 Shan Li1†
Shan Li1† Yixuan Cao1†
Yixuan Cao1† Han Wang1†
Han Wang1† Lulu Li1
Lulu Li1 Xiuzhi Ren2Huan Mi1Yanzhou Wang3
Xiuzhi Ren2Huan Mi1Yanzhou Wang3 Yun Guan4Feiyue Zhao1
Yun Guan4Feiyue Zhao1 Bin Mao1Tao Yang1Yi You1Xin Guan1Yujiao Yang1
Bin Mao1Tao Yang1Yi You1Xin Guan1Yujiao Yang1 Xue Zhang1*
Xue Zhang1* Xiuli Zhao1*
Xiuli Zhao1*- 1Department of Medical Genetics, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and School of Basic Medicine, Peking Union Medical College, Beijing, China
- 2The People’s Hospital of Wuqing District, Tianjin, China
- 3Shandong Provincial Hospital Affiliated to Shandong University, Jinan, China
- 4Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Osteogenesis imperfecta (OI) is a rare heritable skeletal disorder which is mainly caused by defected type I collagen. Autosomal recessive OI (AR-OI) is caused by mutations of genes that are responsible for type I collagen modification and folding, and is often associated with more severe phenotypes. Due to the limited number of recessive OI patients, it has been difficult to study the mutation spectrum as well as the correlation of genotype and phenotype. This study recruited a Chinese cohort of 74 AR-OI families, aiming to establish the mutation spectrum and to examine the genotypic and phenotypic correlation. We identified 82 variants including 25 novel variants and 57 HGMD reported variants in these AR-OI patients, using whole exome sequencing/panel sequencing combined with Sanger sequencing. Pathogenic mutations were found at WNT1 (n = 30, 40.54%), SERPINF1 (n = 22, 29.73%), FKBP10 (n = 10, 13.51%), CRTAP (n = 3, 4.05%), P3H1 (n = 3, 4.05%), SERPINH1 (n = 2, 2.70%), SEC24D (n = 3, 4.05%), and PLOD2 (n = 1, 1.35%) respectively. Thus, WNT1 represents the most frequent pathogenic gene of AR-OI in Chinese population. The most common clinical manifestations of AR-OI patients include walking problem (72.86%), scoliosis (65.28%) and frequent fractures (fractures ≥2/year) (54.05%). Interestingly, ptosis represents a unique phenotype of patients carrying WNT1 variants, and it was rare in patients harboring other pathogenic genes. Our study expanded the mutation spectrum of AR-OI and enriched the knowledge of genotypic and phenotypic correlation in Chinese cohort with AR-OI.
Introduction
Osteogenesis imperfecta (OI), also known as brittle bone disease, is a genetically and clinically heterogeneous skeletal disorder. Typical clinical manifestations include low bone mass, frequent fractures, short stature, blue sclerea, and bone deformity. More than 90% of OI cases are caused by a defect of type I collagen, and therefore OI is known as an autosomal dominant inherited disease due to a mutation in COL1A1 or COL1A2, which encodes alpha 1 or alpha 2 chains of type I collagen (Marini et al., 2007). Multiple genes have been reported to contribute to the development of autosomal recessive OI (AR-OI), including SERPINF1, LEPRE1, CRTAP, PPIB, FKBP10, BMP1, SP7, PLOD2, TMEM38B, P4HB, SERPINH1, SEC24D, WNT1, CREB3L1, and SPARC (Kang et al., 2017). Although the incidence rate of AR-OI is < 10% of the whole OI population, the clinical manifestations of AR-OI are much more severe than the dominant OI patients (Liu et al., 2017; Li et al., 2019).
Synthesis of type I collagen is a sophisticated process. With the development of next generation sequencing, remarkable progress has been made in identifying new genes associated with the modification and folding of type I collagen. These genes have formed a unique biological network, and deficiency in any individual gene may lead to recessive OI. Propeptides encoded by COL1A1 and COL1A2 undergo post-translational modifications in the endoplasmic reticulum, followed by transportation to Golgi and cleavage of N-/C- terminal propeptides. Consequently, collagen fibers are formed to further build up the collagen matrix. Defect in any step during the type I collagen synthesis can lead to the development of OI (Kang et al., 2017). For example, deficiency of CRTAP, P3H1 or PPIB, the components of collagen prolyl 3-hydroxylation complex, will lead to defects in post translational modification of unfolded collagen alpha-chains (Morello et al., 2006). Mutations in FKBP10, SERPINH1 or BMP1 cause defects in collagen folding and crosslinking (Koide et al., 2006; Schwarze et al., 2013; Kang et al., 2017). Alterations in SP7 (Azetsu et al., 2017), WNT1 (Keupp et al., 2013; Laine et al., 2013; Pyott et al., 2013) or CREB3L1 (Murakami et al., 2009) affect osteoblast differentiation (Forlino and Marini, 2016). Dysfunction of PEDF, which is encoded by SERPINF1, will damage the bone homeostasis (Akiyama et al., 2010).
Although mutation spectrums on autosomal dominant OI have been established in large cohorts of Chinese (Li et al., 2019), Swedish (Lindahl et al., 2015) and Canadian/American populations (Bardai et al., 2016), mutation spectrum on AR-OI remains unclear due to its rare incidence. There was only one report about the molecular spectrum based on findings from a small cohort of 19 recessive OI patients from Mediterranean (Rauch et al., 2010). Here, we examined the mutation spectrum in 74 AR-OI families, the largest cohort worldwide to our knowledge. Current findings expand our knowledge about the gene spectrum and phenotypic spectrum of AR-OI, and provide important information for genetic diagnosis of AR-OI patients.
Materials and Methods
Subjects
We recruited 74 AR-OI probands from a cohort of 1095 OI patients (646 families) in mainland China. These patients displayed typical clinical manifestations of OI: recurrent fractures, short stature, bone malformation, with or without extra-skeletal manifestations such as blue sclera, hearing loss and dentinogenesis imperfecta. After obtaining approval from Institutional Review Board (IRB) of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing, China (015-2015) and informed consent from all participants/legal guardians of children under 18, peripheral blood samples were collected from all available family members of AR-OI patients. Skin samples were collected from some of the patients based on the availability.
Clinical parameters of probands were recorded at their first visit, including age, gender, height, times of fractures, presence of ptosis, scoliosis, blue sclerae, dentinogenesis imperfecta, and ability of walking. Height was also converted to Z-score calculated based on the age and medium height of Chinese population (Li et al., 2019).
Variant Nomenclature
Variants were named according to the nomenclature provided by Human Genome Variation Society1. Genomic DNA and cDNA sequences of SERPINF1 (NC_000017.10), CRTAP (NC_000003.11), SERPINH1 (NC_000011.9), FKBP10 (NC_000017.10), PLOD2 (NC_000003.11), WNT1 (NC_000012.11), SEC24D (NC_000004.11), P3H1 (NC_000001.11) were obtained from National Center for Biotechnology Information (NCBI) reference sequence and University of California, Santa Cruz (UCSC) Genome browser database2.
Whole Exome Sequencing (WES)
Samples from 59 probands underwent WES process. Genomic DNA was extracted from leukocytes using a standard sodium dodecyl sulfate-proteinase K-phenol/chloroform extraction method. 1–3 μg genomic DNA was used for WES as described previously (Li et al., 2019). DNA was fragmented as 150 bp, and the primers and adapters were then ligated to the DNA fragments to construct libraries. The analysis was performed on singletons, looking on previously known genes as filtering criteria. Sequencing was carried out on HiSeq 4000 System (Illumina). SAMtools mpileup and bcftools were used for variant calling and SNP/Indels identification. Control-FREEC was utilized for CNV detection.
Genomic Panel Sequencing
Fifteen probands were examined by genomic panel sequencing. Customized panel sequencing including 184 genes related to the monogenic disorders focused in our lab was conducted as described previously (Li et al., 2019). 21 OI-related genes were included with 18 recessive OI genes. Sequencing was performed on the HiSeq 2500 System (Illumina, San Diego, CA, United States). All reference sequences were based on the GRCh37/hg19 assembly of the human genome.
Confirmation of Pathogenic Mutation by Sanger Sequencing
To verify the candidate mutations detected by WES or Panel Sequencing, genomic DNA was amplified following the PCR program: 95°C for 3 min; 94°C for 30 s, 58°C for 30 s, 72°C for 50 s (35 cycles); 72°C for 8 min. PCR amplified fragments was subjected to Sanger Sequencing based on Applied Biosystems 3730xl DNA Analyzer (Thermo Fisher Scientific, Waltham, MA, United States). Sequence was analyzed using CodonCode Aligner (version 6.0.2.6; CodonCode, Centerville, MA, United States).
Isolation and Culturing of Dermal Fibroblasts
To test the splicing effect caused by two variants in FKBP10 in proband PUMC-121 on endogenous level, skin samples of the proband and an ethnically matched healthy individual were collected from fresh skin biopsies. Human fibroblasts were maintained at 37°C and 5% CO2 and supplied with F-12 supplemented with 1% L-glutamine, 20% fetal bovine serum, 1% sodium pyruvate, and 1% penicillin-streptomycin.
RNA Level Analysis
When a mutation was located in an intron, RNA level analysis would be conducted. Total RNA was isolated from peripheral blood or skin fibroblasts using Trizol reagent (Invitrogen, Cat. No. 15596018) followed by cDNA preparation using GoScriptTM Reverse Transcription System (Promega, Cat. No. A5001), according to the manufacturer’s instructions. cDNA was used for sequencing or for quantitative PCR analysis.
Quantitative PCR (qPCR)
qPCR was carried out to detect large fragment deletion and mRNA expression level of FKBP10 gene. PCR amplification was carried out with SYBR Premix ExTaq (Takara, Bio., Dalian, China) and primer pairs (Supplementary Table S1) according to the manufacturer’s protocol. The reactions were monitored continuously in a Rotor-Gene 6000 instrument (Qiagen, Hilden, Germany) according to the following program: 95°C for 3 min followed by 40 cycles of 95°C for 10 s, 60°C for 15 s, and 72°C for 20 s. The relative copy number (RCN) of the targeted sequence was normalized to the expression levels of GAPDH by calculating the ΔCt (Ctgene of interest - CtGAPDH), and RCN = 2–ΔΔCt.
T-Clone Sequencing
T-Clone sequencing was used when Sanger sequencing results showed interlaced alleles. Briefly, The PCR product of target samples with disrupted signals was linked to the pMD19-T vector. The ligation reaction contained 4 μl Solution I (Takara, Shiga, Japan), 1 μl pMD19-T vector and 5 μl purified PCR product. DNA Sanger sequencing was performed followed by vector ligation, E. coli transformation and bacterial culturing.
Statistical Analysis
Statistical analysis was conducted for age of first onset, times of fractures, frequency of fractures, height, height Z-score and ptosis for the patients with pathogenic genes WNT1, SERPINF1, and FKBP10. GraphPad Prism (version 6.00; GraphPad Software, La Jolla, CA, United States) was used for statistical analysis and was performed for n ≥ 9. Differences between three or more groups were analyzed by one-way ANOVA, and comparisons between two groups were analyzed by student t-test. Graphs were presented as average ± standard deviation, and p < 0.05 was considered as significant difference.
Results
Phenotypic Characterization of AR-OI
We recruited 74 probands (39 males, 34 females, and one gender unknown fetus) with AR-OI from 60 non-consanguineous families and 14 consanguineous families. Except for the fetus, their ages ranged from 6 months to 35 years old and their parents were confirmed to be unaffected. The clinical manifestations of the 74 probands, including frequent fractures, scoliosis, short stature, blue sclerae, ptosis, disability to walk and dentinogenesis imperfecta (DI) were recorded and analyzed (Figure 1). Phenotypes of all probands were summarized in Supplementary Table S2. These patients presented severe phenotypes: most of them showed disability to walk (72.86%, Figure 1C) and scoliosis (65.28%, Figure 1D). Nearly half of them experienced more than 2 fracture times per year (54.05%, Figure 1F), 38.03% of them had dentinogenesis imperfecta (Figure 1B), and 38.03% of patients presented extremely low short statures with a Z score less than −4 SD (corresponding to type III OI). Compared to dominant inherited OI, blue sclerae was less frequent (32.43%) in AR-OI individuals. A unique phenotype, ptosis (23.88%), was observed in recessive inherited OI patients (Figure 1A), but was absent in dominant OI patients. None of AR-OI individuals displayed hearing loss or intellectual disability.
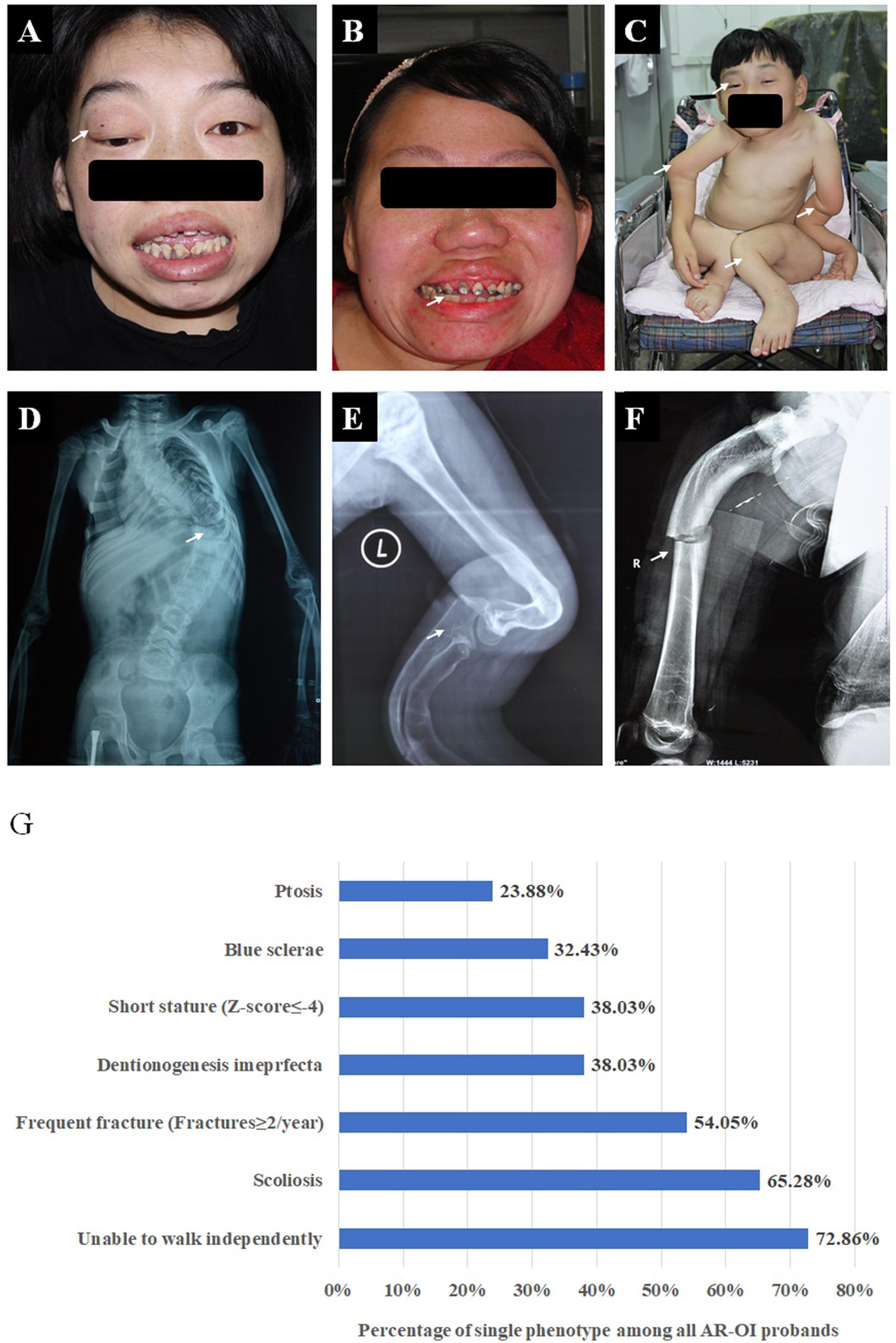
Figure 1. Phenotypic characteristics of AR-OI patients (A–F). AR-OI patients had severe clinical manifestations including ptosis (A), dentinogenesis imperfecta (B), severe limb deformity and disability to walk independently (C), scoliosis (D), skeleton deformity (E), and frequent fractures (F). (G) Percentage of the presence of each clinical manifestation in AR-OI probands.
Genotypic Characterization of Recessive OI
Variants Identification of Recessive OI
Variants of the 74 probands were identified in WNT1 (n = 30), SERPINF1 (n = 22), FKBP10 (n = 10), SERPINH1 (n = 2), CRTAP (n = 3), PLOD2 (n = 1), P3H1 (n = 3), and SEC24D (n = 3) (Table 1 and Figure 2B). Mutations in WNT1 showed the highest percentage (40.54%) in this cohort, followed by mutations in SERPINF1 (29.73%) and FKBP10 (13.51%, Figure 2A).
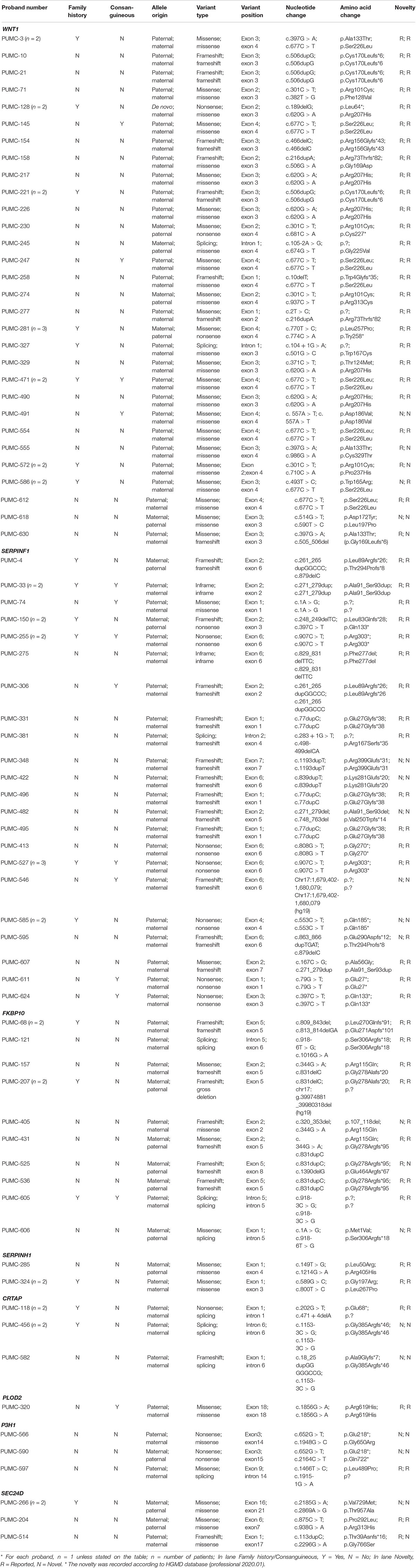
Table 1. Identification of mutations of recessive osteogenesis imperfecta in Chinese population.
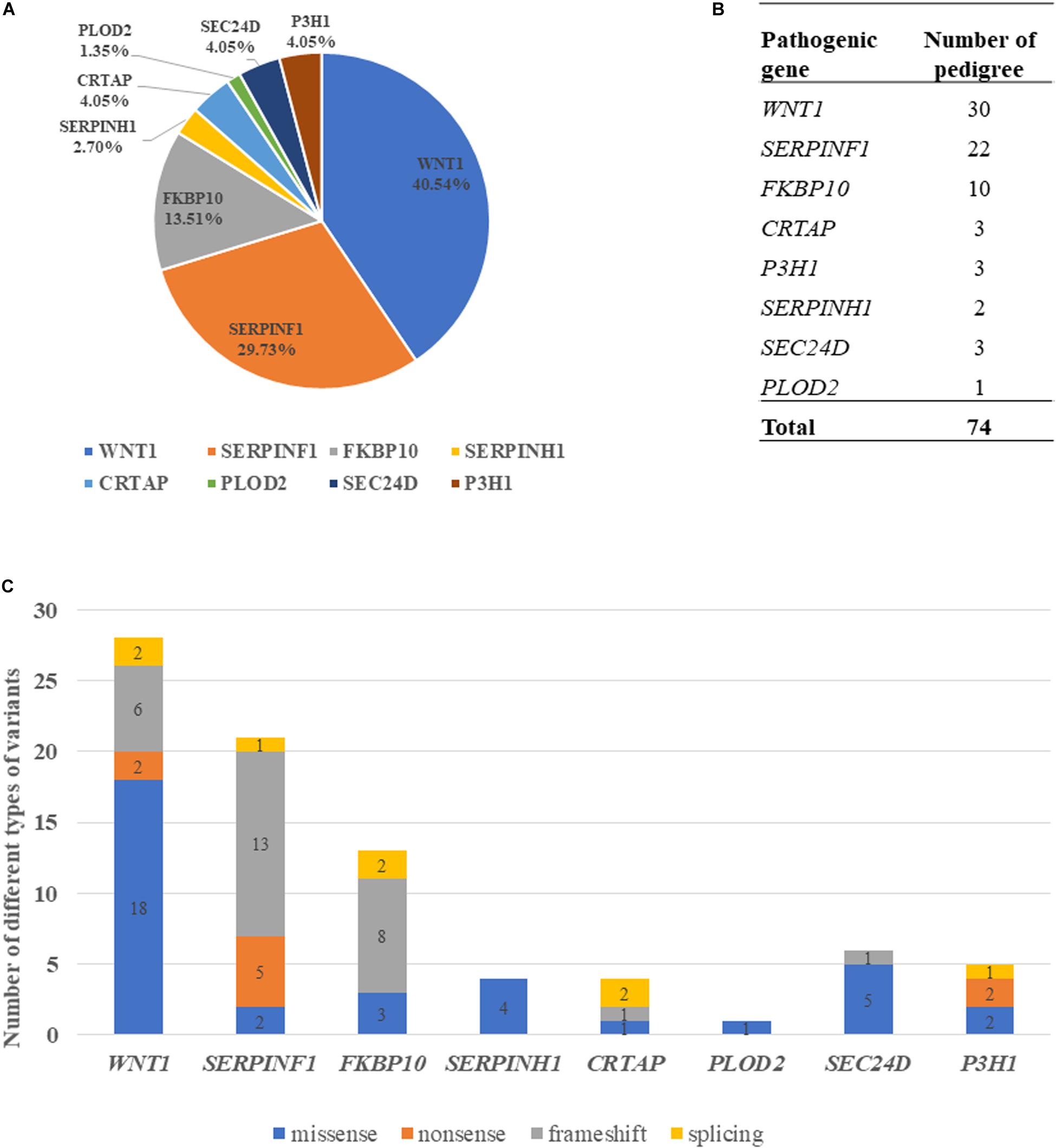
Figure 2. Genotypic characterization of AR-OI patients. (A) Mutation distribution in Chinese AR-OI patients. (B) The number of pedigrees in each pathogenic gene. (C) Distribution of mutation types in Chinese AR-OI patients.
Four major types of variants were found in this cohort, including missense, nonsense, frameshift and splicing mutations. We analyzed the number of each type of variant in each pathogenic gene and found that missense mutation was the main variant type in WNT1, SERPINH1 and SEC24D, and frameshift was frequently observed in SERPINF1 and FKBP10 (Figure 2C). Most variants were located at exons, but a homozygous intronic variant c.1153-3C > G was found in CRTAP (PUMC-456) (Supplementary Figure S1). Sequencing analysis of RT-PCR product of RNA isolated from dermal fibroblasts of this proband and his father confirmed that variant c.1153-3C > G led to an insertion of AG in the mutant transcript (Supplementary Figure S1C).
Establishment of Mutation Spectrums of WNT1, SERPINF1, and FKBP10
We identified 82 variants including 25 novel variants and 57 HGMD (professional 2020.01) reported variants (Table 1). Note that there were 36 variants reported in our previous study (Li et al., 2019). Because most variants are located in WNT1, SERPINF1 and FKBP10, the mutation spectrums of these genes were established (Figure 3).
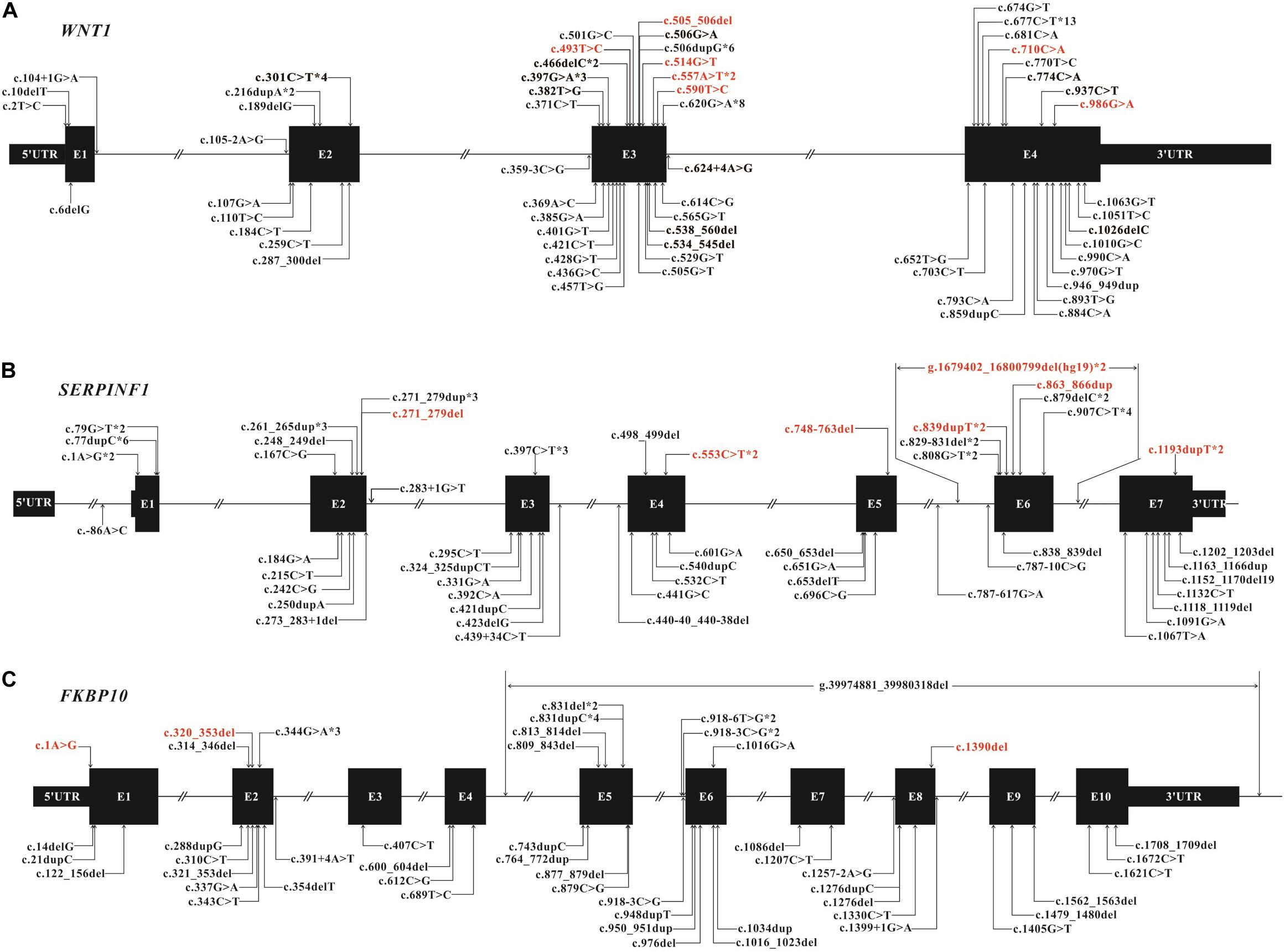
Figure 3. Mutation spectrum of WNT1 (A), SERPINF1 (B), and FKBP10 (C) of Chinese AR-OI patients. Black boxes represent the exons in each gene and lines represent the introns. Variants found in this study are listed above the boxes with novel variants in red and reported variants in black. Variants that have been reported previously are also listed under the boxes. Notation *digit indicates the number of probands carrying the same variant.
WNT1
We found 28 variants in WNT1 from 30 probands, including 21 reported variants and 7 novel variants (Table 1 and Figure 3A). There were some hotspot variants in WNT1: homozygous mutation c.506dupG(p.Cys170Leufs∗6) was found in three unrelated non-consanguineous families (PUMC-10, 21, and 221), leading to the production of truncated protein; Mutation c.677C > T(p.Ala133Thr) was observed in PUMC-3, 145, 247, 258, 471, and 554; Mutation c.620G > A (p.Arg207His) occurred in two probands (PUMC-128 and 329) and three non-consanguineous families (PUMC-217, 226, and 490), representing a hotspot in this Chinese cohort.
SERPINF1
Eight consanguineous and 14 non-consanguineous families were identified with mutations in SERPINF1, including 21 variants (14 reported variants and 7 novel variants) (Table 1 and Figure 3B). A known homozygous mutation, c.77dupC (p.Glu27Glyfs∗38), was found in three non-consanguineous families, which resulted in frameshift and truncated protein. Interestingly, there were three siblings from a consanguineous family (PUMC-527) identified with same homozygotes of a novel variant, c.907C > T in SERPINF1, but they exhibited extremely different phenotypes (Supplementary Figure S2). The proband had severe scoliosis and unable to walk independently with more than 45 times of fracture. However, his elder sister, harboring the same genotype, did not present any OI phenotype at all with only once fracture lifetime.
FKBP10
Thirteen variants in FKBP10, including 10 known variants and 3 novel variants, were identified in 10 non-consanguineous families (Table 1 and Figure 3C). Hotspot c.831indelC was identified in PUMC-157, 207, 431, 525 and 536, and these variants led to a premature termination codon. Hotspot c.344G > A was identified in PUMC-157, 405, and 431. Among the patients with variants in FKBP10, three special cases needed to be highlighted. Case I, Patient PUMC-405 was a compound heterozygote with mutations in FKBP10. Sanger sequencing analysis by software Chromas showed disrupted signals in exon 2 in FKBP10 (Supplementary Figure S4A), and two alleles were separated by T clone sequencing of the mutant region: c.320_353del(p.107_118del) with a 34 bp deletion (Supplementary Figure S4B), and a missense mutation c.344G > A(p.Arg115Gln) within the deleted region (Supplementary Figure S4C); Case II, Patient PUMC-207 harbored a gross deletion and an indel variant in FKBP10. The compound mutations found in proband PUMC-207 were inherited from the parents: the indel of c.831delC was inherited from the mother and the gross deletion of chr17: g.39974881_39980318del (hg19) was inherited from the father, which was confirmed in breakpoint analysis using Gap-PCR and Sanger sequencing (Supplementary Figure S5); Case III, Patient PUMC-121 was previously reported that an intronic variant c.918-6T > G and a variant in adjacent exon c.1016G > A in FKBP10 similarly drove to skipping of exon 6, indicated by a minigene assay (Supplementary Figures S6A,B) (Li et al., 2019). Based on the sequencing results from cDNA (Supplementary Figure S6C) and RNA isolated from the patient’s dermal fibroblasts in the follow up study, we found that variant c.918-6T > G caused skipping of exon 6 and variant c.1016G > A caused partial skipping of exon 6. This was confirmed by qPCR analysis (Supplementary Figure S6D).
Correlation Between Genotypic and Phenotypic Changes
In order to answer whether there is a correlation between genotypes and observed phenotypes in AR-OI patients, we first analyzed the sites of fractures in patients harboring different pathogenic genes. We did not find significant difference in fracture locations among patients with different causative genes (Figure 4A). We then analyzed the phenotypes including fracture times, scoliosis, height, blue sclerae, DI, walking ability, and ptosis in these patients. Statistical analysis was conducted for WNT1, SERPINF1 and FKBP10, with > 3 probands (Supplementary Table S3), and only ptosis was significantly different between groups.
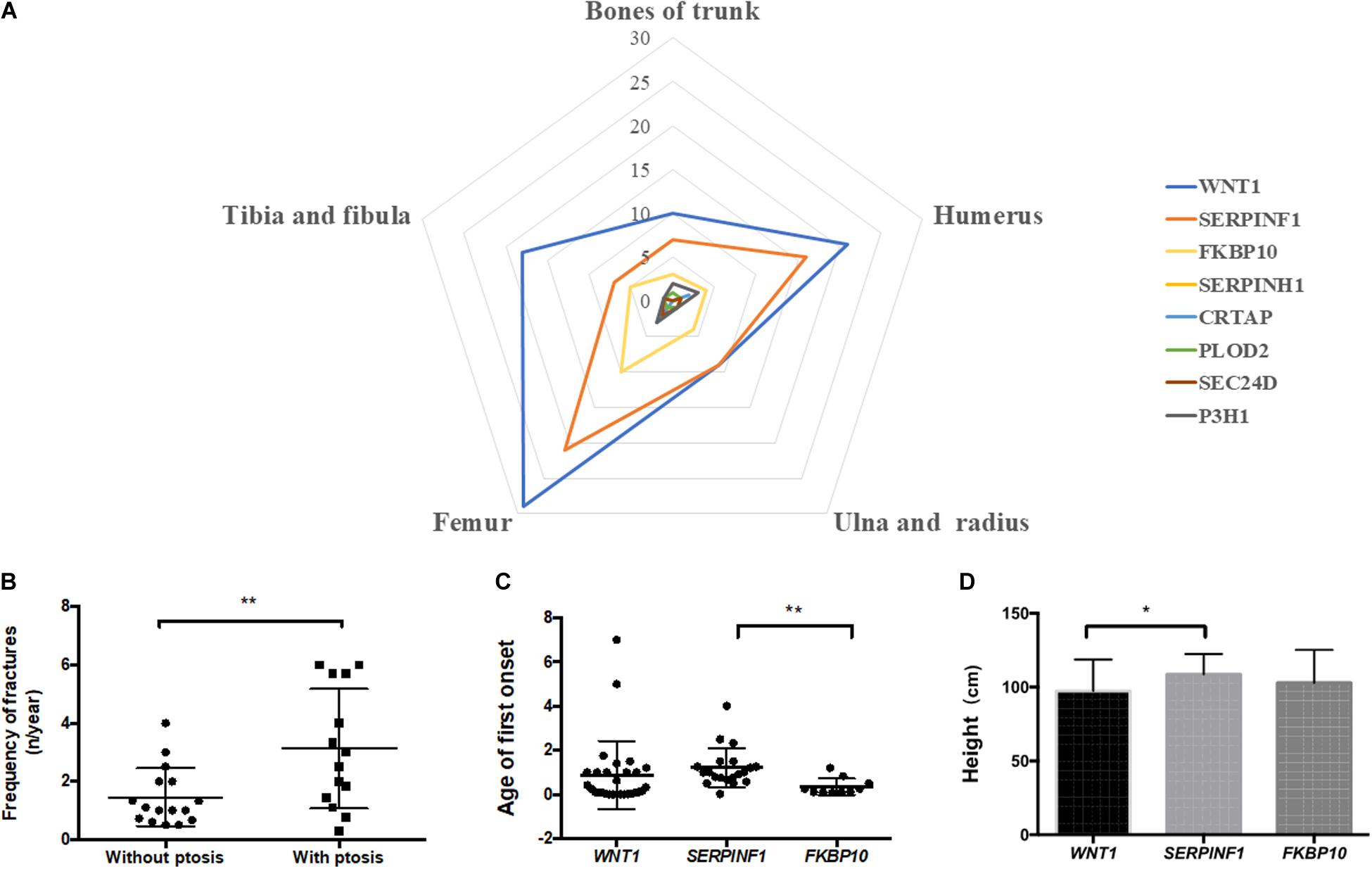
Figure 4. Correlation analysis of genotype and phenotype of Chinese AR-OI patients. (A) Analysis of fracture sites in patients with variants in WNT1, SERPINF1, FKBP10, SERPINH1, CRTAP, PLOD2, P3H1, and SEC24D. (B). Fracture frequencies in patients with and without ptosis phenotype. Comparison of age of first onset (C) and height (D) of patients with variants in WNT1, SERPINF1, and FKBP10 (*p < 0.05, **p < 0.01).
Patients With WNT1 Mutation Had Ptosis Phenotype
We noticed that AR-OI patients carrying WNT1 mutations were prone to developing ptosis, with 46.67% penetrance (Supplementary Table S3). This manifestation was common in patients with a homozygous function loss of WNT1, but was rare in patients with a loss of SERPINF1. Ptosis distribution displayed significant difference in different genes (p = 0.0042). In patients with WNT1 mutations, those with ptosis also showed more severe skeletal dysfunctions including higher fracture frequency (Figure 4B) and unable to walk independently (80%), as compared to those without ptosis.
Patients With Variants in SERPINF1 Presented a Distinct Phenotype
Compared to patients with mutations in WNT1 and FKBP10, patients with variants in SERPINF1 showed significantly delayed first fracture age (p = 0.0072, Figure 4C) and taller height (p = 0.0460, Figure 4D).
Discussion
With the development of next generation sequencing, genes associated with OI have been well-studied and identified. We examined mutation spectrum of AR-OI in a large cohort of 74 Chinese OI families. Current study unraveled 25 novel variants from 82 identified variants, and demonstrated WNT1 mutation as the most frequent mutation in all AR-OI genes. Furthermore, these AR-OI patients showed a unique phenotype, ptosis.
Type I collagen is a heterotrimer structure with two alpha 1 chains and one alpha 2 chain. Dominant OI is caused by mutations in COL1A1 or COL1A2, and phenotypes can vary from mild to moderate/severe depending on the whether the collagen defect was caused by haplo-insufficiency or helical mutations that led to dominant negative effect (Rauch et al., 2010; Lindahl et al., 2015). In AR-OI, alteration occurs during post translational modification progress when a triple helix pro-collagen is already formed, and hence most of these mutations affect both the synthesis of the whole type I collagen protein and bone homeostasis. Accordingly, AR-OI patients often exhibit more severe phenotypes than dominant OI, including significantly earlier first fracture age, higher fracture frequency, higher percentage of scoliosis and higher proportion of disability to walk (Li et al., 2019). Consistent with previous findings, more severe clinical manifestations were also observed in a cohort of Chinese patients with recessive OI in current study, including a high rate of scoliosis, disability of independent walking, extreme short stature, and high facture frequency (Figure 1G).
Current study in Chinese AR-OI patients suggests that WNT1 is the most frequently involved gene, followed by SERPINF1 and FKBP10. These findings differ from that in Caucasian population, which showed that SERPINF1 was the highest, followed by CRTAP (Bardai et al., 2016). The most distinct phenotypic characteristic in Chinese AR-OI patients was a unique ptosis phenotype, which was presented almost exclusively in patients associated with WNT1 variants (Supplementary Table S3). This result is consistent with recent finding that ptosis was a hallmark for OI patients with WNT1 mutations from Indian and Turkish families (Nampoothiri et al., 2019), and that from seven Chinese families (Lu et al., 2018). WNT1 signaling was critical for the cross talk between osteoblastic lineage and osteoclastic-lineage cells (Laine et al., 2013). In addition to its vital importance in bone homeostasis (Luther et al., 2018), WNT1 also plays an essential role in neurological development (Faqeih et al., 2013; Aldinger et al., 2016). Because patients displayed ptosis all suffered neurological dysfunctions (Nampoothiri et al., 2019), it was postulated that ptosis phenotype may be associated with impaired brain or nerve development. Although we did not observe any intellectual problem in AR-OI patients with ptosis phenotype, these patients developed more severe skeletal phenotypes than those without ptosis (Figure 4D). No correlation was found between ptosis and mutation type or variant location. The mechanism of the development of ptosis in patients with WNT1 variant-induced AR-OI remains to be elucidated in future studies.
The severity of skeletal phenotype was compared in patients with WNT1, SERPINF1, and FKBP10 variants. Patients with variants in SERPINF1 had the highest number of average fracture times (30.77 ± 6.626) and highest frequency of fractures (4.193 ± 0.7986 n/year). However, their stature is the tallest (108.7 ± 3.154 cm, Supplementary Table S3). Mrosk et al. (2018) reported that the phenotypic severity of genes associated with OI is: WNT1 > SERPINF1 > FKBP10, based on findings from 50 OI families, including 24 recessive OI families. In the current study, patients with WNT1 variants presented the shortest height (97.40 ± 3.889) and highest percentage of scoliosis 80.00%, but did not show any significant difference in other parameters such as height Z-score, blue sclerae, dentinogenesis imperfecta and disability of independent walking from patients with variants in SERPINF1 and FKBP10 (Supplementary Table S3). It needs to be noted that patients with FKBP10 variants all presented a short neck phenotype and severe spinal malformation (Supplementary Figure S3). FKBP10 is known to function as a molecular chaperone that interacts with collagen (Ishikawa et al., 2008), but how it affects the cervical vertebra development remains unclear.
Recurrent variants were found in WNT1 (c.506dupG, c.677C > T, and c.620G > A), SERPINF1 (c.77dupC, c.907C > T) and FKBP10 (c.831indelC and c.344G > A). All the homozygous of these recurrent variants corresponded to a severe phenotype (Supplementary Table S2). In particularly, OI patients with the novel variant c.907C > T in SERPINF1 presented the most severe phenotypes: with 45–100 times of fractures, severe scoliosis and extremely short stature (Z-score < −8) (Supplementary Table S2). Previous studies also identified some recurrent variants, such as c.506dupG in WNT1, but did not find a clear correlation with the severity of phenotype (Liu et al., 2016). Variant c.506dupG in WNT1 was found to be a hotspot region in 5 families among 10 total families examined (Nampoothiri et al., 2019). Kelley et al. (2011) reported the recurrent variant, c.344G > A in FKBP10 from a Caucasian child, who presented multiple fractures and a short stature. Further studies are needed to establish a causal-relationship between hotspot variant sites and phenotypes in these patients.
Frameshift occupied a high proportion in SERPINF1 and FKBP10 mutations (Figure 2C), suggesting that the two genes may have a distinct mechanism for OI development. In particular, most of variants located in SERPINF1 may be involved in occurrence of nonsense/frameshift mediated pre-termination codon (Table 1), and these patients had very severe skeletal phenotypes, which is in line with previous findings (Becker et al., 2011). SERPINF1 encodes pigment epithelium-derived factor (PEDF), which can inhibit osteoclastogensis by regulating osteoprotegerin expression (Akiyama et al., 2010) and enhancing pre-osteoblast differentiation (Toru Akiyama et al., 2003). The truncated PEDF protein which mainly results in the absence of C terminal of PEDF, led to the malfunction of protein (Shao et al., 2003). Therefore, nonsense/frameshift mutations in SERPINF1 may lead to truncated PEDF and severe OI phenotype.
In summary, we examined the largest cohort (n = 74) of Chinese AR-OI probands and established the mutation spectrum of the three most frequent pathogenic genes (WNT1, SERPINF1, FKBP10) in this population. We also identified 25 novel variants and 7 hotspot variants. Genotypic and phenotypic analysis unraveled ptosis as a unique phenotype in patients with WNT1 variants. Furthermore, patients with ptosis or hotspot variants showed more severe skeletal phenotypes than others. Current findings may enrich our understanding of the genetic basis of AR-OI, and our study provides new knowledge for a precise diagnosis of this disease in Chinese population.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board (IRB) of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing, China (015-2015). Written informed consent to participate in this study was provided by all participants/legal guardians of children under 18.
Author Contributions
SL, YC, and HW performed the experiment and wrote the manuscript. XR and YW collected the samples of patients. HM, YG, FZ, LL, BM, TY, YYo, XG, and YYa conducted data analysis. XZho and XZhn designed and supervised this research. All authors performed critical reading and approved the final version of manuscript.
Funding
This study was supported by grants from National Key Research and Development Program of China (2016YFE0128400 and 2016YFC0905100), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-3-003), and National Natural Science Foundation of China (81472053).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the OI patients and their families for their participation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00984/full#supplementary-material
FIGURE S1 | Dermal fibroblast analysis confirmed the sequencing result of intronic variant c.1153-3C > G in CRTAP. (A) Pedigree information was shown in the family tree. (B) Results from genomic DNA sequencing showed an intronic mutation of c.1153-3C > G in CRTAP in the proband II1. Parents of the proband were heterozygous carrier of the same variant. (C) Sequencing of dermal fibroblasts of I1 and II1 confirmed an insertion of AG in the mutant transcript.
FIGURE S2 | A consanguineous family that had the same variant but presented variable expressivity. (A) The pedigree of consanguineous family PUMC-527. (B) A photo of patients II1 (mild) and II2 (severe) with a wide difference in their phenotypes. (C) Genotype of all the familial members revealed by DNA sequencing.
FIGURE S3 | Recessive OI patients with variants in FKBP10 exhibited a unique short neck phenotype (A–C). Photo records and X-ray examinations of three individuals with FKBP10 variants. (A) Short neck (PUMC-68), (B) short neck and severe deformity of spinal (PUMC-68), (C) compressible vertebrae (PUMC-121).
FIGURE S4 | A case of micro-deletion and missense mutation in the same region in FKBP10. (A) Sanger DNA sequencing of proband PUMC-405 showed disrupted signal in exon 2 in FKBP10. Two mutations was separated after T-clone sequencing: c.320_353del(p.107_118del) (B) and c.344G > A(p.Arg115Gln) which was located within the deletion region (C).
FIGURE S5 | A case of gross deletion in FKBP10. (A) Sanger sequencing results of the proband and his parents. (B) An intragenic deletion was found in the proband and his father indicated by quantitative real-time PCR. The values presented as triplicate determinations ± SD. RCN, relative copy number. (C) Breakpoint analysis showed that breakpoint was found in proband and his father (640 bp), while absence in the mother of proband because of the existence of original fragment. M: Marker, 640 bp: fragment length of the deletion junction product, 304 bp: control. (D) Sequence chromatograms of the g.41818632_41824069del breakpoint.
FIGURE S6 | Identification of the splicing effect of compound mutations in FKBP10. (A) Sanger sequencing indicated a missense variant c.1016G > A(p.Arg339Gln) in exon 6 and an intronic variants c.918-6T > G in adjacent intron 5 in FKBP10 in patient PUMC-121. (B) Minigene assay showed that both c.918-6T > G and c.1016G > A led in skipping of exon 6. (C) Sequencing result of RT-PCR product from RNA extracted from the patient’s dermal fibroblasts. (D) Relative normalized expression of exon 6, exons 4–5, and exons 7–8 comparing the patient and control. Gene expression in normal control was normalized as 1.
Footnotes
References
Akiyama, T., Dass, C. R., Shinoda, Y., Kawano, H., Tanaka, S., and Choong, P. F. (2010). PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem. Biophys. Res. Commun. 391, 789–794. doi: 10.1016/j.bbrc.2009.11.139
Aldinger, K. A., Mendelsohn, N. J., Chung, B. H., Zhang, W., Cohn, D. H., Fernandez, B., et al. (2016). Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. J. Med. Genet. 53, 427–430. doi: 10.1136/jmedgenet-2015-103476
Azetsu, Y., Inohaya, K., Takano, Y., Kinoshita, M., Tasaki, M., and Kudo, A. (2017). The sp7 gene is required for maturation of osteoblast-lineage cells in medaka (Oryzias latipes) vertebral column development. Dev. Biol. 431, 252–262. doi: 10.1016/j.ydbio.2017.09.010
Bardai, G., Moffatt, P., Glorieux, F. H., and Rauch, F. (2016). DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos. Int. 27, 3607–3613. doi: 10.1007/s00198-016-3709-3701
Becker, J., Semler, O., Gilissen, C., Li, Y., Bolz, H. J., Giunta, C., et al. (2011). Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 88, 362–371. doi: 10.1016/j.ajhg.2011.01.015
Faqeih, E., Shaheen, R., and Alkuraya, F. S. (2013). WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J. Med. Genet. 50, 491–492. doi: 10.1136/jmedgenet-2013-101750
Forlino, A., and Marini, J. C. (2016). Osteogenesis imperfecta. Lancet 387, 1657–1671. doi: 10.1016/s0140-6736(15)00728-x
Ishikawa, Y., Vranka, J., Wirz, J., Nagata, K., and Bachinger, H. P. (2008). The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 283, 31584–31590. doi: 10.1074/jbc.M802535200
Kang, H., Aryal, A. C. S., and Marini, J. C. (2017). Osteogenesis imperfecta: new genes reveal novel mechanisms in bone dysplasia. Transl. Res. 181, 27–48. doi: 10.1016/j.trsl.2016.11.005
Kelley, B. P., Malfait, F., Bonafe, L., Baldridge, D., Homan, E., Symoens, S., et al. (2011). Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone. Miner. Res. 26, 666–672. doi: 10.1002/jbmr.250
Keupp, K., Beleggia, F., Kayserili, H., Barnes, A. M., Steiner, M., Semler, O., et al. (2013). Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 92, 565–574. doi: 10.1016/j.ajhg.2013.02.010
Koide, T., Nishikawa, Y., Asada, S., Yamazaki, C. M., Takahara, Y., Homma, D. L., et al. (2006). Specific recognition of the collagen triple helix by chaperone HSP47. II. the HSP47-binding structural motif in collagens and related proteins. J. Biol. Chem. 281, 11177–11185. doi: 10.1074/jbc.M601369200
Laine, C. M., Joeng, K. S., Campeau, P. M., Kiviranta, R., Tarkkonen, K., Grover, M., et al. (2013). WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 368, 1809–1816. doi: 10.1056/NEJMoa1215458
Li, L., Bin, M., Li, S., Xiao, J., Wang, H., Zhang, J., et al. (2019). Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum. Mutat. 40, 588–600. doi: 10.1002/humu.23718
Lindahl, K., Astrom, E., Rubin, C. J., Grigelioniene, G., Malmgren, B., Ljunggren, O., et al. (2015). Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet. 23, 1042–1050. doi: 10.1038/ejhg.2015.81
Liu, Y., Asan, Ma, D., Lv, F., Xu, X., Wang, J., et al. (2017). Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos. Int. 28, 2985–2995. doi: 10.1007/s00198-017-4143-4148
Liu, Y., Song, L., Ma, D., Lv, F., Xu, X., Wang, J., et al. (2016). Genotype-phenotype analysis of a rare type of osteogenesis imperfecta in four Chinese families with WNT1 mutations. Clin. Chim. Acta 461, 172–180. doi: 10.1016/j.cca.2016.07.012
Lu, Y., Ren, X., Wang, Y., Bardai, G., Sturm, M., Dai, Y., et al. (2018). Novel WNT1 mutations in children with osteogenesis imperfecta: clinical and functional characterization. Bone 114, 144–149. doi: 10.1016/j.bone.2018.06.018
Luther, J., Yorgan, T. A., Rolvien, T., Ulsamer, L., Koehne, T., and Liao, N. (2018). Wnt1 is an Lrp5-independent bone-anabolic Wnt ligand. Sci. Transl. Med. 10:eaau7137. doi: 10.1126/scitranslmed.aau7137
Marini, J. C., Forlino, A., Cabral, W. A., Barnes, A. M., San Antonio, J. D., Milgrom, S., et al. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–221. doi: 10.1002/humu.20429
Morello, R., Bertin, T. K., Chen, Y., Hicks, J., Tonachini, L., Monticone, M., et al. (2006). CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127, 291–304. doi: 10.1016/j.cell.2006.08.039
Mrosk, J., Bhavani, G. S., Shah, H., Hecht, J., Kruger, U., Shukla, A., et al. (2018). Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone 110, 368–377. doi: 10.1016/j.bone.2018.02.029
Murakami, T., Saito, A., Hino, S., Kondo, S., Kanemoto, S., Chihara, K., et al. (2009). Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 11, 1205–1211. doi: 10.1038/ncb1963
Nampoothiri, S., Guillemyn, B., Elcioglu, N., Jagadeesh, S., Yesodharan, D., Suresh, B., et al. (2019). Ptosis as a unique hallmark for autosomal recessive WNT1-associated osteogenesis imperfecta. Am. J. Med. Genet. A 179, 908–914. doi: 10.1002/ajmg.a.61119
Pyott, S. M., Tran, T. T., Leistritz, D. F., Pepin, M. G., Mendelsohn, N. J., Temme, R. T., et al. (2013). WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 92, 590–597. doi: 10.1016/j.ajhg.2013.02.009
Rauch, F., Lalic, L., Roughley, P., and Glorieux, F. H. (2010). Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J. Bone. Miner. Res. 25, 1367–1374. doi: 10.1359/jbmr.091109
Schwarze, U., Cundy, T., Pyott, S. M., Christiansen, H. E., Hegde, M. R., Bank, R. A., et al. (2013). Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum. Mol. Genet. 22, 1–17. doi: 10.1093/hmg/dds371
Shao, H., Schvartz, I., and Shaltiel, S. (2003). Secretion of pigment epithelium-derived factor. Mutagenic Study Eur. J. Biochem. 270, 822–831. doi: 10.1046/j.1432-1033.2003.03374.x
Toru Akiyama, P. B., Tsuyoshi Miyazaki, Y. K., Hirotaka Chikuda, U.-I. C., Akira Fukuda, A. H., Hiroaki Seto, T. O., Toshiya Inaba, A. S., et al. (2003). Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member bim. EMBO J. 22, 6653–6664. doi: 10.1093/emboj/cdg635
Keywords: autosomal recessive osteogenesis imperfecta, mutation spectrum, phenotype, WNT1, Chinese cohort
Citation: Li S, Cao Y, Wang H, Li L, Ren X, Mi H, Wang Y, Guan Y, Zhao F, Mao B, Yang T, You Y, Guan X, Yang Y, Zhang X and Zhao X (2020) Genotypic and Phenotypic Analysis in Chinese Cohort With Autosomal Recessive Osteogenesis Imperfecta. Front. Genet. 11:984. doi: 10.3389/fgene.2020.00984
Received: 31 October 2019; Accepted: 04 August 2020;
Published: 15 September 2020.
Edited by:
Oliver Semler, Universitätsklinikum Köln, GermanyReviewed by:
Annalisa Vetro, Meyer University Hospital, University of Florence, ItalyHao Zhang, Shanghai Sixth People’s Hospital, China
Copyright © 2020 Li, Cao, Wang, Li, Ren, Mi, Wang, Guan, Zhao, Mao, Yang, You, Guan, Yang, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuli Zhao, eGl1bGl6aGFvQGlibXMucHVtYy5lZHUuY24=; Xue Zhang, eHVlemhhbmdAcHVtYy5lZHUuY24=
†These authors have contributed equally to this work