 Xiao-Rong Liu
Xiao-Rong Liu Wen-Jun Bian
Wen-Jun Bian Jie Wang
Jie Wang Ting-Ting Ye
Ting-Ting Ye Bing-Mei LiDe-Tian LiuBin TangWei-Wen Deng
Bing-Mei LiDe-Tian LiuBin TangWei-Wen Deng Yi-Wu ShiTao Su
Yi-Wu ShiTao Su Yong-Hong YiWei-Ping Liao* on behalf of the China Epilepsy Gene 1.0 Project
Yong-Hong YiWei-Ping Liao* on behalf of the China Epilepsy Gene 1.0 Project- Key Laboratory of Neurogenetics and Channelopathies of Guangdong Institute, Department of Neurology of the Second Affiliated Hospital of Guangzhou Medical University, of Neuroscience, Province and the Ministry of Education of China, Guangzhou, China
Introduction: Idiopathic focal epilepsy (IFE) is a group of self-limited epilepsies. The etiology for the majority of the patients with IFE remains elusive. We thus screened disease-causing variants in the patients with IFE.
Methods: Whole-exome sequencing was performed in a cohort of 323 patients with IFE. Protein modeling was performed to predict the effects of missense variants. The genotype–phenotype correlation of the newly defined causative gene was analyzed.
Results: Four novel heterozygous variants in PGM3, including two de novo variants, were identified in four unrelated individuals with IFE. The variants included one truncating variant (c.1432C > T/p.Q478X) and three missense variants (c.478C > T/p.P160S, c.1239C > G/p.N413K, and c.1659T > A/p.N553K), which had no allele frequency in the gnomAD database. The missense variants were predicted to be damaging and affect hydrogen bonds with surrounding amino acids. Mutations Q478X, P160S, and N413K were associated with benign childhood epilepsy with centrotemporal electroencephalograph (EEG) spikes. P160S and N413K were located in the inner side of the enzyme active center. Mutation N553K was associated with benign occipital epilepsy with incomplete penetrance, located in the C-terminal of Domain 4. Further analysis demonstrated that previously reported biallelic PGM3 mutations were associated with severe immunodeficiency and/or congenital disorder of glycosylation, commonly accompanied by neurodevelopmental abnormalities, while monoallelic mutations were associated with milder symptoms like IFE.
Conclusion: The genetic and molecular evidence from the present study implies that the PGM3 variants identified in IFE patients lead to defects of the PGM3 gene, suggesting that the PGM3 gene is potentially associated with epilepsy. The genotype–phenotype relationship of PGM3 mutations suggested a quantitative correlation between genetic impairment and phenotypic severity, which helps explain the mild symptoms and incomplete penetrance in individuals with IFE.
Introduction
Idiopathic focal epilepsy (IFE), also named as localization-related idiopathic epilepsy (G40.0 in ICD-10 2016, WHO), is a common group of self-limited childhood-onset epilepsies. It contains two major subtypes, i.e., benign childhood epilepsy with centrotemporal electroencephalograph (EEG) spikes (BECTS, OMIM# 117100) and benign occipital epilepsy (BOE, OMIM# 132090). BECTS is affecting 0.2% of the population (Strug et al., 2009), while the prevalence of BOE is unknown but is considered to affect around 6% of children aged 1–15 years (Weir et al., 2018). Although both BECTS and BOE are generally regarded as autosomal dominant inheritance with age-dependent penetrance, rare causative genes are identified in the patients. Recent studies have shown that several genes, including GRIN2A, ELP4, SRPX2, and DEPDC5, are associated with BECTS (Strug et al., 2009; Lemke et al., 2013; Lal et al., 2014; Reinthaler et al., 2014) GRIN2A mutations were regarded as the most relevant causative gene for BECTS but accounted for only 4.9% of the patients with BECTS (Lemke et al., 2013). So far, no causative gene has been reported to be associated with BOE. Thus, the etiology for majority of patients with IFE remains unknown.
In this study, we performed whole-exome sequencing (WES) in a cohort of patients with IFE. Four novel heterozygous variants in PGM3 were identified. Previously, PGM3 mutations were associated with immunodeficiency-23 (IMD23, OMIM# 615816) and/or congenital disorder of glycosylation (CDG) that is occasionally accompanied by seizures. Our further analysis showed that previously reported homozygous and compound heterozygous mutations in PGM3 were associated with severe immunodeficiency and glycosylation disturbance, while monoallelic mutations were potentially associated with mild phenotypes like IFE, suggesting that PGM3 is potentially a candidate causative gene of epilepsy.
Materials and Methods
Subjects
A total of 323 subjects with IFE were recruited in the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University in China from January 2014 to 2019. These subjects were epilepsy patients with partial seizures and/or focal discharges but without acquired causes. Their EEGs showed focal abnormalities with features of idiopathic epilepsies, including shifting, bilateral, or multiple focal discharges with normal backgrounds. Eligible subjects had a clinical diagnosis of IFE according to the International League Against Epilepsy (ILAE-1989, 2006, and 2017) (Commission on Classification and Terminology of the International League Against Epilepsy, 1989; Engel, 2006; Scheffer et al., 2017) after appropriate investigations, including long-term video electroencephalography (VEEG), brain MRI, cognitive and behavioral evaluation, and neurometabolic testing. All of the subjects were followed up for at least 1 year.
The studies adhered to the guidelines of the International Committee of Medical Journal Editors with regard to patient consent for research or participation and received approval from local ethics committees of the participating hospitals. The Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University provided ethics approval.
Whole-Exome Sequencing
Genomic DNA was extracted from peripheral blood using a QuickGene DNA whole blood kit (Fujifilm, Tokyo, Japan). Blood samples of parents and siblings were used for linkage and segregation analysis. Trio-based whole-exome sequencing was conducted on the Illumina HiSeq 2500/4000 platform by BGI-Shenzhen (Shenzhen, China). Paired-end reads with a length of 90bp were generated by massive parallel sequencing with more than 125 times average depth and more than 98% coverage of the target region. The raw data were aligned to the reference human genome (GRCh37) using the Burrows–Wheeler Alignment (BWA). Potential pathogenic variants were derived by stepwise filtering. Population-based filtration removed common variants presenting a minor allele frequency ≥0.005 in the gnomAD database, except for those previously reported to be associated with disease in the Human Gene Mutation Database (HGMD) and/or the Online Mendelian Inheritance in Man database. Potential pathogenic variants were flagged if predicted as damaging by multiple in silico programs (Supplementary Table 1). Inheritance-based filtration filtered variants based on family history and possible inheritance models. Assessment of pathogenicity of the variants was performed following the ACMG guidelines (Richards et al., 2015). Conservation of mutated positions was evaluated using sequence alignment of different species. All PGM3 variants were annotated based on the transcript ENST00000512866.1. Sanger sequencing was used to validate the positive findings and the variant origination.
Mutation Analysis
To evaluate the damaging effect of candidate variants, protein modeling was performed using the Iterative Threading ASSEmbly Refinement (I-TASSER) software (Zhang, 2009; Yang and Zhang, 2015). The confidence of each modeling was quantitatively measured by a C-score of 0.14. The three-dimensional structures were shown using PyMOL 1.7.
In an attempt to evaluate the genotype–phenotype correlation, PGM3 mutations and their related phenotypes were systematically retrieved on the PubMed database till November 2019.
Results
Identification of PGM3 Variants
Four variants in the PGM3 gene (OMIM∗ 172100) were identified, including one nonsense variant (c.1432C > T/p.Q478X) and three missense variants (c.478C > T/p.P160S, c.1239C > G/p.N413K, and c.1659T > A/p.N553K). Three variants (P160S, N413K, and Q478X) were identified in the cases with BECTS, and one variant (N553K) was in the case with BOE. Variants N413K and Q478X were de novo, while variants P160S and N553K originated from their asymptomatic parents, respectively (Figures 1A,B). These variants in PGM3 had no allele frequency in the gnomAD database. Variant P160S was predicted to be damaging or probably damaging by all used in silico programs, variant N413K was predicted to be damaging or probably damaging by 11 out of 16 prediction tools, and variant N553K was predicted to be damaging by SIFT and fitCons (Supplementary Table 1). The amino acid sequence alignment showed that residues P160 and N413 were highly conserved across vertebrates. Residue N553 was less conserved in vertebrates and did not appear in the transcript of several species (Figure 1C). Variant Q478X was estimated as a pathogenic variant, and variant N413K was estimated as a likely pathogenic variant by ACMG assessment, while variants P160S and N553K were uncertain significant variants. All cases had no other pathogenic or likely pathogenic variants in genes known to be associated with seizure disorders (Wang et al., 2017).
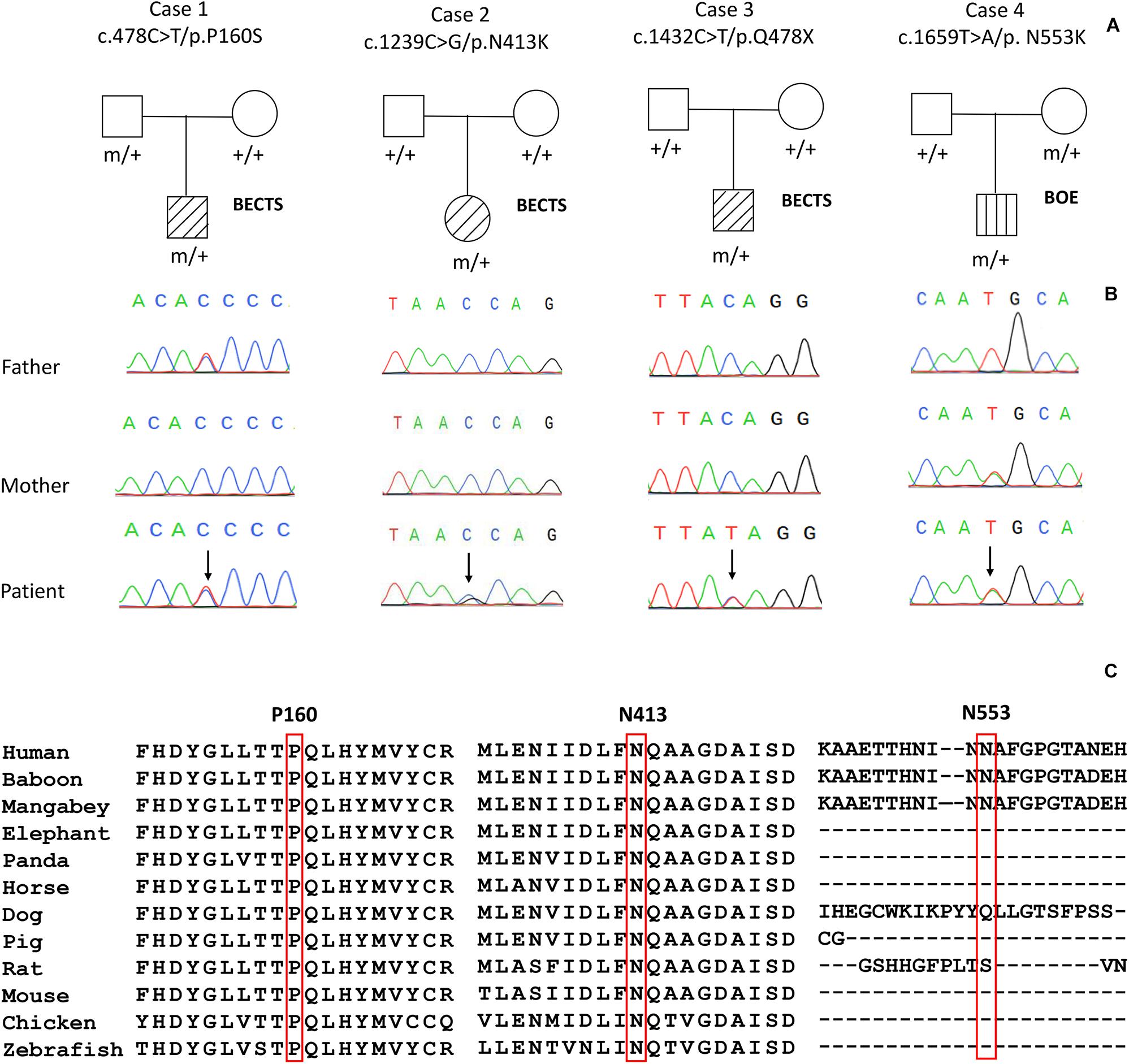
Figure 1. Genetic data on the patients with idiopathic focal epilepsy with PGM3 mutations. (A) Pedigrees of the four cases with PGM3 mutations and their corresponding phenotypes. (B) DNA sequence chromatogram of the PGM3 mutations. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the three missense mutations shows that residues P160 and N413 are highly conserved across vertebrates. Residue N553 is less conserved and does not appear in the transcript of several species.
Structural Alteration of PGM3 Protein
PGM3 contains four domains (Domains 1–4) (Figure 2A). The catalytic cleft is formed by the active serine loop in Domain 1, the metal-binding loop in Domain 2, the sugar-binding loop in Domain 3, and the phosphate-binding loop in Domain 4 (Nishitani, 2006). The truncating variant Q478X is located in Domain 4 and would lead to haploinsufficiency. Variant P160S is located in Domain 1, and variant N413K is located in Domain 3. The three-dimensional structural model of PGM3 indicated that residues P160 and N413 lay in the inner side of the enzyme active center and were expected to be more “deleterious.” Variant N553K, located in Domain 4, which lay in the C-terminal of Domain 4 and a distance away from the enzyme active center (Figure 2B), potentially led to mild damage effect. The molecular effects of the missense variants analyzed by protein modeling using I-TASSER showed that all the three missense variants led to the alterations of hydrogen bonds and may affect the protein steric configuration. Originally, residue P160 formed a hydrogen bond with Y164. When proline was replaced by serine at residue 160, additional two hydrogen bonds were formed with Y183 and T158 (Figure 2C). Residual N413 formed five hydrogen bonds with T376, A415, A416, D418, and S421, respectively. When asparagine was replaced by lysine, the hydrogen bonds with A415, A416, and D418 were destroyed, and only the hydrogen bonds with T376 and S421 were kept (Figure 2D). Residue N553 originally formed hydrogen bonds with I551, N552, A554, G558, and T559, respectively. When the asparagine was replaced by lysine at 553, the hydrogen bonds with N552, A554, and G558 were destroyed, and a new hydrogen bond with F555 was formed instead (Figure 2E).
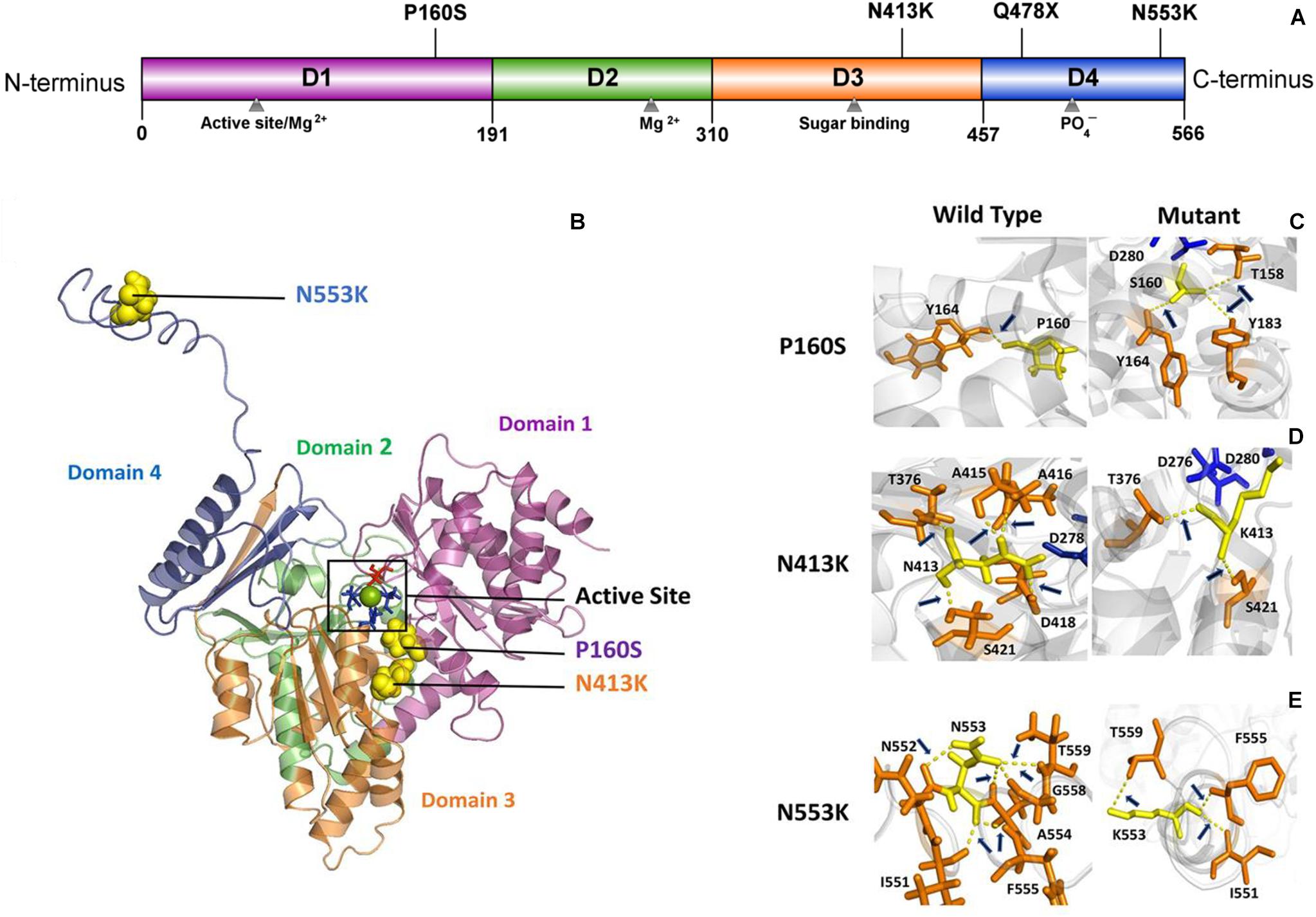
Figure 2. Schematic illustration of mutation and interactions with surrounding amino acids. (A) Linear schematic of the PGM3 structure and the location of identified PGM3 mutations. PGM3 is composed of Domains 1–4. Domain 1 is colored purple, Domain 2 in green, Domain 3 in yellow, and Domain 4 in blue. Mutation P160S locates in Domain 1, mutation N413K locates in Domain 3, and mutations Q478X and N553K in Domain 4. (B) Schematic illustration of the location of mutations in the three-dimensional structure of PGM3. The active site is depicted in black frame. The residue S64 (colored red) locates in Domain 1. Residues D276, D278, and D280 (colored blue) locate in Domain 3. The green sphere represents the magnesium (Mg) ion. The yellow spheres represent the mutations. Mutations P160S and N413K locate within the active center. Mutation N553K is a distance away from the active center. (C–E) Hydrogen bond changes of mutants P160, N413, and N553. Arrows index the hydrogen bonds.
Clinical Features of the Cases With PGM3 Variants
All affected cases showed childhood-onset focal epilepsy. Table 1 summarized the main clinical features of the four cases with PGM3 variants. The cases with variants P160S, N413K, and Q478X were diagnosed as BECTS. The three cases started seizures at preschool or school age with generalized tonic–clonic seizure (GTCS) during sleep. The GTCSs occurred with approximately one to two attacks per year. No developmental delay, intellectual or speech disability, or visual abnormalities were observed. Their electroencephalograph (EEG) showed unilateral or bilateral independent centrotemporal discharges, dominantly during sleep (Figures 3A–C). The brain MRIs were normal. They all got seizure-free by low-dose monotherapy of valproic acid or lamotrigine. The case with variant N553K was diagnosed as BOE. He was a 3-year-old boy with normal development and had two GTCSs at the age of 2 years. The EEG obtained at the age of 3 years showed right occipital discharges (Figure 3D). His brain MRI was normal. He was seizure-free for 9 months without any treatment of antiepileptic drugs. Except seizures, no manifestation of immunodeficiency and CDG was observed in the four cases.
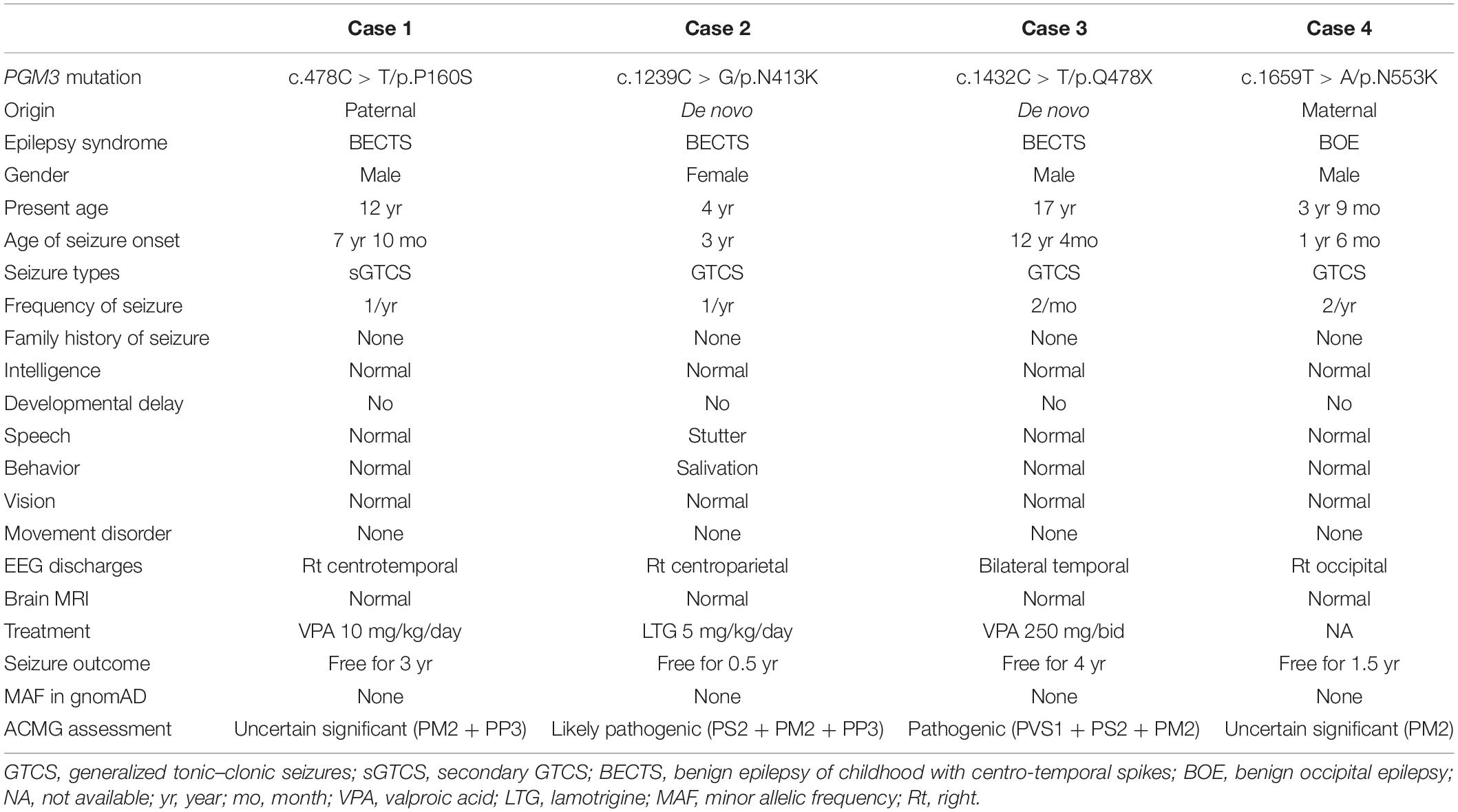
Table 1. Clinical and genetic features of the cases with PGM3 mutations.
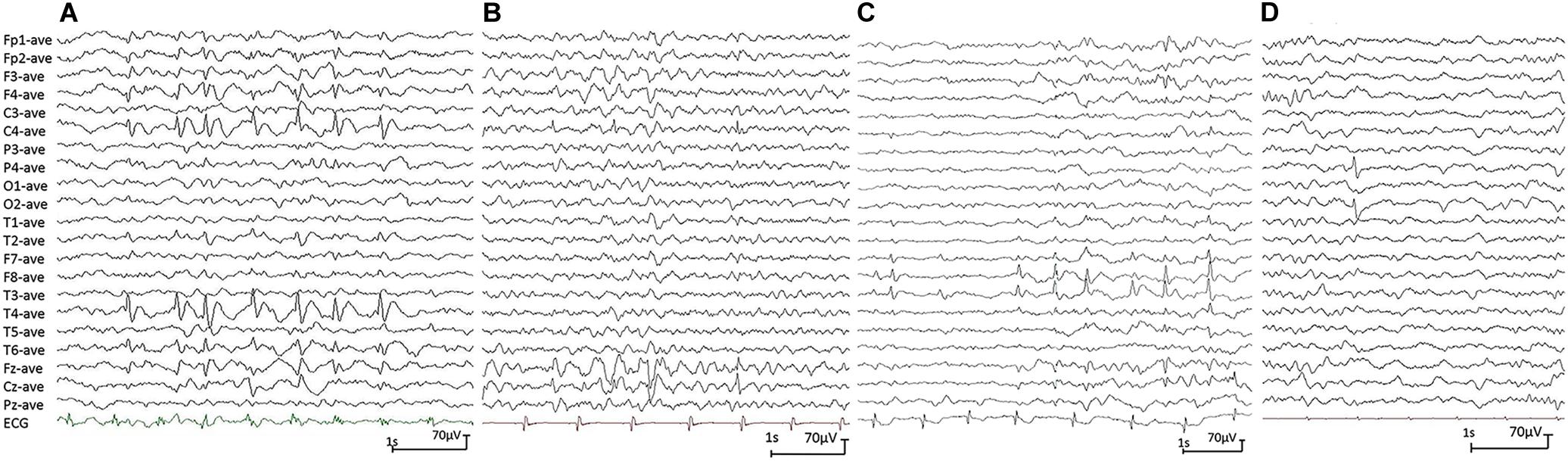
Figure 3. Electroencephalograph (EEG) changes during non-rapid-eye-movement sleep in the cases with idiopathic focal epilepsy with PGM3 mutations (referential montage with average reference was used in all EEGs). (A) Interictal EEG of case 1 showed right centrotemporal spike and slow waves (obtained at the age of 8 years). (B) Interictal EEG of case 2 showed right central spike and slow waves (at the age of 3 years). (C) Interictal EEG of case 3 showed bilateral temporal spike and slow waves (at the age of 12 years). (D) Interictal EEG of case 4 showed right occipital spike waves (at the age of 3 years).
Genotype–Phenotype Correlation of PGM3 Mutations
Previously, 18 mutations in PGM3 were identified (Table 2). Sixteen out of 18 mutations, including 10 homozygous mutations and 3 compound heterozygous mutations, were identified in 16 families, involved 28 individuals (Sassi et al., 2014; Stray-Pedersen et al., 2014; Zhang et al., 2014; Lundin et al., 2015; Bernth-Jensen et al., 2016; Pacheco-Cuellar et al., 2017; Lundin et al., 2018; Abolhassani et al., 2019). All the individuals carrying biallelic mutations had the symptoms of immunodeficiency and/or congenital disorder of glycosylation (CDG). The neurological features included cognitive disability (12/28), developmental delay (12/28), seizures (10/28), characteristic facies (8/28), and hypotonia (4/28). The mutations included 11 missense, two small deletions, one small insertion, one splicing mutation, and one gross deletion including the whole gene of PGM3.
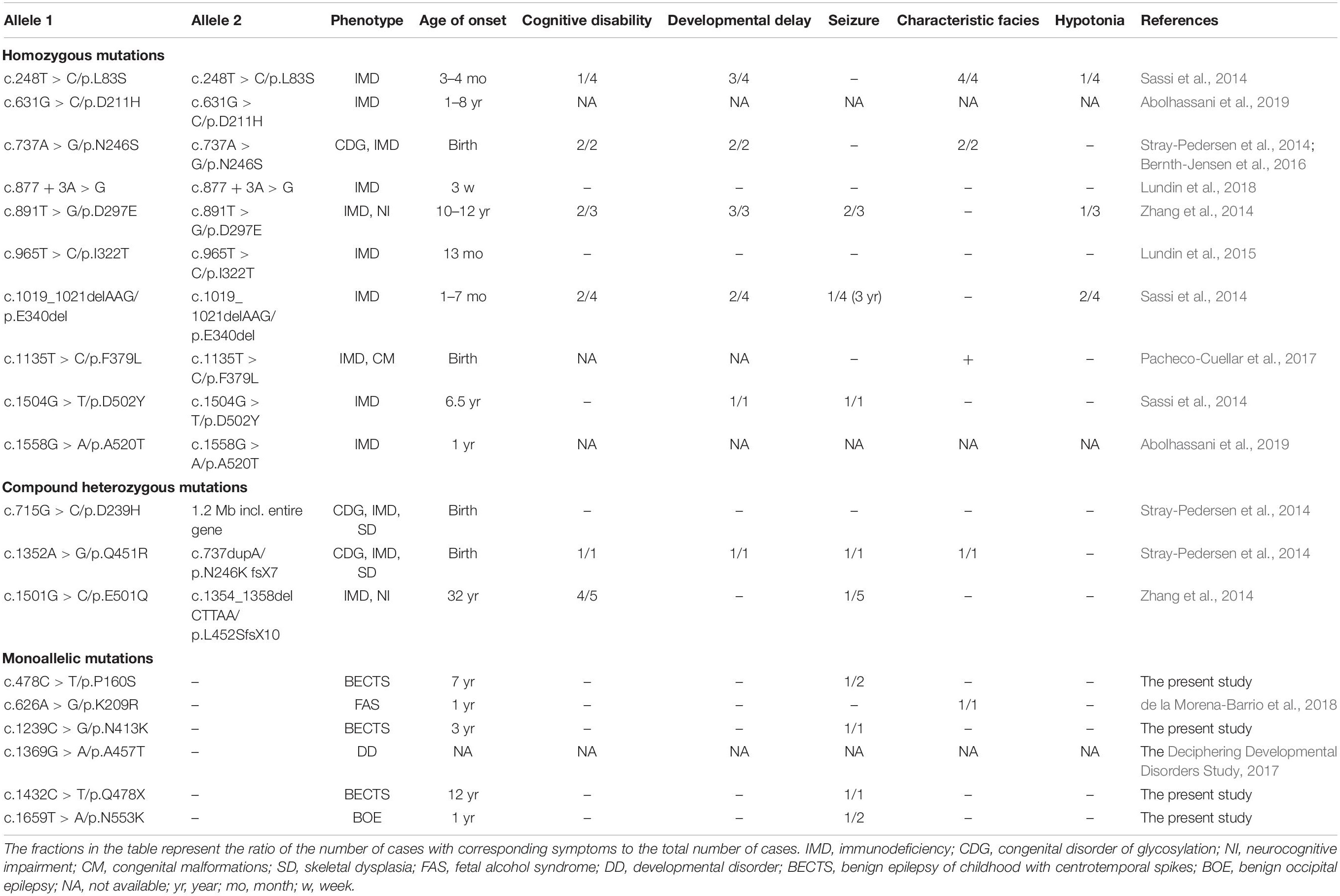
Table 2. Summary of neurological features in the cases/families with PGM3 mutations.
In addition, one heterozygous mutation c.626A > G/p.K209R was identified in a patient with fetal alcohol syndrome (de la Morena-Barrio et al., 2018); the other heterozygous mutation c.1369G > A/p.A457T was identified in a patient from a big cohort of developmental disorders by WES (Deciphering Developmental Disorders Study, 2017). No manifestation about immunodeficiency and glycosylation disturbance was mentioned.
Discussion
PGM3 gene is located on chromosome 6q14.1–q15 (chr6:83164873-83193936) and encodes phosphoglucomutase 3 (PGM3). Previous studies have demonstrated that PGM3 mutations were associated with severe immunodeficiency and glycosylation disorder, commonly accompanied by neurodevelopmental disorders and occasionally by seizures. In this study, four novel heterozygous PGM3 variants, including two de novo variants, were identified in the cases with IFE and without the manifestation of immunodeficiency or glycosylation disorders. Further analysis indicated that biallelic mutations were associated with severer immunodeficiency or glycosylation disorders, whereas heterozygous mutations were potentially associated with mild phenotypes like IFE. The evidence suggests that PGM3 gene is potentially a candidate pathogenic gene of epilepsy.
PGM3, which catalyzes the conversion of N-acetyl-glucosamine (GlcNAc)-6-phosphate into GlcNAc-1-phosphate, is universally required for the synthesis of UDP-GlcNAc, a sugar nucleotide critical to multiple glycosylation pathways (Stray-Pedersen et al., 2014). The PGM3 gene is ubiquitously expressed across the whole lifespan (including embryonic period)1. In PGM3-deficiency mice, homozygous or compound heterozygous hypomorphic alleles caused trilineage cytopenias, even embryonic lethality (Greig et al., 2007), suggesting that PGM3 is a vital gene for fetal development. In contrast, the heterozygous PGM3+/gt and PGM3+/mld1 mice presented subclinical alterations like reduction of PGM3 activity in multiple tissues (Greig et al., 2007), indicating a genetic dose effect (quantitative correlation). In humans, PGM3 mutations were initially associated with IMD-23 characterized by recurrent respiratory, skin infections beginning in early childhood, and notably increased serum IgE in laboratory studies. Recently, PGM3 mutations have also been identified as a cause of CDG, usually exhibiting severe skeletal dysplasia, congenital malformations, and developmental delay. Among the patients with IMD-23 and CDG, neurodevelopmental abnormalities, including developmental delay, cognitive disability, seizures, and hypotonia, were common. Many patients with PGM3 gene deficiency need hematopoietic stem cell therapy to save their life. Our analysis demonstrated that the cases with severe immunodeficiency and/or glycosylation disturbance all carried homozygous or compound heterozygous mutations, consistent with the results from biallelic deficiency animal models. Previously, two monoallelic mutations (K209R and A457T) were reported in the patients with fetal alcohol syndrome and developmental disorder, respectively (Deciphering Developmental Disorders Study, 2017; de la Morena-Barrio et al., 2018), and no immunodeficiency was mentioned. In the present study, four monoallelic mutations, including two de novo mutations, were identified in patients with pure IFE. One truncating mutation led to haploinsufficiency of PGM3. The other three missense mutations were highly conserved across the vertebrates and were predicted to alter hydrogen bond formation with surrounding amino acids. All mutations had no allele frequency in the gnomAD database and were predicted to be damaging by multiple software. Taking together the evidence that PGM3 gene is expressed in brain and associated with neurodevelopmental disturbance, the PGM3 gene is suggested to be potentially a candidate pathogenic gene of epilepsy. The genotype–phenotype correlations indicate that biallelic mutations in the PGM3 gene are associated with severe immunodeficiency and glycosylation disturbance, while heterozygous mutations are potentially associated with mild phenotypes like IFE, in accordance with the findings in the PGM3 deficiency mice.
PGM3 protein has four domains arranged in an overall heart shape similar to phosphoglucomutase 1 (PGM1) (Stiers et al., 2017). The catalytic cleft is formed by four loops from each domain. The active site of PGM3 is located in a large central cleft, formed at the confluence of the four structural domains. The cleft contains (i) phosphoserine 64 that participates in phosphoryl transfer, (ii) the metal-binding loop including the three coordinating aspartates (residues 276, 278, and 280), (iii) a sugar-binding loop, and (iv) the phosphate-binding site (residue 505) (Uniprot database) (Stiers et al., 2016). In the present study, besides the truncating mutation Q478X, all missense mutations were predicted to change the hydrogen bonds with their surrounding amino acids and affect the protein structural stability, potentially impairing enzyme catalytic activity. Mutations P160S and N413K were located within the active center and were associated with typical BECTS. Instead, mutation N553K was identified in a family with BOE with incomplete penetrance. N553K was a distance away from the active center and was less conserved, suggesting less damage effect. Our recent study showed that molecular subregional location was a critical factor to determine the damaging effect that presented a continuous distribution and correlated with the severity of phenotypes (Tang et al., 2019), providing one of the explanations for the less damaging effect and atypical symptoms/incomplete penetrance associated with N553K. However, further studies are required to determine its pathogenicity and association with BOE.
IFE is a group of self-limited epilepsies with infrequent partial seizures and a normal background of EEG. Although both BECTS and BOE are regarded as genetically determined epileptic syndromes, big pedigrees are rare. Majority of patients with BECTS and BOE are sporadic. Incomplete penetrance is potentially one of the explanations. The present study demonstrated that less impaired functions of the PGM3 gene, i.e., heterozygous mutations, were associated with IFE. Additionally, heterozygous mutations with a less damaging effect were potentially associated with the incomplete penetrance. These findings were consistent with the evidence of quantitative correlation from PGM3 deficiency models, in which the heterozygous damaging resulted in subclinical abnormalities, such as decrease in enzyme activity and hematological alterations (Greig et al., 2007). In the literature, clinical information about parents in the biallelic cases was not available. Generally, in the patients with BECTS and BOE, epileptiform discharges in EEG will disappear, and epileptic seizures will stop in adulthood. Even in childhood, the epileptic seizures in BECTS and BOE are infrequent and usually occur during sleep, which are usually overlooked. These could be the reasons of incomplete penetrance in the parents with PGM3 heterozygous mutations. It was also found that most early-onset patients with immunodeficiency had no seizure, while the late-onset patients had seizure manifestations, suggesting an age-dependent penetrance. The early-onset patients usually had more severe symptoms and rarely survived to the age of epilepsy occurrence. This may be one of the explanations for a portion of the patients with biallelic mutations having no seizure.
Clinically, the features of BECTS and BOE are often overlapped. Up to 20% patients with BOE have rolandic spikes as those with BECTS (Lada et al., 2003). BECTS and BOE may appear in the same patient at different life periods. In the present study, PGM3 mutations were identified in both BECTS and BOE, suggesting that BECTS and BOE potentially have similar etiologies. To date, multiple genes have been implicated in IFE. The role of multiple rare heterozygous variants in IFE warrants further study.
In conclusion, we identified four novel heterozygous PGM3 mutations in patients with IFE without apparent manifestation of immunodeficiency and glycosylation disorder. The genetic and molecular evidence implies that the mutations lead to defect of the PGM3 gene. Taking together the data from the gene expression profile and the PGM3 deficiency animal model, it is suggested that PGM3 gene is potentially a candidate pathogenic gene of epilepsy. Further analysis revealed that previously reported biallelic mutations in the PGM3 gene were associated with severe immunodeficiency and glycosylation disturbance, while monoallelic mutations were associated with mild phenotypes like IFE, suggesting a quantitative correlation between genetic impairment and phenotypic severity, which help explain the mild symptoms and incomplete penetrance in individuals with IFE.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University provided ethics approval. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individuals and minors’ legal guardian for the publication of any potentially identifiable images or data included in this article.
Author Contributions
X-RL and W-PL contributed to the conception of the study. X-RL contributed to the interpretation of clinical data and drafting of the figures and the manuscript. W-JB, JW, and T-TY examined the patient and participated in drafting of the manuscript. B-ML, D-TL, BT, and W-WD contributed to the collection and analysis of clinical data. Y-WS and TS helped perform the analysis with constructive discussions. W-PL provided critical review and substantially revised the manuscript. All the authors read and approved the manuscript before sending the manuscript to the journal for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81871015), China Association Against Epilepsy-UCB Pharma Ltd., Joint Science Research Foundation (2018024), Science and Technology Project of Guangdong Province (2017B09090436 and 2017B030314159), Natural Science Foundation of Guangdong Province (2020A1515010108), and Science and Technology Project of Guangzhou (201904020028).
Conflict of Interest
This study received funding from UCB Pharma Ltd. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are deeply grateful to the patients and clinicians who participated in this work and appreciate the supporting of the China Epilepsy Gene 1.0 Project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.559080/full#supplementary-material
Footnotes
References
Abolhassani, H., Aghamohammadi, A., Fang, M., Rezaei, N., Jiang, C., Liu, X., et al. (2019). Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet. Med. 21, 243–251. doi: 10.1038/s41436-018-0012-x
Bernth-Jensen, J. M., Holm, M., and Christiansen, M. (2016). Neonatal-onset T(-)B(-)NK(+) severe combined immunodeficiency and neutropenia caused by mutated phosphoglucomutase 3. J. Allergy Clin. Immunol. 137, 321–324. doi: 10.1016/j.jaci.2015.07.047
Commission on Classification and Terminology of the International League Against Epilepsy. (1989). Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 30, 389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x
de la Morena-Barrio, M. E., Ballesta-Martinez, M. J., Lopez-Galvez, R., Anton, A. I., Lopez-Gonzalez, V., Martinez-Ribot, L., et al. (2018). Genetic predisposition to fetal alcohol syndrome: association with congenital disorders of N-glycosylation. Pediatr. Res. 83, 119–127. doi: 10.1038/pr.2017.201
Deciphering Developmental Disorders Study, (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Engel, J. (2006). ILAE classification of epilepsy syndromes. Epilepsy Res. 70, 5–10. doi: 10.1016/j.eplepsyres.2005.11.014
Greig, K. T., Antonchuk, J., Metcalf, D., Morgan, P. O., Krebs, D. L., Zhang, J. G., et al. (2007). Agm1/Pgm3-mediated sugar nucleotide synthesis is essential for hematopoiesis and development. Mol. Cell. Biol. 27, 5849–5859. doi: 10.1128/mcb.00802-07
Lada, C., Skiadas, K., Theodorou, V., Loli, N., and Covanis, A. (2003). A Study of 43 patients with panayiotopoulos syndrome, a common and benign childhood seizure susceptibility. Epilepsia 44, 81–88. doi: 10.1046/j.1528-1157.2003.32602.x
Lal, D., Reinthaler, E., Schubert, J., Muhle, H., Riesch, E. G. K., Kluger, G., et al. (2014). DEPDC5 mutations in genetic focal epilepsies of childhood. Ann. Neurol. 75, 788–792.
Lemke, J. R., Lal, D., Reinthaler, E. M., Steiner, I., Nothnagel, M., Alber, M., et al. (2013). Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072. doi: 10.1038/ng.2728
Lundin, K. E., Hamasy, A., Backe, P. H., Moens, L. N., Falk-Sorqvist, E., Elgstoen, K. B., et al. (2015). Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clin. Immunol. 161, 366–372. doi: 10.1016/j.clim.2015.10.002
Lundin, K. E., Wang, Q., Hamasy, A., Marits, P., Uzunel, M., Wirta, V., et al. (2018). Eleven percent intact PGM3 in a severely immunodeficient patient with a novel splice-site mutation, a case report. BMC Pediatr. 18:285. doi: 10.1186/s12887-018-1258-9
Pacheco-Cuellar, G., Gauthier, J., Desilets, V., Lachance, C., Lemire-Girard, M., Rypens, F., et al. (2017). A novel PGM3 mutation is associated with a severe phenotype of bone marrow failure, severe combined immunodeficiency, skeletal dysplasia, and congenital malformations. J. Bone Miner. Res. 32, 1853–1859. doi: 10.1002/jbmr.3173
Reinthaler, E. M., Lal, D., Jurkowski, W., Feucht, M., Steinbock, H., Gruber-Sedlmayr, U., et al. (2014). Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 55, e89–e93. doi: 10.1111/epi.12712
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sassi, A., Lazaroski, S., Wu, G., Haslam, S. M., Fliegauf, M., Mellouli, F., et al. (2014). Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J. Allergy Clin. Immunol. 133, 1410–9, 1419.e1–e13. doi: 10.1016/j.jaci.2014.02.025
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 512–521. doi: 10.1111/epi.13709
Stiers, K. M., Graham, A. C., Kain, B. N., and Beamer, L. J. (2017). Asp263 missense variants perturb the active site of human phosphoglucomutase 1. FEBS J. 284, 937–947. doi: 10.1111/febs.14025
Stiers, K. M., Kain, B. N., Graham, A. C., and Beamer, L. J. (2016). Induced structural disorder as a molecular mechanism for enzyme dysfunction in phosphoglucomutase 1 deficiency. J. Mol. Biol. 428, 1493–1505. doi: 10.1016/j.jmb.2016.02.032
Stray-Pedersen, A., Backe, P. H., Sorte, H. S., Mørkrid, L., Chokshi, N. Y., Erichsen, H. C., et al. (2014). PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am. J. Hum. Genet. 95, 96–107. doi: 10.1016/j.ajhg.2014.05.007
Strug, L. J., Clarke, T., Chiang, T., Chien, M., Baskurt, Z., Li, W., et al. (2009). Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur. J. Hum. Genet. 17, 1171–1181. doi: 10.1038/ejhg.2008.267
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2019). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief. Bioinform. 2019, bbz115. doi: 10.1093/bib/bbz115
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Weir, E., Gibbs, J., and Appleton, R. (2018). Panayiotopoulos syndrome and benign partial epilepsy with centro-temporal spikes: a comparative incidence study. Seizure 57, 66–69. doi: 10.1016/j.seizure.2018.03.002
Yang, J., and Zhang, Y. (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342
Zhang, Y. (2009). I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77, 100–113. doi: 10.1002/prot.22588
Zhang, Y., Yu, X., Ichikawa, M., Lyons, J. J., Datta, S., Lamborn, I. T., et al. (2014). Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J. Allergy Clin. Immunol. 133, 1400–9, 1409.e1–e5. doi: 10.1016/j.jaci.2014.02.013
Keywords: whole-exome sequencing, congenital disorder of glycosylation, immunodeficiency, PGM3 gene, idiopathic focal epilepsy
Citation: Liu X-R, Bian W-J, Wang J, Ye T-T, Li B-M, Liu D-T, Tang B, Deng W-W, Shi Y-W, Su T, Yi Y-H and Liao W-P (2020) Heterozygous PGM3 Variants Are Associated With Idiopathic Focal Epilepsy With Incomplete Penetrance. Front. Genet. 11:559080. doi: 10.3389/fgene.2020.559080
Received: 05 May 2020; Accepted: 17 September 2020;
Published: 15 October 2020.
Edited by:
Patrick E. Williams, University of Kansas, United StatesReviewed by:
Daniele Ghezzi, Carlo Besta Neurological Institute (IRCCS), ItalyAlysia Vrailas-Mortimer, Illinois State University, United States
Copyright © 2020 Liu, Bian, Wang, Ye, Li, Liu, Tang, Deng, Shi, Su, Yi and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Ping Liao, d3BsaWFvQDE2My5uZXQ=