 Sateesh Maddirevula1
Sateesh Maddirevula1 Hanan E. Shamseldin1
Hanan E. Shamseldin1 Amy Sirr2
Amy Sirr2 Lama AlAbdi1,3Russell S. Lo2Nour Ewida1Mashael Al-Qahtani1Mais Hashem1Firdous Abdulwahab1Omar Aboyousef1
Lama AlAbdi1,3Russell S. Lo2Nour Ewida1Mashael Al-Qahtani1Mais Hashem1Firdous Abdulwahab1Omar Aboyousef1 Namik Kaya1Dorota Monies1May H. Salem4Naffaa Al Harbi4Hesham M. Aldhalaan5
Namik Kaya1Dorota Monies1May H. Salem4Naffaa Al Harbi4Hesham M. Aldhalaan5 Hamad Alzaidan6,7Hadeel M. Almanea8
Hamad Alzaidan6,7Hadeel M. Almanea8 Abrar K. Alsalamah9Fuad Al Mutairi10Samira Ismail11
Abrar K. Alsalamah9Fuad Al Mutairi10Samira Ismail11 Ghada M. H. Abdel-Salam11
Ghada M. H. Abdel-Salam11 Amal Alhashem7,12Ali Asery13
Amal Alhashem7,12Ali Asery13 Eissa Faqeih14Amal AlQassmi15
Eissa Faqeih14Amal AlQassmi15 Waleed Al-Hamoudi16
Waleed Al-Hamoudi16 Talal Algoufi17Mohammad Shagrani7,17
Talal Algoufi17Mohammad Shagrani7,17 Aimée M. Dudley2
Aimée M. Dudley2 Fowzan S. Alkuraya1,7*
Fowzan S. Alkuraya1,7*- 1Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
- 2Pacific Northwest Research Institute, Seattle, WA, United States
- 3Department of Zoology, College of Science, King Saud University, Riyadh, Saudi Arabia
- 4Pediatric Nephrology Service, Department of Pediatrics, King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia
- 5Department of Neuroscience, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
- 6Department of Medical Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
- 7College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 8Anatomic Pathology, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
- 9Vitreoretinal and Uveitis Divisions, King Khaled Eye Specialist Hospital, Riyadh, Saudi Arabia
- 10Medical Genetics Division, Department of Pediatrics, King Abdullah International Medical Research Centre, King Abdulaziz Medical City, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
- 11Human Genetics & Genome Research Division, Clinical Genetics Department, Center of Excellence of Human Genetics, National Research Centre, Cairo, Egypt
- 12Department of Pediatric, Prince Sultan Medical Military City, Riyadh, Saudi Arabia
- 13Section of Pediatric Gastroenterology, Children’s Specialist Hospital, King Fahad Medical City, Riyadh, Saudi Arabia
- 14Department of Pediatric Subspecialties, Children’s Hospital, King Fahad Medical City, Riyadh, Saudi Arabia
- 15Pediatric Neurology, King Saud Medical City, Riyadh, Saudi Arabia
- 16Department of Medicine, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 17King Faisal Specialist Hospital and Research Center, Organ Transplant Centre, Riyadh, Saudi Arabia
There is a growing interest in standardizing gene-disease associations for the purpose of facilitating the proper classification of variants in the context of Mendelian diseases. One key line of evidence is the independent observation of pathogenic variants in unrelated individuals with similar phenotypes. Here, we expand on our previous effort to exploit the power of autozygosity to produce homozygous pathogenic variants that are otherwise very difficult to encounter in the homozygous state due to their rarity. The identification of such variants in genes with only tentative associations to Mendelian diseases can add to the existing evidence when observed in the context of compatible phenotypes. In this study, we report 20 homozygous variants in 18 genes (ADAMTS18, ARNT2, ASTN1, C3, DMBX1, DUT, GABRB3, GM2A, KIF12, LOXL3, NUP160, PTRHD1, RAP1GDS1, RHOBTB2, SIGMAR1, SPAST, TENM3, and WASHC5) that satisfy the ACMG classification for pathogenic/likely pathogenic if the involved genes had confirmed rather than tentative links to diseases. These variants were selected because they were truncating, founder with compelling segregation or supported by robust functional assays as with the DUT variant that we present its validation using yeast model. Our findings support the previously reported disease associations for these genes and represent a step toward their confirmation.
Introduction
Mendelian (aka monogenic) diseases are collectively common despite the rarity of most individual entities. Their diagnostic landscape has been fundamentally changed by massively parallel sequencing technologies. These technological advances have enabled the rapid and high throughput screening of a large number of genes thus circumventing the historical bottleneck of sequential sequencing of genes deemed relevant to the clinical phenotype. In the case of exome or genome sequencing, an additional advantage lies in their potential to detect causal variants in genes with no established disease links in humans, i.e., novel candidates (Alkuraya, 2016). The latter scenario poses a major challenge because variants in such genes cannot be classified according to the current ACMG guidelines as pathogenic or likely pathogenic so these patient’s molecular diagnosis remains ambiguous until such time that sufficient evidence is established in the literature to confirm the disease-gene association (Richards et al., 2015). The widespread use of exome and genome sequencing clinically has made this problem more acute.
As noted by the ClinGen guidance, there are two major lines of evidence to support gene-disease association: genetic (e.g., case series) and experimental (e.g., animal model) (Strande et al., 2017). To reflect the superiority of genetic evidence, the maximum allowed score for the experimental evidence (6 points) is only half of that allowed for genetic evidence (12 points). This underscores the importance of reporting additional cases with compatible phenotypes to increase the confidence of gene-disease associations. In the case of candidate genes for autosomal recessive phenotypes, consanguineous populations offer a unique opportunity to accelerate the confirmation of these candidates (Alkuraya, 2012, 2016). The enrichment of autozygosity in consanguineous populations, as measured by the inbreeding coefficient, translates into a rich supply of homozygous variants including deleterious variants (Alkuraya, 2010). If these deleterious variants involve previously reported candidate genes and the observed phenotype is compatible, this provides an important genetic evidence especially when the variant is predicted null or segregates strongly in a multiplex family as stipulated by ClinGen.
We have previously implemented this approach to support the candidacy of dozens of previously reported candidate genes (Maddirevula et al., 2019b). This study is a continuation of that effort where we present supportive genetic evidence of 18 additional genes with only tentative gene-disease associations.
Materials and Methods
Human Subjects
Informed consent was obtained from all subjects included in this analysis in accordance with the local IRB guidelines (KFSHRC RAC# 2070 023, 2080 006, 2121 053). Phenotypic information was collected, and segregation analysis was performed where applicable among available relatives.
Autozygome Mapping, Exome Analysis, and Variant Calling
Exome analysis and variant filtering of variants by autozygome analysis was as described before (Anazi et al., 2017; Monies et al., 2019). Briefly variants were retained only if coding/splice, within autozygome, novel or very rare (MAF <0.001) in our internal database (SHGP database with 2,379 exomes) and gnomAD. HGMD reported variants and genes with OMIM entry were prioritized in the analysis. In silico (CADD, PolyPhen, SIFT, and TraP) pathogenicity was considered for the all variants.
We only included in this report variants that met both of the following criteria:
1- Variant that would have met the ACMG guidelines for pathogenic/likely pathogenic if the involved genes were to have an established association with the phenotype.
2- Variant in a gene with less than definitive link to phenotype because (a) gene has no listed OMIM phenotype, (b) gene has a listed OMIM phenotype but with a question mark, (c) gene has a listed OMIM phenotype based on a single study, or (d) variant with incompatible mode of inheritance to that reported for the respective gene in OMIM.
Yeast Experimental Methods
Unless noted, all Saccharomyces cerevisiae strains were grown at 30°C using standard media conditions and methods (Rose et al., 1990). Strains are derived from a prototrophic diploid strain (Winston et al., 1995) modified to bear a single copy of the DUT ortholog, DUT1, which is an essential yeast gene (dut1Δ0/DUT1) (Gadsden et al., 1993; Winzeler et al., 1999; Supplementary Table S1).
The allele yDUT refers to sequence encompassing 293 bp of the S. cerevisiae DUT1 promoter region, the human DUT protein coding sequence (NP_001939.1), and 202 bp of the yeast DUT1 terminator sequence. The protein coding sequence was codon optimized for expression in yeast (IDT). gBlocks of the yDUT and yDUT-R128Q alleles were synthesized (IDT) with added homology to plasmid AB523. Constructs were then assembled (NEB HiFi Assembly) into AfeI digested AB523 to generate plasmids AB527 and AB531 (Supplementary Table S2). For control strains, the native S. cerevisiae DUT1 allele, including the same regulatory regions as yDUT was PCR amplified from yeast DNA using primers with added homology to AB523 using primers DUT1_HO_F and DUT1_natNT1_R (Supplementary Table S3) and assembled into AfeI digested AB523 to create plasmid AB525. All plasmids contain targeting sequence to direct integration of the construct to a neutral location (the HO locus) in the yeast genome on chromosome IV of the yeast genome (Voth et al., 2001).
NotI digested AB525, AB527, and AB531 were transformed into a dut1Δ0/DUT1 diploid (YAD714 and YAD715). Spores bearing drug markers for both the dut1Δ0 locus and the desired yDUT or DUT1 construct were isolated by standard tetrad dissection and confirmed by PCR and Sanger’s sequencing (Supplementary Table S1). Six isolates of each genotype were grown overnight in liquid cultures (YPD 2% glucose) and then spotted onto agar plates (YPD 2% glucose). Plates were imaged after 2 days of growth at 30°C. Yeast growth was quantified as described previously (Sirr et al., 2020).
Plasmid sequences are available in GenBank (Accession numbers in Supplementary Table S1). All yeast strains and plasmids are available upon request (YWltZWUuZHVkbGV5QGdtYWlsLmNvbQ==).
Results
We report 24 patients who harbor 20 homozygous variants that met our inclusion criteria and spanned 18 genes (Table 1). Clinical details of all included families in this study are provided in Supplementary Table S4. Detailed pedigrees of the families with the segregation data are provided in Supplementary Figure S1. These variants can be grouped into three classes listed below along with all the cases contained therein:
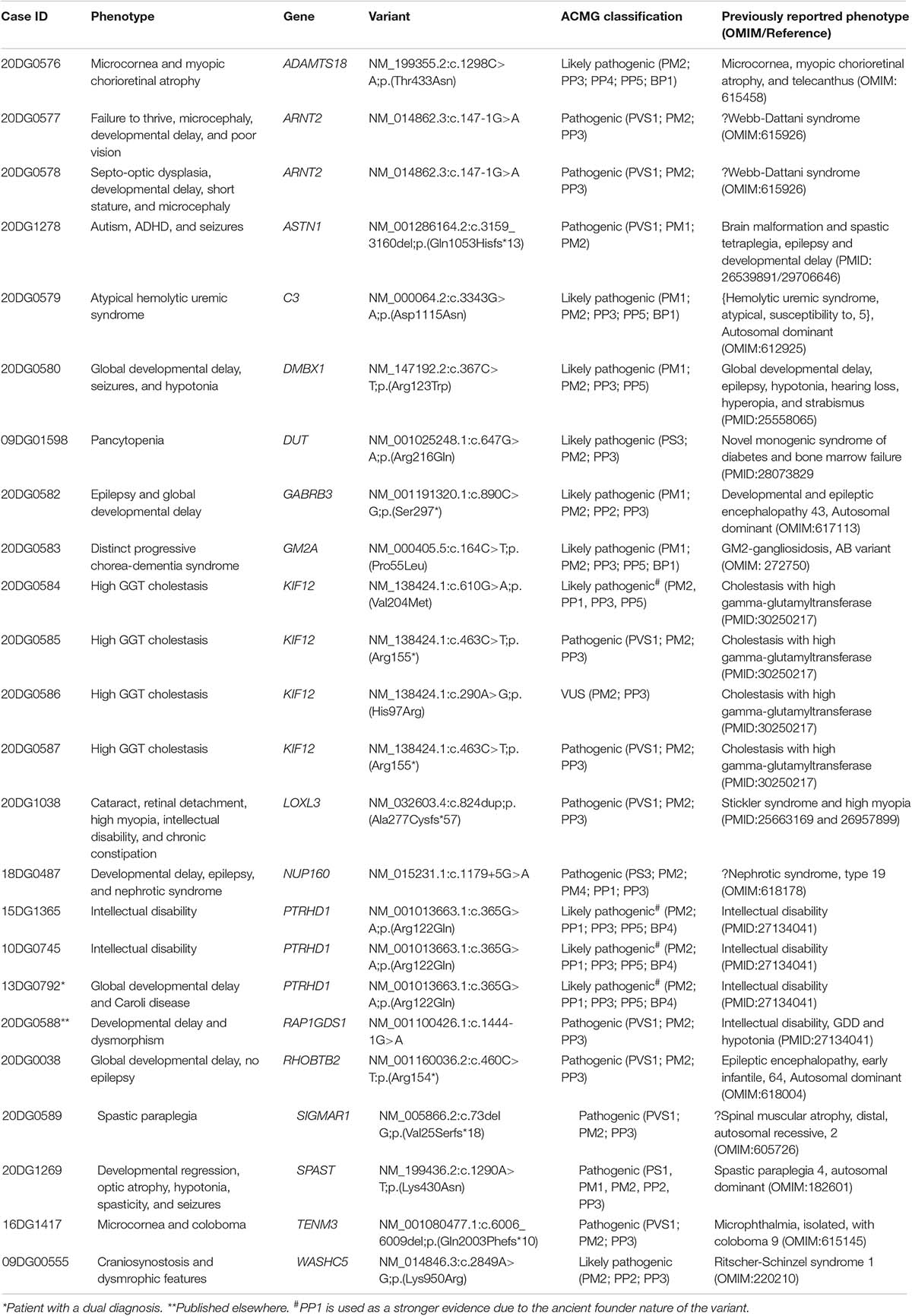
Table 1. List of the identified homozygous variants in 18 genes.
Class 1: variants that are identical to the ones that were the basis for the original report of candidacy. Since these are different patients from those originally reported, they serve as a strong line of segregation-based evidence according to the ACMG guidelines. For example, PTRHD1:NM_001013663.1:c.365G>A;p.(Arg122Gln) was reported in 2017 (Reuter et al., 2017) in an Egyptian family with mild intellectual disability (ID) prompting the authors to propose PTRHD1 as a novel candidate gene. Here we report three unrelated Saudi patients with the same variant and confirm its founder nature based on haplotype analysis. All three patients had ID with no facial dysmorphism (Figure 1A). Of note, a dual molecular diagnosis is observed in the patient 13DG0792, who is homozygous for the founder variant PTRHD1 and a ciliopathy phenotype (Caroli disease) caused by the variant WDR35 [NM_001006657.1:c.206G>A; p.(Gly69Asp)] as described before (Shaheen et al., 2016).
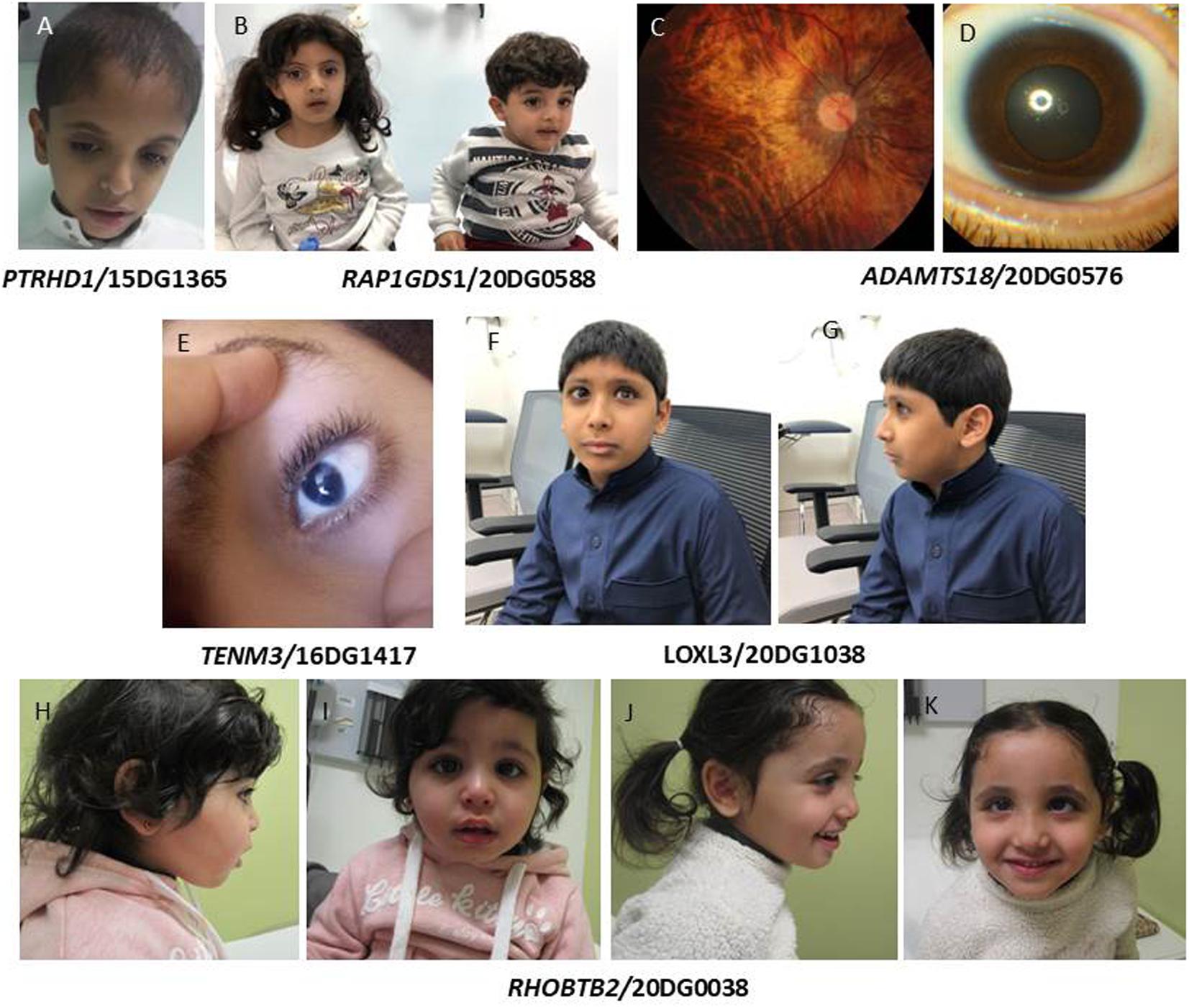
Figure 1. (A) Subject with PTRHD1 mutation lacking gross facial dysmorphism. (B) Siblings with RAP1GDS1 mutation showing small mouth, prominent nasal bridge, mild synophrys, and micrognathia. (C,D) Color fundus photo of the right eye showing myopic chorioretinal atrophy and slit-lamp photograph of the left eye showing microcornea of the patient with ADAMTS18 mutation. (E) Patient with mutation in TENM3 showing microcornia. (F,G) Facial images showing high myopia in a patient with mutation in LOXL3. (H–K) Facial images of siblings with homozygous mutation in RHOBTB2 showing depressed nasal bridge and strabismus.
Another example is the founder KIF12 variants NM_138424. 1:c.610G>A;p.(Val204Met) and NM_138424.1:c.463C>T;p. (Arg155∗) that we previously published in patients with congenital hepatic fibrosis/sclerosing cholangitis and high gamma-glutamyltransferase (GGT)-cholestasis (Maddirevula et al., 2019a), which we identified in another patient with the same phenotype. Of note, we also identified in this study a novel homozygous variant [KIF12:NM_ 138424.1:c.290A>G;p.(His97Arg)] in a patient with a late-onset phenotype (17 years) although we emphasize that this remains VUS at this point. The founder variant we identified in RAP1GDS1 (NM_001100426.1:c.1444-1G>A) is another example. This variant was reported in an apparently new syndrome of dysmorphic facies and intellectual disability (Asiri et al., 2020). We identified the same variant in the two siblings with the same phenotype (Figure 1B) (Bertoli-Avella et al., submitted).
ADAMTS18 has a listed OMIM phenotype (microcornea, myopic chorioretinal atrophy, and telecanthus, MMCAT) based on a previous study in which we reported several families with different homozygous variants (Aldahmesh et al., 2013); however, our finding has not been confirmed by follow up studies. Here, we report the identification of a new case with the same founder variant [NM_001326358.2:c.782C>A;p.(Thr261Asn)] who, in addition to the classical findings of microcornea, myopic chorioretinal atrophy and telecanthus (Figures 1C,D), also has epilepsy and hypertension that may or may not be related to congenital renal anomalies. Interestingly, we have previously argued in support of the involvement of DMBX1 in the etiology of autosomal recessive intellectual disability based on the original family and a follow up family with the same founder (NM_147192.2:c.367C>T:p.(Arg123Trp) (Maddirevula et al., 2019b). However, this gene remains with no OMIM phenotype. Here, we report a new family with the same founder and same phenotype (Table 1).
GM2A is an established gene for GM2-gangliosidosis, AB variant (MIM: 272750). An extended multiplex family was reported with a surprisingly different phenotype that lacks organomegaly and cherry red macula and instead comprises childhood onset progressive chorea-dementia syndrome, which was proposed as a novel GM2A-related phenotype (Alazami et al., 2015;Salih et al., 2015). We identified the same founder variant [NM_000405.5:c.164C>T;p.(Pro55Leu)] in a new patient with an identical phenotype (Table 1). The last example in this category is C3:NM_000064.2:c.3343G>A;p.(Asp1115Asn), which we report here for the first time in homozygosity even though it was reported twice in the heterozygous state in patients with atypical hemolytic uremic syndrome (Fremeaux-Bacchi et al., 2008;Schramm et al., 2015), just as observed in the patient we report here. Unfortunately, the ancestry of the two previously reported patients has not been described so we are unable to speculate on its potential founder nature.
Class 2: variants that expand the allelic heterogeneity of the gene-disease association. These include two unrelated patients who share a novel founder variant (as revealed by haplotype sharing) ARNT2:NM_014862.3:c.147-1G>A. We had proposed ARNT2 as a novel candidate gene for a condition characterized by hypothalamo-pituitary-frontotemporal hypoplasia with visual and renal anomalies based on a single frameshift variant in a multiplex family (Webb et al., 2013). Brain MRI for one of these two patients showed hypoplasia of the pituitary gland with no demonstrable posterior lobe or pituitary stalk and brain atrophic changes with delayed myelination (Figures 2A,B), which is consistent with the previously reported family. The canonical splicing founder variant in ARNT2 in the two siblings we report here lends further support to the original report.
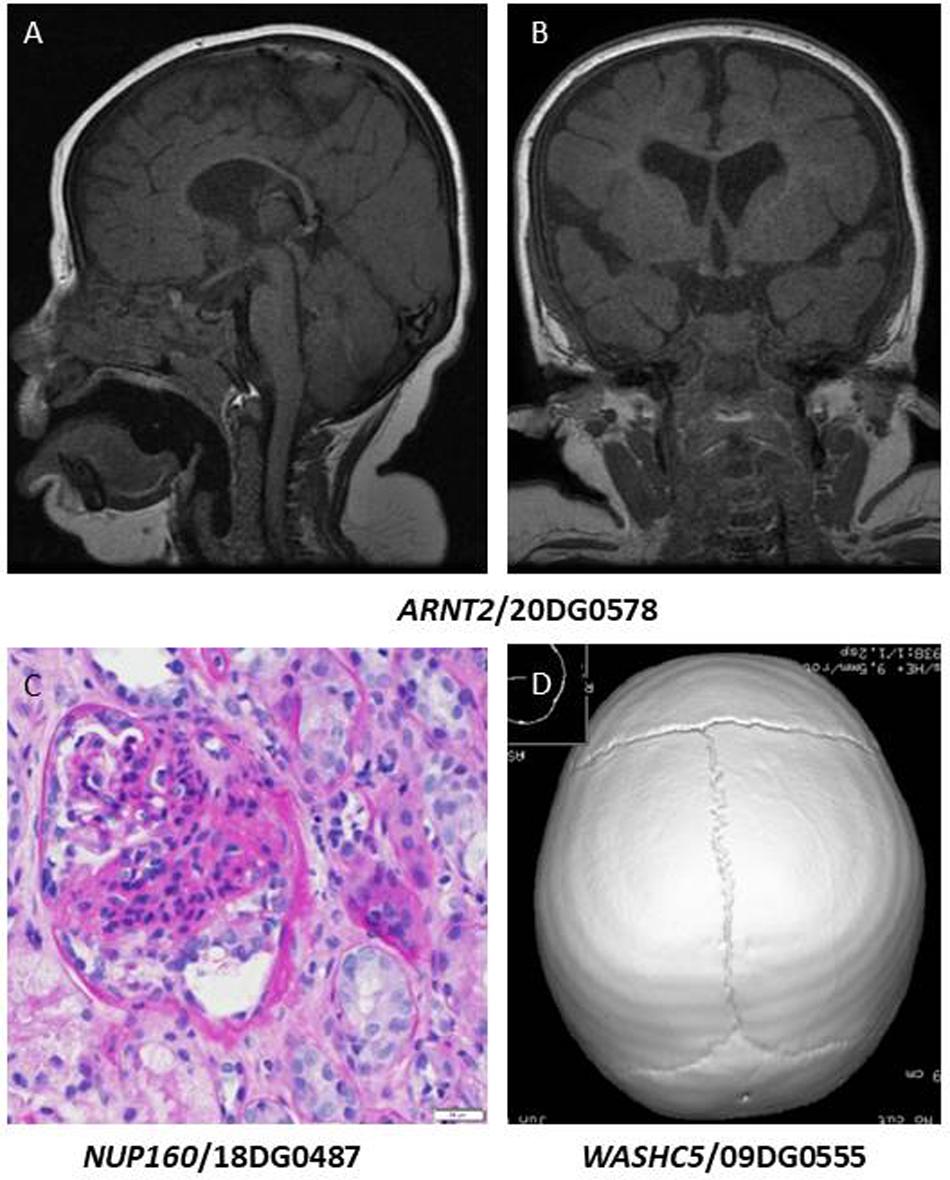
Figure 2. (A,B) Brain MRI images of the patient with homozygous ARNT2 mutation showing thinning of the corpus callosum, brain atrophic changes, and hypoplasia of the pituitary gland with no demonstrable posterior lobe bright spot or pituitary stalk. (C) Renal biopsy of the patient with NUP160 mutation showing segmental solidification of the glomerular tuft (classical segmental scar). (D) CT scan image of the patient with a variant in WASHC5 showing partial premature closure of the lambdoid suture.
Another example is TENM3 (formally ODZ3), which we had proposed as a novel gene for colobomatous microphthalmia based on a single frameshift variant in a multiplex family (Aldahmesh et al., 2012). Here, we report a novel homozygous frameshift variant [NM_001080477.1:c.6006_6009del;p.(Gln2003Phefs∗10)] in a patient with a similar phenotype (Figure 1E).
Similarly, NUP160 is a gene we had proposed as a novel candidate for steroid-resistant nephrotic syndrome based on two siblings who were compound heterozygous for missense and nonsense variants (Braun et al., 2018). The variant we report here (NM_015231.1:c.1179+5G>A) was confirmed by RTPCR to cause abnormal splicing [r.1102_1179del;p.(Phe368_Gln393del)] (Maddirevula et al., 2020). The patient has steroid-resistant nephrotic syndrome and chronic kidney disease, which is consistent with the previously reported patients (Figure 2C). However, we note the additional neurological features of intellectual disability and epilepsy, which co-segregated with the steroid-resistant nephrotic syndrome in both siblings.
Other variants in this class include SIGMAR1:NM_005866.2:c.73delG;p.(Val25Serfs∗18), which supports the involvement of this gene in autosomal recessive juvenile amyotrophic lateral sclerosis (Table 1). ASTN1 was reported as a candidate gene for brain malformation (Karaca et al., 2015), and spastic tetraplegia, epilepsy and developmental delay (Wiszniewski et al., 2018). Here we report a homozygous truncating variant [ASTN1:NM_001286164.2:c.3159_3160del;p.(Gln1053Hisfs∗13)] in a patient with autism, ADHD and seizures. LOXL3 was reported in patients with high myopia (Li et al., 2016) and in Sticklers syndrome (Alzahrani et al., 2015). Interestingly, we report a truncating variant in LOXL3 [NM_032603.4:c.824dup;p.(Ala277Cysfs∗57)] in a patient with high myopia (Figures 1F,G), retinal detachment with mild ID (IQ 62). It is unclear, however, if ID is related to the LOXL3 variant. To date a single report was published linking WASHC5 to variable craniofacial features, cerebellar and cardiac anomalies (Ritscher-Schinzel syndrome 1/MIM: 220210). We report a homozygous exonic splicing variant in WASHC5 [NM_014846.3:c.2849A>G;p.(Lys950Arg)] in a patient with a distinct clinical feature of lambdoid synostosis (Figure 2D) along with dysmrophic features like malar hypoplasia, micrognathia, mild turricephaly, and broad hallux.
The last variant in this class is worth highlighting to justify its inclusion despite being missense. DUT was published as a candidate for a novel syndromic form of diabetes involving bone marrow failure based on a single missense variant (Dos Santos et al., 2017). The patient we present here has the following novel missense variant DUT:NM_001025248.1:c.647G>A;p.(Arg216Gln), which we included because of additional functional evidence in yeast. Arginine at 216 is highly conserved from human to yeast (Figure 3A). The yeast and human orthologs are highly conserved (55% amino acid identity across the length of the protein) (Tchigvintsev et al., 2011) and a previous publication had shown that the human ortholog is able to functionally replace (genetically complement) a deletion of the yeast gene (Hamza et al., 2015; Kachroo et al., 2015). Because the yeast Dut1 protein localizes to the nucleus and cytoplasm (Huh et al., 2003), we used the DUT protein sequence corresponding to the nuclear isoform DUT-N (NP_001939.1), which lacks the 93 amino acid mitochondrial leader sequence (Uniprot: P33316). As such, our allele yDUT-R128Q, harbors the same amino acid change as NM_001025248.1:c.647G>A;p.(Arg216Gln). Consistent with previous studies (Hamza et al., 2015; Kachroo et al., 2015), our yeast codon optimized version of the DUT protein coding sequence integrated into the yeast genome and expressed from the yeast DUT1 transcriptional promoter (section “Materials and Methods”) was able to completely (100%) complement loss of the yeast gene (Figure 3B). In contrast, the same construct harboring the Arg216Gln allele, exhibited a significant loss of function phenotype, with 63% growth relative to wildtype (Figure 3B).
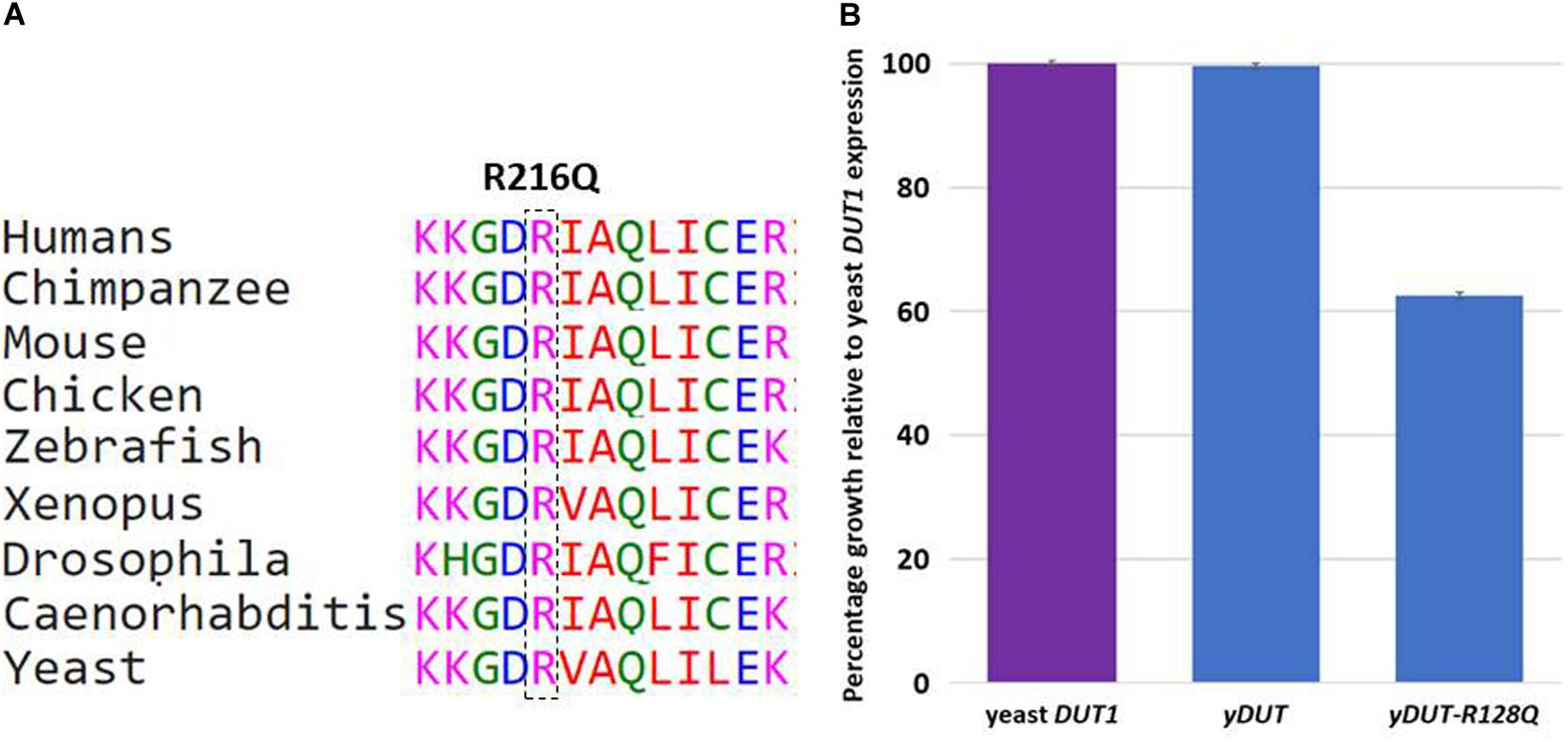
Figure 3. (A) Conservation of variant (p.Arg216Gln) in DUT. (B) Yeast assays of human DUT variant suggest partial loss of function for the human Arg216Gln equivalent, yDUT-R128Q. Values represent the mean and standard error of the mean for each genotype, rescaled to make the wildtype yDUT mean value equal to 1. The purple bar is the growth value of the control strains expressing the yeast ortholog DUT1. Blue bars are growth values of yeast strains expressing either the wildtype yDUT construct or the same construct harboring the R128Q variant. Error bars are standard error of the mean. Assays were performed in replicate with 6 independently constructed clones each assayed 12 times.
Class 3: These are variants identified in homozygosity in genes with associations only to dominant phenotypes in OMIM. We opted to include these variants given their important implications in the interpretation of the lack of phenotype among the obligate carrier parents as described before (Monies et al., 2017). The first example is RHOBTB2, a gene only linked to autosomal dominant epileptic encephalopathy, early infantile, 64 (MIM: 618004). Here, we report the identification of a homozygous nonsense variant [NM_001160036.2:c.460C>T:p.(Arg154∗)] in three siblings with global developmental delay, facial dysmorphism (Figures 1H–K), normal brain MRI and no epilepsy, thus expanding the phenotype in addition to expanding the mode of inheritance of RHOBTB2-related neurodevelopmental disease.
GABRB3 is another example since this is a gene only associated with autosomal dominant epileptic encephalopathy, early infantile, 43. We identified a homozygous nonsense variant [NM_001191320.1:c.890C>G;p.(Ser297∗)] in a patient with dystonia and infantile spasm with profound global developmental delay (Table 1).
Another example is SPAST, an established gene for autosomal dominant spastic paraplegia 4 (MIM: 182601). We report three siblings with developmental regression, optic atrophy, central hypotonia, peripheral spasticity and seizures who have a pathogenic homozygous missense variant [NM_199436.2:c.1290A>T;p.(Lys430Asn)]. In these three examples, the parents were normal clinically, consistent with the bona fide recessive inheritance we propose here.
Discussion
The determination of whether a given Mendelian gene-disease association is established has been largely subjective until the publication of an evidence-based framework by ClinGen, which represents a major step toward standardization (Strande et al., 2017). For example, the framework proposes replacing the binary “candidate” vs. “established” with a much more nuanced labeling system that more truly reflects the spectrum of evidence. The supporting evidence, when present, is classified into definitive, strong, moderate and limited. Importantly, the framework also accounts for evidence that contradicts previously reported gene-disease associations rendering the latter disputed or even refuted. Indeed, we have previously shown the power of the autozygome to generate pathogenic variants in individuals that lack phenotypes previously associated with the respective genes (Shamia et al., 2015). However, our focus in this study is on variants that lend supportive evidence.
The current OMIM listing of Mendelian phenotypes does not necessarily reflect the ClinGen framework since it fails to include genes with definitive evidence e.g., LBR, MYO9A, and VPS8 while including others with only limited evidence even without the cautionary question mark e.g., PCK2 has a listed OMIM phenotype even though no single patient with PCK2 mutation has been reported to date (Stark and Kibbey, 2014). Nonetheless, we have opted to use OMIM as a starting point given its authoritative standing in the clinical genetics community, and the genes we report in this study, as originally intended, only have “moderate” (e.g., TENM3, SIGMAR1, ARNT2, and C3) or “limited” (e.g., NDUFA12 and NUP160) evidence supporting their involvement in human diseases leaving room for further support as we hope to have accomplished through our analysis. Because our goal in this paper is to enhance the interpretation of variants, we opted to also include recessive variants in genes with links only to dominant phenotypes. Establishing bona fide recessive inheritance of these genes greatly influences the interpretation of apparently pathogenic variants in asymptomatic heterozygous individuals who would otherwise be considered as non-penetrant individuals. The impact this change of interpretation has on recurrence risk estimates cannot be overemphasized.
It should be noted that the variants we report do not necessarily make the respective gene-disease associations “definitive.” Instead, they corroborate the previously published association and can be considered collectively with future reported evidence in an iterative process as outlined by the ClinGen framework. Nonetheless, clinical molecular laboratories will find this and similar reports helpful in their determination of genes that are relevant to the tested patient’s phenotype. One should also note that a given gene may have several disease associations and the evidence for each must be weighted differently. For example, while the association between C3 and C3 deficiency has “definitive” supportive evidence, C3-atypical hemolytic uremic syndrome association has “limited” supportive evidence; hence the value of the variant presented in this study.
Although the supportive evidence presented in this study is overwhelmingly genetic in nature, we also share experimental evidence in support of the pathogenesis of the missense variant identified in DUT. Since the case reported by Dos Santos et al. (2017), no follow up studies have been published on the involvement of DUT in the pathogenesis of the syndrome of diabetes with bone marrow failure. Without functional validation, the novel missense variant we encountered in a patient with that syndrome will not have much weight due to lack of compelling segregation. However, the strong conservation of this gene in yeast presented an opportunity to validate its deleterious effect on the gene function, which strengthens its relevance as a supportive evidence. Indeed, the use of yeast as a model organism as a high throughput system for testing VUS in genes that are conserved has seen increasing use in recent years (Hamza et al., 2015; Sun et al., 2016; Sirr et al., 2020).
In summary, we present data that support 18 previously reported gene-disease associations. This approach also allowed us to identify homozygous pathogenic variants in autosomal dominant genes like RHOBTB3, SPAST, and GABRB3. The identification of these variants in the homozygous state despite their rarity is an obvious advantage of conducting this kind of analysis in a highly consanguineous population. The benefits of this approach, however, extend to the global clinical genetics community.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the FSA RkFsS3VyYXlhQGtmc2hyYy5lZHUuc2E=.
Ethics Statement
The studies involving human participants were reviewed and approved by the Office of Research Ethics at the King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
SM performed the analysis, collected and organized the data, and wrote the manuscript. HS, LA, NK, and DM performed analysis of exomes and validation of variants. MH, OA and FA coordinated with the patient for sampling. NE and MA-Q performed segregation analysis. MHS, NA, HMAld, HA, HMAlm, AKA, FAM, SI, GA-S, AAl, AAs, EF, AA, WA, TA, and MS referred the patients for genetic test and provided phenotypic data. AS performed the yeast validation work. AD supervised the yeast validation work and wrote the manuscript. FSA designed, supervised the project, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LA was funded by Researchers Supporting Project number (RSP-2020/181), King Saud University, Riyadh, Saudi Arabia. AD was funded by NIH/NIGMS R01GM134274.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the study families for their enthusiastic participation. We also thank the Genotyping and Sequencing Core Facilities at KFSHRC for their technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.580484/full#supplementary-material
Supplementary Figure S1 | Pedigrees and segregation data of all the study families.
Supplementary Table S1 | S. cerevisiae strains used in this study.
Supplementary Table S2 | Plasmids used in this study.
Supplementary Table S3 | Oligonucleotides and gBlocks (IDT) used in this study.
Supplementary Table S4 | Clinical synopsis of the 24 patients described in this study. ACMG, American College of Medical Genetics; SHGP, Saudi Human Genome Program; HGMD, Human Gene Mutation Database.
References
Alazami, A. M., Patel, N., Shamseldin, H. E., Anazi, S., Al-Dosari, M. S., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10, 148–161. doi: 10.1016/j.celrep.2014.12.015
Aldahmesh, M. A., Alshammari, M. J., Khan, A. O., Mohamed, J. Y., Alhabib, F. A., and Alkuraya, F. S. (2013). The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum. Mutat. 34, 1195–1199. doi: 10.1002/humu.22374
Aldahmesh, M. A., Mohammed, J. Y., Al-Hazzaa, S., and Alkuraya, F. S. (2012). Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genet. Med. 14, 900–904. doi: 10.1038/gim.2012.71
Alkuraya, F. S. (2010). Autozygome decoded. Genet. Med. 12, 765–771. doi: 10.1097/gim.0b013e3181fbfcc4
Alkuraya, F. S. (2012). Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. Chapter 6:Unit612.
Alkuraya, F. S. (2016). Discovery of mutations for Mendelian disorders. Hum. Genet. 135, 615–623. doi: 10.1007/s00439-016-1664-8
Alzahrani, F., Al Hazzaa, S. A., Tayeb, H., and Alkuraya, F. S. (2015). LOXL3, encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Hum. Genet. 134, 451–453. doi: 10.1007/s00439-015-1531-z
Anazi, S., Maddirevula, S., Faqeih, E., Alsedairy, H., Alzahrani, F., Shamseldin, H. E., et al. (2017). Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatry 22, 615–624. doi: 10.1038/mp.2016.113
Asiri, A., Aloyouni, E., Umair, M., Alyafee, Y., Al Tuwaijri, A., Alhamoudi, K. M., et al. (2020). Mutated RAP1GDS1 causes a new syndrome of dysmorphic feature, intellectual disability & speech delay. Ann. Clin. Transl. Neurol. 7, 956–964. doi: 10.1002/acn3.51059
Braun, D. A., Lovric, S., Schapiro, D., Schneider, R., Marquez, J., Asif, M., et al. (2018). Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Invest. 128, 4313–4328.
Dos Santos, R. S., Daures, M., Philippi, A., Romero, S., Marselli, L., Marchetti, P., et al. (2017). dUTPase (DUT) is mutated in a novel monogenic syndrome with diabetes and bone marrow failure. Diabetes 66, 1086–1096. doi: 10.2337/db16-0839
Fremeaux-Bacchi, V., Miller, E. C., Liszewski, M. K., Strain, L., Blouin, J., Brown, A. L., et al. (2008). Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112, 4948–4952.
Gadsden, M. H., Mcintosh, E. M., Game, J. C., Wilson, P. J., and Haynes, R. H. (1993). dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 12, 4425–4431. doi: 10.1002/j.1460-2075.1993.tb06127.x
Hamza, A., Tammpere, E., Kofoed, M., Keong, C., Chiang, J., Giaever, G., et al. (2015). Complementation of yeast genes with human genes as an experimental platform for functional testing of human genetic variants. Genetics 201, 1263–1274. doi: 10.1534/genetics.115.181099
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., et al. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691.
Kachroo, A. H., Laurent, J. M., Yellman, C. M., Meyer, A. G., Wilke, C. O., and Marcotte, E. M. (2015). Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925. doi: 10.1126/science.aaa0769
Karaca, E., Harel, T., Pehlivan, D., Jhangiani, S. N., Gambin, T., Coban Akdemir, Z., et al. (2015). Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88, 499–513. doi: 10.1016/j.neuron.2015.09.048
Li, J., Gao, B., Xiao, X., Li, S., Jia, X., Sun, W., et al. (2016). Exome sequencing identified null mutations in LOXL3 associated with early-onset high myopia. Mol. Vis. 22, 161–167.
Maddirevula, S., Alhebbi, H., Alqahtani, A., Algoufi, T., Alsaif, H. S., Ibrahim, N., et al. (2019a). Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 21, 1164–1172. doi: 10.1038/s41436-018-0288-x
Maddirevula, S., Alzahrani, F., Al-Owain, M., Al Muhaizea, M. A., Kayyali, H. R., Alhashem, A., et al. (2019b). Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 21, 736–742. doi: 10.1038/s41436-018-0138-x
Maddirevula, S., Kuwahara, H., Ewida, N., Shamseldin, H. E., Patel, N., Alzahrani, F., et al. (2020). Analysis of transcript-deleterious variants in Mendelian disorders: implications for RNA-based diagnostics. Genome Biol. 21:145.
Monies, D., Abouelhoda, M., Assoum, M., Moghrabi, N., Rafiullah, R., Almontashiri, N., et al. (2019). Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 104, 1182–1201.
Monies, D., Maddirevula, S., Kurdi, W., Alanazy, M. H., Alkhalidi, H., Al-Owain, M., et al. (2017). Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet. Med. 19, 1144–1150. doi: 10.1038/gim.2017.22
Reuter, M. S., Tawamie, H., Buchert, R., Hosny Gebril, O., Froukh, T., Thiel, C., et al. (2017). Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74, 293–299. doi: 10.1001/jamapsychiatry.2016.3798
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rose, M. D., Winston, F. M., and Hieter, P. (1990). Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Salih, M. A., Seidahmed, M. Z., El Khashab, H. Y., Hamad, M. H., Bosley, T. M., Burn, S., et al. (2015). Mutation in GM2A leads to a progressive chorea-dementia syndrome. Tremor. Other. Hyperkinet. Mov. 5:306. doi: 10.5334/tohm.246
Schramm, E. C., Roumenina, L. T., Rybkine, T., Chauvet, S., Vieira-Martins, P., Hue, C., et al. (2015). Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 125, 2359–2369. doi: 10.1182/blood-2014-10-609073
Shaheen, R., Patel, N., Shamseldin, H., Alzahrani, F., Al-Yamany, R., ALMoisheer, A., et al. (2016). Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet. Med. 18, 686–695. doi: 10.1038/gim.2015.147
Shamia, A., Shaheen, R., Sabbagh, N., Almoisheer, A., Halees, A., and Alkuraya, F. S. (2015). Revisiting disease genes based on whole-exome sequencing in consanguineous populations. Hum. Genet. 134, 1029–1034. doi: 10.1007/s00439-015-1580-3
Sirr, A., Lo, R. S., Cromie, G. A., Scott, A. C., Ashmead, J., Heyesus, M., et al. (2020). A yeast-based complementation assay elucidates the functional impact of 200 missense variants in human PSAT1. J. Inherit. Metab. Dis. 43, 758–769. doi: 10.1002/jimd.12227
Stark, R., and Kibbey, R. G. (2014). The mitochondrial isoform of phosphoenolpyruvate carboxykinase (PEPCK-M) and glucose homeostasis: has it been overlooked? Biochim. Biophys. Acta Gen. Sub. 1840, 1313–1330. doi: 10.1016/j.bbagen.2013.10.033
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., Distefano, M., Dwight, S. S., et al. (2017). Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100, 895–906. doi: 10.1016/j.ajhg.2017.04.015
Sun, S., Yang, F., Tan, G., Costanzo, M., Oughtred, R., Hirschman, J., et al. (2016). An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 26, 670–680. doi: 10.1101/gr.192526.115
Tchigvintsev, A., Singer, A. U., Flick, R., Petit, P., Brown, G., Evdokimova, E., et al. (2011). Structure and activity of the Saccharomyces cerevisiae dUTP pyrophosphatase DUT1, an essential housekeeping enzyme. Biochem. J. 437, 243–253. doi: 10.1042/bj20110304
Voth, W. P., Richards, J. D., Shaw, J. M., and Stillman, D. J. (2001). Yeast vectors for integration at the HO locus. Nucleic Acids Res. 29:e59.
Webb, E. A., Almutair, A., Kelberman, D., Bacchelli, C., Chanudet, E., Lescai, F., et al. (2013). ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 136, 3096–3105. doi: 10.1093/brain/awt218
Winston, F., Dollard, C., and Ricupero-Hovasse, S. L. (1995). Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11, 53–55. doi: 10.1002/yea.320110107
Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B., et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. doi: 10.1126/science.285.5429.901
Keywords: autozygosity, diabetes, yeast, founder mutation, disease-gene associations
Citation: Maddirevula S, Shamseldin HE, Sirr A, AlAbdi L, Lo RS, Ewida N, Al-Qahtani M, Hashem M, Abdulwahab F, Aboyousef O, Kaya N, Monies D, Salem MH, Al Harbi N, Aldhalaan HM, Alzaidan H, Almanea HM, Alsalamah AK, Al Mutairi F, Ismail S, Abdel-Salam GMH, Alhashem A, Asery A, Faqeih E, AlQassmi A, Al-Hamoudi W, Algoufi T, Shagrani M, Dudley AM and Alkuraya FS (2020) Exploiting the Autozygome to Support Previously Published Mendelian Gene-Disease Associations: An Update. Front. Genet. 11:580484. doi: 10.3389/fgene.2020.580484
Received: 06 July 2020; Accepted: 30 November 2020;
Published: 31 December 2020.
Edited by:
Katrina Tatton-Brown, St George’s, University of London, United KingdomReviewed by:
Roddy Walsh, Amsterdam University Medical Center (UMC), NetherlandsDiana Baralle, University of Southampton, United Kingdom
Copyright © 2020 Maddirevula, Shamseldin, Sirr, AlAbdi, Lo, Ewida, Al-Qahtani, Hashem, Abdulwahab, Aboyousef, Kaya, Monies, Salem, Al Harbi, Aldhalaan, Alzaidan, Almanea, Alsalamah, Al Mutairi, Ismail, Abdel-Salam, Alhashem, Asery, Faqeih, AlQassmi, Al-Hamoudi, Algoufi, Shagrani, Dudley and Alkuraya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fowzan S. Alkuraya, RkFsS3VyYXlhQGtmc2hyYy5lZHUuc2E=