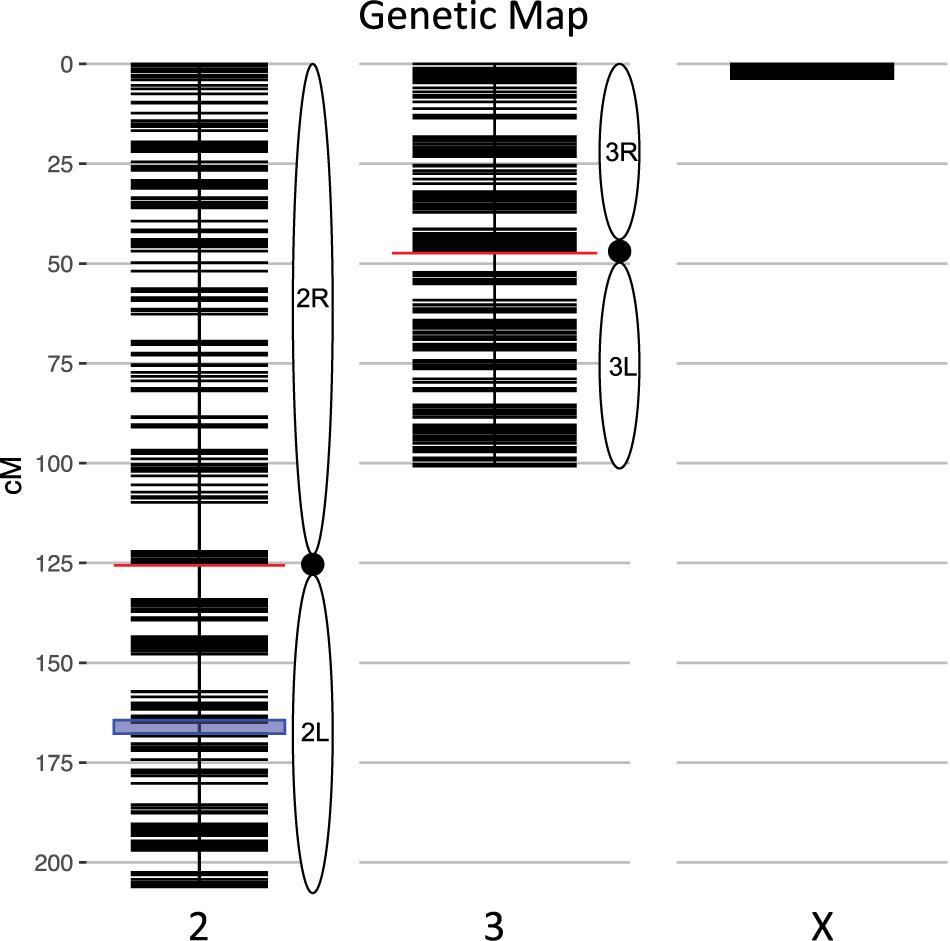
Figure 1. Genetic map of the An. coluzzii × An. quadriannulatus backcross showing centromere locations (red horizontal lines) and marker locations (black horizontal lines). The centromere of the telocentric X chromosome is opposite zero cM. The blue highlighted area on chromosome 2 indicates the location of the 2La inversion in An. coluzzii.
The genetic length of the X chromosome was expected to be low due to the presence of the Xag inversion, which comprises 80% of the X and is fixed for alternate arrangements between these species. The majority of hybrids (70.3%, N = 568) inherited a non-recombinant An. quadriannulatus X chromosome, 26.1% (N = 211) inherited a non-recombinant An. coluzzii X, and 3.5% (N = 29) a recombinant X. The mean sterility score (ranging from 1 to 7, normal to completely sterile) was 2.62 (SD = 2.34). The majority of backcross mosquitoes (64.23%, N = 519) were fully fertile (sterility score = 1).
Segregation Distortion and Inviability
Segregation distortion was previously documented in male and female hybrids between member species of this complex (Slotman et al., 2004, 2005; Smith et al., 2015). Furthermore, the complete absence of specific genotypes can indicate their inviability. We tested for significant segregation distortion of autosomal loci in the An. coluzzii X and An. quadriannulatus X datasets, respectively, be comparing the proportion of An. quadriannulatus homozygosity at each locus to the genome-wide average in the full dataset. It is important to note that this analysis was performed on a per locus basis, and is agnostic of interactions between loci.
In the An. quadriannulatus X dataset, 67.3% (N = 296) of chromosome 2 loci and 18.4% (N = 72) of chromosome 3 loci showed significant segregation distortion (Figure 2). Loci on the chromosome 2 exhibit both homozygote excess and deficit (heterosis), while only homozygote excess is exhibited on chromosome 3. In the An. coluzzii X dataset, 23.9% (N = 105) of chromosome 2 loci and 69.6% (N = 273) of chromosome 3 loci showed significant segregation distortion. Interestingly, 40.8% of chromosome 3 loci (spanning a 18.9 Mb region) lack homozygotes in all genotyped individuals, indicating individuals that inherited the An. coluzzii X and were An. quadriannulatus homozygous at loci inside this region were completely inviable (Figure 2). No autosomal heterozygote excess was observed in the An. coluzzii X dataset.
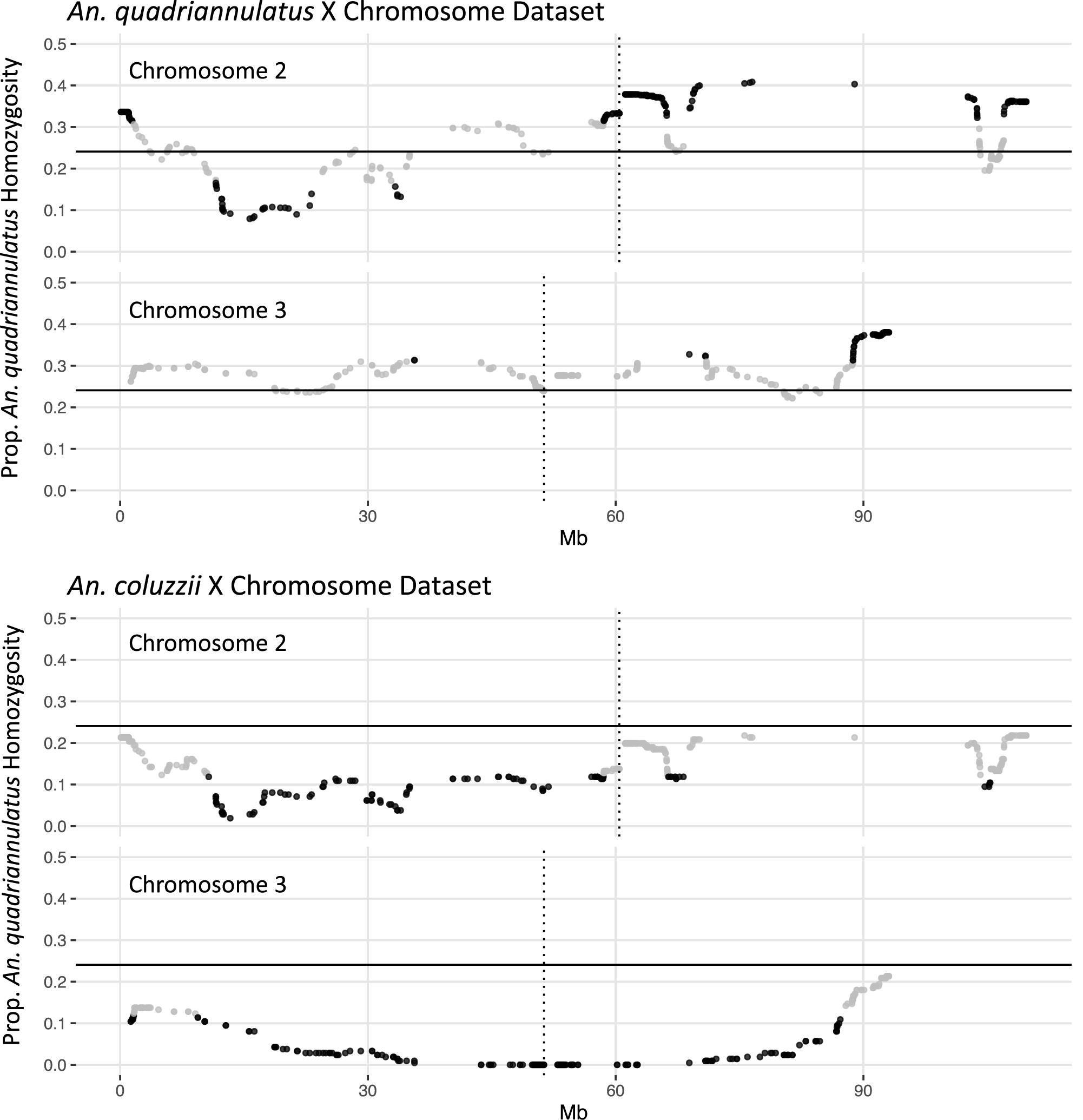
Figure 2. Segregation distortion at chromosome 2 and 3 loci for the An. quadriannulatus X chromosome (top) and An. coluzii X chromosome dataset (bottom). Gray dots indicate SNPs that do not deviate significantly from the expected proportion of homozygosity (solid horizontal line), while black dots deviate significantly (chi-square p < 0.05). Vertical dotted lines indicate centromere locations.
Composite Interval Mapping
Due to minimal recombination events, the X chromosome is effectively one large QTL with a LOD score of 203.4 (Table 2). By comparison, none of the autosomal QTL reach a LOD score of 4.0. No autosomal QTL fall above the CIM 5% significance threshold (LOD = 4.31, 1,000 permutations). Despite this, we annotated three QTL peaks on the second chromosome (1.63, 20.87, and 134.26 cM) (Figure 3 and Table 2) that contributed significantly to the sterility phenotype when included in the multiple imputation QTL model (see Multiple Imputation, below, and Table 4). The second chromosome peak at 1.63 cM (QTL “2R-A”) was present in the full and An. quadriannulatus X datasets, but not the An. coluzzii X dataset (Figure 3). QTL “2L” at 134.26 cM was present in the full and An. coluzzii X datasets, but not the An. quadriannulatus X dataset. Finally, QTL “2R-B” (20.87 cM) was found in the full and both X chromosome split datasets, suggesting that it contains loci involved in autosomal-autosomal incompatibilities (Figure 3). While additional LOD peaks are present in the An. coluzzii X dataset, these were non-significant when included in the multiple imputation QTL model (see section “Multiple Imputation,” below) (Figure 3). None of the second chromosome QTL overlap with those found in the An. coluzzii × An. arabiensis cross (Slotman et al., 2004; Figure 3 and Table 2).
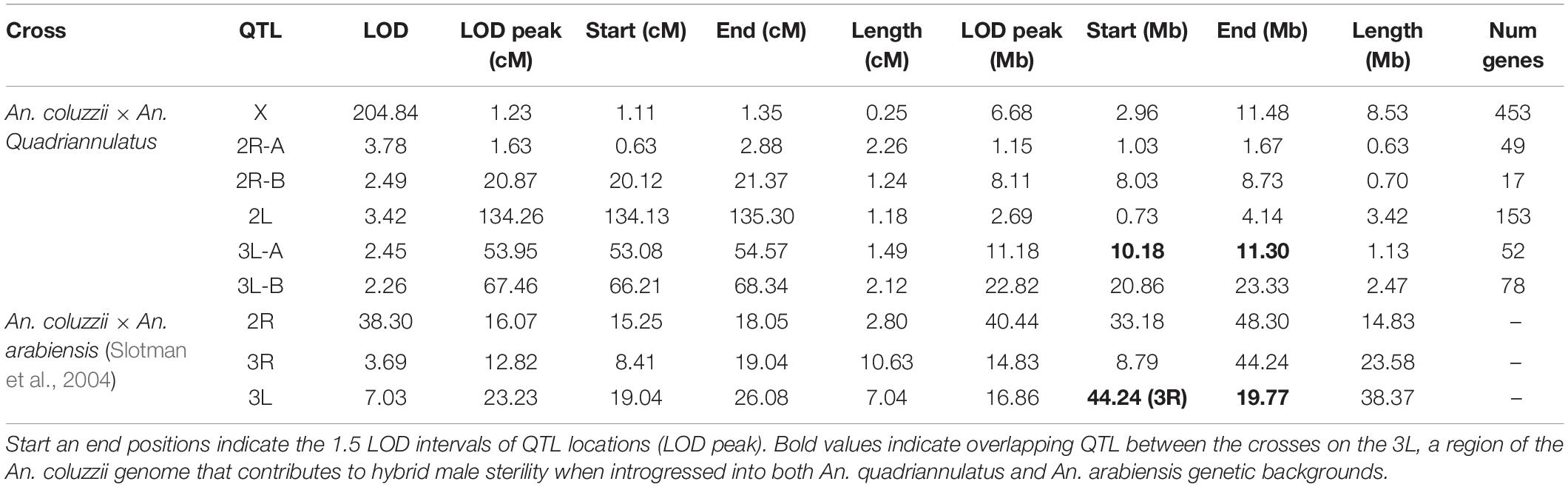
Table 2. Genomic regions of QTL in physical (Mb) and map (cM) distances.
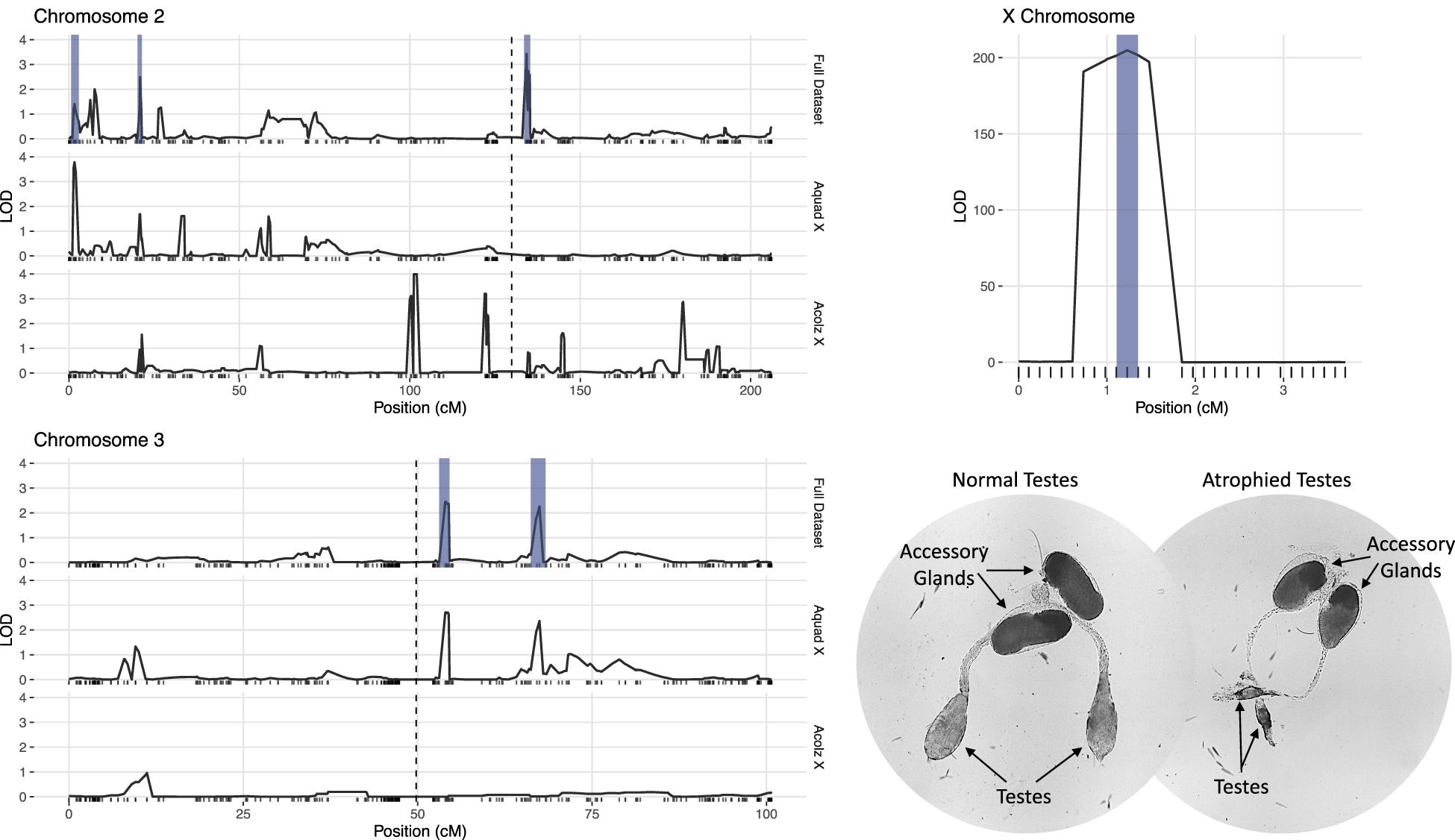
Figure 3. LOD plots for hybrid male sterility QTL identified with composite interval mapping in the An. coluzzii × An. quadriannulatus cross. Note the differences in the y-axis scales between the chromosome 2 (top left) and 3 (bottom left) panels, and the X chromosome panel (top right). Vertical, dashed lines indicate centromere locations on chromosomes 2 and 3. Second and third chromosome right arms are left of the centromere, and left arms are to the right. Vertical blue bars indicate 1.5 LOD intervals of QTL that explain a significant proportion of phenotypic variation in multiple imputation QTL model of the full dataset. The bottom right panel illustrates the gross morphological differences between hybrid testes with a normal sterility score (1) and a atrophied hybrid testes (sterility score = 6) which contain no sperm or only a few sperm with abnormal morphology.
Two QTL were identified on the third chromosome at 53.95 (QTL “3L-A”) and 67.46 cM (QTL “3L-B”) (Figure 3 and Table 2). These are both present in the full and An. quadriannulatus X dataset, but not the An. coluzzii X dataset, indicating that An. coluzzii introgressions into a predominantly An. quadriannulatus genetic background are driving this signal. QTL “3L-A” in the An. coluzzii × An. quadriannulatus dataset overlaps with a QTL in the An. coluzzii × An. arabiensis dataset (Slotman et al., 2004; Figure 3 and Table 2). The 3L-A and 3L-B QTL have seemingly additive-only effects on sterility (see section “Multiple Imputation,” below); these QTL were not identified by CIM in the An. coluzzii X dataset, suggesting that they result only from An. coluzzii introgressions into predominantly An. quadriannulatus genetic backgrounds. However, only three hybrid males were sampled that are An. quadriannulatus 3L-B homozygous and inherited the An. coluzzii X (Figure 4), and each had a sterility score of five or six. No hybrid males were sampled that are An. quadriannulatus 3L-A homozygous and inherited the An. coluzzii X. The reduction (or absence) of homozygous genotypes at the 3L-A and 3L-B QTL differs significantly from expectation (the percentage of homozygous genotypes across all autosomal loci, assuming HWE) (Table 3). Thus, these regions of the An. quadriannulatus genome interact epistatically with the X and result in almost complete male inviability when introgresesed into an An. coluzzii genetic background.
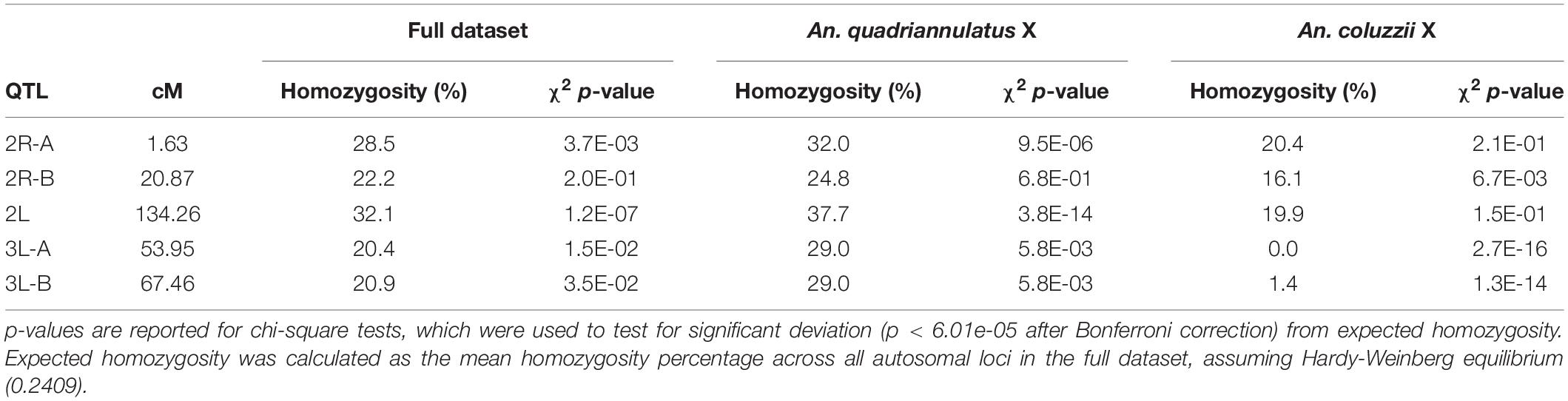
Table 3. Percentage of An. quadriannulatus homozygosity at hybrid male sterility autosomal QTL peaks in the full, An. quadriannulatus X chromosome, and An. coluzzii X chromosome datasets.
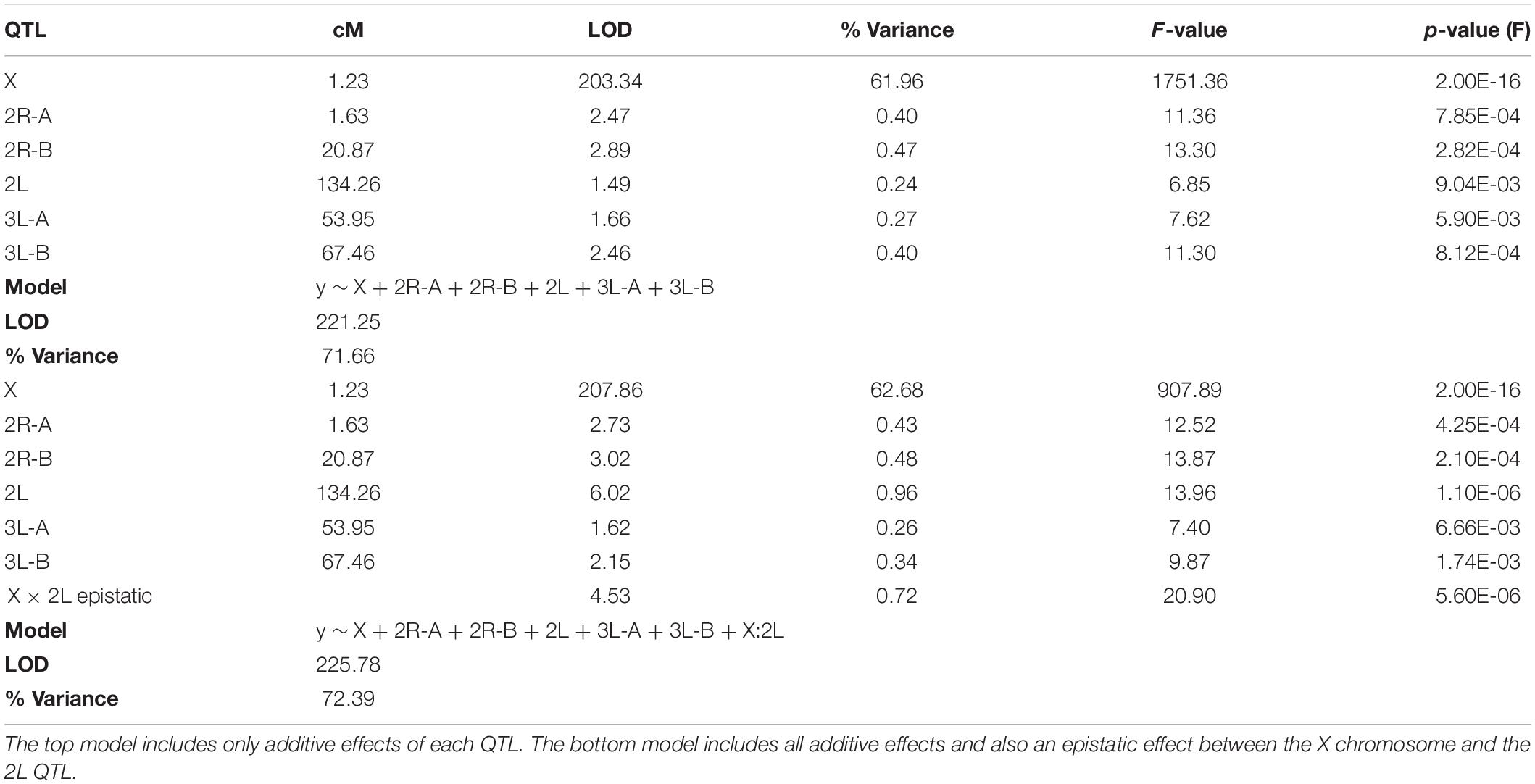
Table 4. Results of two multiple imputation QTL models (ANOVA).
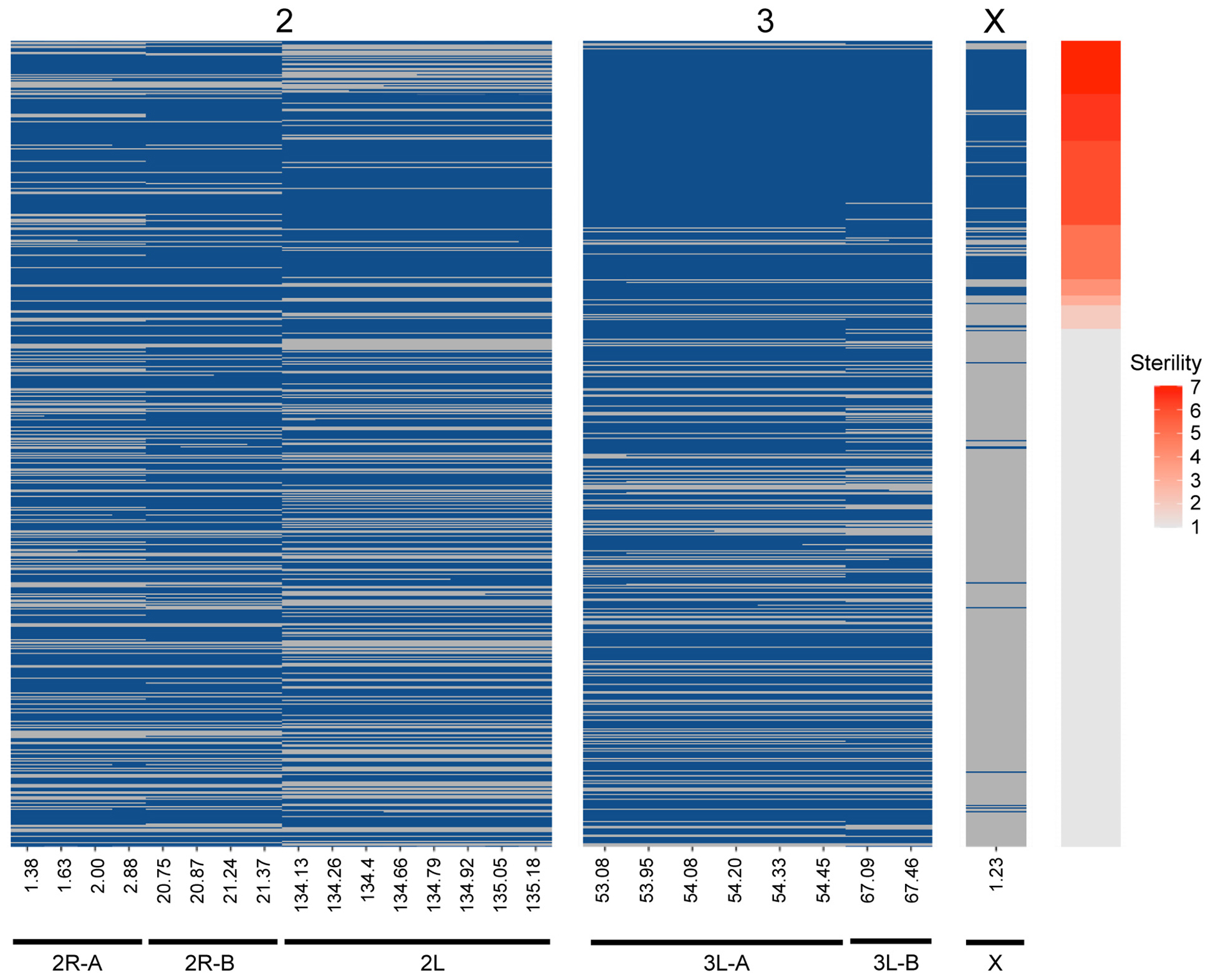
Figure 4. Genotype heat map for each An. coluzzii × An. quadriannulatus backcross male hybrid (y-axis) at the six QTL (x-axis) identified using CIM and multiple imputation. All markers within the 1.5 LOD interval of each QTL peak (Table 2) are included (x-axis, positions in cM). Individuals are sorted on the y-axis by sterility, with the individuals with the highest sterility score (7) at the top. Blue cells indicate Aquad/Acolz heterozygosity on the autosomes, and Acolz hemizygosity on the X chromosome. Gray cells indicate Aquad/Aquad homozygosity on the autosomes, and Aquad hemizygosity on the X chromosome.
Multiple Imputation
CIM detects QTL by calculating LOD scores at each marker or map location along each chromosome and does this agnostic of other QTL contributions to the phenotype and potential epistatic interactions between QTL. Due to this limitation, we built multiple imputation QTL models to detect the significance of all QTL LOD peaks found during CIM of the full (i.e., not split by X chromosome) dataset. The QTL discussed above, and those reported in Table 2, are only those that contributed significantly to the variance in sterility in the multiple imputation models described herein. We report two QTL models, one that considers only additive effects of QTL on sterility, and one which includes significant epistatic effects between QTL. We took an iterative process during testing QTL models and added QTL locations identified through CIM in a stepwise fashion. After we had identified those that had significant additive effects, we tested all pairwise epistatic effects between these loci. The final “additive only” and “epistasis allowed” models explain 71.66 and 72.29% of the variance in sterility, respectively (Table 4). The vast majority of this variance is explained by the inheritance of the X chromosome. As expected, individuals harboring the An. coluzzii X chromosome have on average higher sterility scores because in these backcrosses there are more opportunities for An. quadriannulatus autosomal – An. coluzzii X incompatibilities than vice versa (Figure 4).
Each of the QTL described had a significant additive effect in both the “additive only” and “epistasis allowed” models (F-test p < 0.01) (Table 4). After testing all possible pair-wise interactions between these loci, we found that only the X and 2L (134.26 cM) QTL pair had a significant interaction (F-test p = 5.60E-06). In both models, additive effects of autosomal loci, and the epistatic interaction between the X and 2L QTL, each explained less than 1% of the variance in hybrid sterility. The “epistasis allowed” model explained a higher proportion of the variance and had a higher model LOD (225.78) when compared to the “additive only” model (221.25) (Table 4).
To better understand the direction of the effect of each QTL in the full dataset, we compared the phenotypes of each individual given their genotype [An. quadriannulatus homozygous (QQ) vs. An. quadriannulatus/An. coluzzii heterozygous (QC)] at autosomal QTL and the X chromosome (Figure 5). Each autosomal locus increases sterility in the heterozygous state, and individuals with the An. coluzzii X are significantly more sterile. The interaction plot (Figure 5, “X vs. 2L,” right panel) between the X and 2L QTL demonstrates that heterozygosity at the 2L QTL alone does not impact sterility when an individual has the An. quadriannulatus X, but reduces sterility when inherited with the An. coluzzii X. The evidence suggests that each autosomal QTL indicate the location of a (partially) dominant An. coluzzii sterility gene that causes incompatibilities with the An. quadriannulatus background.
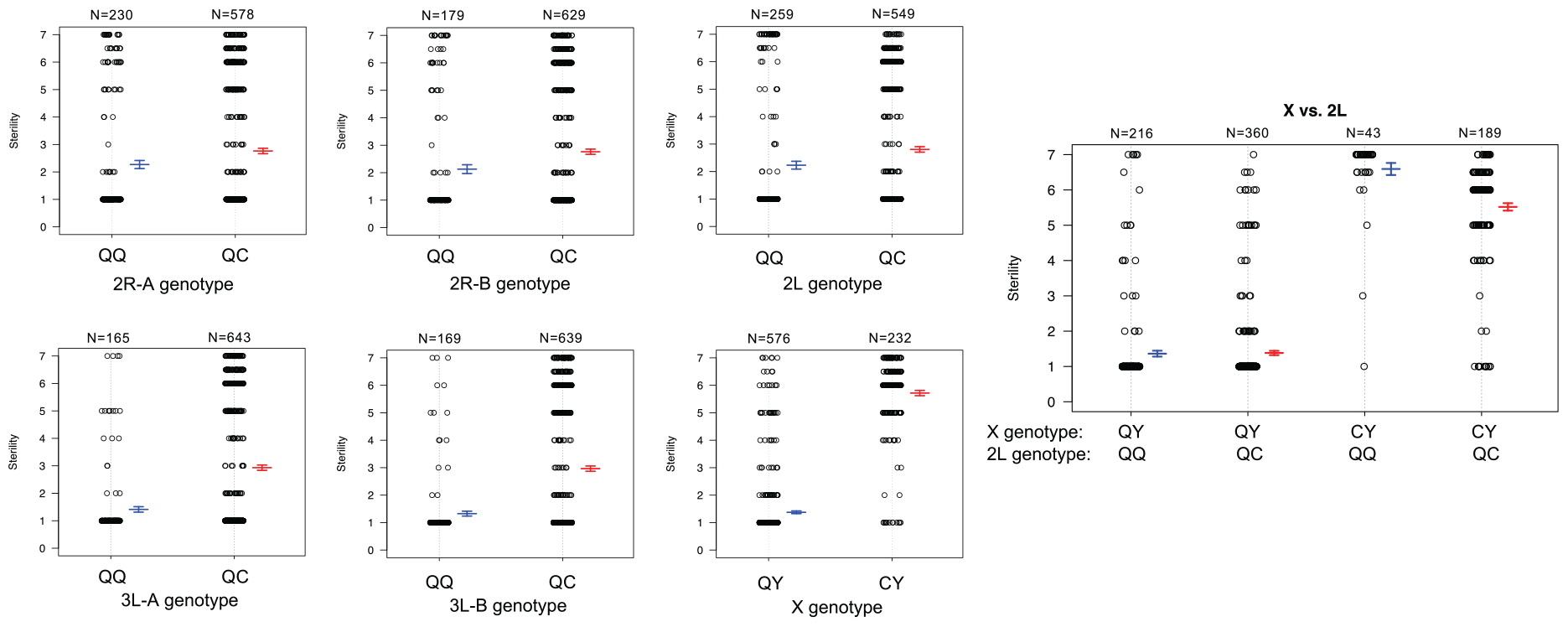
Figure 5. Genotype (x-axis) by phenotype (sterility score, y-axis) plots for the six QTL identified using CIM and multiple imputation, and an interaction plot (right) for the epistatic effect between the X chromosome and 2L QTL. Each individual is represented as a dot, which are jittered to avoid overlap. Box plots illustrate the mean (middle line) of each phenotype distribution and whiskers illustrate ± 1 standard error of the mean. QTL names and locations correspond to those in Table 2. QQ, An. quadriannulatus homozygous; QC, An quadriannulatus/An. coluzzii heterozygous.
We determined the 1.5 LOD interval of each QTL and, using the base pair positions of our SNP markers, determined the physical map span of each (Table 2). Next, we compared QTL locations to male sterility QTL found in a An. coluzzii × An. arabiensis cross (Figure 3; Slotman et al., 2004). We re-analyzed this data using CIM with three marker covariates. No significant QTL on the second chromosome overlap between these crosses. However, the 3L-A QTL in our An. coluzzii × An. quadriannulatus cross falls within the 3L QTL from the An. coluzzii × An. arabiensis cross, and the 3L-B QTL overlaps it. In the Slotman et al. (2004) data the third chromosome QTLs are also only present when the hybrid has the backcross (An. arabiensis in this case) parent X chromosome (Figure 3). Thus, interestingly this represents a region of the An. coluzzii that contributes to sterility when introgressed into both An. quadriannulatus and An. arabiensis genetic backgrounds.
Candidate Speciation Genes
We identified all annotated genes in the AgamP4 genome within the 1.5 LOD intervals of each QTL using VectorBase2 and found the protein family (if available) of each gene using PANTHER. Due to the lack of resolution on the X chromosome, we excluded this chromosome from this analysis. We identified 349 genes within all autosomal QTL (2.47 Mb total, Table 2). We identified ten genes with protein functions related to mating, reproduction, testes, and microtubule morphogenesis and function (Table 5) in the full gene set (Supplementary Table S1).
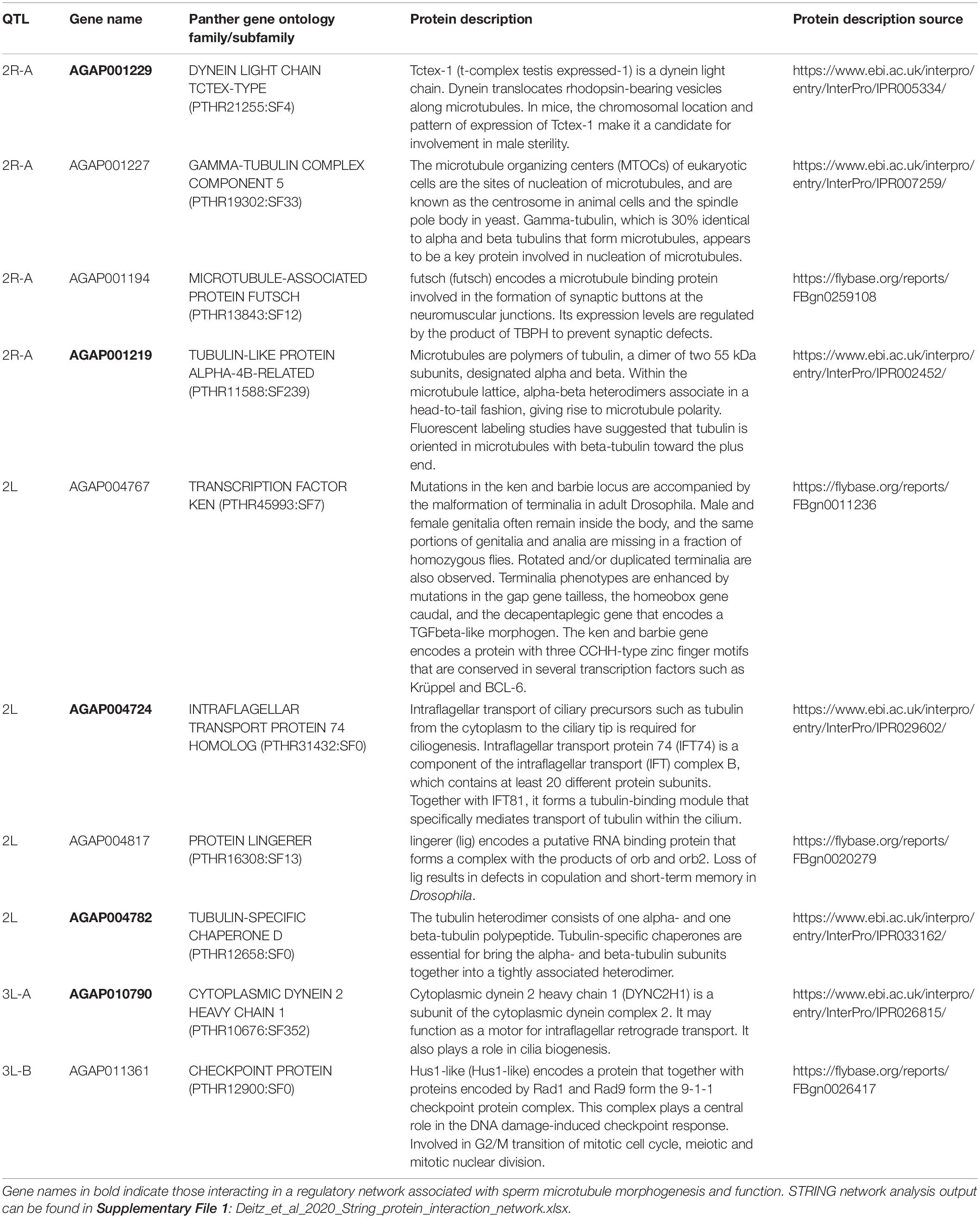
Table 5. Annotated genes in the AgamP4 genome within autosomal QTL with functions related to mating, reproduction, and sperm motility and morphogenesis.
We performed a protein network analysis on the entire gene list using STRING (Szklarczyk et al., 2019). The full STRING protein interaction results can be found in Supplementary File 1. Many protein products of genes in autosomal QTL regions have direct interactions, and four proteins with functions related to reproduction (bold in Table 5) interact directly in an insular protein network: dynein light chain Tctex-type, intra-flagellar transport protein 74 homolog, tubulin-specific chaperone D, and cytoplasmic dynein 2 heavy chain 1.
Discussion
One or more loci on the X chromosomes of both species have a large impact on hybrid male sterility in the An. coluzzii × An. quadriannulatus hybrids. In both multiple imputation QTL models presented (with and without X by 2L epistasis) the cumulative effect of the X chromosome accounted for greater than 60% of the phenotypic variation. These results support the theory of large X effect on hybrid male sterility due to its hemizygosity in the heterogametic sex (Anopheles males) where recessive sterility genes are expressed, but cannot discount the role of faster X evolution, which we did not test. The large proportion of variance explained by the X in our An. coluzzii × An. quadriannulatus backcross contrasts with the An. coluzzii × An. arabiensis backcross (to An. arabiensis) performed by Slotman et al. (2004). Slotman et al. (2004) found that the X chromosome accounted for only 4.7% of the variance in the hybrid male sterility phenotype while each of five autosomal QTL accounted for 3.4–20.3% of the variance (Table 4 and Slotman et al., 2004). However, the relatively small proportion of the variance explained by the X in the An. coluzzii × An. arabiensis backcross resulted, at least in part, from the inviability observed when hybrids inherited the An. coluzzii X (only 9.7% of hybrid males inherited the An. coluzzii X, Table 1 of Slotman et al., 2004).
When An. coluzzii × An. arabiensis F1 hybrids were backcrossed to An. coluzzii, the X accounted for 39.5% of the variance and additive effects of autosomal QTL accounted for 4.2–7.9% of the variance (Slotman et al., 2004). The differences in the effect size of the X between crosses, and directions of a back cross between two species (Slotman et al., 2004) is not unexpected. First, the X chromosome has different effects on inviability in each direction of a cross. Second, the genetic architecture of DMIs are unique to each species pair. Additionally, DMIs accumulate over time faster than linearly, and the genetic architecture of individual DMIs becomes more complex as additional mutations accumulate (Orr and Turelli, 2001; Matute et al., 2010; Moyle and Nakazato, 2010; Matute and Gavin-Smyth, 2014).
Unfortunately, due to the Xag inversion, the entire X chromosome is effectively one QTL, and we have limited resolution to identify candidate genes on this chromosome. While the 1.5 LOD interval of the X chromosome QTL is 8.53 Mb, the entire span of the QTL is 14.2 Mb, or 58% of the X chromosome. However, we determined which autosomal An. coluzzii loci contributed to sterility when introgressed into a predominantly An. quadriannulatus genomic background, and vice versa, by dividing the dataset by the parental origin of the X chromosome. This analysis revealed that the epistatic interaction between the 2L and the X QTL is driven by the interaction between An. quadriannulatus QTL 2L homozygotes and the hemizygous An. coluzzii X chromosome (Figures 3, 4). Individuals with this genotype combination are among the most sterile in the cross (Figure 4), and the 2L QTL is absent in the An. quadriannulatus X chromosome CIM results (Figure 3).
The 3L-A QTL sterility QTL in our An. coluzii × An. quadriannulatus cross overlaps entirely with the 3L QTL identified in the An. coluzzii × An. arabiensis cross (backcrossed to An. arabiensis, Figure 2D of Slotman et al., 2004). No An. arabiensis homozygotes were sampled in this region when the hybrid inherited the An. coluzzii X chromosome. Thus, the An. coluzzii X chromosome interacts epistatically with this same region of 3L in both An. quadriannulatus and An. arabiensis to contribute to hybrid male sterility. We hypothesize that the 3L-A QTL harbors an ancestral allele that is shared between An. quadriannulatus and An. arabiensis that has not been subjected to introgression between An. coluzzii and An. arabiensis.
Due to the rapid radiation of the Anopheles gambiae species complex, incomplete lineage sorting, and ongoing introgression between member species via fertile F1 hybrid females, the topology of the species complex phylogeny, which is based upon the X chromosome, is discordant with the whole-genome topology (Figure 1 of Fontaine et al., 2015). Bi-directional introgression between An. arabiensis and the ancestor of An. gambiae s.s. + An. coluzzii has reduced the genetic distance between the autosomes of these species relative to the X chromosome. As a result, An. arabiensis is sister to An. quadriannulatus in the X chromosome phylogeny but is sister to An. gambiae s.s. + An. coluzzii in the whole genome phylogeny.
While gene tree phylogenies vary across the genome due to the reasons outlines above, on the 3L, where we find overlap of sterility QTL between An. quadriannulatus and An. arabiensis crosses, the most commonly found gene phylogenies are those that group An. arabiensis closer to An. coluzzii than An. quadriannulatus (Figure 3 of Fontaine et al., 2015). If we assume that genes involved in post-zygotic reproductive isolation between this species trio evolved first on the X chromosome between the ancestor of An. gambiae s.s. + An. coluzzii and the ancestor of An. arabiensis + An. quadriannulatus, we can hypothesize that the 3L-A QTL shared between these crosses may have an X chromosome-like topology despite the homogenizing effect of introgression between the An. coluzzii and An. arabiensis autosomes. This topology is found in ∼12.6% of 50 kb genomics windows on the 3L (Figure 3 of Fontaine et al., 2015; see “other, A + Q trees”).
Our hypothesis that the 3L-A by X chromosome DMI may have been the first to arise between the ancestor of An. gambiae s.s. + An. coluzzii and the ancestor of An. arabiensis + An. quadriannulatus can be tested in the following ways. First, this sterility QTL should also exist in a cross between An. gambiae s.s. and An. arabiensis or An. quadriannulatus, and second, it should be absent in a cross between An. arabiensis and An. quadriannulatus. No hybrid sterility or inviability exists between An. coluzzii and its sister species An. gambiae s.s., so we expect that the same loci will be responsible for reproductive isolation between both of these species and either An. arabiensis or An. quadriannulatus. A cross between An. arabiensis and An. quadriannulatus may yield more interesting results; it would help us to understand the order by which individual DMI arose between these species pairs, and how this effects our understanding of the “snowball effect” of DMI evolution in the context of rapidly radiating species. For example, while the 3L-A by X chromosome DMI has been maintained between An. coluzzii and both An. arabiensis and An. quadriannulatus, additional DMI may have evolved in a “snowball effect” type scenario between An. coluzzii and An. quadriannulatus, where there is no evidence historical autosomal introgression (Fontaine et al., 2015), whereas, DMI may have been lost as a result of autosomal introgression between An. coluzzii and An. arabiensis, or their relative rate of evolution may have been slower.
Our candidate sterility gene analysis identified seven genes (out of 349) within autosomal An. coluzzii × An. quadriannulatus hybrid male sterility QTL that had protein functions related to tubulin or microtubule biogenesis, structure, or function. Four of these (AGAP004782, AGAP001219, AGAP010790, and AGAP001229) have experimentally determined, direct interactions with each other, while another interaction is predicted due to co-expression (AGAP004724 and AGAP010790) (Table 5 and Supplementary File 1). While these genes are found within autosomal QTL that contribute to a small proportion of variation in the sterility phenotype, one or more could have an interaction with an X chromosome locus that is disrupted during hybrid testes morphogenesis and/or spermatogenesis. In Drosophila, spermatogenesis progresses through mitosis of germ cells, meiosis, and finally the differentiation of spermatids into mature sperm (White-Cooper et al., 2009). Infertility in Drosophila hybrid males is associated with spermiogenic (post- rather than pre-meiotic) failure associated with sperm individualization and maturation (Wu et al., 1992). Recent work in the Anopheles gambiae complex demonstrates that hybrid sterility results in part from pre-meiotic arrest in degenerate testes (Liang and Sharakhov, 2019).
Sperm flagellum elongate from the spermatid head after meiosis. The axoneme is the central component of the sperm flagellum and has a 9 + 2 structure of a central pair of microtubules surrounded by nine doublet microtubules. Doublet microtubules have an inner and outer dynein arm that serve to attach and detach to neighboring doublet microtubules, and function in the motor activity of the flagellum. The axoneme is surrounded by a mitochondrial sheath that serves to provide ATP and power motor activity. Tektins are located near the junction points of dynein arms and neighboring microtubules (Gagon, 1995; Linck et al., 2016). Seven of the ten reproduction genes have PANTHER protein functions related to tubulin or microtubule biogenesis, structure, or function: gamma-tubulin complex component 5, microtubule-associated protein futsch, tubulin-like protein alpha-4B-related, dynein light chain Tctex-type, intra-flagellar transport protein 74 homolog, tubulin-specific chaperone D, and cytoplasmic dynein 2 heavy chain 1. The latter five genes (bold, Table 5) in this list have direct interactions in the STRING protein interaction network, and may represent a candidate genetic pathway whose disruption results in hybrid male sterility in the An. coluzzii × An. quadriannulatus cross. Interestingly, another tubulin-specific chaperone, E-like, is required for sperm individualization and male fertility in Drosophila (Nuwal et al., 2012).
The three genes without tubulin/microtubule functions are: transcription factor ken, protein lingerer, and a checkpoint protein Hus1-like. Mutations in the ken and barbie transcription factor result in malformed genitalia in Drosophila (Lukacsovich et al., 2003). lingerer encodes a putative RNA binding protein and Drosophila lingerer mutant males fail to withdraw their genitalia after copulation (Kuniyoshi et al., 2002). Finally, Hus1-like is a component of the 9-1-1 checkpoint protein complex, which coordinates DNA damage sensing, cell cycle progression, and DNA repair (Cotta-Ramusino et al., 2010). Future analyses of the molecular evolution of these genes and their regulatory regions between member species of the An. gambiae complex may yield insight into the specific alleles responsible for hybrid male sterility between An. coluzzii, An. quadriannulatus, and An. arabiensis.
In this study we have determined that the X chromosome accounts for that majority of the variance in the hybrid male sterility phenotype in a cross between An. coluzzii and An. quadriannulatus. We have also narrowly defined autosomal QTL which have a statistically significant contribution to sterility despite their smaller effect sizes in comparison to the X. The shared, autosomal sterility QTL between the An. coluzzii × An. quadriannulatus and An. coluzzii × An. arabiensis backcrosses suggest that these may represent important autosomal regions involved in the post-zygotic reproductive isolation of An. coluzzii from its sister species that diverged early in the radiation of the An. gambiae species complex. Future analyses into factors contributing to female hybrid sterility in the An. coluzzii × An. quadriannulatus cross will add to our knowledge of the genetics of speciation in this evolutionarily and medically important group of human malaria mosquitoes.
Data Availability Statement
Raw genome sequencing data can be found on the NCBI SRA here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA647477 Genotype and phenotype data can be found on Dryad here: https://doi.org/10.5061/dryad.bvq83bk6d, with a a direct download of the dataset available here: https://datadryad.org/stash/share/kw5d5Pio8DMRJuogJtE5x1kf5zC81ViDnH9FNKOAxxs.
Author Contributions
KD, WT, and MS designed the experiment. KD and MS performed the experiment and wrote the manuscript. KD analyzed the data. WT edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Science Foundation Doctoral Dissertation Improvement Grant (award # 1601675) to KD and MS, the Texas A&M University Genomics Seed Grant to KD and MS, and the Texas A&M University Dissertation Fellowship to KD.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Patrick Reilly and Andrew Taverner for helpful discussions regarding the analysis of the dataset and Leon Westerd for assistance with mosquito rearing. We are also thankful to BEI Resources for providing mosquito strains.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00925/full#supplementary-material
Footnotes
References
Andolfatto, P., Davison, D., Erezyilmaz, D., Mast, J., Sunayama-Morita, T., and Stern, D. L. (2011). Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 21, 610–617. doi: 10.1101/gr.115402.110
Bakker, E. G., Toomajian, C., Kreitman, M., and Bergelson, J. (2006). A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell 18, 1803–1818. doi: 10.1105/tpc.106.042614
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bomblies, K., Lempe, J., Epple, P., Warthmann, N., Lanz, C., Dangle, J. L., et al. (2007). Autoimmune response as a mechanism for Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 5:e236. doi: 10.1371/journal.pbio.0050236
Bomblies, K., and Weigel, D. (2007). Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 8, 382–393. doi: 10.1038/nrg2082
Broman, K. W., Wu, H., Sen, S., and Churchill, G. A. (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. doi: 10.1093/bioinformatics/btg112
Charlesworth, B., Coyne, J. A., and Barton, N. H. (1987). The relative raes of evolution of sex chromosomes and autosomes. Am. Natura. 130, 113–146. doi: 10.1086/284701
Cotta-Ramusino, C., McDonald, E. R., Hurov, K., Sowa, M. E., Harper, J. W., and Elledge, S. J. (2010). A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317. doi: 10.1126/science.1203430
Dekker, T., and Takken, W. (1998). Responses of Anopheles arabiensis patton and Anopheles quadriannulatus theobald to carbon dioxide, a man and a calf. Med. Vet. Entomol. 12, 136–140. doi: 10.1046/j.1365-2915.1998.00073.x
Dobzhansky, T. (1936). Studies on hybrid sterility, II. Localization of sterility factors in Drosophila Pseudoobscura hybrids. Genetics 146, 239–244.
Fontaine, M. C., Pease, J. B., Steele, A., Waterhouse, R. M., Neafsey, D. E., Sharakhov, I. V., et al. (2015). Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347:1258524. doi: 10.1126/science.1258524
Gagon, C. (1995). Regulation of sperm motility at the axonemal level. Reprod. Fertil. Dev. 7, 847–855.
Gibson, G. (1996). “Genetics, ecology and behaviour of anophelines. Olfaction in Mosquito–Host Interactions,” in Ciba Foundation Symposium 200, eds G. R. Bock and G. Cardew (Chichester: John Wiley & Sons), 22–45.
Graze, R. M., Novelo, L. L., Amin, V., Fear, J. M., Casella, G., Nuzhdin, S. V., et al. (2012). Allelic imbalance in Drosophila hybrid heads: exons, isoforms, and evolution. Mol. Biol. Evol. 29, 1521–1532. doi: 10.1093/molbev/msr318
Hackett, C., and Broadfoot, L. (2003). Effects of genotyping errors, missing values, and segregation distortion in molecular marker data on the construction of linkage maps. Heredity 90, 33–38. doi: 10.1038/sj.hdy.6800173
Hollocher, H., and Wu, C. I. (1996). The genetics of reproductive isolation in the Drosophila simulans clade: X vs. autosomal effects and male vs. female effects. Genetics 143, 1243–1255.
Holt, R. A., Subramanian, G. M., Halpern, A., Sutton, G. G., Charlab, R., Nusskern, D. R., et al. (2002). The genome sequence of the malaria mosquito Anopheles gambiae. Science 298, 129–149.
Hunt, R. H., Coetzee, M., and Fettene, M. (1998). The Anopheles gambiae complex: a new species from Ethiopia. Trans. R. So. Trop. Med. Hygiene 92, 231–235. doi: 10.1016/s0035-9203(98)90761-1
Kalirad, A., and Azevedo, R. B. R. (2017). Spiraling complexity: a test of the snowball effect in a computational model of RNA folding. Genetics 206, 377–388. doi: 10.1534/genetics.116.196030
Kirkpatrick, M., and Hall, D. W. (2004). Male-biased mutation, sex linkage, and the rate of adaptive evolution. Evolution 58, 437–440. doi: 10.1111/j.0014-3820.2004.tb01659.x
Kosambi, D. D. (1943). The estimation of map distances from recombination values. Ann. Eugen. 12, 172–175. doi: 10.1111/j.1469-1809.1943.tb02321.x
Kuniyoshi, H., Baba, K., Ueda, R., Kondo, S., Awano, W., Juni, N., et al. (2002). lingerer, a Drosophila gene involved in initiation and termination of copulation, encodes a set of novel cytoplasmic proteins. Genetics 162, 1775–1789.
Kyrou, K., Hammond, A., Galizi, R., Kranjc, N., Burt, A., Beaghton, A. K., et al. (2018). A CRISPR–Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 36, 1062–1066. doi: 10.1038/nbt.4245
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM). format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liang, J., and Sharakhov, I. V. (2019). Premeiotic and meiotic failures lead to hybrid male sterility in the Anopheles gambiae complex. Proc. B 286:20191080. doi: 10.1098/rspb.2019.1080
Linck, R. Q., Chemes, H., and Albertini, D. (2016). The axoneme: the propultive engine of spermatozoa and cilia and associated ciliopathies leading to infertility. J. Assist. Reprod. Genet. 33, 141–156. doi: 10.1007/s10815-016-0652-1
Lucchesi, J. C., and Suzuki, D. T. (1968). The interchromosomal control of recombination. Ann. Rev. Genet. 2, 53–86. doi: 10.1146/annurev.ge.02.120168.000413
Lukacsovich, T., Yuge, K., Awano, W., Asztalos, Z., Kondo, S., Juni, N., et al. (2003). The ken and barbie gene encoding a putative transcription factor with a BTB domain and three zinc finger motifs functions in terminalia development of Drosophila. Arch. Insect Biochem. Physiol. 54, 77–94. doi: 10.1002/arch.10105
MacNair, M. R. (1983). The genetic control of copper tolerance in the yellow monkey flower, Mimulus guttatus. Heredity 50, 283–293. doi: 10.1038/hdy.1983.30
MacNair, M. R., and Christie, P. (1983). Reproductive isolation as a pleiotropic effect of copper tolerance in Mimulus guttatus? Heredity 50, 295–302. doi: 10.1038/hdy.1983.31
Maheshwari, S., and Barbash, D. A. (2011). The genetics of hybrid incompatabilities. Annu. Rev. Genet. 45, 331–355.
Mani, G. S., and Clarke, B. C. (1990). Mutational order: a major stochastic process in evolution. Proc. R. Soc. Lon. Biol. 240, 29–37. doi: 10.1098/rspb.1990.0025
Masley, J. P., Jones, C. D., Noor, M. A. F., Locke, J., and Orr, H. A. (2006). Gene transposition as a cause of hybrid sterility in Drosophila. Science 313, 1448–1450. doi: 10.1126/science.1128721
Matute, D. R., Butler, I. A., Turissini, D. A., and Coyne, J. A. (2010). A test of the snowball theory for the rate of evolution of hybrid incompatibilities. Science 329, 1518–1521. doi: 10.1126/science.1193440
Matute, D. R., and Gavin-Smyth, J. (2014). Fine mapping of dominant X-linked incompatibility alleles in Drosophila hybrids. PLoS Genet. 11:e1005558. doi: 10.1371/journal.pgen.1005558
Mi, H., Muruganujan, A., Huang, X., Ebert, D., Mills, C., Guo, X., et al. (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721. doi: 10.1038/s41596-019-0128-8
Moyle, L. C., and Nakazato, T. (2010). Hybrid incompatibility “snowballs” between Solanum species. Science 329, 1521–1523. doi: 10.1126/science.1193063
Müller, H. J. (1940). “Bearing of the Drosophila work on systematics,” in The New Systematics, ed. J. S. Huxley (Oxford: Clarendon Press), 185–268.
Nosil, P., and Flaxman, S. M. (2011). Conditions for mutation-order speciation. Proc. R. Soc. Lon. Biol. 278, 399–407. doi: 10.1098/rspb.2010.1215
Nuwal, T., Kropp, M., Wegener, S., Racic, S., Montalban, I., and Buchner, E. (2012). The Drosophila homologue of tubulin-specific chaperon E-like protein is required for synchronous sperm individualization and normal male fertility. J. Neurogenet. 26, 374–381. doi: 10.3109/01677063.2012.731119
Orr, H. A. (1995). The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139, 1805–1813.
Orr, H. A., and Coyne, J. A. (1989). The genetics of postzygotic isolation in the Drosophila virilis group. Genetics 121, 527–537.
Orr, H. A., and Irving, S. (2005). Segregation distortion in hybrids between the Bogota and USA subspecies of Drosophila pseudoobscura. Genetics 169, 671–682. doi: 10.1534/genetics.104.033274
Orr, H. A., and Turelli, M. (2001). The evolution of postzygotic isolation: accumulating dobzhansky-muller incompatibilities. Evolution 55, 1085–1094. doi: 10.1111/j.0014-3820.2001.tb00628.x
Pates, H. V., Takken, W., Curtis, C. F., Huisman, W., Akinpelu, O., and Gill, G. S. (2001a). Unexpected anthropophagic behavior in Anopheles quadriannulatus. Med. Vet. Entomol. 15, 293–298. doi: 10.1046/j.0269-283x.2001.00310.x
Pates, H. V., Takken, W., Stuke, K., and Curtis, C. F. (2001b). Differential behaviour of Anopheles gambiae sensu stricto (Diptera: Culicidae) to human and cow odours in the laboratory. Bull. Entomol. Res. 4, 289–296. doi: 10.1079/ber200198
Phadnis, N., and Orr, H. A. (2009). A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science 323, 376–379. doi: 10.1126/science.1163934
Presgraves, D. C., and Orr, H. A. (1998). Haldane’s rule in taxa lacking a hemizygous X. Science 282, 952–954. doi: 10.1126/science.282.5390.952
Schultz, J., and Redfield, H. (1951). Interchromosomal effects on crossing over in Drosophila. Cold Spring Harb. Symp. Quantitative Biol. 16, 175–197. doi: 10.1101/sqb.1951.016.01.015
Sen, S., and Churchhill, G. A. (2001). A statistical framework for quantitative trait mapping. Genetics 159, 371–387.
Slotman, M. A., Della Torre, A., and Powell, J. R. (2004). The genetics of inviability and male sterility in hybrids between Anopheles gambiae and Anopheles arabiensis (2004). Evolution 59, 1016–1026. doi: 10.1111/j.0014-3820.2005.tb01040.x
Slotman, M. A., della Torre, A., and Powell, J. R. (2005). Female sterility in hybrids between Anopheles gambiae and A. arabiensis, and the cause of Haldane’s Rule. Evolution 59, 1016–1026. doi: 10.1111/j.0014-3820.2005.tb01040.x
Smith, H. A., White, B. J., Kundert, P., Cheng, C., Romero-Severson, J., Andolfatto, P., et al. (2015). Genome-wide QTL mapping of saltwater tolerance in sibling species of Anopheles (malaria vector). mosquitoes. Heredity 115, 471–479. doi: 10.1038/hdy.2015.39
Stevison, L. S., Hoehn, K. B., and Noor, M. A. (2011). Effects of inversions on within- and between-species recombination divergence. Genome Biol. Evol. 3, 830–841. doi: 10.1093/gbe/evr081
Sun, S., Ting, C. T., and Wu, C. I. (2004). The normal function of a speciation gene. Odysseus, and its hybrid sterility effect. Science 305, 81–83. doi: 10.1126/science.1093904
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, 607–613.
Takken, W., and Knols, B. G. J. (1990). Flight behaviour of Anopheles gambiae Giles (Diptera: Culicidae) in response to host stimuli: a windtunnel study. Proc. Exp. Appl. Entomol. 1, 121–128.
Tao, Y., and Hartl, D. L. (2003). Genetic dissection of hybrid incompatibilities between Drosophila simulans and D. mauritiana. III Heterogeneous accumulation of hybrid incompatibilities, degree of dominance, and implications for Haldane’s rule. Evolution 57, 2580–2598. doi: 10.1111/j.0014-3820.2003.tb01501.x
Ting, C. T., Tsaur, S. C., Sun, S., Browne, W. E., Chen, Y. C., Patel, N. H., et al. (2004). Gene duplication and speciation in Drosophila: evidence from the Odysseus locus. Proc. Natl. Acad. Sci. U.S.A. 101, 12232–12235. doi: 10.1073/pnas.0401975101
True, J. R., Weir, B. S., and Laurie, C. C. (1996). A genome-wide survey of hybrid incompatibility factors by the introgression of marked segments of Drosophila mauritiana chromosomes into Drosophila simulans. Genetics 142, 819–837.
White, G. B. (1974). Anopheles gambiae complex disease transmission in Africa. Trans. R. Soc. Trop. Med. Hygiene 68, 278–298. doi: 10.1016/0035-9203(74)90035-2
White-Cooper, H., Doggett, K., and Ellis, R. E. (2009). “The evolution of spermatogenesis,” in Sperm Biology: An Evolutionary Perspective, eds T. R. Birkhead, D. J. Hosken, and S. Pitnick (Burlington, MA: Academic Press), 151–183. doi: 10.1016/b978-0-12-372568-4.00004-5
Wu, C. I., and Beckenback, A. T. (1983). Evidence for extensive genetic differentiation between the sex-ratio and the standard arrangement of Drosophila pseudoobscura and Drosophila persimilis and identification of hybrid sterility factors. Genetics 105, 71–86.
Wu, C. I., Perez, D. E., Davis, A. W., Johnson, N. A., Cabot, E. L., Palopoli, M. F., et al. (1992). “Molecular genetic studies of post-mating reproductive isolation in Drosophila,” in Molecular Paleo-population Biology, eds N. Takahata and A. G. Clark (Berlin: Springer-Verlag), 191–212.
Keywords: Anopheles gambiae complex, speciation, hybrid, sterility, inviability, Dobzhansky–Muller incompatibilities, post-zygotic reproductive isolation
Citation: Deitz KC, Takken W and Slotman MA (2020) The Genetic Architecture of Post-Zygotic Reproductive Isolation Between Anopheles coluzzii and An. quadriannulatus. Front. Genet. 11:925. doi: 10.3389/fgene.2020.00925
Received: 19 May 2020; Accepted: 24 July 2020;
Published: 14 August 2020.
Edited by:
Jacob A. Tennessen, Harvard University, United StatesReviewed by:
Michelle Marie Riehle, Medical College of Wisconsin, United StatesAlberto Civetta, University of Winnipeg, Canada
Copyright © 2020 Deitz, Takken and Slotman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin C. Deitz, a2NkZWl0ekBnbWFpbC5jb20=