 Chengwen Gao
Chengwen Gao Chuanhong Wu
Chuanhong Wu Xia Zhao
Xia Zhao- Laboratory of Medical Biology, Medical Research Center, The Affiliated Hospital of Qingdao University, Qingdao, China
Salvia species have been widely used as medicinal plants and have played an important role in the treatment and recovery of individuals with COVID-19. In this study, we reported two newly identified whole chloroplast genome sequences of Salvia medicinal plants (Salvia yangii and Salvia miltiorrhiza f. alba) and compared them with those of seven other reported Salvia chloroplast genomes. These were proven to be highly similar in terms of overall size, genome structure, gene content, and gene order. We identified 10 mutation hot spots (trnK-rps16, atpH-atpI, psaA-ycf3, ndhC-trnV, ndhF, rpl32-trnL, ndhG-ndhI, rps15-ycf1, ycf1a, and ycf1b) as candidate DNA barcodes for Salvia. Additionally, we observed the transfer of nine large-sized chloroplast genome fragments, with a total size of 49,895 bp (accounting for 32.97% of the chloroplast genome), into the mitochondrial genome as they shared >97% sequence similarity. Phylogenetic analyses of the whole chloroplast genome provided a high resolution of Salvia. This study will pave the way for the identification and breeding of Salvia medicinal plants and further phylogenetic evolutionary research on them as well.
Introduction
Salvia L. species constitute the largest genus in the family Lamiaceae and comprise approximately 1,000 species worldwide. Of these, 84 Salvia species originated from southwestern China, especially from the Hengduan Mountains region (Li et al., 2013). The name Salvia comes from the Latin word “salvare,” which means “healing.” Salvia species have been widely used as medicinal plants, and as an important component of the Xuebijing injection, they have played an important role in the treatment and recovery of individuals with COVID-19 (Pan et al., 2020; Wen et al., 2020; Zhang Y.L. et al., 2020). For thousands of years in China, more than 40 Salvia species have been used for treating common cold, tuberculosis, bronchitis, hemorrhages, and menstrual disorders (Topcu, 2006). Among these, Salvia miltiorrhiza Bunge (Danshen), a characteristic traditional Chinese medicine (TCM) (Li et al., 2018), has been used for treating cerebrovascular and cardiovascular diseases effectively (Chen and Chen, 2017; Wang et al., 2017). However, compared with the purple-flowered S. miltiorrhiza variety, the white-flowered landrace of Salvia miltiorrhiza f. alba exerts superior medicinal qualities (Zhang et al., 2016). Salvia yangii also possesses high medicinal value (Mazandarani and Ghaemi, 2010). However, the difficulty in identifying these plants leads to an uneven quality of Salvia medicinal materials (Hu G.X. et al., 2016; Xiang et al., 2016; Drew et al., 2017). With the development of sequencing technology, the sequencing of chloroplast genomes can be fulfilled in a short time at a relatively low cost currently. Whole chloroplast genome sequences can provide more genetic information and higher species resolution ability than other molecular data, and they provide a possible resolution to these issues. However, the chloroplast genomes of most Salvia plants remain unknown.
Chloroplasts are specialized organelles where photosynthesis occurs and are composed of stacked thylakoids interconnected by lamellae (Douglas, 1998; Brunkard et al., 2015). The highly conserved chloroplast genomes possess a quadripartite structure including small single-copy (SSC) and large single-copy (LSC) regions along with dual copies of inverted repeat (IR) regions (Wicke et al., 2011; Wang et al., 2015). Whole chloroplast genomes provide crucial information, which can be used for understanding evolutionary differences, exploring genetic diversity, and generating highly resolved phylogenies, especially in complex/low taxonomic levels (Hu H. et al., 2016; Xu et al., 2019; Huang et al., 2020).
Here, we assembled the chloroplast genomes of S. miltiorrhiza f. alba and S. yangii, followed by comparing them with seven previously reported Salvia chloroplast genomes from GenBank. This study aimed to (1) perform a comparative analysis of the chloroplast genomes of these nine Salvia species; (2) ascertain the highly divergent sequences in the Salvia chloroplast genomes; (3) identify chloroplast gene insertion in the mitochondria; and (4) explore the evolutionary differences and similarities in the genus Salvia and lamiids. Thus, the information generated in this study would expand our knowledge on the evolution of plastome as well as the phylogenies of Salvia species.
Materials and Methods
Plant Material Sampling
The Salvia accessions in this study were from the Salvia nuclear genome sequencing project, and all plant materials were conserved in China Academy of Chinese Medical Sciences, Beijing, China (Xu et al., 2016; Boachon et al., 2018). The DNA was extracted from fresh leaves, followed by the use of Illumina sequencing technology to generate libraries. Post-sequencing, the paired-end sequence reads were obtained through next-generation sequencing.
Chloroplast Genome Assembly and Annotation
Trimmomatic v0.38 was used to filter the raw sequencing data (Bolger et al., 2014). N-containing sequences and adapter sequences were removed. Sequences with a Q value less than 20 were also removed. Then, the clean data were used to perform de novo assembly using SPAdes 3.61 with varying K-mer parameters (Bankevich et al., 2012). We ordered de novo scaffolds that were positively correlated to the chloroplasts on the reference chloroplast genome of S. miltiorrhiza (NC_020431). Next, the Geneious Prime software v2020.0.4 (Kearse et al., 2012) was used to remap the paired-end reads to fill gaps in the final consensus sequence with multiple iterations.
GeSeq was used to conduct chloroplast genome annotation to predict transfer RNA (tRNA), gene-encoding proteins, and ribosomal RNA (rRNA), with manual adjustments as required (Tillich et al., 2017). We manually examined the IR junctions of Salvia species. A circular map of Salvia chloroplast genomes was subsequently drawn using OGDraw (Greiner et al., 2019).
Genome Comparative Analysis and Identification of Hypervariable Regions
MAFFT 7.221 was used to align the chloroplast genome sequences of S. miltiorrhiza f. alba and S. yangii (Katoh and Standley, 2013). Next, DnaSP 6.12 was used to perform a sliding window analysis with a step size of 200 bp and window length of 600 bp in order to detect the rapidly evolving molecular markers for performing phylogenetic analysis (Librado and Rozas, 2009).
Tree-based methods were utilized to assess the hypervariable barcodes and to compare the chloroplast genes matK and rbcL. MEGA 7.0 software was used to build maximum likelihood (ML) trees with 1,000 bootstrap replicates for each hypervariable marker (Kumar et al., 2016).
Identification of Chloroplast Gene Insertion in the Mitochondria
The mitochondrial genome of S. miltiorrhiza was retrieved from GenBank (NC_023209), followed by a homology search between chloroplast and mitochondria genomes using BLAST (with default parameters) to identify the transferred genes between the mitochondrial and chloroplast genomes. Circos was used to draw the chloroplast and mitochondrial maps from Salvia species, along with the fragments of gene transfers (Krzywinski et al., 2009).
Phylogenetic Analysis
We used the following three methods to perform phylogenetic analyses of Salvia species: Bayesian inference (BI) with a GTR + I + G model using MrBayes 3.2 [the Markov chain Monte Carlo (MCMC) algorithm was run for 1 million generations and sampled every 100 generations] (Ronquist et al., 2012); maximum likelihood (ML) using MEGA 7.0 with 1,000 bootstrap replicates (Kumar et al., 2016); and maximum parsimony (MP) with a heuristic search in PAUP 4.0 with 1,000 random taxon stepwise addition sequences (Swofford, 1993).
Results
Chloroplast Genome Organization and Features
The chloroplast genomes of S. yangii and S. miltiorrhiza f. alba were 151,473 and 151,389 bp long, respectively, and exhibited a quadripartite structure with dual IR regions (25,603 and 25,521 bp), an SSC region (17,566 and 17,572 bp), and an LSC region (82,701 and 82,775 bp) (Figure 1). S. yangii and S. miltiorrhiza f. alba contained 116 and 115 unique genes in their chloroplast genomes, respectively, of which, 82 and 81 were protein-coding genes, four were rRNAs, and 30 were tRNAs (Table 1). Eighteen genes exhibited introns, of which six tRNAs (trnV-UAC, trnG-UCC, trnK-UUU, trnL-UAA, trnI-GAU, and trnA-UGC) and nine protein-coding genes (accD, rps16, rpl16, ndhB, petB, rpl2, atpF, rpoC1, and ndhA) had a single intron and the remaining three genes displayed two introns (ycf3, rps12, and clpP). For both S. yangii and S. miltiorrhiza f. alba, the trnK-UUU intron, comprising the matK gene, had the largest intron for both species (2,515 and 2,522 bp, respectively). Furthermore, the Salvia chloroplast genomes possessed similar GC contents (38.0–38.1%). IR expansion was considered the main cause for the size differences in the Salvia chloroplast genomes. Both Salvia species were found to be generally conserved in terms of gene order and genome structure (Table 1). The GenBank accession numbers for the complete chloroplast genome sequences of the two Salvia species were MT012420 and MT012421.

Figure 1. Comparison of inverted repeat (IR)/small single-copy (SSC) expansions in Salvia species. Colored boxes above the horizontal line indicate the genes and gene segments across the IRa/b junctions. IR segments and genes are not to scale.
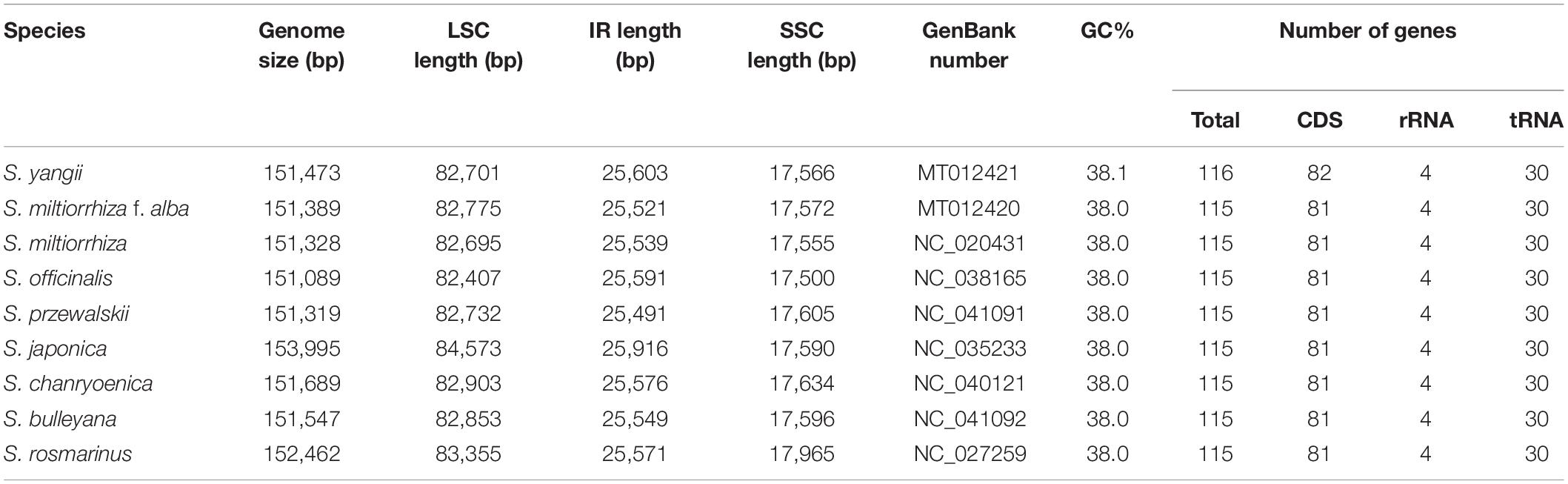
Table 1. Summary of the features of the chloroplast genome of nine Salvia species.
Comparisons of the fully annotated IR/SC junction regions were found to exhibit almost the same relative positions among the nine Salvia chloroplast genomes (Figure 1). The rps19 gene contained all LSC/IRb junctions, resulting in the partial expansion (42–134 bp) of the IRb region toward the rps19 gene. The IRb/SSC boundary positions were located on the ycf1 and ndhF genes, and the SSC/IRa borders in the nine chloroplast genomes were located on the ycf1 gene.
Comparative Salvia Chloroplast Genomes and Divergence Hot Spot Regions
The results of the comprehensive sequence divergence of the two newly assembled and the seven previously reported Salvia chloroplast genomes with S. miltiorrhiza as the control displayed high sequence similarity (Figure 2). As expected, the LSC and SSC regions exhibited comparatively higher sequence divergence than did the IR regions. A search for nucleotide substitutions identified 5,833 variable sites (3.69%), including 2,486 parsimony-informative sites (1.57%), across the nine Salvia chloroplast genomes.
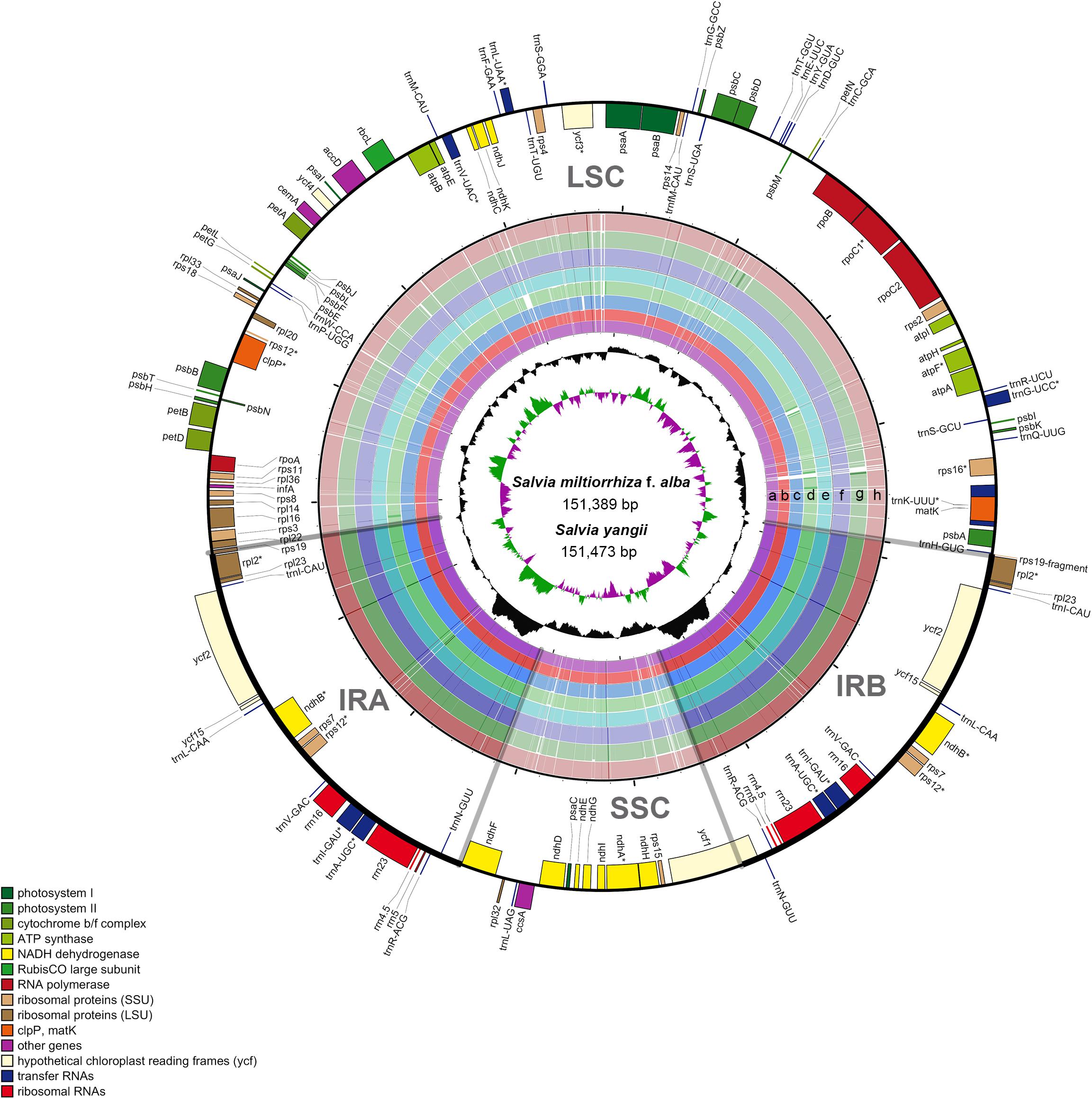
Figure 2. Map of the aligned Salvia chloroplast genomes. Gene map of the Salvia chloroplast genomes, sequence alignment of Salvia species chloroplast genomes [S. miltiorrhiza f. alba (a); S. yangii (b); S. officinalis (c); S. japonica (d); S. chanryoenica (e); S. bulleyana (f); S. rosmarinus (g); S. przewalskii (h), with S. miltiorrhiza as the reference], GC content, and GC skew from outside to inside (the genes that are transcribed clockwise are depicted on the outside of the circle, while those transcribed counterclockwise are depicted inside). *Indicate genes exhibited introns.
Next, in the Salvia chloroplast genomes, we calculated the nucleotide diversity (π) values within 600-bp windows to detect the sequence divergence hot spots. The π values were in the range of 0–0.119, with extremely high values (π > 0.002) in the following 10 regions (trnK-rps16, atpH-atpI, psaA-ycf3, ndhC-trnV, ndhF, rpl32-trnL, ndhG-ndhI, rps15-ycf1, ycf1a, and ycf1b) (Figure 3 and Table 2). Divergence hot spot regions could be the ideal molecular markers to distinguish Salvia species. Two conventional candidate DNA barcodes (matK and rbcL) were used to compare the marker divergence. The results revealed a lower variability in these barcodes compared with the newly identified markers (Table 2). The ndhC-trnV region exhibited the highest variability (38.75%). Supplementary Figure S1 presents the graphical representation of these results using the ML method.
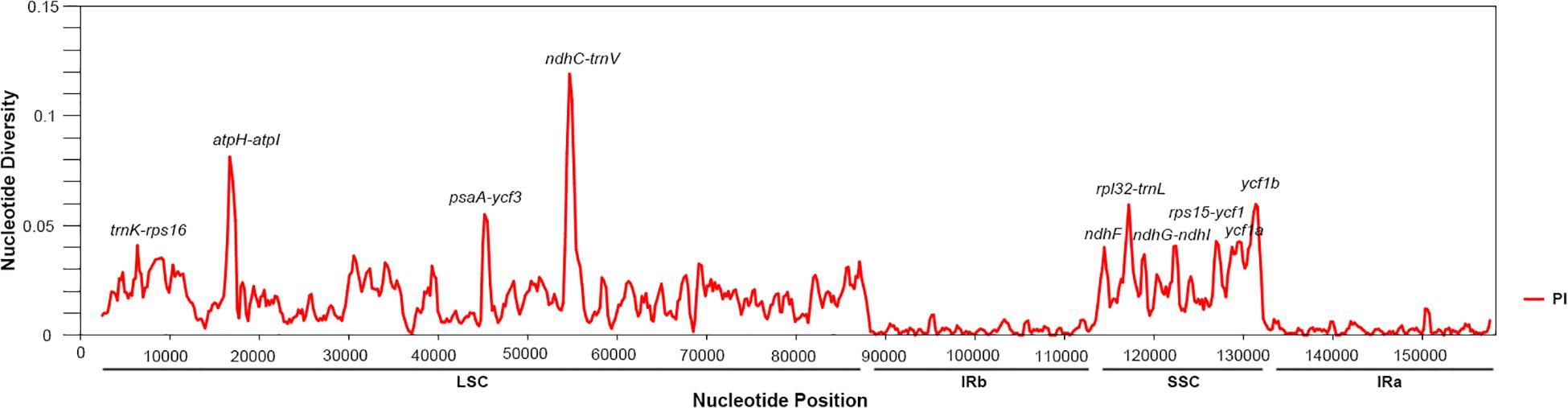
Figure 3. Sliding window analysis of the Salvia chloroplast genomes (step size, 200 bp; window length, 600 bp). X-axis: midpoint position of the window; Y-axis: nucleotide diversity in each window.
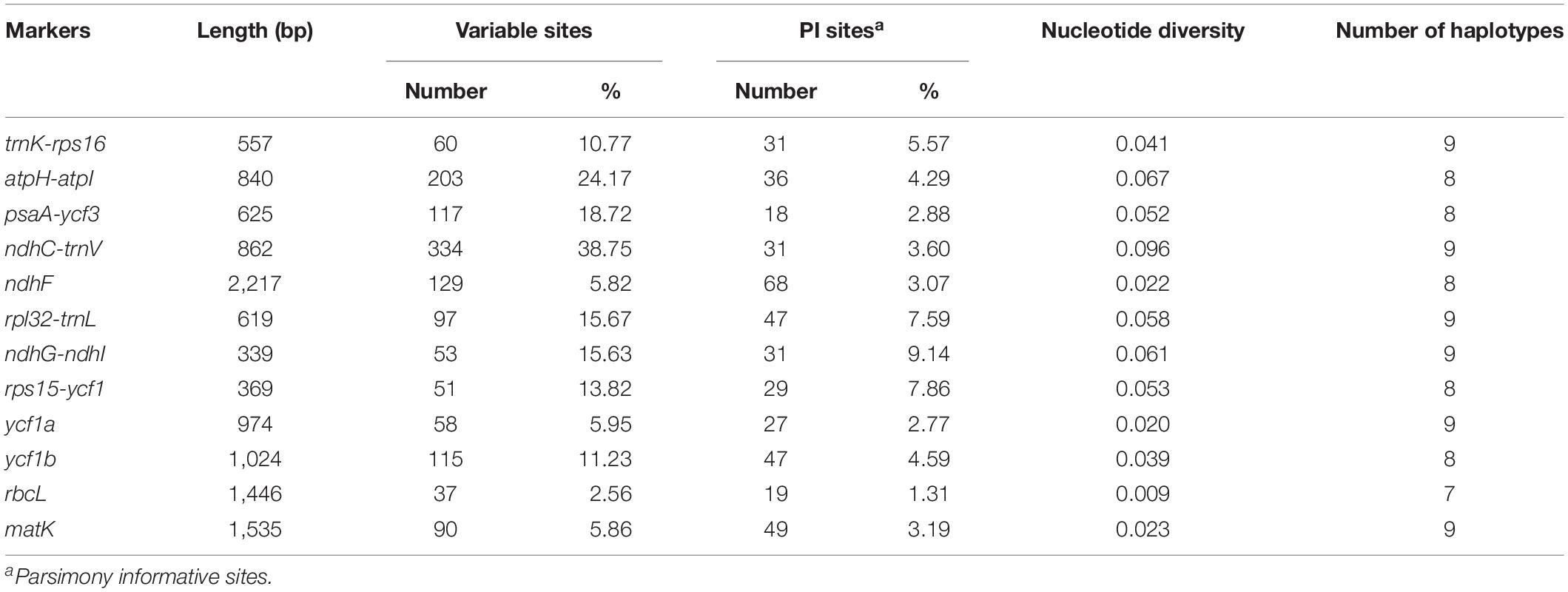
Table 2. Variability in the two universal chloroplast DNA barcodes and the 10 novel markers in Salvia species.
Characterization of Salvia Chloroplast Genome Transfer Into the Mitochondrial Genome
The length of the GenBank mitochondrial genome sequence of S. miltiorrhiza (499,236 bp) was found to be approximately 3.3 times longer than that of the chloroplast genome. We identified nine large chloroplast genome fragments in the mitochondrial genome, including both genes and intergenic regions. The fragments ranged from 1,737 to 9,734 bp and retained >97% of their sequence identity with their original chloroplast counterparts. These fragments had a total length of 49,895 bp, accounting for ∼32.97% of the chloroplast genome (Figure 4 and Supplementary Table S1). Eleven intact chloroplast genes (ndhB, rps7, rps12, ycf2, rpl23, atpE, atpB, rbcL, psbB, petL, and petG), six tRNAs (trnL-CAA, trnV-GAC, tRNA-Ile, trnM-CAU, trnW-CCA, and trnP-UGG), one rRNA (rrn23), and numerous partial genes and intergenic spacer regions were identified.
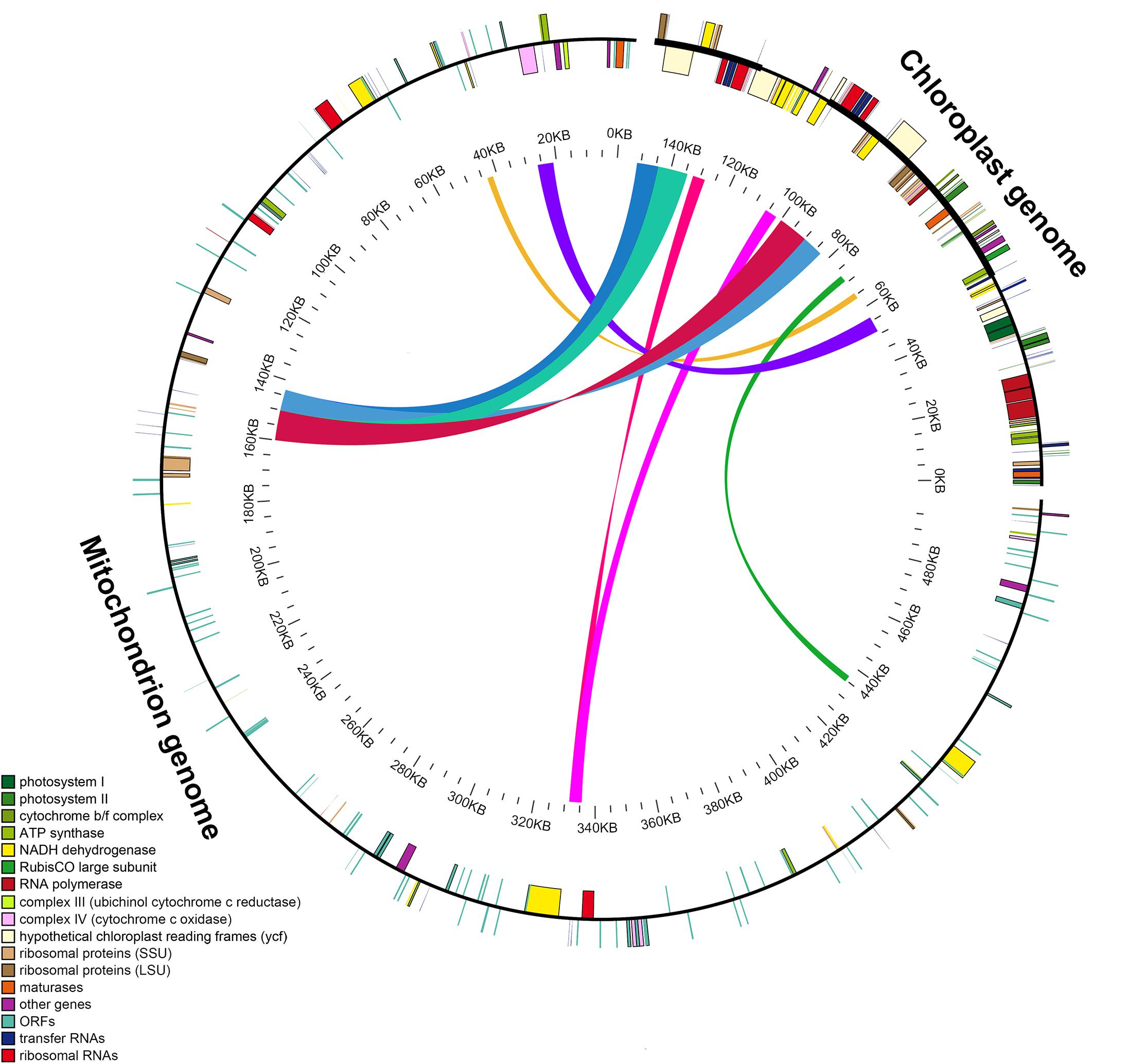
Figure 4. Schematic for the chloroplast-to-mitochondrial gene transfer in Salvia species. A colored line within the circle represents the areas of the chloroplast genome that were transferred across the specified position in the mitochondrial genome. Genes shown inside and outside the circle have been transcribed in a clockwise and counterclockwise manner, respectively.
Phylogenetic Analysis
In this study, the phylogenetic position of Salvia in the lamiids was standardized using chloroplast genomes (Figure 5). Three separate methods were used for conducting the phylogenetic analyses of the chloroplast genomes: ML, BI, and MP. Approximately identical topologies were generated by the ML and BI methods, with a high support for the majority of the branches (Supplementary Figure S2). However, marginally different positions of a few taxa were revealed by the MP trees compared with those by ML and BI (Supplementary Figure S3). Despite these variations, a majority of taxonomical relationships were highly supported and well resolved, which supported that the use of chloroplast genomic data promoted the resolution of phylogenetic analyses. Thus, the phylogenetic tree showed that S. miltiorrhiza f. alba, S. miltiorrhiza, Salvia bulleyana, and Salvia przewalskii were clustered on a single terminal branch.
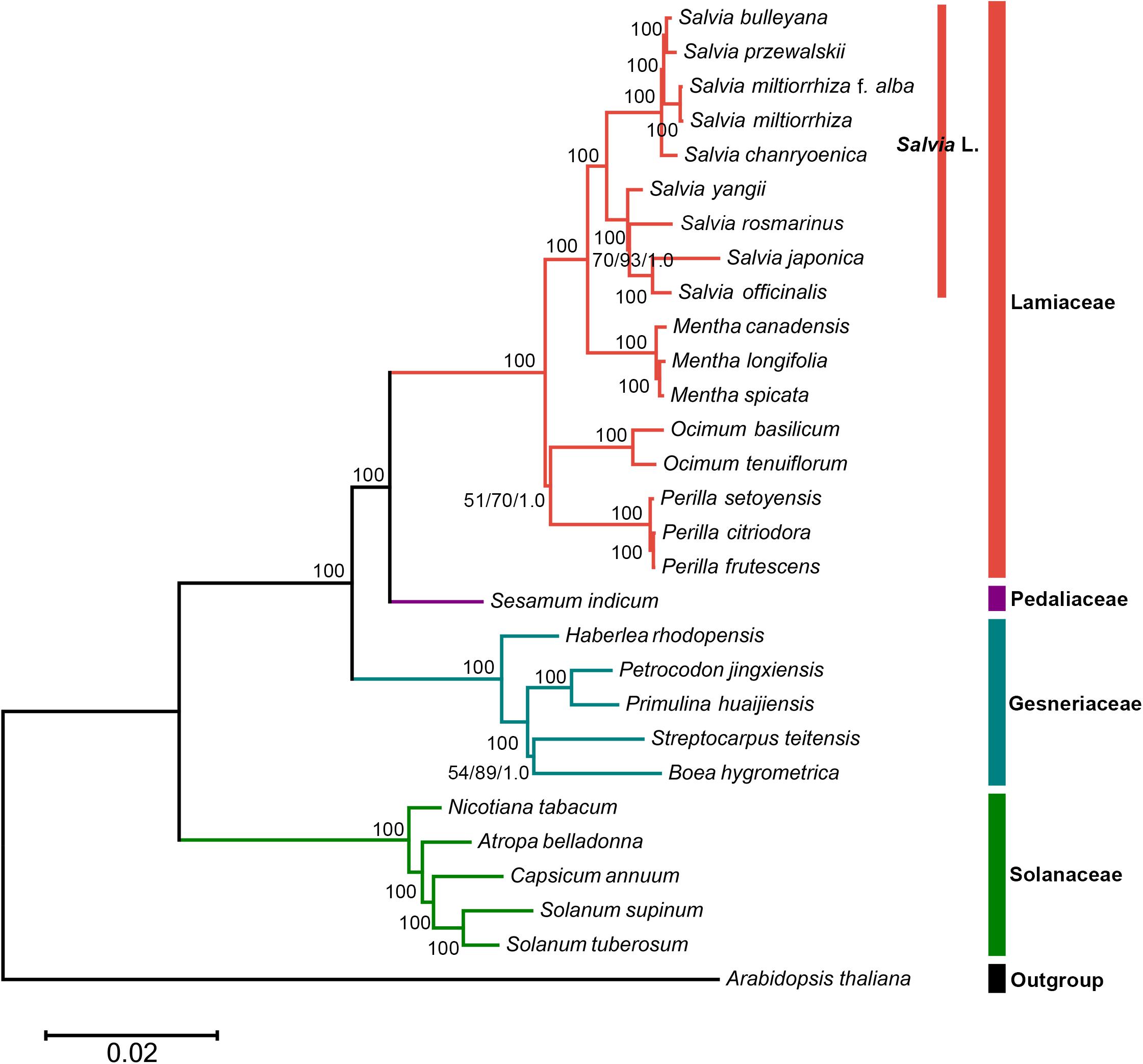
Figure 5. The whole chloroplast genome sequence-based maximum likelihood (ML) phylogenetic tree of the nine Salvia species with 19 related species in the lamiids. Numbers posted with branches are the ML bootstrap values, maximum parsimony (MP) bootstrap values, and Bayesian posterior probabilities, respectively. 100 indicate 100% ML and MP bootstrap support and 1.0 Bayesian posterior probability.
Discussion
Characterization of Complete Chloroplast Genome Structures
This is the first study to report the complete chloroplast genome of S. yangii and S. miltiorrhiza f. alba. We found the genomic structures along with gene type, number, and order to be fairly identical among the nine Salvia chloroplast genomes, with the exception of variations in the numbers of SNPs and small Indels (Qian et al., 2013; He et al., 2017; Du et al., 2019; Liang et al., 2019; Tao et al., 2019; Zhang X.J. et al., 2020). Thus, the study of the complete chloroplast genomes would afford useful genetic information to study the authentication, breeding, and evolutionary similarities/differences between Salvia species.
Comparative Analysis of Salvia Chloroplast Genomes and DNA Barcodes
The chloroplast genomes were found to be fairly conserved across the nine taxa, except for a few variable regions, based on the results of the comparative analysis. The coding regions contained most of the conserved sequences, whereas the non-coding regions had most of the variable sequences. Consistent with the results of similar studies on other plants, the LSC and SSC regions were less conserved than the IR region (Figure 1; Song et al., 2019).
DNA barcoding was first proposed in 2003 (Hebert et al., 2003). DNA barcoding is a new technique that is widely used as a biological tool for breeding, species identification, and evolutionary research (Techen et al., 2014; Mishra et al., 2016). The herbal medicine industry relies on the identification of novel plant species. Due to their high medicinal value and cost-effective processing, it has become necessary to develop easy and safe methods for the identification and development of Salvia species.
The chloroplast genomes possess a smaller size and multiple copies in a cell compared with the nuclear genome. Furthermore, due to the presence of adequate interspecific divergence, in chloroplast genomes, the best species authentication methods are based on chloroplast genome-based DNA barcodes (Hollingsworth et al., 2011).
Here, we found an increase in the number of variable sites in the following 10 specific regions based on the results of pairwise chloroplast genomic alignment and SNP analysis: trnK-rps16, atpH-atpI, psaA-ycf3, ndhC-trnV, ndhF, rpl32-trnL, ndhG-ndhI, rps15-ycf1, ycf1a, and ycf1b. Thus, Salvia species may be detected using these regions as novel candidate fragments. However, further experiments are required to support this Salvia chloroplast sequence data.
Chloroplast Genome Fragments Were Found in the Mitochondrial Genome
Specific information pertaining to the intracellular gene transfer between different genomes (mitochondrial, nuclear, and chloroplast) has been disclosed through sequencing analysis (Timmis et al., 2004; Nguyen et al., 2020). Previous research has detected high amounts of transfer of nuclear DNA from the organelle in angiosperms (Hazkani-Covo et al., 2010; Smith, 2011; Park et al., 2014). Additionally, a characteristic feature of long-term evolution has been identified as chloroplast-to-mitochondrial gene transfer (Gui et al., 2016; Nguyen et al., 2020). Here, in Salvia species, we identified nine large chloroplast genome (32.97% of the chloroplast genome) fragments in the mitochondrial genomes.
Phylogenetic Analysis of Salvia Species
The whole chloroplast genome sequence-based phylogenetic tree was built to explore the evolutionary similarities/differences between Salvia species and between genera in the lamiids. We found that S. przewalskii, S. miltiorrhiza, S. bulleyana, and S. miltiorrhiza f. alba were clustered on a single terminal branch. Several studies have revealed similarities in appearance and characteristics between these species (Li et al., 2008; Wang et al., 2014). Regarding their compositions, common substituents of S. miltiorrhiza (S. bulleyana, S. przewalskii, and S. miltiorrhiza f. alba) shared the maximum chemical composition of S. miltiorrhiza (Danshen) (Wang et al., 2018).
Data Availability Statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT012420 and MT012421. Chloroplast genome raw sequencing data have been deposited at the NCBI Sequence Read Archive (SRA) under accession PRJNA646330.
Author Contributions
ZL and CG conceived the study and acquired the funding. CG performed the data analyses and drafted the earlier version of the manuscript. CW, QZ, XZ, MW, RC, and YZ revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Clinical Medicine +X, The Affiliated Hospital of Qingdao University, and Qingdao University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing. We finally thank the reviewers for reviewing the manuscript and providing valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.574962/full#supplementary-material
References
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Boachon, B., Buell, C. R., Crisovan, E., Dudareva, N., Garcia, N., Godden, G., et al. (2018). Phylogenomic mining of the mints reveals multiple mechanisms contributing to the evolution of chemical diversity in lamiaceae. Mol. Plant 11, 1084–1096. doi: 10.1016/j.molp.2018.06.002
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brunkard, J. O., Runkel, A. M., and Zambryski, P. C. (2015). Chloroplasts extend stromules independently and in response to internal redox signals. Proc. Natl. Acad. Sci. U.S.A. 112, 10044–10049. doi: 10.1073/pnas.1511570112
Chen, W., and Chen, G. X. (2017). Danshen (Salvia miltiorrhiza Bunge): a prospective healing sage for cardiovascular diseases. Curr. Pharm. Des. 23, 5125–5135. doi: 10.2174/1381612823666170822101112
Douglas, S. E. (1998). Plastid evolution: origins, diversity, trends. Curr. Opin. Genet. Dev. 8, 655–661. doi: 10.1016/s0959-437x(98)80033-6
Drew, B. T., Gonzalez-Gallegos, J. G., Xiang, C. L., Kriebel, R., Drummond, C. P., Walker, J. B., et al. (2017). Salvia united: the greatest good for the greatest number. Taxon 66, 133–145. doi: 10.12705/661.7
Du, Y., Wang, Y. Y., Xiang, C. L., and Yang, M. Q. (2019). Characterization of the complete chloroplast genome of salvia przewalskii Maxim (Lamiaceae), a substitute for Dan-Shen Salvia miltiorrhiza Bunge. Mitochond. DNA Part B Resour. 4, 981–982. doi: 10.1080/23802359.2019.1581107
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Gui, S. T., Wu, Z. H., Zhang, H. Y., Zheng, Y. Z., Zhu, Z. X., Liang, D. Q., et al. (2016). The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 6:11. doi: 10.1038/srep30158
Hazkani-Covo, E., Zeller, R. M., and Martin, W. (2010). Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6:834. doi: 10.1371/journal.pgen.1000834
He, Y., Han, L., Liu, Y., Tian, N., Su, X., and Wang, Z. (2017). Complete sequence analysis of chloroplast genome of salvia japonica. Bull. Bot. Res. 37, 572–578.
Hebert, P. D. N., Ratnasingham, S., and Dewaard, J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. B Biol. Sci. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Hollingsworth, P. M., Graham, S. W., and Little, D. P. (2011). Choosing and using a plant DNA barcode. PLoS One 6:e019254. doi: 10.1371/journal.pone.0019254
Hu, G. X., Xiang, C. L., Liu, E. D., Dong, H. J., and Funamoto, T. (2016). Karyotypic study of eighteen taxa of Salvia (Lamiaceae) from China. Caryologia 69, 50–57. doi: 10.1080/00087114.2015.1109955
Hu, H., Hu, Q. J., Al-Shehbaz, I. A., Luo, X., Zeng, T. T., Guo, X. Y., et al. (2016). Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 7:1826. doi: 10.3389/fpls.2016.01826
Huang, J., Yu, Y., Liu, Y. M., Xie, D. F., He, X. J., and Zhou, S. D. (2020). Comparative chloroplast genomics of Fritillaria (Liliaceae), inferences for phylogenetic relationships between Fritillaria and Lilium and plastome evolution. Plants Basel 9:133. doi: 10.3390/plants9020133
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, M. H., Li, Q. Q., Zhang, C. H., Zhang, N., Cui, Z. H., Huang, L. Q., et al. (2013). An ethnopharmacological investigation of medicinal Salvia plants (Lamiaceae) in China. Acta Pharm. Sin. B 3, 273–280. doi: 10.1016/j.apsb.2013.06.001
Li, Z., Zaijun, Y., Xia, H., Jin, L. I, and Deguang, W. A. N. (2008). Study on the leaf epidermal structural characters of Salvia miltiorrhiza and Salvia from Sichuan. J. Sichuan Univer. Nat. Sci. Edn. 45, 674–680.
Li, Z. M., Xu, S. W., and Liu, P. Q. (2018). Salvia miltiorrhiza Burge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 39, 802–824. doi: 10.1038/aps.2017.193
Liang, C. L., Wang, L., Lei, J., Duan, B. Z., Ma, W. S., Xiao, S. M., et al. (2019). A Comparative analysis of the chloroplast genomes of four salvia medicinal plants. Engineering 5, 907–915. doi: 10.1016/j.eng.2019.01.017
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Mazandarani, M., and Ghaemi, E. (2010). Ethnopharmacological investigation of different parts of Perovskia abrotanoides Karel. Planta Med. 76, 1223–1223.
Mishra, P., Kumar, A., Nagireddy, A., Mani, D. N., Shukla, A. K., Tiwari, R., et al. (2016). DNA barcoding: an efficient tool to overcome authentication challenges in the herbal market. Plant Biotechnol. J. 14, 8–21. doi: 10.1111/pbi.12419
Nguyen, V. B., Giang, V. N. L., Waminal, N. E., Park, H. S., Kim, N. H., Jang, W., et al. (2020). Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. Ginseng Res. 44, 135–144. doi: 10.1016/j.jgr.2018.06.003
Pan, X., Dong, L., Yang, L., Chen, D., and Peng, C. (2020). Potential drugs for the treatment of the novel coronavirus pneumonia (COVID-19) in China. Virus Res. 286:198057. doi: 10.1016/j.virusres.2020.198057
Park, S., Ruhlman, T. A., Sabir, J. S. M., Mutwakil, M. H. Z., Baeshen, M. N., Sabir, M. J., et al. (2014). Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genom. 15:405. doi: 10.1186/1471-2164-15-405
Qian, J., Song, J. Y., Gao, H. H., Zhu, Y. J., Xu, J., Pang, X. H., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8:e057607. doi: 10.1371/journal.pone.0057607
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian Phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Song, Y., Zhang, Y. J., Xu, J., Li, W. M., and Li, M. F. (2019). Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Sci. Rep. 9:20401. doi: 10.1038/s41598-019-56727-x
Swofford, D. L. (1993). PAUP – a Computer-Program for phylogenetic inference using maximum parsimony. J. General Physiol. 102, A9–A9.
Tao, A., Zhao, F. Y., and Qian, J. F. (2019). The complete chloroplast genome sequence of the medicinal plant Salvia yunnanensis C. H. Wright (Lamiaceae). Mitochond. DNA Part B Resour. 4, 3603–3605. doi: 10.1080/23802359.2019.1677523
Techen, N., Parveen, I., Pan, Z. Q., and Khan, I. A. (2014). DNA barcoding of medicinal plant material for identification. Curr. Opin. Biotechnol. 25, 103–110. doi: 10.1016/j.copbio.2013.09.010
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–U116. doi: 10.1038/nrg1271
Topcu, G. (2006). Bioactive triterpenoids from Salvia species. J. Nat. Prod. 69, 482–487. doi: 10.1021/np0600402
Wang, L. L., Ma, R. F., Liu, C. Y., Liu, H. X., Zhu, R. Y., Guo, S. Z., et al. (2017). Salvia miltiorrhiza: a potential red light to the development of cardiovascular diseases. Curr. Pharm. Des. 23, 1077–1097. doi: 10.2174/1381612822666161010105242
Wang, M. X., Cui, L. C., Feng, K. W., Deng, P. C., Du, X. H., Wan, F. H., et al. (2015). Comparative analysis of Asteraceae chloroplast genomes: structural organization. RNA Editing and Evolution. Plant Mol. Biol. Rep. 33, 1526–1538. doi: 10.1007/s11105-015-0853-2
Wang, T., Liu, S.-Y., Wang, L., Wang, H.-Y., and Zhang, L. (2014). Anatomical characteristics of laminae and petioles of 11 species of Salvia and their taxonomic significance. China J. Chin. Mater. Med. 39, 2629–2634.
Wang, Y., Jiang, K., Wang, L. J., Han, D. Q., Yin, G., Wang, J., et al. (2018). Identification of Salvia species using high-performance liquid chromatography combined with chemical pattern recognition analysis. J. Separ. Sci. 41, 609–617. doi: 10.1002/jssc.201701066
Wen, L., Zhou, Z., Jiang, D., and Huang, K. (2020). Effect of Xuebijing injection on inflammatory markers and disease outcome of coronavirus disease 2019. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 32, 426–429. doi: 10.3760/cma.j.cn121430-20200406-00386
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xiang, C. L., Hu, G. X., and Peng, H. (2016). Salvia wuana (Lamiaceae), a new name for S-pauciflora E. Peter. Phytotaxa 255, 99–100. doi: 10.11646/phytotaxa.255.1.10
Xu, H. B., Song, J. Y., Luo, H. M., Zhang, Y. J., Li, Q. S., Zhu, Y. J., et al. (2016). Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant 9, 949–952. doi: 10.1016/j.molp.2016.03.010
Xu, W. Q., Losh, J., Chen, C., Li, P., Wang, R. H., Zhao, Y. P., et al. (2019). Comparative genomics of figworts (Scrophularia, Scrophulariaceae), with implications for the evolution of Scrophularia and Lamiales. J. Syst. Evol. 57, 55–65. doi: 10.1111/jse.12421
Zhang, L. L., Liu, Y. Y., Liu, Z. L., Wang, C., Song, Z. Q., Liu, Y. X., et al. (2016). Comparison of the roots of Salvia miltiorrhiza Bunge (Danshen) and its variety S. miltiorrhiza Bge f. Alba (Baihua Danshen) based on multi-wavelength HPLC-fingerprinting and contents of nine active components. Analyt. Methods 8, 3171–3182. doi: 10.1039/c6ay00185h
Zhang, X. J., Chen, C., Wang, R., Yao, Y., Liu, L. X., and Zhang, L. Y. (2020). Characterization of the complete chloroplast genome of Salvia hispanica (Lamiaceae). Mitochondr. DNA Part B Resour. 5, 1748–1750. doi: 10.1080/23802359.2020.1749162
Keywords: Salvia, chloroplast genome, DNA barcodes, gene transfer, phylogenetic
Citation: Gao C, Wu C, Zhang Q, Zhao X, Wu M, Chen R, Zhao Y and Li Z (2020) Characterization of Chloroplast Genomes From Two Salvia Medicinal Plants and Gene Transfer Among Their Mitochondrial and Chloroplast Genomes. Front. Genet. 11:574962. doi: 10.3389/fgene.2020.574962
Received: 22 June 2020; Accepted: 23 September 2020;
Published: 22 October 2020.
Edited by:
Rong Zhou, Aarhus University, DenmarkReviewed by:
Cheng Qin, Zunyi Vocational and Technical College, ChinaRajesh Kumar Gazara, Indian Institute of Technology Roorkee, India
Copyright © 2020 Gao, Wu, Zhang, Zhao, Wu, Chen, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengwen Gao, Z2FvY2hlbmd3ZW42QDEyNi5jb20=; Zhiqiang Li, bGl6cXNqdHVAMTYzLmNvbQ==