 Ryan J. Corbett1*
Ryan J. Corbett1* Marinus F. W. te Pas2
Marinus F. W. te Pas2 Henry van den Brand3
Henry van den Brand3 Martien A. M. Groenen2
Martien A. M. Groenen2 Richard P. M. A. Crooijmans2
Richard P. M. A. Crooijmans2 Catherine W. Ernst4
Catherine W. Ernst4 Ole Madsen2*
Ole Madsen2*- 1Genetics and Genome Sciences Graduate Program, Michigan State University, East Lansing, MI, United States
- 2Animal Breeding and Genomics, Wageningen University & Research, Wageningen, Netherlands
- 3Adaptation Physiology Group, Wageningen University & Research, Wageningen, Netherlands
- 4Department of Animal Science, Michigan State University, East Lansing, MI, United States
Temperature and CO2 concentration during incubation have profound effects on broiler chick development, and numerous studies have identified significant effects on hatch heart weight (HW) as a result of differences in these parameters. Early life environment has also been shown to affect broiler performance later in life; it has thus been suggested that epigenetic mechanisms may mediate long-term physiological changes induced by environmental stimuli. DNA methylation is an epigenetic modification that can confer heritable changes in gene expression. Using reduced-representation bisulfite sequencing (RRBS), we assessed DNA methylation patterns in cardiac tissue of 84 broiler hatchlings incubated at two egg shell temperatures (EST; 37.8°C and 38.9°C) and three CO2 concentrations (0.1%, 0.4%, and 0.8%) from day 8 of incubation onward. We assessed differential methylation between EST treatments and identified 2,175 differentially methylated (DM) CpGs (1,121 hypermethylated, 1,054 hypomethylated at 38.9° vs. 37.8°) in 269 gene promoters and 949 intragenic regions. DM genes (DMGs) were associated with heart developmental processes, including cardiomyocyte proliferation and differentiation. We identified enriched binding motifs among DM loci, including those for transcription factors associated with cell proliferation and heart development among hypomethylated CpGs that suggest increased binding ability at higher EST. We identified 9,823 DM CpGs between at least two CO2 treatments, with the greatest difference observed between 0.8 and 0.1% CO2 that disproportionately impacted genes involved in cardiac muscle development and response to low oxygen levels. Using HW measurements from the same chicks, we performed an epigenome-wide association study (EWAS) for HW, and identified 23 significantly associated CpGs, nine of which were also DM between ESTs. We found corresponding differences in transcript abundance between ESTs in three DMGs (ABLIM2, PITX2, and THRSP). Hypomethylation of an exonic CpG in PITX2 at 38.9°C was associated with increased expression, and suggests increased cell proliferation in broiler hatchlings incubated at higher temperatures. Overall, these results identified numerous epigenetic associations between chick incubation factors and heart development that may manifest in long-term differences in animal performance.
Introduction
Early life environmental parameters have profound effects on broiler chick development and post-hatch performance. Incubation temperature is one of the most important factors influencing embryonic growth, development, and physiology (Ricklefs, 1987; French, 1994; Christensen et al., 1999; Lourens et al., 2005). In studies assessing the effects of incubation egg shell temperature (EST) on organ growth, heart weight (HW) is consistently observed to be negatively correlated with EST (Leksrisompong et al., 2007; Molenaar et al., 2010, 2011; Maatjens et al., 2014, 2016). Mechanisms linking incubation EST and heart development have been proposed; studies have shown that increased incubation temperature decreases the mitotic index of cardiomyocytes (Romanoff, 1960), as well as the concentration of circulating T3, the metabolically active form of thyroid hormone (Kühn et al., 1984; Yahav et al., 1996). Additionally, phenotypic differences in adult broilers associated with differences in early life temperature have been observed. Chicks incubated at a higher EST from embryonic day 7 (E7) to hatch were found to have a higher incidence of ascites mortality at 6 weeks of age (Molenaar et al., 2011; Sözcü and Ipek, 2015). However, broilers exposed to increased temperature early in life exhibited decreased mortality when exposed to heat stress again at 6 weeks of age relative to unexposed animals, suggesting that early heat stress confers thermotolerance that can improve performance upon subsequent exposures (Yahav and Hurwitz, 1996).
In addition to temperature, CO2 concentration during the incubation period has been suggested to be important for regulating chick growth and physiology. Embryos incubated at increased CO2 concentration have been found to exhibit numerous phenotypes; in addition to a significantly greater average HW at hatch (Maatjens et al., 2014), lower blood oxygen levels relative to control chicks have also been observed (Hassanzadeh et al., 2010). Increased incubator CO2 concentration was associated with decreased ascites mortality in broilers later in life (Buys et al., 1998), suggesting long-term effects of early life CO2 exposure. Alterations made to the epigenome have been proposed as one mechanism by which early life environmental differences can manifest in phenotypic differences in adult precocial birds (Tzschentke and Basta, 2002; Nichelmann, 2004), and it has recently been shown that embryonic thermal manipulation induces epigenetic modifications in the hypothalamus of 35-day old broilers (David et al., 2019).
Numerous epigenetic molecular mechanisms including DNA methylation, histone tail modifications, and non-coding RNA interactions have been observed to induce mitotically heritable changes in gene expression without altering DNA sequence (Jaenisch and Bird, 2003; Ibeagha-Awemu and Zhao, 2015). DNA methylation is an epigenetic modification involving the enzymatic addition of a methyl group to the 5-carbon of cytosine rings, producing 5-methylcytosine. Methylation occurs almost exclusively at CpG dinucleotides in vertebrates and has context-specific associations with gene expression. In gene promoters, methylation generally functions to decrease levels of transcription through the alteration of transcription factor (TF) binding sites, the recruitment of transcriptional repressors, or changes in chromatin conformation (Bird and Wolffe, 1999; Greenberg and Bourc’his, 2019). Conversely, gene body methylation is generally associated with increased levels of transcription (Lorincz et al., 2004; Ball et al., 2009; Maunakea et al., 2010), although negative associations have been identified in the context of genic enhancers (Varley et al., 2013). Epigenetic processes are known to be altered in response to environmental perturbations, and this phenomenon has been studied in livestock species in response to heat stress (Hao et al., 2016; Skibiel et al., 2018; Vinoth et al., 2018). Additionally, numerous epigenome-wide association studies (EWAS) have been performed in humans and mice to identify loci at which methylation level is significantly associated with complex and disease traits (Orozco et al., 2015; reviewed in Flanagan, 2015). Bisulfite-sequencing approaches allow for genome-wide assessment of DNA methylation patterns, yet this approach has been underutilized in the chicken thus far (Li et al., 2015; Lee et al., 2017; Zhang et al., 2017, 2018).
In the current study, we assessed DNA methylation patterns in cardiac tissue of broiler chicks incubated at normal or high EST (37.8 or 38.9°C, respectively) and low, medium, or high CO2 concentration (0.1%, 0.4%, 0.8%, respectively) from embryonic day 8 (E8) until hatch. Using reduced representation bisulfite sequencing (RRBS), we tested for differences in CpG methylation rates between EST and CO2 treatments and identified thousands of loci exhibiting differential methylation that are associated with heart development. Additionally, by integrating phenotypic records from the same samples, we performed an EWAS and identified loci at which methylation rate was significantly associated with HW, and many of these sites were within genes with known roles in heart development. Our results provide evidence that EST and CO2 may be influencing heart growth and physiology through changes in cardiac DNA methylation patterns.
Materials and Methods
Experimental Design and Sample Collection
Heart tissue samples were collected from a 2 × 3 factorial study. Briefly, Cobb 500 broiler eggs were obtained from commercial broiler breeder farms and incubated in six consecutive batches. From day 0 to 8 of incubation (E8), all eggs were incubated at an EST of 37.8°C and 0.1% CO2. At E8 eggs were divided into two ESTs (37.8°C and 38.9°C) and three CO2 concentrations (0.1%, 0.4%, and 0.8% CO2) until hatch. Both EST treatments were applied in all batches, but CO2 treatment application varied between batches. Time until hatch was recorded for each chick, and animals were removed from the incubator 6 h post-hatch. Chicks were then killed by cervical dislocation followed by decapitation. Hearts were removed and stored at −20°C until further analysis. Heart samples derived from the left ventricle of 84 chicks (13 to 15 samples per EST-CO2 combination) were utilized for DNA isolation and RRBS.
Statistical Analyses of Heart Weight
We tested for the significance of EST, CO2 concentration, and their interaction on HW using analysis of variance (ANOVA). To account for the significantly shorter incubation time of 38.9°C chicks, we constructed a linear model with HW as the response variable and incubation time as a fixed effect, and subsequently used the model residuals as a response variable to test the significance of the environmental parameter main effects and their interaction.
Sample Processing and Bisulfite Sequencing
DNA and RNA from heart samples were isolated using the AllPrep DNA/RNA Mini Kit (Qiagen) following manufacturer’s instructions. The RRBS libraries were prepared with the Ovation RRBS Library construction kit from NuGEN following manufacturer’s instructions. Isolated DNA was cut with MspI and a fragment size selection of 20–250 bases was applied. Samples were pooled across 9 flow cell lanes with each pool containing no more than 10 samples, and all pools were sequenced for 161 cycles on a HiSeq 2500 using the TruSeq SBS sequencing kit version 4. Fastq files were generated and demultiplexed with the bcl2fastq v2.17.1.14 Conversion Software (Illumina).
Bioinformatics Analyses
Raw RRBS FASTQ files were trimmed of adapter sequences using Trim Galore v0.5.0 with Cutadapt v1.8.1 (Babraham Bioinformatics) and the parameters: -a AGATCGGAAGAGC. Reads were subsequently filtered for only those beginning with the expected YGG trinucleotide sequence using a python script provided by NuGEN Technologies1.
Read alignment was performed using BS-seeker2 v2.1.3 (Guo et al., 2013). An RRBS bowtie2 index was generated from the Gallus gallus GRCg6a reference genome assembly using bs_seeker_build.py with the following parameters: –aligner bowtie2 –rrbs –low 10 –up 280. Trimmed reads were subsequently aligned to the RRBS index using bs_seeker2-align.py with the following parameters: –rbbs –low 10 –up 280 –mismatches 0.05 –aligner = bowtie2 –bt2-p 10 –bt–local –bt2-N 1. Binary alignment map (BAM) files were converted to CGmap files using CGmapTools v0.1.2 (Guo et al., 2018), which reports methylation rates for all covered CpGs.
CGmap files were used to calculate sample global methylation rates, and to assess their associations with EST, CO2, and individual CpG methylation rates. We identified an overrepresentation of CpG sites where methylation was significantly correlated with sample global CpG methylation rate (data not shown). We therefore corrected for this factor in subsequent site-specific differential methylation analyses.
Differential Methylation Analyses
We filtered CGmap files to only include CpGs with coverage of at least 10 reads within a sample. Filtered CGmap files were subsequently converted to CpG report text files for methylKit analysis using a custom python script.
Differential methylation analyses were performed using the methylKit R package v1.8.1 (Akalin et al., 2012). Briefly, we removed CpGs in the 99.5th percentile of coverage for each sample to remove potential PCR duplicates. For the EST differential methylation analysis (38.9 vs. 37.8°C), CpGs were retained if they were covered in at least 25 samples per EST treatment over all CO2 treatments. A logistic regression model was fitted for each CpG accounting for the covariates of CO2 level and sample mean CpG methylation rate to test if EST had a significant effect on the log odds ratio of the CpG methylation rate. For CO2 differential methylation analyses, CpGs were retained if they were covered in at least 16 samples per CO2 treatment over both EST treatments. Three different analyses were performed to contrast the three different CO2 treatment pairs (0.4 vs. 0.1%, 0.8 vs. 0.4%, and 0.8 vs. 0.1% CO2). Again, a logistic regression model was fitted for each CpG, accounting for covariates of EST and sample mean CpG methylation rate, and testing for the effect of CO2 level. A CpG site was considered significantly differentially methylated (DM) if the difference in methylation rate between treatments was greater than 10% and the corresponding q-value was less than 0.05 in both EST and CO2 comparisons.
CpG Annotation
All analyzed CpGs were annotated with respect to overlap with genes using the genomation R package v1.14.0 (Akalin et al., 2015). We defined promoter CpGs as those within 2 kb upstream or 200 bp downstream of a gene transcription start site (TSS). Intragenic CpGs were defined as CpGs within any other region of a gene, while all remaining CpGs were classified as intergenic. Promoter- and intragenic-DM genes (DMGs) were defined as those with a DM promoter and intragenic CpG, respectively.
To determine if DMGs were disproportionately involved in certain biological processes, gene lists were submitted for gene set enrichment analysis (GSEA) using the Panther database (Thomas et al., 2003, 2006). We submitted promoter- and intragenic-DMG lists separately, and a background list composed of all genes with CpGs covered by our RRBS data was applied. We considered GO terms significantly enriched at FDR < 0.05.
Motif Enrichment Analysis
We extracted 100 bp genomic sequences centered around EST hyper- and hypomethylated CpGs, as well as 100 bp sequences at 10 k random CpGs included in the DM analysis to use as control sequences. Sequences were uploaded to the MEME suite (Bailey et al., 2009; McLeay and Bailey, 2010) and the Analysis of Motif Enrichment (AME) tool was applied to identify enriched motifs in the Vertebrates (in vivo and in silico) database among our hyper- and hypomethylated sequences. We utilized the average odds score scoring method and Fisher’s exact test to calculate motif enrichment. Motifs were considered significantly enriched at adjusted p-value < 0.05 and E-value < 0.05. We identified motifs that were uniquely enriched among hyper- or hypomethylated sequences, as well as their corresponding TF. The genes encoding these TFs were submitted to GSEA using the PANTHER database to identify enriched GO terms.
Epigenome-Wide Association Study
We performed an EWAS to identify significant associations between CpG methylation rates and HW. CpG sites with a methylation rate variance of at least 1 across samples with coverage ≥10 were retained for further analysis. For each CpG site, a linear model was fitted with methylation rate as a response variable and including the fixed effects of sample mean methylation rate and CO2 level. Residual methylation rates were extracted and used for subsequent association analyses. For each CpG site, a linear model was fitted with log2-transformed HW as a response variable, and CO2 concentration, sample mean methylation rate, and CpG methylation rate as fixed effects. Analysis of variance was performed to assess the effect of the CpG methylation rate on HW, and this process was repeated for all sites. Associations were considered significant at p < 1e-5.
Quantitative Reverse-Transcription PCR
We performed quantitative reverse-transcription PCR (RT-qPCR) on eight DMGs (TBX1, TBX4, THRSP, PITX2, ABLIM2, NGB, GSC, and DAG1). Total RNA from six samples per EST treatment was reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen). Custom primers were designed for the eight DMGs and two stably methylated genes (MEAF6 and SRRM1), as well as for YWHAZ and RPL13 which have been cited as suitable reference genes in chicken heart (Hassanpour et al., 2018; Supplementary Table S1). All assays were performed in singleton on a QuantStudio 5 Real-Time PCR System (Thermo Fisher) using 3.75 ul cDNA template (10 ng total), 1.25 ul of forward and reverse primers, and 6.25 ul MESA BLUE qPCR Master Mix (Eurogentec). Cycling conditions were 50°C for 2 min. and 95°C for 10 min., followed by 40 cycles of 95°C for 15 s and 60°C for 1 min., followed by a melt curve stage of 95°C for 15 s and 60°C for 1 min. and a dissociation step of 95°C for 15 s. Delta Cts (dCts) were obtained for each gene per sample by subtracting the test gene Ct from the geometric mean of the reference gene Cts, and analyses of variance were performed to assess the significance of EST on dCts correcting for sample CO2 concentrations. Fold changes in abundance at 38.9°C relative to 37.8°C were calculated using the 2^-ddCt method.
Results
Broiler Chicks Differ Significantly in HW Between ESTs
We measured HW in all broiler chicks 6 h post-hatch. Chicks incubated at 38.9°C exhibited significantly lower HW relative to 37.8°C chicks (Figure 1; F = 37.53, p = 3.19E-08). There was no significant effect of CO2 concentration (F = 0.254, p = 0.62) or EST-CO2 interaction (F = 0.257, p = 0.61) on HW. When correcting for the significantly shorter incubation time of 38.9°C chicks (F = 52.79, p = 2.18E-10), the difference in HW between ESTs remained significant (F = 12.75, p = 6.05E-04). These results recapitulate the well-established negative association between incubation EST and HW that has been found in numerous studies (Leksrisompong et al., 2007; Molenaar et al., 2010, 2011; Maatjens et al., 2014, 2016), and provided strong support for the assessment of associated differences in cardiac methylation between ESTs.
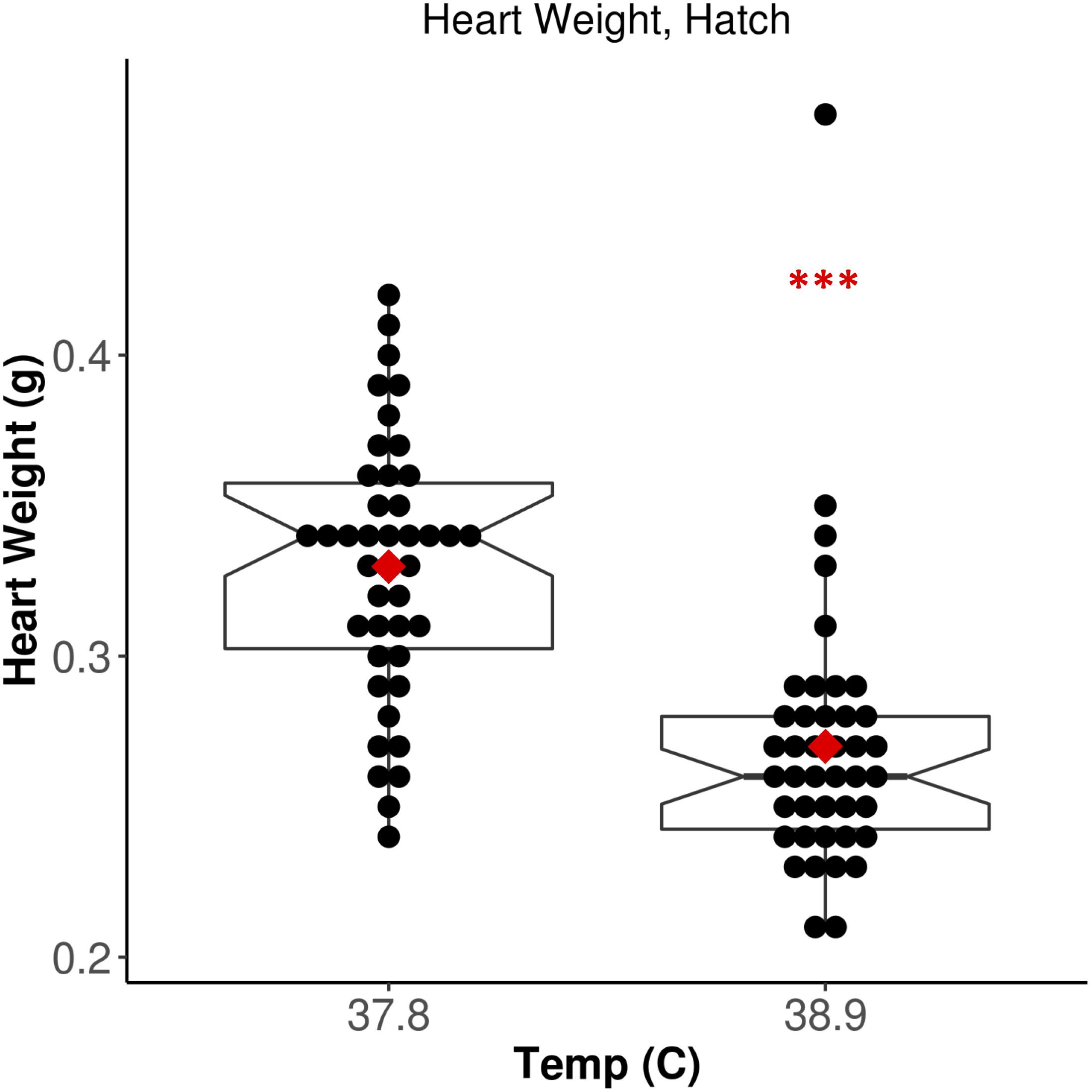
Figure 1. Increased EST is associated with significantly lower heart weight. Notched horizontal lines and red dots indicate treatment median and mean values, respectively. ***p < 0.001.
RRBS Reads Capture Predominantly Lowly Methylated Regions of the Chicken Genome
An average of 18.9 million RRBS reads were generated per sample, of which 18.3 million remained after trimming (Table 1). Mapping of RRBS reads to the G. gallus reference genome resulted in an average unique alignment rate of 73.8%, and an average of 3.1 million CpGs covered at a read depth ≥10X. Global CpG methylation rates across all samples were low on average (17.7%), likely due in part to the fact that RRBS targets CpG-rich regions of the genome which are lowly methylated (Meissner et al., 2008). Average CHG and CHH methylation rates were 0.55% and 0.56%, respectively; given the inherently low prevalence of methylation in non-CpG contexts in vertebrates (Ziller et al., 2011), this suggests high bisulfite conversion efficiency of sequenced reads across samples.
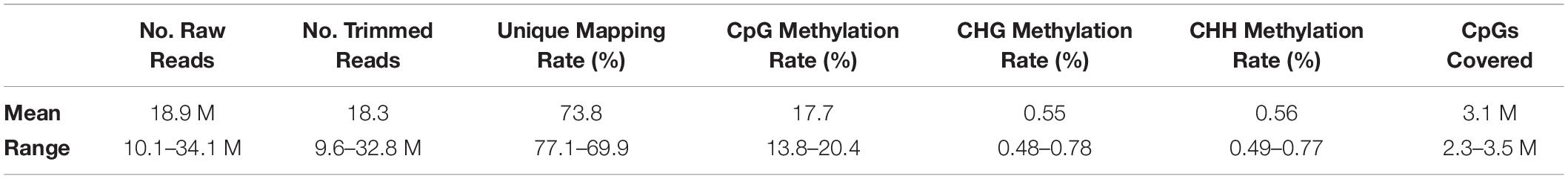
Table 1. Summary of RRBS mapping.
A total of 1,874,588 CpG sites were shared across at least 25 samples per EST treatment, and the majority of these (40.56%) were within gene promoters. 35.18% of CpGs were within gene bodies (25.67% in introns and 12.51% in exons), while 21.26% were intergenic. Promoter CpGs exhibited the lowest methylation rate on average (7.76%), with 85% of sites having an average methylation rate less than 10% (Supplementary Figure S1A). The percentage of lowly methylated (<10%) CpGs in exons, introns, and intergenic regions was significantly lower – 42.74%, 54.52%, and 51.77%, respectively (Supplementary Figures S1B–D). The low methylation observed at a majority of CpGs indicates that many loci covered by our RRBS data are not dynamically methylated in response to the applied environmental conditions, but remain relatively unmethylated regardless of EST or CO2 treatment.
EST Impacts the Methylation State of Genes Involved in General and Heart-Specific Developmental Processes
We identified a total of 2,175 DM CpGs between EST treatments, of which 1121 were hypermethylated and 1054 hypomethylated at 38.9°C relative to 37.8°C (Figure 2 and Supplementary Table S2). Differential methylation was more likely to occur outside of promoter regions; of the 760,332 promoter CpGs tested, only 285 (0.05%) were DM, while 0.14%, 0.18%, and 0.15% of exonic, intronic, and intergenic CpGs, respectively, were DM. In total 269 genes were promoter-DM (promoter-DMGs; 123 hyper- and 146 hypomethylated), while 949 genes were intragenic-DM (intragenic-DMGs; 526 hyper- and 423 hypomethylated).
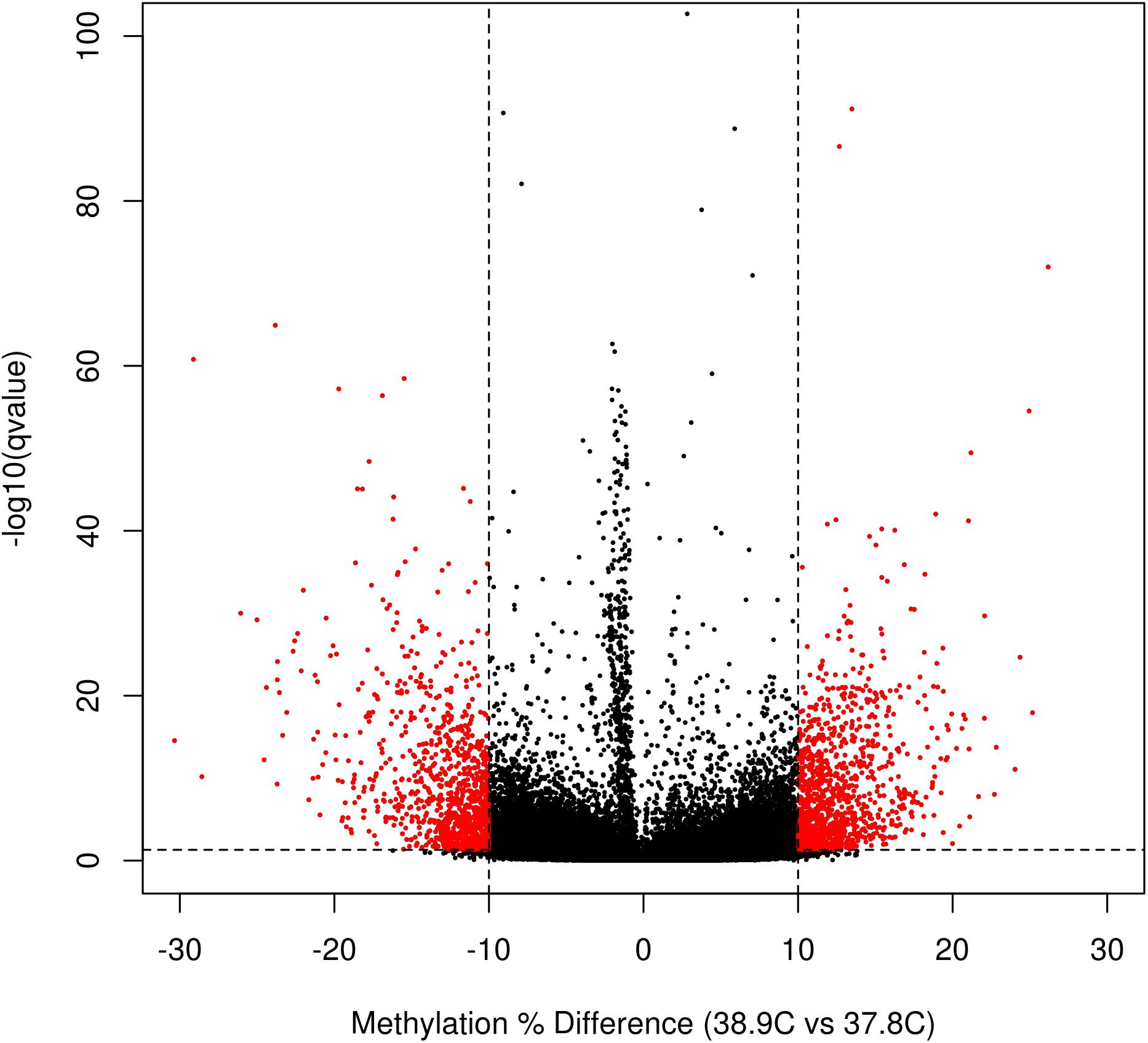
Figure 2. Volcano plot of CpG differential methylation between incubation ESTs. Horizontal line indicates the q-value threshold for significance (0.05), and vertical lines indicate methylation difference threshold for significance (10% difference between EST treatments).
DM gene lists were submitted to Panther for GSEA, and we identified uniquely enriched biological processes among different gene sets (Supplementary Table S3). We did not identify enriched GO terms when considering hyper- and hypomethylated promoter-DMGs separately, however, when combining the two groups we identified enrichment of the terms “Multicellular Organism Development,” “Animal Organ Development,” and “Anatomical Structure Development” (Table 2). Among promoter-DMGs, several are known to play roles in heart development, including T-box transcription factors 1 and 4 (TBX1 and TBX4) that were hypomethylated at 38.9°C, and two genes involved in thyroid hormone signaling: thyroid hormone receptor alpha (THRA) and thyroid hormone responsive (THRSP), which were hypo- and hypermethylated, respectively (Supplementary Figures S2A,B). As promoter methylation is generally inversely correlated with gene expression, hypomethylation of these gene promoters at 38.9°C is indicative of increased potential for gene activation, while hypermethylation suggests decreased activation.
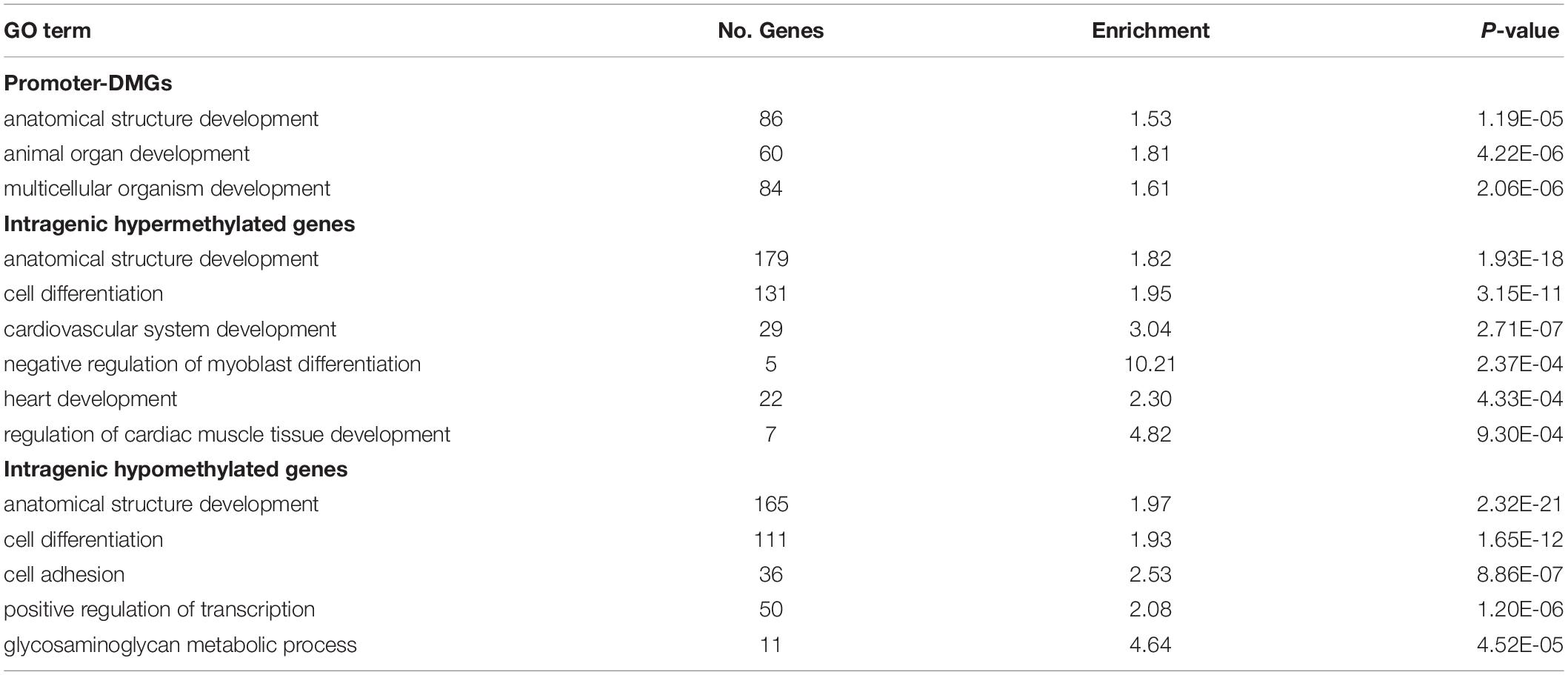
Table 2. Enriched GO terms among DMGs between EST treatments.
Intragenic-DMGs were enriched for processes related to cardiac muscle development (Table 2). Specifically, hypermethylated intragenic-DMGs were enriched for GO terms such as “Heart Development,” “Regulation of Myoblast Differentiation,” and “Cardiovascular System Development,” while hypomethylated intragenic-DMGs were enriched for terms including “Cell Differentiation,” “Cell Adhesion,” and “Glycosaminoglycan Metabolic Process.” Among genes associated with these GO terms are many involved in cardiomyocyte proliferation and differentiation, including: dystroglycan 1 (DAG1, hyper- and hypomethylated; Supplementary Figure S2C), paired like homeodomain 2 (PITX2, hypomethylated; Supplementary Figure S2D), and Erb-B2 receptor tyrosine kinase 2 (ERBB2, hypomethylated) and 4 (ERBB4, hyper- and hypomethylated). Associations between intragenic methylation and gene expression are context-specific (Varley et al., 2013). Nevertheless, temperature-associated differential methylation within genes involved in heart development demonstrates the potential for corresponding differences in gene expression.
EST Differential Methylation Impacts Binding Sites for TFs Involved in Cell Proliferation
As methylation is known to affect gene regulation through the alteration of TF binding sites, we sought to identify enriched motifs in the vicinity of DM CpGs between EST treatments. We submitted 100-bp sequences centered on the 2,175 DM CpGs for motif enrichment analysis, and were specifically interested in identifying motifs that were uniquely enriched among hyper- or hypomethylated sequences. We identified 71 motifs for 49 TFs uniquely enriched among hypermethylated regions, and 120 motifs for 81 TFs enriched among hypomethylated regions (Supplementary Table S4). Among the most uniquely enriched binding motifs for hypermethylated regions were numerous members of the zinc finger and BTB domain containing (ZBTB) family (ZBTB7C, ZBTB7A, ZBTB7B, and ZBTB49), many of which have been shown to negatively regulate cell proliferation (Jeon et al., 2014; Liu et al., 2014). Among hypermethylated regions, the most uniquely enriched binding motif was for musculin (MSC), which functions as a repressor of the myogenic factor MYOD (Zhao and Hoffman, 2006).
We submitted the gene IDs for TFs of hyper- and hypomethylated motifs to the PANTHER database for GO enrichment analysis. Uniquely enriched terms among TFs of hypermethylated motifs were not directly related to heart development or function; however, TFs of hypomethylated motifs were uniquely enriched for the GO terms “cardiovascular system development” and “positive regulation of cell population proliferation” (Table 3). Genes associated with these terms included several with known roles in positively regulating cardiomyocyte proliferation, including retinoic acid receptor alpha (RARA) (Xavier-Neto et al., 2015), T-box transcription factor 20 (TBX20) (Lu et al., 2017), lymphoid enhancer binding factor 1 (LEF1) (Ye et al., 2019) and PITX2, which was also found to be hypomethylated at 38.9°C. Overall, these results reveal the potential for decreased binding ability (via hypermethylation) of TFs involved in negatively regulating cell proliferation, and increased binding ability (via hypomethylation) of TFs involved in positively regulating cardiomyocyte proliferation at 38.9 vs. 37.8°C.
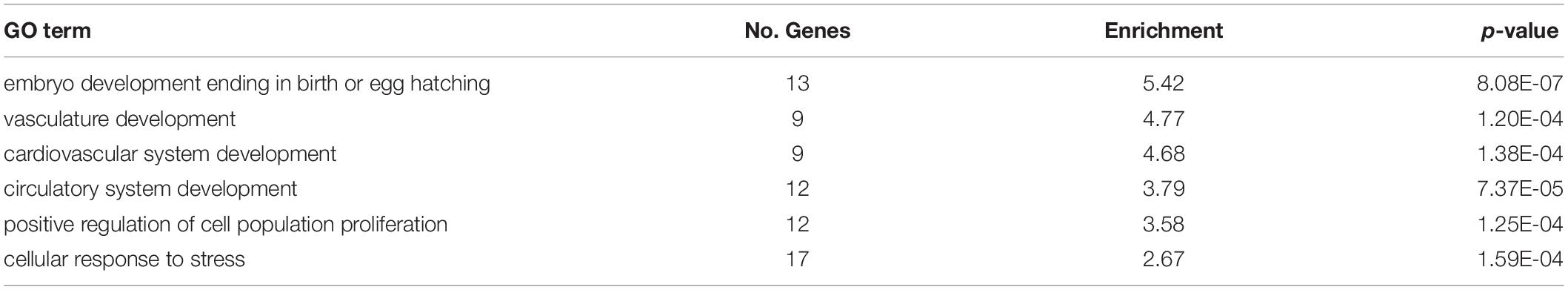
Table 3. Uniquely enriched GO terms among TFs of hypomethylated motifs.
CO2 Differential Methylation Impacts Genes Involved in Heart Development and Response to Hypoxia
We identified a total of 9,823 DM CpG between at least one pair of CO2 treatments. Among these, 3,652 were DM between 0.4 and 0.1% CO2 chicks, 4,695 between 0.8 and 0.4% CO2, and 4,482 between 0.8 and 0.1% CO2 (Table 4 and Supplementary Table S5). In each contrast, the higher CO2 treatment was associated with a higher proportion of hypermethylated CpGs, especially when comparing 0.8% CO2 to the other treatments.

Table 4. Summary of CO2 differential methylation analyses.
DM genes were enriched for more heart- and muscle-specific GO terms when contrasting 0.8% CO2 to either of the other two CO2 treatments (Table 5 and Supplementary Table S6) Among hypermethylated intragenic-DMGs for each of the three contrasts, 18 such GO terms were enriched among the 0.8 vs. 0.1% contrast, while only five and six terms were enriched at 0.4 vs. 0.1% and 0.8 vs. 0.4%, respectively. Among hypomethylated intragenic-DMGs, the 0.8 vs. 0.4% CO2 contrast yielded the greatest number of enriched heart/muscle-related terms (n = 23), while the 0.4 vs. 0.1% and 0.8 vs. 0.1% yielded only three and seven, respectively. Intragenic-DMGs at 0.8% CO2 vs. 0.4% and 0.1% included: PDLIM7, whose resulting protein regulates valve annulus size and hemostasis (Krcmery et al., 2013); TGFB1, which regulates postnatal cardiomyocyte differentiation (Engelmann et al., 1992); and ILK, which has been shown to exhibit protective effects against cardiomyopathy (Hannigan et al., 2007). Additionally, we also identified an enrichment of DMGs associated with response to low oxygen only when comparing 0.8 to 0.1% CO2 (Table 5). Hypomethylated intragenic-DMGs were enriched for the GO terms “response to decreased oxygen levels” and “response to hypoxia,” and these were not enriched among the DMGs in the other two contrasts.
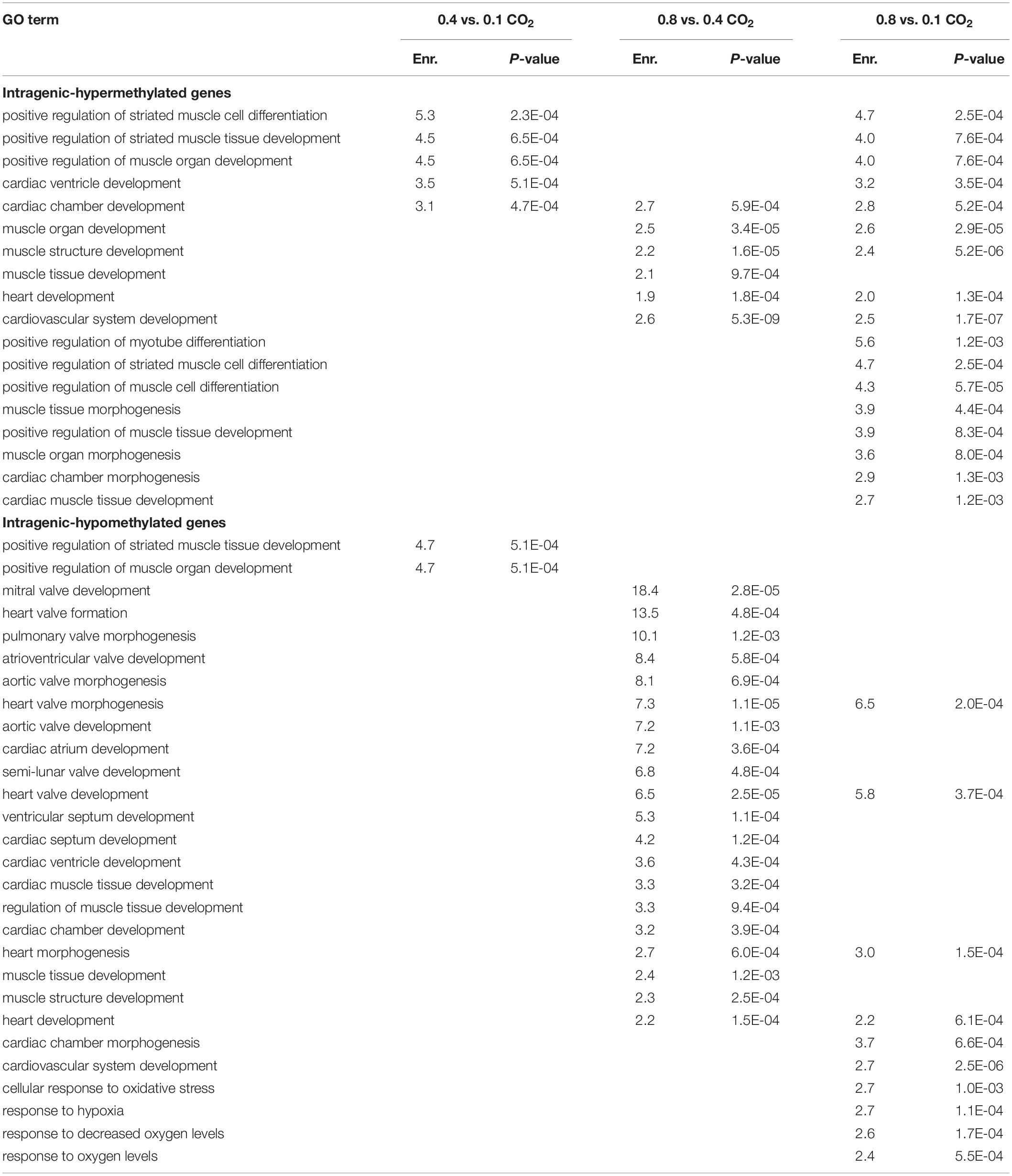
Table 5. Enriched GO terms among CO2 intragenic-DMGs related to heart/muscle development and hypoxic response.
EWAS Identifies Methylation Signatures Significantly Associated With HW
Given the significant difference in HW between chicks of different incubation ESTs, we sought to identify CpG sites at which methylation rate was significantly associated with HW. We tested 1,521,346 CpG sites and identified 23 significant associations (Figure 3 and Supplementary Table S7), which represented a modest enrichment in significant p-values compared to a random distribution (Supplementary Figure S3). Three of these CpGs were within gene promoter regions (in GFI1, SP9, and ST6GALNAC4), 13 were within gene bodies (exons or introns), and seven were in intergenic regions.
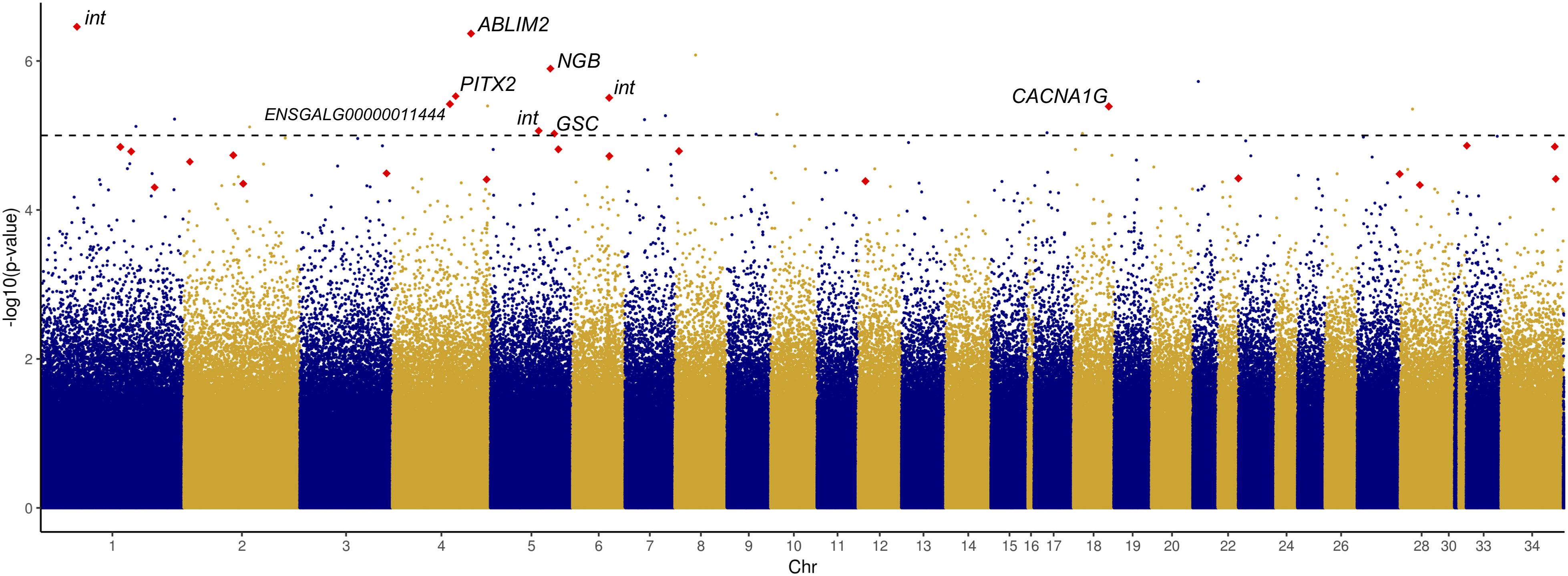
Figure 3. Manhattan plot displaying –log10 p-values of association between CpG methylation rate and log2-transformed relative heart weight, ordered by chromosome. Red points indicate DM CpGs; significantly associated and DM CpGs are labeled with their corresponding gene ID (int, intergenic).
Among the 23 significantly associated CpG sites, nine were also significantly DM between EST treatments, and only one site – the hypomethylated CpG in PITX2 – had a methylation difference greater than 10%. This site was found to be significantly positively correlated with HW (r = 0.60, p = 2.97E-06), along with four other sites: two exonic CpGs in neuroglobin (NGB) and goosecoid homeobox (GSC), an intronic CpG in calcium voltage-gated channel subunit alpha1 G (CACNA1G), and an intergenic CpG. Four hypermethylated CpGs were negatively associated with HW: an exonic CpG in ENSGALG00000011444, an intronic CpG in actin binding LIM protein family member 2 (ABLIM2), and two intergenic CpGs.
RT-qPCR Identifies Genes With Coordinated Differences in Gene Methylation and Expression Between EST Treatments
We assessed transcript abundance of eight genes exhibiting differential methylation and/or significant associations with HW via RT-qPCR, to determine if differential methylation was associated with differences in gene expression. We detected quantifiable transcript abundance for all genes except NGB, which was undetectable in multiple samples in both EST treatments. Three genes – ABLIM2, PITX2, and THRSP – exhibited significant differences in transcript abundance between EST treatments (Figure 4). In all three cases, abundance was higher in the treatment with lower methylation. We observed significantly lower THRSP abundance in 38.9°C chicks (p = 0.046), which is consistent with the promoter hypermethylation observed. Abundance of PITX2, which contained a hypomethylated CpG in its last exon, was significantly higher in 38.9°C chicks (p = 7.59E-03). ABLIM2, containing a hypermethylated intronic CpG, exhibited lower abundance at 38.9°C (p = 1.37E-03). Among the remaining five DMGs, the differences in expression were negatively associated with the differences in methylation for DAG1, while the differences were positively associated for TBX1, TBX4, and GSC. We also assessed transcript abundance in two genes that did not exhibit differential methylation (MEAF6 and SRRM1), and did not detect differences in abundance between ESTs. These results have identified candidate genes exhibiting coordinated differences in methylation and expression in the developing chick heart that are associated with incubation EST.
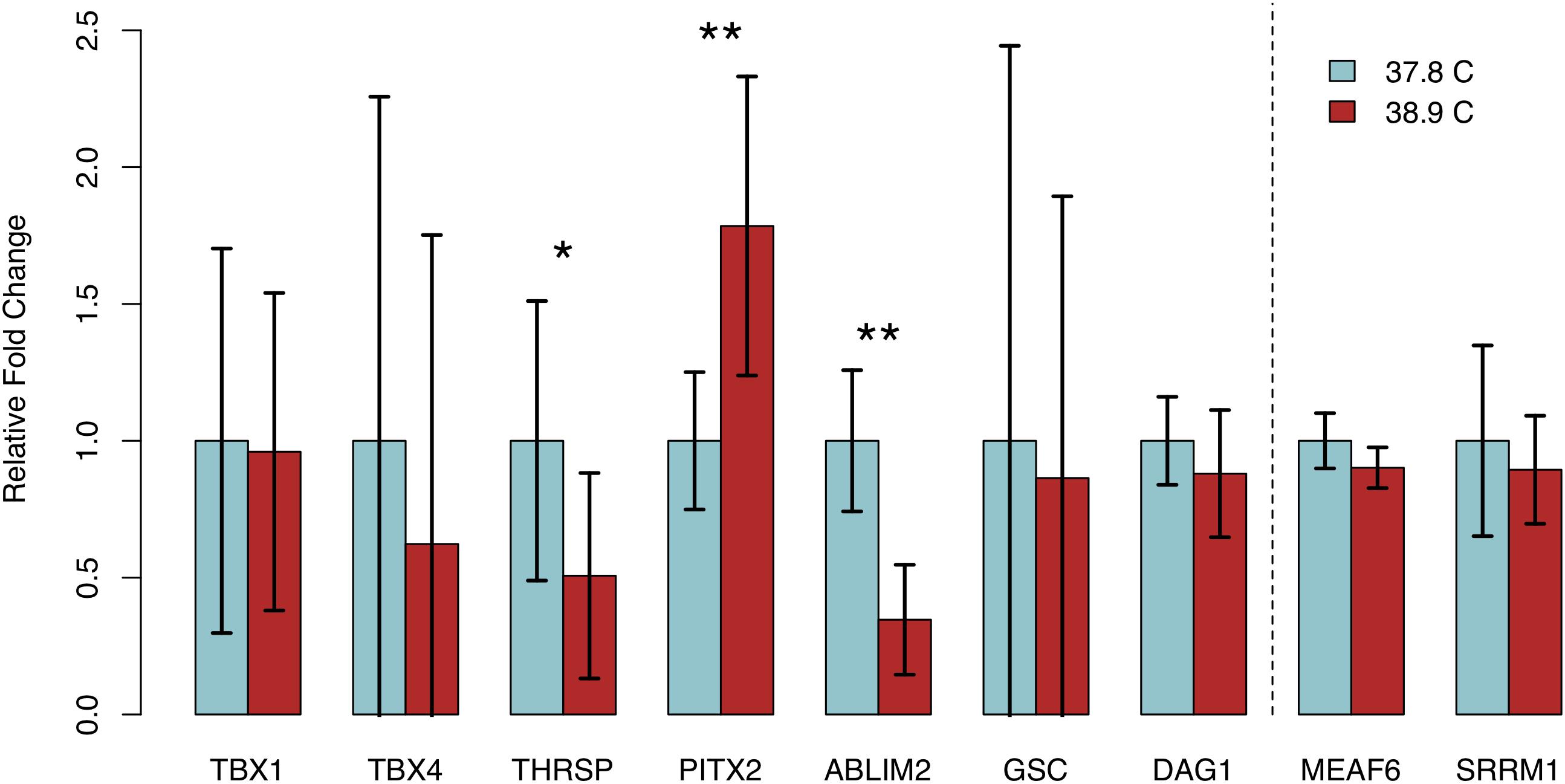
Figure 4. Relative fold changes in transcript abundance (38.9°C relative to 37.8°C) for seven expressed genes exhibiting differential methylation and two stably methylated genes between temperature treatments. Error bars indicate standard deviation. *p < 0.05, **p < 0.01.
Discussion
To the best of our knowledge, this study represents the largest epigenome-wide analysis in livestock species, the first EWAS in broiler chickens, and the first study assessing genome-wide DNA methylation between chickens incubated in different environments. RRBS was performed in cardiac tissue collected from broiler chicks in a study that recapitulated the established negative correlation between incubation EST and HW. We observed significantly lower HW in chicks incubated at 38.9 vs. 37.8°C, which was independent of differences in incubation length. The heart appears to be uniquely impacted by incubation EST, a finding that has been observed in numerous studies showing up to 30% lower HW in 38.9°C chicks with no significant differences in organs such as the liver, intestines or stomach (Molenaar et al., 2011; Maatjens et al., 2014, 2016; Nangsuay et al., 2016).
Using RRBS, we assessed methylation at over 3 million CpGs across 84 heart samples. Global methylation rates in these samples were low (13.8–20.4%), due to the fact that a majority of sites exhibited methylation rates less than 10%. RRBS reads are disproportionally derived from CpG dense regions of the genome, which are known to be less methylated than the genome at large (Meissner et al., 2008). However, RRBS data from eight pig tissues revealed an average methylation rate of 41% (Schachtschneider et al., 2011). Thus the low methylation observed in our data may be specific to the species, tissue, or stage of development. To date, whole-genome bisulfite sequencing studies in G. gallus and other avian species have reported global CpG methylation rates between 50 and 65% (Li et al., 2015; Derks et al., 2016; Lee et al., 2017; Zhang et al., 2017, 2018), which is lower than the 60–80% that has been reported in mammalian studies. It is possible that the low levels of CpG methylation detected in our data can be attributed to lower CpG methylation rates in avian genomes.
We identified thousands of DM CpGs between ESTs, and annotation of DMGs revealed an enrichment for general and heart-specific developmental processes. While promoter-DMGs were only significantly enriched for processes related to general development, many of these genes are known to be involved in heart development. Among these were two T-box transcription factors, TBX1 and TBX4, that were promoter-hypomethylated at 38.9°C. TBX1 has been shown to promote cell proliferation and inhibit differentiation of heart cells (Xu et al., 2004; Chen et al., 2009). TBX4 has been shown to be expressed in the developing heart, and has been hypothesized to play a role in regulating tissue-specific gene expression (Krause et al., 2004). Also among promoter-DMGs were two genes involved in response to thyroid hormone (THRA, hypomethylated and THRSP, hypermethylated at 38.9°C), which has previously been hypothesized to mediate temperature-driven differences in heart growth. THRA is the major thyroid hormone receptor expressed in the heart (Gloss et al., 2001), and THRA mutations in zebrafish have been linked to defects in heart development (Marelli et al., 2017). THRSP is expressed in chicken cardiac tissue (Ren et al., 2017), and research has shown that overexpression of THRSP results in decreased levels of genes involved in heart development (Wellberg et al., 2014). Intragenic-DMGs were enriched for GO terms related to heart development. Among these, DAG1 possessed hypo- and hypermethylated CpGs, and encodes a glycoprotein that has been shown to play an important role in the inhibition of cardiomyocyte proliferation in mice (Morikawa et al., 2017). PITX2 contained an exonic CpG that was significantly hypomethylated at 38.9°C. This gene encodes a homeobox TF with roles in heart development that have been reviewed extensively (Franco et al., 2017); of note, it has been shown to promote cardiomyocyte proliferation in mice (Tao et al., 2016). ERBB2 and ERBB4 encode receptor kinases that have been shown to promote cardiomyocyte proliferation and survival (Zhao et al., 1998). The observed differential epigenetic regulation of genes involved in cardiomyocyte proliferation and differentiation may be contributing to observed differences in HW between chicks of different incubation ESTs.
Furthermore, assessment of enriched binding motifs among DM loci provides evidence that temperature-differential methylation is impacting the developmental trajectories of the chick heart. Methylation is known to reduce the binding potential of TFs by altering binding motif sequences (Yin et al., 2017) At 38.9°C, hypomethylated binding motifs – which would be predicted to be more accessible due to lower methylation levels – were enriched for TFs involved in positive regulation of cardiomyocyte proliferation. Conversely, the binding sites for TFs involved in negative regulation of cell proliferation – namely TFs in the ZBTB family – were the most enriched motifs among hypermethylated regions, which would be expected to be less accessible. The more favorable binding of pro-proliferation TFs in the cardiac methylome of chicks at higher temperatures suggests that observed differences in HW may be associated with differences in rates of differentiation and maturation.
We also identified thousands of DM CpGs between CO2 treatments. Increasing CO2 concentrations above 0.8% during late stages of incubation have benefits in commercial settings such as reduced hatching time variability (van Roovert-Reijrink, 2016). However, continuous incubation in a hypercapnic environment has been shown to increase ascites incidence later in life (Hassanzadeh et al., 2002). Not only did differential methylation at 0.8% CO2 relative to the lower treatments disproportionately affect genes involved in heart development, but contrasting 0.8% with 0.1% CO2 revealed differential methylation of genes that were enriched for hypoxia response GO terms. Such DMGs associated with hypoxia included: ETS1, a TF that has been shown to activate hypoxia-inducible genes (Salnikow et al., 2008); SRC, encoding a tyrosine kinase that is upregulated in rat cardiomyocytes by hypoxia to activate MAPK signaling pathways (Seko et al., 1996); and DNM2, encoding a GTPase and actin-binding protein that has been shown to be downregulated in cardiomyocytes subject to hypoxic conditions (Shi et al., 2014). Hypoxia-inducible genes being specifically overrepresented when comparing extremes for CO2 indicate a cardiac response to differences in gas exchange via differences in gene regulation. Incubation CO2 level was not associated with differences in HW in this study, which has been shown previously (Maatjens et al., 2014). However, CO2 concentration may have observable effects on physiological processes that were not assessed here – including angiogenesis – that may contribute to adverse conditions later in life including increased ascites risk.
Our EWAS revealed a small but significant enrichment of association p-values between CpG methylation and HW, suggesting a relationship between gene regulation in the heart and its size at hatch. At the 23 significantly associated CpGs, epigenetic regulation of gene expression by DNA methylation may directly or indirectly affect heart growth. A DM CpG in an exon of PITX2 was also the most significantly positively correlated with HW. We identified additional significant CpGs in our EWAS that were also DM, albeit at a difference less than 10%. An intragenic CpG in ABLIM2 was significantly hypermethylated at 38.9°C and negatively correlated with HW; this gene encodes a actin binding protein that has been shown to be highly expressed in striated muscle and is thought to regulate cytoskeletal signaling pathways (Barrientos et al., 2007). Additionally, an intergenic CpG in NGB was significantly hypomethylated and positively correlated with HW. The gene, encoding neuroglobin, was found to be differentially expressed in response to water temperature in Whiteleg shrimp (Wu et al., 2017). However, we did not detect NGB transcript abundance at appreciable levels in our samples, suggesting a limited role at this stage in chick heart development and temperature response. CpGs in these genes, along with those identified in other genes and in intergenic regions, are strong candidates for identifying epigenetic-mediated associations between temperature and heart development.
We identified coordinated changes in methylation and expression for three of the eight tested genes: ABLIM2, PITX2, and THRSP. In all three cases, direction of the difference in gene expression was opposite that of the difference in methylation between ESTs. This inverse relationship was expected at THRSP, since the DM CpG was immediately upstream of the TSS in the likely promoter region. Intragenic methylation in ABLIM2 and PITX2 were also negatively associated with expression, and this is in contrast to findings in other animal studies that the majority of CpGs in such regions are positively correlated with gene expression. Cases of intragenic methylation being negatively associated with gene expression have been reported by ENCODE (Varley et al., 2013), and they found these CpGs to be enriched in intragenic enhancers. The presence of CpGs in ABLIM2 and PITX2 at which methylation rate is inversely correlated with gene expression suggests that these CpGs may be located within gene regulatory elements. To explore this idea, we searched for binding motifs in the genomic regions flanking in DM CpGs in ABLIM2 and PITX2. The PITX2 DM regions had strong sequence similarity to the Kruppel Like Factor 5 (KLF5) binding motif, and this TF has been shown to be upregulated during heat stress in chickens previously (Sun et al., 2015). Additionally, the ABLIM2 DM regions showed strong sequence similarity to the binding motif of Achaete-Scute Family BHLH Transcription Factor 2 (ASCL2), which is a known inhibitor of myogenic differentiation (Wang et al., 2017). While differential expression was not observed for five of the eight tested DMGs, this could be due in part to the limited number of samples that were assayed via qPCR (n = 6/trt) relative to the number assayed via RRBS (n = 42/trt).
Among these three DM and differentially expressed genes, PITX2 has the most well-established role in heart development, specifically in promoting cardiomyocyte proliferation. In this study, increased EST was associated with both decreased methylation at an exonic CpG in PITX2 as well as increased PITX2 transcript abundance. As PITX2 is known to promote cardiomyocyte proliferation, this suggests an increased proliferative capacity in the hearts of 38.9°C chicks. This conflicts with results reported in Romanoff, 1960, in which increased incubation temperature was associated with a reduced mitotic index from E8 to hatch. However, our results indicate that proliferation in high EST chicks may be increased relative to lower EST chicks as a partial compensatory mechanism immediately following hatch, and that developmental trajectories of hearts at different ESTs continue to be affected even after incubation.
In conclusion, this study has identified differential methylation patterns in the post-hatch chick heart associated with differences in incubation EST and CO2 level, and such differences may be impacting heart growth and development through associated changes in TF binding and gene expression. Future studies should seek to further assess differences in methylation between incubation treatments at later stages of post-hatch life. Additionally, characterizing epigenetic patterns in additional organs responsible for regulating body temperature, including the hypothalamus and thyroid gland, may help in characterizing the mechanisms regulating temperature-driven differences in heart development. Knowledge of such epigenetic signatures influenced by early life environment may benefit animal breeding by serving as predictors of future animal performance.
Data Availability Statement
The RRBS data is available by the following SRA/ENA accession number PRJEB39999.
Ethics Statement
The animal study was reviewed and approved by the Central Animal Experiments Committee (The Hague, Netherlands; approval no. 2016.W-0087).
Author Contributions
OM, MG, and RPC conceived of the study design. HB conducted the incubation study from which heart samples were collected. RJC performed all of the bioinformatics, differential methylation, GO enrichment analyses, as well as the EWAS, and wrote the manuscript. MP and CE contributed intellectually to the interpretation of results. All authors read and approved of the manuscript.
Funding
This study was financially supported by the Dutch Ministry of Economic Affairs (TKI-Toeslag) and the Breed4Food partners Cobb Europe, CRV, Hendrix Genetics, and Topigs Norsvin. RJC was funded under a USDA NIFA NNF Training Grant (#2015-38420-23697). The authors declare that the funders Breed4Food partners Cobb Europe, CRV, Hendrix Genetics and Topigs Norsvin were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Breed4Food EWAS committee for useful discussion during the project and Bert Dibbits for technical laboratory support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.558189/full#supplementary-material
Footnotes
References
Akalin, A., Franke, V., Vlahoviček, K., Mason, C. E., and Schübeler, D. (2015). Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 31, 1127–1129. doi: 10.1093/bioinformatics/btu775
Akalin, A., Kormaksson, M., Li, S., Garrett-Bakelman, F. E., Figueroa, M. E., Melnick, A., et al. (2012). MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13:R87. doi: 10.1186/gb-2012-13-10-R87
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Ball, M. P., Li, J. B., Gao, Y., Lee, J.-H., Leproust, E., Park, I.-H., et al. (2009). Targeted and genome-scale methylomics reveals gene body signatures in human cell lines HHS Public access author manuscript. Nat. Biotechnol. 27, 361–368. doi: 10.1038/nbt.1533
Barrientos, T., Frank, D., Kuwahara, K., Bezprozvannaya, S., Pipes, G. C. T., Bassel-Duby, R., et al. (2007). Two novel members of the ABLIM protein family, ABLIM-2 and -3, associate with STARS and directly bind F-actin. J. Biol. Chem. 282, 8393–8403. doi: 10.1074/jbc.M607549200
Bird, A. P., and Wolffe, A. P. (1999). Methylation-induced repression-belts, braces, and chromatin. Cell 99, 451–454. doi: 10.1016/S0092-8674(00)81532-81539
Buys, N., Dewil, E., Gonzales, E., and Decuypere, E. (1998). Avian pathology different CO2 levels during incubation interact with hatching time and ascites susceptibility in two broiler lines selected for different growth rate. Avian. Pathol. 27, 605–612. doi: 10.1080/03079459808419391
Chen, L., Fulcoli, F. G., Tang, S., and Baldini, A. (2009). Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circ. Res. 105, 842–851. doi: 10.1161/CIRCRESAHA.109.200295
Christensen, V. L., Donaldson, W. E., and Nestor, K. E. (1999). Length of the plateau and pipping stages of incubation affects the physiology and survival of turkeys. Br. Poult. Sci. 40, 297–303. doi: 10.1080/00071669987737
David, S.-A., Vitorino Carvalho, A., Gimonnet, C., Brionne, A., Hennequet-Antier, C., Piégu, B., et al. (2019). Thermal manipulation during embryogenesis impacts H3K4me3 and H3K27me3 histone marks in chicken hypothalamus. Front. Genet. 10:1207. doi: 10.3389/fgene.2019.01207
Derks, M. F. L., Schachtschneider, K. M., Madsen, O., Schijlen, E., Verhoeven, K. J. F., and van Oers, K. (2016). Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics 17:332. doi: 10.1186/s12864-016-2653-y
Engelmann, G. L., Boehm, K. D., Birchenall-Roberts, M. C., and Ruscetti, F. W. (1992). Transforming growth factor-beta1 in heart development. Mech. Dev. 38, 85–97. doi: 10.1016/0925-4773(92)90001-Z
Flanagan, J. M. (2015). Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol. Biol. 1238, 51–63. doi: 10.1007/978-1-4939-1804-1_3
Franco, D., Sedmera, D., and Lozano-Velasco, E. (2017). Multiple roles of Pitx2 in cardiac development and disease. J. Cardiovasc. Dev. Dis. 4:16. doi: 10.3390/jcdd4040016
French, N. A. (1994). Effect of incubation temperature on the gross pathology of turkey embryos. Br. Poult. Sci. 35, 363–371. doi: 10.1080/00071669408417701
Gloss, B., Trost, S. U., Bluhm, W. F., Swanson, E. A., Clark, R., Winkfein, R., et al. (2001). Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor α or β∗∗This work was supported by grant HL-25022 (to W.H.D.) and by operating grants from the Canadian medical research council and the heart and stroke foundation of Canada (to W.G. and R.C.). Endocrinology 142, 544–550. doi: 10.1210/endo.142.2.7935
Greenberg, M. V. C., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-156
Guo, W., Fiziev, P., Yan, W., Cokus, S., Sun, X., Zhang, M. Q., et al. (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 14:774. doi: 10.1186/1471-2164-14-774
Guo, W., Zhu, P., Pellegrini, M., Zhang, M. Q., Wang, X., and Ni, Z. (2018). CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 34, 381–387. doi: 10.1093/bioinformatics/btx595
Hannigan, G. E., Coles, J. G., and Dedhar, S. (2007). Integrin-linked kinase at the heart of cardiac contractility. Repair Dis. Circ. Res. 100, 1408–1414. doi: 10.1161/01.RES.0000265233.40455.62
Hao, Y., Cui, Y., and Gu, X. (2016). Genome-wide DNA methylation profiles changes associated with constant heat stress in pigs as measured by bisulfite sequencing. Sci. Rep. 6:27507. doi: 10.1038/srep27507
Hassanpour, H., Bahadoran, S., Farhadfar, F., Fallahi Chamali, Z., Nazari, H., and Kaewduangta, W. (2018). Identification of reliable reference genes for quantitative real-time PCR in lung and heart of pulmonary hypertensive chickens. Poult. Sci. 97, 4048–4056. doi: 10.3382/ps/pey258
Hassanzadeh, M., Buyse, J., and Decuypere, E. (2002). Further evidence for the involvement of cardiac β-adrenergic receptors in right ventricle hypertrophy and ascites in broiler chickens. Avian. Pathol. 31, 177–181. doi: 10.1080/03079450120118676
Hassanzadeh, M., Maddadi, M. S., Mirzaie, S., Assasie, K., and Moayyedian, H. (2010). Partial pressure of carbon dioxide in the venous blood of young birds as a predictor of ascites susceptibility in broiler chickens. Acta Vet. Hung. 58, 221–230. doi: 10.1556/AVet.58.2010.2.8
Ibeagha-Awemu, E. M., and Zhao, X. (2015). Epigenetic marks: regulators of livestock phenotypes and conceivable sources of missing variation in livestock improvement programs. Front. Genet. 6:302. doi: 10.3389/fgene.2015.00302
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33, 245–254. doi: 10.1038/ng1089
Jeon, B. N., Kim, M. K., Yoon, J. H., Kim, M. Y., An, H., Noh, H. J., et al. (2014). Two ZNF509 (ZBTB49) isoforms induce cell-cycle arrest by activating transcription of p21/CDKN1A and RB upon exposure to genotoxic stress. Nucleic Acids Res. 42, 11447–11461. doi: 10.1093/nar/gku857
Krause, A., Zacharias, W., Camarata, T., Linkhart, B., Law, E., Lischke, A., et al. (2004). Tbx5 and Tbx4 transcription factors interact with a new chicken PDZ-LIM protein in limb and heart development. Dev. Biol. 273, 106–120. doi: 10.1016/J.YDBIO.2004.05.024
Krcmery, J., Gupta, R., Sadleir, R. W., Ahrens, M. J., and Misener, S. (2013). Loss of the cytoskeletal protein pdlim7 predisposes mice to heart defects and hemostatic dysfunction. PLoS One 8:80809. doi: 10.1371/journal.pone.0080809
Kühn, E. R., Decuypere, E., Colen, L. M., Chadwick, A., Heyns, W., and Michels, H. (1984). “59 Orcadian rhythm of Corticosterone, Prolactin and thyroid hormones in serum and endocrine glands of post-hatch chicks, incubated at different temperatures,” in Proceeding of the 15th International Conference of the International Society for Chronobiology 1982-1983, Minneapolis, MI.
Lee, I., Rasoul, B. A., Holub, A. S., Lejeune, A., Enke, R. A., and Timp, W. (2017). Whole genome DNA methylation sequencing of the chicken retina, cornea and brain. Sci. Data 4:170148. doi: 10.1038/sdata.2017.148
Leksrisompong, N., Romero-Sanchez, H., Plumstead, P. W., Brannan, K. E., and Brake, J. (2007). Broiler Incubation. 1. Effect of elevated temperature during late incubation on body weight and organs of chicks. Poult. Sci. 86, 2685–2691. doi: 10.3382/ps.2007-2170
Li, J., Li, R., Wang, Y., Hu, X., Zhao, Y., Li, L., et al. (2015). Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genom. 16:851. doi: 10.1186/s12864-015-2098-2098
Liu, X. S., Haines, J. E., Mehanna, E. K., Genet, M. D., Ben-Sahra, I., Asara, J. M., et al. (2014). ZBTB7A acts as a tumor suppressor through the transcriptional repression of glycolysis. Genes Dev. 28, 1917–1928. doi: 10.1101/gad.245910.114
Lorincz, M. C., Dickerson, D. R., Schmitt, M., and Groudine, M. (2004). Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11, 1068–1075. doi: 10.1038/nsmb840
Lourens, A., van den Brand, H., Meijerhof, R., and Kemp, B. (2005). Effect of eggshell temperature during incubation on embryo development, hatchability, and posthatch development. Poult. Sci. 84, 914–920. doi: 10.1093/ps/84.6.914
Lu, F., Langenbacher, A., and Chen, J. N. (2017). Tbx20 drives cardiac progenitor formation and cardiomyocyte proliferation in zebrafish. Dev. Biol. 421, 139–148. doi: 10.1016/j.ydbio.2016.12.009
Maatjens, C. M., Reijrink, I. A. M., Molenaar, R., van der Pol, C. W., Kemp, B., and van den Brand, H. (2014). Temperature and CO2 during the hatching phase. I. Effects on chick quality and organ development. Poult. Sci. 93, 645–654. doi: 10.3382/ps.2013-03490
Maatjens, C. M., Van Roovert-Reijrink, I. A. M., Engel, B., Van Der Pol, C. W., Kemp, B., and Van Den Brand, H. (2016). Temperature during the last week of incubation. I. Effects on hatching pattern and broiler chicken embryonic organ development. Poult. Sci. 95, 956–965. doi: 10.3382/ps/pev447
Marelli, F., Carra, S., Rurale, G., Cotelli, F., and Persani, L. (2017). In vivo functional consequences of human THRA variants expressed in the Zebrafish. Thyroid 27, 279–291. doi: 10.1089/thy.2016.0373
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., Dsouza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
McLeay, R. C., and Bailey, T. L. (2010). Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinform. 11:165. doi: 10.1186/1471-2105-11-165
Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. doi: 10.1038/nature07107
Molenaar, R., Hulet, R., Meijerhof, R., Maatjens, C. M., Kemp, B., and van den Brand, H. (2011). High eggshell temperatures during incubation decrease growth performance and increase the incidence of ascites in broiler chickens. Poult. Sci. 90, 624–632. doi: 10.3382/ps.2010-2970
Molenaar, R., Meijerhof, R., van den Anker, I., Heetkamp, M. J. W., van den Borne, J. J. G. C., Kemp, B., et al. (2010). Effect of eggshell temperature and oxygen concentration on survival rate and nutrient utilization in chicken embryos. Poult. Sci. 89, 2010–2021. doi: 10.3382/ps.2010-2787
Morikawa, Y., Heallen, T., Leach, J., Xiao, Y., and Martin, J. F. (2017). Dystrophin glycoprotein complex sequesters yap to inhibit cardiomyocyte proliferation HHS public access. Nature 547, 227–231. doi: 10.1038/nature22979
Nangsuay, A., Meijerhof, R., Van Den Anker, I., Heetkamp, M. J. W., Morita, V. D. S., Kemp, B., et al. (2016). Effects of breeder age, broiler strain, and eggshell temperature on development and physiological status of embryos and hatchlings. Poult. Sci. 95, 1666–1679. doi: 10.3382/ps/pew080
Nichelmann, M. (2004). Perinatal epigenetic temperature adaptation in avian species: comparison of turkey and Muscovy duck. J. Therm. Biol. 29, 613–619. doi: 10.1016/j.jtherbio.2004.08.032
Orozco, L. D., Morselli, M., Rubbi, L., Guo, W., Go, J., Shi, H., et al. (2015). Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab. 21, 905–917. doi: 10.1016/j.cmet.2015.04.025
Ren, J., Xu, N., Zheng, H., Tian, W., Li, H., Li, Z., et al. (2017). Expression of thyroid hormone responsive SPOT 14 gene is regulated by estrogen in chicken (Gallus gallus). Sci. Rep. 7:10243. doi: 10.1038/s41598-017-08452-8456
Ricklefs, R. E. (1987). Comparative analysis of avian embryonic growth. J. Exp. Zool. Suppl. Publ. 1, 309–323.
Romanoff, A. L. (1960). The Avian Embryo: Structure and Functional Development. New York, NY: Macmillan.
Salnikow, K., Aprelikova, O., Ivanov, S., Tackett, S., Kaczmarek, M., Karaczyn, A., et al. (2008). Regulation of hypoxia-inducible genes by ETS1 transcription factor. Carcinogenesis 29, 1493–1499. doi: 10.1093/carcin/bgn088
Schachtschneider, K. M., Madsen, O., Park, C., Rund, L. A., Groenen, M. A. M., and Schook, L. B. (2011). Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model. BMC Genom. 16:743. doi: 10.1186/s12864-015-1938-x
Seko, Y., Tobe, K., Takahashi, N., Kaburagi, Y., Kadowaki, T., and Yazaki, Y. (1996). Hypoxia and hypoxia/reoxygenation activate Src family tyrosine kinases and p21(ras) in cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 226, 530–535. doi: 10.1006/bbrc.1996.1389
Shi, D., Xie, D., Zhang, H., Zhao, H., Huang, J., Li, C., et al. (2014). Reduction in dynamin-2 is implicated in ischaemic cardiac arrhythmias. J. Cell. Mol. Med. 18, 1992–1999. doi: 10.1111/jcmm.12335
Skibiel, A. L., Peñagaricano, F., Amorín, R., Ahmed, B. M., Dahl, G. E., and Laporta, J. (2018). In utero heat stress alters the offspring epigenome. Sci. Rep. 8:14609. doi: 10.1038/s41598-018-32975-1
Sözcü, A., and Ipek, A. (2015). Acute and chronic eggshell temperature manipulations during hatching term influence hatchability, broiler performance, and ascites incidence. Poult. Sci. 94, 319–327. doi: 10.3382/ps/peu080
Sun, L., Lamont, S. J., Cooksey, A. M., McCarthy, F., Tudor, C. O., Vijay-Shanker, K., et al. (2015). Transcriptome response to heat stress in a chicken hepatocellular carcinoma cell line. Cell Stress Chaper. 20, 939–950. doi: 10.1007/s12192-015-0621-620
Tao, G., Kahr, P. C., Morikawa, Y., Zhang, M., Rahmani, M., Heallen, T. R., et al. (2016). Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123. doi: 10.1038/nature17959
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. doi: 10.1101/GR.772403
Thomas, P. D., Kejariwal, A., Guo, N., Mi, H., Campbell, M. J., Muruganujan, A., et al. (2006). Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 34, W645–W650. doi: 10.1093/nar/gkl229
Tzschentke, B., and Basta, D. (2002). Early development of neuronal hypothalamic thermosensitivity in birds: influence of epigenetic temperature adaptation. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 131, 825–832. doi: 10.1016/S1095-6433(02)00020-X
van Roovert-Reijrink, I. (2016). Incubation Affects Chick Quality. Available online at: https://www.poultryworld.net/Genetics/Articles/2013/5/Incubation-affects-chick-quality-1183725W/ (accessed April 20, 2019).
Varley, K. E., Gertz, J., Bowling, K. M., Parker, S. L., Reddy, T. E., Pauli-Behn, F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567. doi: 10.1101/gr.147942.112
Vinoth, A., Thirunalasundari, T., Shanmugam, M., Uthrakumar, A., Suji, S., and Rajkumar, U. (2018). Evaluation of DNA methylation and mRNA expression of heat shock proteins in thermal manipulated chicken. Cell Stress Chaper. 23, 235–252. doi: 10.1007/s12192-017-0837-832
Wang, C., Wang, M., Arrington, J., Shan, T., Yue, F., Nie, Y., et al. (2017). Ascl2 inhibits myogenesis by antagonizing the transcriptional activity of myogenic regulatory factors. Development 144, 235–247. doi: 10.1242/dev.138099
Wellberg, E. A., Rudolph, M. C., Lewis, A. S., Padilla-Just, N., Jedlicka, P., and Anderson, S. M. (2014). Modulation of tumor fatty acids, through overexpression or loss of thyroid hormone responsive protein spot 14 is associated with altered growth and metastasis. Breast Cancer Res. 16:481. doi: 10.1186/s13058-014-0481-z
Wu, M., Chen, N., Huang, C. X., He, Y., Zhao, Y. Z., Chen, X. H., et al. (2017). Effect of low temperature on globin expression, respiratory metabolic enzyme activities, and gill structure of Litopenaeus vannamei. Biochemistry 82, 844–851. doi: 10.1134/S0006297917070100
Xavier-Neto, J., Sousa Costa, ÂM., Carolina, A., Figueira, M., Donato, C. C., and Neves Do Amaral, F. (2015). Signalling through retinoic acid receptors in cardiac development: doing the right things at the right times HHS public access author manuscript. Biochim. Biophys. Acta 1849, 94–111. doi: 10.1016/j.bbagrm.2014.08.003
Xu, H., Morishima, M., Wylie, J. N., Schwartz, R. J., Bruneau, B. G., Lindsay, E. A., et al. (2004). Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131, 3217–3227. doi: 10.1242/dev.01174
Yahav, S., and Hurwitz, S. (1996). Induction of thermotolerance in male broiler chickens by temperature conditioning at an early age. Poult. Sci. 75, 402–406. doi: 10.3382/ps.0750402
Yahav, S., Straschnow, A., Plavnik, I., and Hurwitz, S. (1996). Effects of diurnally cycling versus constant temperatures on chicken growth and food intake. Br. Poult. Sci. 37, 43–54. doi: 10.1080/00071669608417835
Ye, B., Li, L., Xu, H., Chen, Y., and Li, F. (2019). Opposing roles of TCF7/LEF1 and TCF7L2 in cyclin D2 and Bmp4 expression and cardiomyocyte cell cycle control during late heart development. Lab. Investig. 99, 807–818. doi: 10.1038/s41374-019-0204-202
Yin, Y., Morgunova, E., Jolma, A., Kaasinen, E., Sahu, B., Khund-Sayeed, S., et al. (2017). Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356:eaaj2239. doi: 10.1126/science.aaj2239
Zhang, M., Yan, F.-B., Li, F., Jiang, K.-R., Li, D.-H., Han, R.-L., et al. (2017). Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 7:45564. doi: 10.1038/srep45564
Zhang, Z., Du, H., Bai, L., Yang, C., Li, Q., Li, X., et al. (2018). Whole genome bisulfite sequencing reveals unique adaptations to high-altitude environments in Tibetan chickens. PLoS One 13:e0193597. doi: 10.1371/journal.pone.0193597
Zhao, P., and Hoffman, E. P. (2006). Musculin isoforms and repression of MyoD in muscle regeneration. Biochem. Biophys. Res. Commun. 342, 835–842. doi: 10.1016/j.bbrc.2006.01.188
Zhao, Y. Y., Sawyer, D. R., Baliga, R. R., Opel, D. J., Han, X., Marchionni, M. A., et al. (1998). Neuregulins promote survival and growth of cardiac myocytes: persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem. 273, 10261–10269. doi: 10.1074/jbc.273.17.10261
Keywords: DNA methylation, epigenetics, temperature, CO2, heart, chicken
Citation: Corbett RJ, te Pas MFW, van den Brand H, Groenen MAM, Crooijmans RPMA, Ernst CW and Madsen O (2020) Genome-Wide Assessment of DNA Methylation in Chicken Cardiac Tissue Exposed to Different Incubation Temperatures and CO2 Levels. Front. Genet. 11:558189. doi: 10.3389/fgene.2020.558189
Received: 01 May 2020; Accepted: 30 September 2020;
Published: 28 October 2020.
Edited by:
George E. Liu, United States Department of Agriculture, United StatesReviewed by:
Shahin Eghbalsaied, Islamic Azad University, Isfahan, IranEd Smith, Virginia Tech, United States
Copyright © 2020 Corbett, te Pas, van den Brand, Groenen, Crooijmans, Ernst and Madsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan J. Corbett, Y29yYmV0MzVAbXN1LmVkdQ==; Ole Madsen, b2xlLm1hZHNlbkB3dXIubmw=