 Shanshan Chen1,2†
Shanshan Chen1,2† Xiao Wang3,4†
Xiao Wang3,4† Yangzi Wang5†
Yangzi Wang5† Guanghui Zhang6Wanling Song5
Guanghui Zhang6Wanling Song5 Xiao Dong5
Xiao Dong5 Michael L. Arnold7Wen Wang8,9,10Jianhua Miao11*
Michael L. Arnold7Wen Wang8,9,10Jianhua Miao11* Wei Chen12,13,14*
Wei Chen12,13,14* Yang Dong5,11,12,14*
Yang Dong5,11,12,14*- 1School of Life Sciences, Zhengzhou University, Zhengzhou, China
- 2BGI College, Zhengzhou University, Zhengzhou, China
- 3State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 4Jiaxing Synbiolab Biotechnology Co., Ltd., Jiaxing, China
- 5College of Biological Big Data, Yunnan Agricultural University, Kunming, China
- 6The Key Laboratory of Medicinal Plant Biology of Yunnan Province, National and Local Joint Engineering Research Center on Germplasm Innovation and Utilization of Chinese Medicinal Materials in Southwest China, Yunnan Agricultural University, Kunming, China
- 7Department of Genetics, University of Georgia, Athens, GA, United States
- 8State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 9Center for Ecological and Environmental Sciences, Northwestern Polytechnical University, Xi’an, China
- 10Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, China
- 11Guangxi Key Laboratory of Medicinal Resources Protection and Genetic Improvement, Guangxi Botanical Garden of Medicinal Plants, Nanning, China
- 12State Key Laboratory of Conservation and Utilization of Bio-Resources in Yunnan, Yunnan Agricultural University, Kunming, China
- 13College of Agronomy and Biotechnology, Yunnan Agricultural University, Kunming, China
- 14Yunnan Research Institute for Local Plateau Agriculture and Industry, Kunming, China
Achlorophyllous plants are full mycoheterotrophic plants with no chlorophyll and they obtain their nutrients from soil fungi. Gastrodia elata is a perennial, achlorophyllous orchid that displays distinctive evolutionary strategy of adaptation to the non-photosynthetic lifestyle. Here in this study, the genome of G. elata was assembled to 1.12 Gb with a contig N50 size of 110 kb and a scaffold N50 size of 1.64 Mb so that it helped unveil the genetic basics of those adaptive changes. Based on the genomic data, key genes related to photosynthesis, leaf development, and plastid division pathways were found to be lost or under relaxed selection during the course of evolution. Thus, the genome sequence of G. elata provides a good resource for future investigations of the evolution of orchids and other achlorophyllous plants.
Introduction
In the autotroph-dominant plant world, the symbiotic relationship between plants and fungi plays an indispensable role in the maintenance of ecosystem (Read, 1991). In an extreme situation, some plants (at least 50 independent origins) have become solely dependent upon their fungal associates for energy source and other nutrients during the course of evolution (Merckx and Freudenstein, 2010). These fully mycoheterotrophic plants often lack a functional photosynthetic mechanism, so they are termed as “non-photosynthetic plants (Merckx et al., 2009).”
The loss of photosynthesis was independently evolved over 40 times in a diverse range of plant families and genera (Bidartondo, 2005). As an evolutionary adaptation to a low-light undergrowth environment, this extreme phenotype was also associated with some quite remarkable parallel evolutionary traits in plant, such as smaller biomass, specialized leaves without stomata, lack of root hair, reduced vascular tissues, and miniatured seeds (Leake, 1994). Even though multiple studies of plastomes from heterotrophic plants have showed reduction in plastome sizes and housekeeping gene numbers (Krause, 2008; Barrett and Davis, 2012; Wicke et al., 2013, 2014), the nuclear genomic basis of the other parallel traits in non-photosynthetic plants remains elusive.
All members of the Orchidaceae family (up to 26,567 species in 880 genera) rely on fungal associates in some or all stages of their life cycle (Leake, 1994; Cai et al., 2015). In particular, more than 200 orchid species from a myriad genera are fully mycoheterotrophic and non-photosynthetic (Leake, 1994; Merckx et al., 2009; Merckx and Freudenstein, 2010). Since Gastrodia is one of the largest genera consisting of fully mycoheterotrophic species from a wide geographic area (Cribb and Killmann, 2010), members in this genus are excellent models to investigate the reconfigured traits in non-photosynthetic plants.
In China, Gastrodia elata is cultivated for medicinal uses. It is a perennial, non-photosynthetic orchid with an enlarged rhizome and vestigial leaves on an upright flower-bearing stem (Figure 1A). The completion of its life cycle requires at least two types of fungi: Armillaria and Mycena (Lan et al., 1994; Ning et al., 2010) (Supplementary Figure 1). In this study, we presented a high quality de novo assembly of the G. elata nuclear genome, which had a higher coverage, contig N50 size, and Benchmarking Universal Single-Copy Ortholog (BUSCO) completeness than that of a previous report (Yuan et al., 2018). Compared with the genomes of photosynthetic orchid species, such as Phalaenopsis equestris (Cai et al., 2015), Apostasia Shenzhenica (Zhang et al., 2017), and Dendrobium officinale (Yan et al., 2015), the new G. elata nuclear genome and plastome assemblies showed key gene loss and relaxed selection related to its non-functional photosystem and retarded leaf development. These data demonstrated that the improved G. elata genome provides more insights into the genomic and evolutionary mechanisms underlying the morphological and physiological adaptations associated with a non-photosynthetic lifestyle.
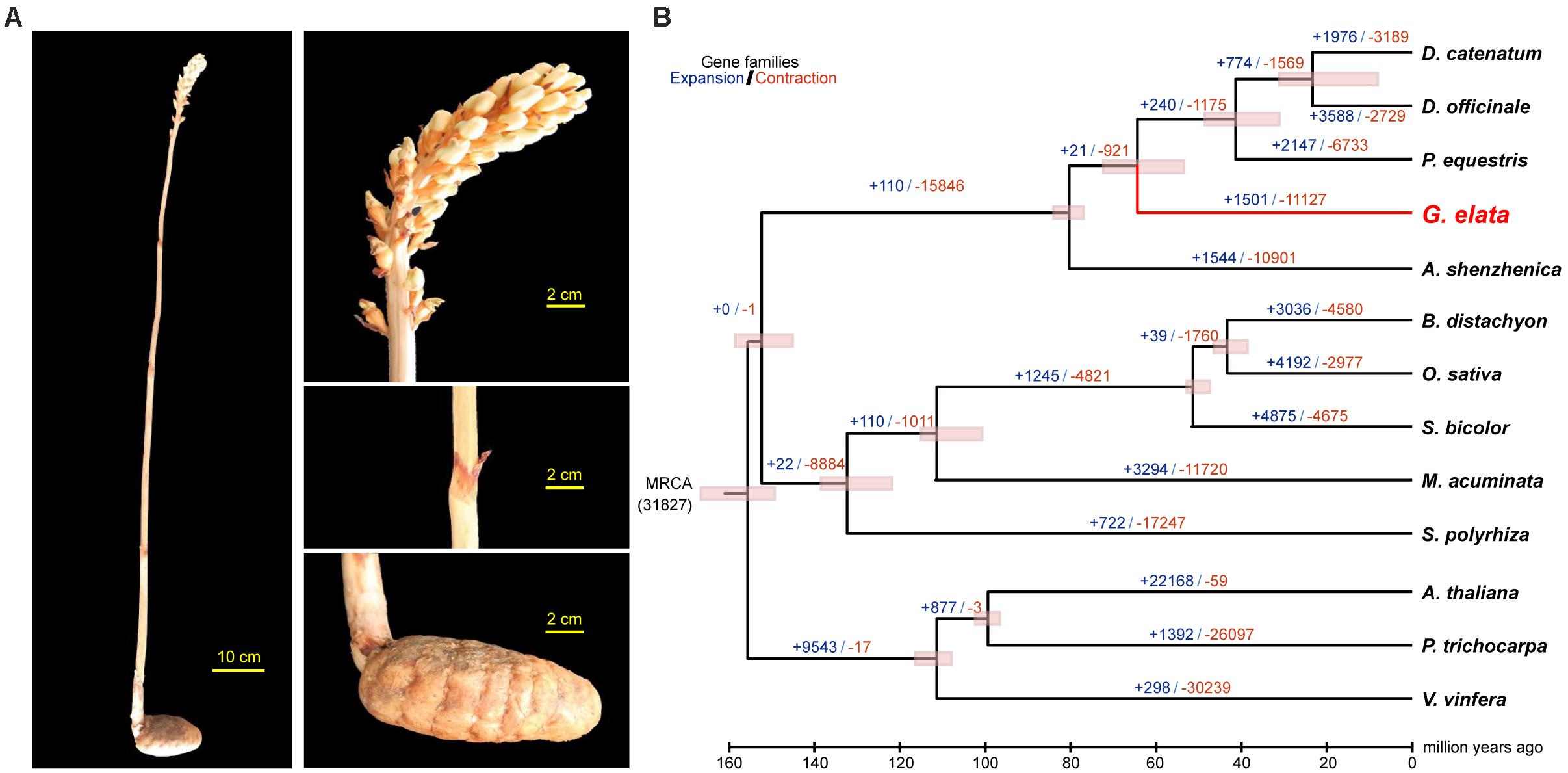
Figure 1. Divergence time and morphology of G. elata. (A) Morphology of G. elata: photos showing a whole plant, flower, stem with degraded blade and tuber. (B) Divergence times are indicated by pink bars, expanded gene family numbers by dark blue letters and contracted gene families by orange letters.
Materials and Methods
Plant Material, Genomic DNA Extraction, and Library Construction
A flowering G. elata Bl. f. glauca S. Chow plant was collected from Zhaotong, Yunnan Province, China (103°43′E, 27°20′N). Genomic DNA was extracted from fresh stems by the Qiagen DNeasy Plant Mini Kit (Cat. No. 69104). After genomic DNA purification and quality validation, 10 paired-end and mate-pair DNA libraries with insert sizes of 169 bp (×2), 300 bp (×2), 374 bp, 545 bp, 753 kb, 2 kb, 5 kb, and 10 kb were constructed using library construction kits (Illumina) as previously described (Liang et al., 2015; Supplementary Table 1).
Genome Sequencing and Assembly
A whole-genome shotgun strategy was adopted to sequence and assemble the genome of G. elata. All ten DNA libraries were sequenced on an Illumina HiSeq 2000 platform and 483.07 Gb of data was obtained. The genome size of G. elata was estimated according to the 17-mer frequency distribution with the formula: Genome size = K-mer_num/Peak_depth. Total of 84.67 Gb data was retained for 17-mer analysis. The 17-mer frequency distribution plot shows the peak at 66 (Supplementary Figure 2), and the total K-mer count is 90,917,019,718, then the genome size is estimated as 1.378 Gb.
The assembly of G. elata genome was carried out using the SOAPdenovo. A de Bruijn graph was constructed using the parameter “−K 83 −d 3”, then the graph was simplified using “−M 3” parameters to clip tips, remove low coverage links, merge bubbles, and solve tiny repeats. Those repeats on the simplified graph were broken at the boundaries and unambiguous DNA fragments were produced as contigs. Filtered sequencing data was then realigned to contigs with the parameters “−k 83”. Parameter “−F” was used to deal with the paired-end information, and unique contigs were joined to construct scaffolds. Gapcloser (v1.12) was used to fill the gaps between scaffolds with the default parameters.1 Contigs short than 100 bp were excluded within the whole assembly process.
The completeness and accuracy of the gap closed assembly was assessed using short insert-sized sequencing reads, conserved genes, and RNA-seq data. Filtered reads from three small-insert libraries (169, 374, and 753 bp) were mapped to the assembled genome using the bowtie2 (version 2.2.92) (Langmead and Salzberg, 2012) with default parameters to estimate the quality of the assembly. The overall mapping rates of these small-insert size reads is higher than 92%, suggesting that most of the G. elata genome has been assembled. BUSCOs approach (Waterhouse et al., 2017) was also used to evaluate the completeness of the G. elata genome. Three hundred and three (303) conserved single-copy orthologs has been used as reference, of which 248 (81.85%) were identified in our genome assembly (Table 1). Over 74.59 millions of cleaned RNA-seq reads from three different G. elata tissues (flower, stem, and tuber) were mapped to the assembled genome with the default settings using the TopHat2 (version 2.0.143) (Trapnell et al., 2009; Supplementary Table 2), and we observed the average mapping ratio of 83%, further validate the high completeness of G. elata genome assembly.
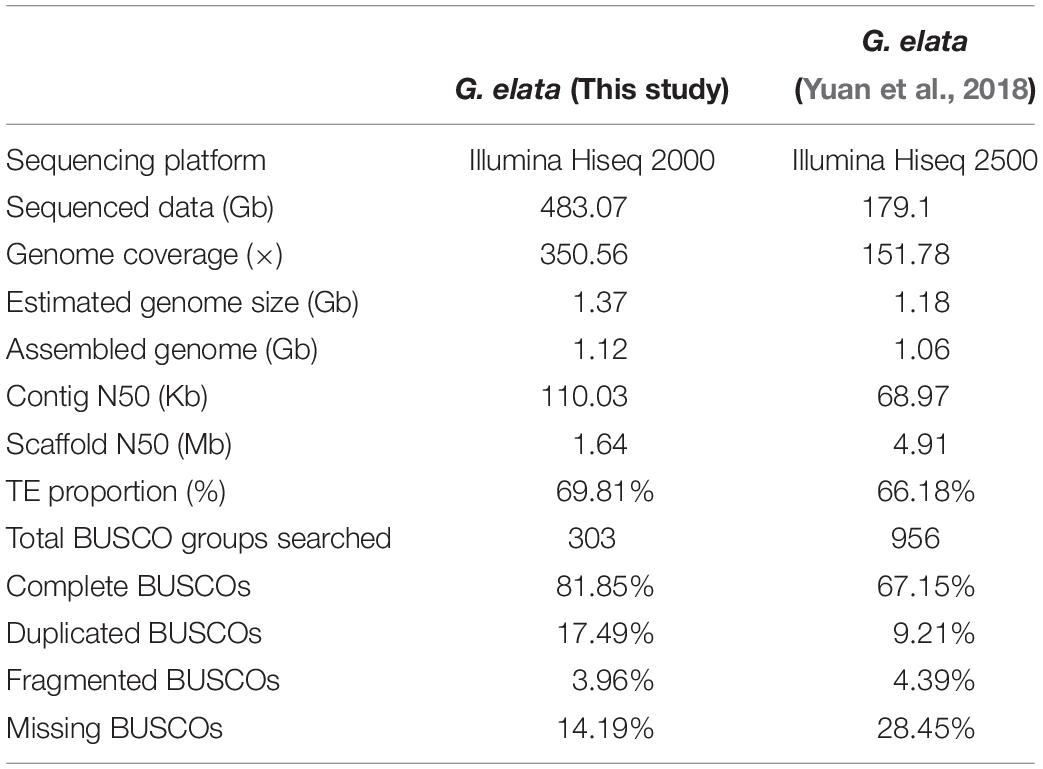
Table 1. Various assembly parameters of the G. elata genome.
RNA-Sequencing Data Analysis
Overall, 82.37 million transcriptome raw reads were obtained from G. elata leaf, stem, and tuber tissues (Supplementary Table 2). First, all RNA-sequencing (RNA-seq) data were filtered by the strict quality control process, then about 74.59 millions cleaned reads were de novo assembled using trinity4 (Grabherr et al., 2011) with default settings to yield transcripts that prepared for the genome annotation. Next, all RNA-seq reads were mapped to the G. elata genome assembly using the TopHat2 (version 2.0.14; see text footnote 3) (Trapnell et al., 2009) with default settings. The fragments per kilobase of exon model per million reads mapped (FPKM) of each predicted protein-coding gene was calculated by the Cufflinks5 using default parameters. FPKM > = 0.05 was set as the threshold to identify expressed genes.
Repeat Sequence Annotation
RepeatMasker (version 3.2.6) with the Repbase TE library was used to identify known transposable elements (TEs) with the default parameters. The library was constructed by generating the consensus sequence of each TE family, which was used for the RepeatMasker to identify additional high and medium copy repeats in the G. elata genome assembly. TRF with parameters set to “Match = 2, Mismatch = 7, Delta = 7, PM = 80, PI = 10, Minscore = 50, and MaxPeriod = 2000” was used to predict tandem repeats.
Protein-Coding Gene Annotation
A combination of de novo-, transcriptome-based prediction, and homology aligning were used to process gene annotation. Gene sets from ten species (Arabidopsis thaliana, Brachypodium distachyon, Oryza sativa, Sorghum bicolor, Solanum tuberosum, Triticum aestivum, Hordeum vulgare, P. equestris, Dendrobium catenatum, and A. shenzhenica) were used for homology-based predictions, one species at a time. We used the TBLASTN to search the non-redundant protein sequences of each gene set with an E-value < 1e-2. Only regions with homologous blocks longer than 80% of the query protein were retained. The best hits were selected. Then, the EVM was used to construct the gene structures. After repeat sequences were masked using the homology-based approach, three softwares, AUGUSTUS (Stanke et al., 2006), SNAP (Korf, 2004), and GlimmerHMM (Majoros et al., 2004), were used for the genes de novo prediction. Information obtained from the homology-based predictions and de novo predictions were then integrated in the GLEAN to generate a consensus gene set. Finally, RNA-seq data from a single G. elata plant’s flower, stem, and tuber tissues were obtained and assembled for the facilitating of the protein-coding gene annotation.
Functional Annotation
Best hits were selected from alignments to the SwissProt and TrEMBL databases to assign gene function information. Gene motifs and domains were identified using the InterProScan56 (Jones et al., 2014) by alignment to databases including the ProDom, PRINTS, Pfam, SMART, PANTHER, and PROSITE. According to the corresponding SwissProt and TrEMBL entries information, Gene Ontology (GO) terms and ID for each gene was obtained. Kyoto Encyclopedia of Genes and Genomes (KEGG) protein database was used as reference for gene alignments to obtain KEGG IDs as well as the corresponding pathways information.
Gene Family Clustering
All proteins from selected 13 species (Gastodia elata, P. equestris, D. officinale, D. catenatum, A. shenzhenica, B. distachyon, O. sativa, S. bicolor, Spirodela polyrhiza, Musa acuminata, A. thaliana, Populus trichocarpa, and Vitis vinifera) were aligned using the BLASTP, then, the gene families were defined using the OrthoMCL (Li et al., 2003). CAFÉ (De Bie et al., 2006) was then used to identify gene family expansions and contractions in G. elata. To identify gene family clusters in these species and G. elata, we performed all-versus-all protein alignments using the BLASTP with the E-value threshold set to “1e-5.” We used the OrthoMCL to process high scoring segment pairs. The MCL module from OrthoMCL was then used to define final paralogous and orthologous genes with the parameter set as “−abc –I = 1.5”.
Phylogenetic Tree Construction and Divergence Time Estimation
Those single-copy orthologs identified from gene family cluster analysis of the aforementioned species were used to construct a phylogenetic tree. MUSCLE version 3.67 (Edgar, 2004) was used with default settings to perform multiple sequence alignments. Fourfold degenerate sites of genes were collected and concatenated into a “super sequence” for each species. We used the MrBayes8 (Huelsenbeck and Ronquist, 2001) to reconstruct phylogenetic trees between species. The “MCMCTREE” module from the PAML package9 (Yang, 2007) was used to estimate the divergence time among species.
Chloroplast Genome (Plastome) Assembly and Annotation
The clean reads were aligned to the plastomes of P. equestris and D. officinale (Jheng et al., 2012; Yang et al., 2016) using the bowtie2 with default settings, respectively. Reads extracted from the “paired-aligned” alignments were merged and assembled using the SOAPdenovo with default parameters. Contigs shorter than 100 bp were excluded. Filtered contigs were joined to scaffolds based on the paired-end information and gaps between scaffolds were closured. DOGMA10 (Wyman et al., 2004) was used to annotate the protein-coding genes and tRNA genes with the cut-off set to 80%. The boundaries of protein-coding genes and plastome structures were manually checked by comparison to the plastomes of P. equestris and D. officinale. The linear plot of G. elata plastome was yielded by the OGDRAW11 (Lohse et al., 2013), followed with some manual adjustments.
Relaxation Selection Analyses and Symbiotic Gene Analysis
These nuclear-encoded photosynthesis-related proteins were identified based on the National Center for Biotechnology Information (NCBI) database. Genome sequences of seven species (G. elata, A. shenzhenica, P. equestris, D. catenatum, D. officinale, O. sativa, and A. thaliana) were searched for all of the known plant nuclear-encoded photosynthesis-related genes. Only genes that were in a one-to-one orthologs for every pair of genomes of the seven species were used in our analyses. For genes that have more than one transcript, we aligned all of the possible transcript pairs to all seven species and retained those that provided the highest alignment scores. Alignments and consensus trees were used for posterior molecular evolutionary analysis. We used a gene-level approach based on the ratio of non-synonymous (Ka) to synonymous (Ks) substitutions rate (ω = Ka/Ks) to identify potential relaxation of selective constraints, using the CODEML likelihood ratio tests (LRTs) algorithm from the PAML package. First, we tested branch models M0, the simplest model, which has a single ω ratio for the entire tree. Subsequently, we used two-ratio models that allow a background ω ratio and a different ω on the branch of interest. For null hypotheses, we used the one-ratio model, two-ratio model, and more models with a fixed ω = 1 on the branch under analysis. The level of significance for these LRTs was calculated using a χ2 approximation, where twice the difference of log likelihood between the models (2ΔlnL) would be asymptotic to a χ2 distribution, with the number of degrees of freedom corresponding to the difference in the number of parameters between the nested models.
These Gastrodia antifungal protein (GAFP) were downloaded from the NCBI database. Genome sequences of seven species (G. elata, A. shenzhenica, P. equestris, D. catenatum, D. officinale, O. sativa, and A. thaliana) were searched for all of the known GAFP genes with blast software. Finally, the genes with E-value ≤ 1e-6 in the comparison result were selected as a candidate GAFP gene. And the protein domain of carotenoid cleavage dioxygenases (CCDs) was downloaded from the pfam website. The hmmersearch software were used for sequence alignment to identify CCDs genes in other species. The identified genes are calculated by Ka/Ks to verify whether positive selection occurred in G. elata.
High Performance Liquid Chromatography Analysis of Photosynthetic Pigments
From each sample of G. elata’s vestigial scalelike leaves and P. equestris’s and D. Officinale’s normal leaves, about 1 g of leave tissue was collected and dried as powder. Then, the dried powder was dealt with 100% 40 mL of methanol for half an hour and sonicated for an hour, and then, methanol diluted to 50 mL. The methanol extract was then filtered by the 0.45 μm membrane filter. Ten microliters of filtrate was prepared for high performance liquid chromatography (HPLC). Quantitative analysis of the photosynthetic pigments of three species was performed using the chromatographic column Inertsil ODS-3 (250 mm × 4.6 mm, 5 μm) and the column temperature was maintained at 25°C with the flow rate set as 1 mL × min–1. The mobile phase was consisted of A: acetonitrile and 0.05 mol/L of Tris–HC1 buffer (70:3); and B: methanol and n-hexane (5:1). Gradient elution was then used with the following system: 100% A at initiation, 100% A at 18 min, 100% B at 20.5 min, and 100% B at 46 min. 445 nm was used to detect photosynthetic pigments.
Results
De novo Assembly of the G. elata Genome
The genome size of G. elata was estimated to be 1.378 Gb using the K-mer distribution analysis (Supplementary Figure 2). All 483.07 Gb clean data (350.56 × genome coverage) were de novo assembled into 69,353 contigs (1.11 Gb in total length) with a contig N50 size of 110.03 kb, and 45,884 scaffolds (1.12 Gb in total length) with a scaffold N50 size of 1.64 Mb (Supplementary Table 3). As a result of high genome coverage, our G. elata assembly had a much longer contig N50 size (110.03 kb) than that from a previous study (68.9 kb, Table 1; Yuan et al., 2018). Preliminary evaluation of the quality of our assembly showed that at least 92% of the clean reads from different insert-sizes of paired-end libraries could be mapped back to the assembled genome (Supplementary Table 4). In addition, over 82.37 million RNA sequencing reads from each of the flower, stem, and tuber tissues were generated to further verify the quality of the assembly. The overall mapping ratios of these reads to the genome assembly were 90.7% for the flower, 93.8% for the stem, and 67.1% for the tuber (Supplementary Table 2), indicating the high quality of the G. elata genome.
The completeness of the genic regions and other genomic elements in our G. elata genome was evaluated by the BUSCOs approach (Waterhouse et al., 2017). The result showed that 248 out of 303 (81.85%) near-universal single-copy orthologs were identified in our G. elata genome. This number was much higher than that reported in the previous study (67.15% BUSCO completeness, Table 1; Yuan et al., 2018). Additionally, 12 BUSCO genes (3.96%) had fragmented matches, and 43 BUSCO genes (14.19%) were missing in our assembly. Both parameters were lower than that reported in the previous study (4.39 and 28.45%, respectively, Table 1; Yuan et al., 2018). These data demonstrated that our G. elata genome had a much higher completeness for subsequent functional analysis.
Gastrodia elata Genome Repeat Analysis
Repetitive DNA elements constituted approximately 68.34% of the acquired genome (Supplementary Table 5). This proportion was higher than those of the other orchids genomes, including P. equestris (62%) (Cai et al., 2015), D. officinale (63.33%) (Liang et al., 2015), and A. shenzhenica (42.05%) (Zhang et al., 2017). Long interspersed nuclear elements (LINEs) and DNA transposons constituted 6.20 and 7.49% of the total genome assembly, respectively (Supplementary Table 5). The long terminal repeats (LTRs) were the most abundant TEs, which constituted 59.95% of the genome (Supplementary Table 5). Notably, Gypsy-type (37.41%) and Copia-type (8.07%) TEs accounted for most of the LTRs (Supplementary Table 6). Sequence divergence line plot of Copia and Gypsy types of TEs shows that both of the two types experienced two recently expansion events during the same periods (Supplementary Figure 3a). Compared to the conserved single copy genes, those duplicated genes (“accessory genes”) in G. elata are located closer to the TEs (Fisher’s exact test, P < 0.005), suggesting their contribution in gene copy number variation and the important role in G. elata evolution (Supplementary Figure 3b).
Orthologous Gene Analysis in the G. elata Genome
Overall, we identified 24,484 protein-coding genes (Supplementary Table 7) with about 10.05 kb average length in the G. elata genome that belonged to 11,065 unique gene families (Supplementary Table 8). Besides, we also identified a myriad of microRNAs (1,125), transfer RNAs (1,123), ribosomal RNAs (1,596), and small nuclear RNAs (868; Supplementary Table 9) in the G. elata genome.
Phylogenetic analysis with divergence time estimation showed that the G. elata diverged from other orchids somewhere between 72.3 and 55.8 million years ago (Mya) (Figure 1B). During the course of evolution, it is apparent that 19 gene families were significantly expanded (P < 0.001; Supplementary Table 10), whereas six gene families were significantly contracted in the G. elata genome (P < 0.001; Supplementary Table 11). KEGG enrichment analyses showed that gene families involved in flavonoid biosynthesis, plant-pathogen interaction, and circadian rhythm were significantly contracted in the G. elata genome (Supplementary Table 12). In comparison, the expanded gene families are mainly involved in the glycan, sphingolipid, and galactose metabolisms, and intriguingly, also plant-pathogen interaction (Supplementary Table 13). Since some members of the plant-pathogen interaction gene families were contracted and other members were expanded, it is possible that G. elata may have rewired its pathogen resistance pathway when adapting to the low-light environment. Indeed, G. elata as a fully mycoheterotrophic plant not only needs to obtain nutrients from the fungi associates, but also have to prevent the fungi from invading into the inner most section of the tuber (Zhang, 1980).
Gastrodia elata Plastome Assembly and Relaxed Selection of Plastid Genes
The conserved plastome structure of most land plants generally is a quadripartite single circular molecule of 100–220 kb. It consists of a small and a large single-copy regions (SSC and LSC) which are separated by a pair of inverted repeats (IRs) (Sandelius and Aronsson, 2009). The plastome of the photosynthetic plant harbors around 100 essential genes that primarily encode RNAs and proteins involved in photosynthesis, transcription, and translation (Daniell et al., 2016). P. equestris and D. officinale are two photosynthetic orchids with fully functional plastomes as other land plants (Jheng et al., 2012; Yang et al., 2016). The plastomes of these two orchids (with sizes of 148,959 and 152,018 bp, respectively) both have the typical quadripartite single circular molecule structure with relatively complete photosynthesis-related gene sets (Figure 2). In comparison, the plastome of G. elata was assembled with the size of 40,037 bp, meaning that more than two thirds of the plastome sequence was lost after the transition to a low-light heterotrophic lifestyle (Figure 2). IRs are considered to play roles in plastome stabilization (Palmer and Thompson, 1982). In the G. elata plastome, one copy of IR was completely lost, and the remaining IR sequence was less than half the size of its counterparts in the other two orchids. The G. elata LSC retained less than a quarter of the sequence size compared to the two orchid plastomes, and the G. elata SSC also lost half of its size.
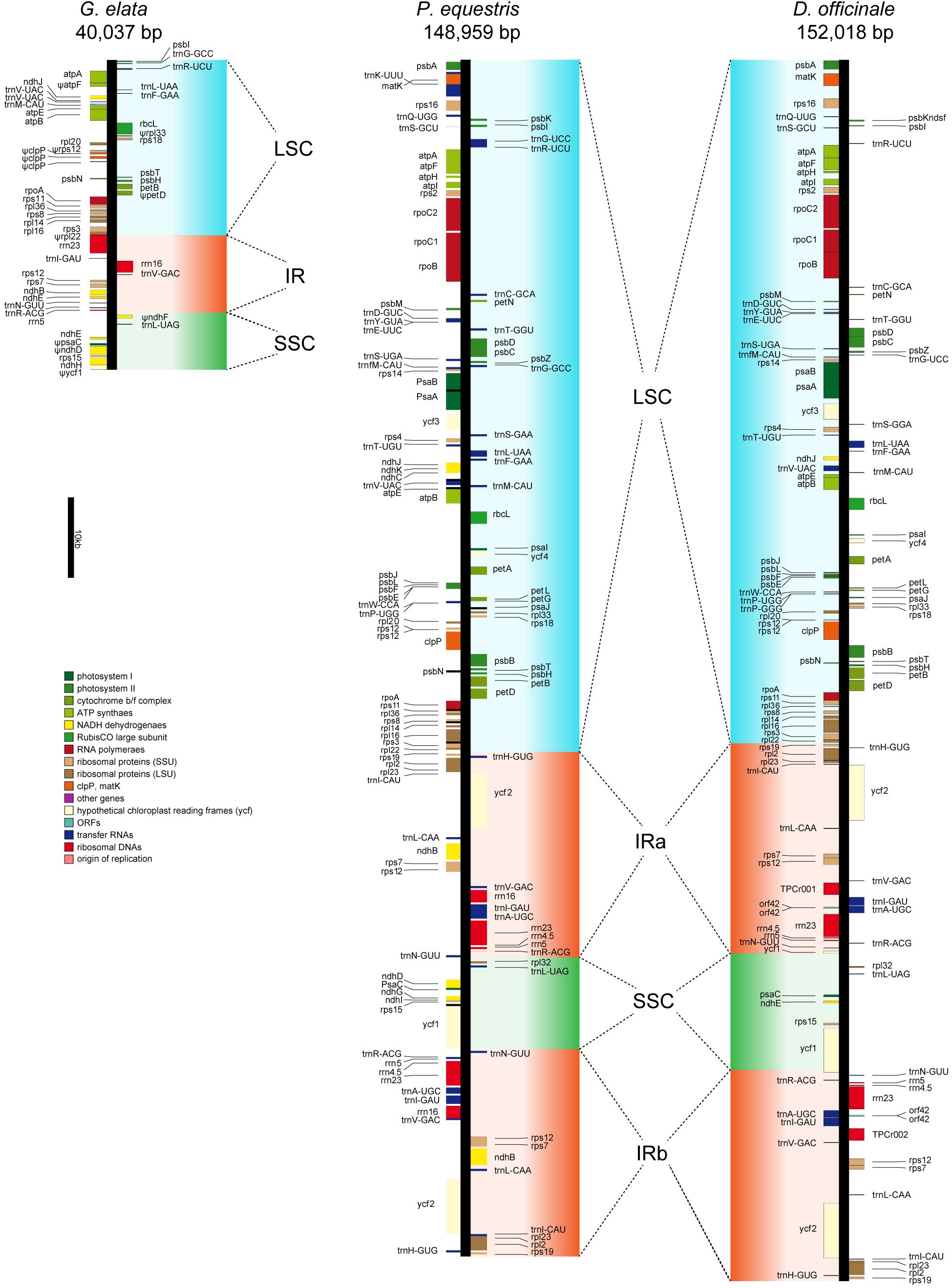
Figure 2. Plastid map of three orchid plants. The plastid of G. elata shows significant degradation comparing with P. equestris and D. officinale. Ψ indicates the pseudogenes.
Only 50 genes were identified in the G. elata plastome, among which 10 genes were manually checked to be pseudogenes (with frameshifts or premature stop codons) (Supplementary Table 14). The photosynthesis-related genes in the G. elata plastome were carefully checked. The result showed that the paralogs of psa and psb (photosystem I and II genes), pet (cytochrome b/f complex genes), rbc (rubisco subunit genes), and atp (ATP synthase genes) were completely lost or were found to have incomplete gene structures due to a reduced LSC. Two of the six remaining ndh (NADH dehydrogenase) genes were found to be non-functional. All these changes concluded that the genomic basis of photosynthesis in the G. elata plastome was highly degenerated. These observations are in accordance with a previous report (Yuan et al., 2018), which includes small assembled plastome sizes (40,026 vs. 35,326 bp), extensively loss of DNA fragments, missing of one copy of IR structure, and loss or non-functional of photosynthesis-related genes.
Natural selection is essential in the maintenance of trait for the plant population. The weakening or disappear of selection strength that leads to trait variations is referred to as “relaxed selection” (Lahti et al., 2009). In its evolutionary history, G. elata experienced the transition to a low-light, undergrowth environment (Bidartondo, 2005) and a fully heterotrophic lifestyle. This transition made photosynthesis dispensable for G. elata to survive. We therefore hypothesized that the remaining photosynthesis-related plastome genes might be under relaxed selection. Indeed, the Ka/Ks values of these genes (includes psa, psb, atp, pet genes, and so on) in G. elata were higher than D. officinale and P. equestris, and non-orchid plant species (Supplementary Table 15). This indicates the reduction of functional constraints, and, thus, these genes were under a significantly different selective regime in G. elata than in those photosynthetically active species.
Relaxation of Selective Constraints of Nuclear-Coded Photosynthesis-Related Genes in G. elata
We define “photosynthesis-related genes” as all known genes that are involved in the structure, development, and normal function of the plastid and leaf in land plants. The plastome only contains a small part of these photosynthesis-related genes, and the rest are within the plant nuclear genome. Based on the aforementioned hypothesis, we also checked the molecular evolution of the photosynthesis-related nuclear genes in the G. elata genome. In brief, they consisted of 4,818 plastid-related genes, 203 leaf development-related genes, and 1,408 genes that were directly involved in the photosynthesis. The Ka/Ks values of these genes in G. elata were significantly higher than those in other orchids and non-orchid plant species, indicating relaxation of selective constraints on the leaf development and the photosynthesis process (t-test and two-way ANOVA test, p < 0.05, Figure 3 and Supplementary Figure 4). We also found that the Ka/Ks values of these searched genes in orchids were higher than those in non-orchid species. It suggested that there might be a partial relaxation of selection on plastid and leave functions in the photosynthetic orchids due to their symbiotic relationships with fungi. These findings also suggested a positive correlation between the degree of heterotrophy in plants and the non-synonymous mutation rates in genes that were involved in the photosynthetic process, plastid and leave functions.
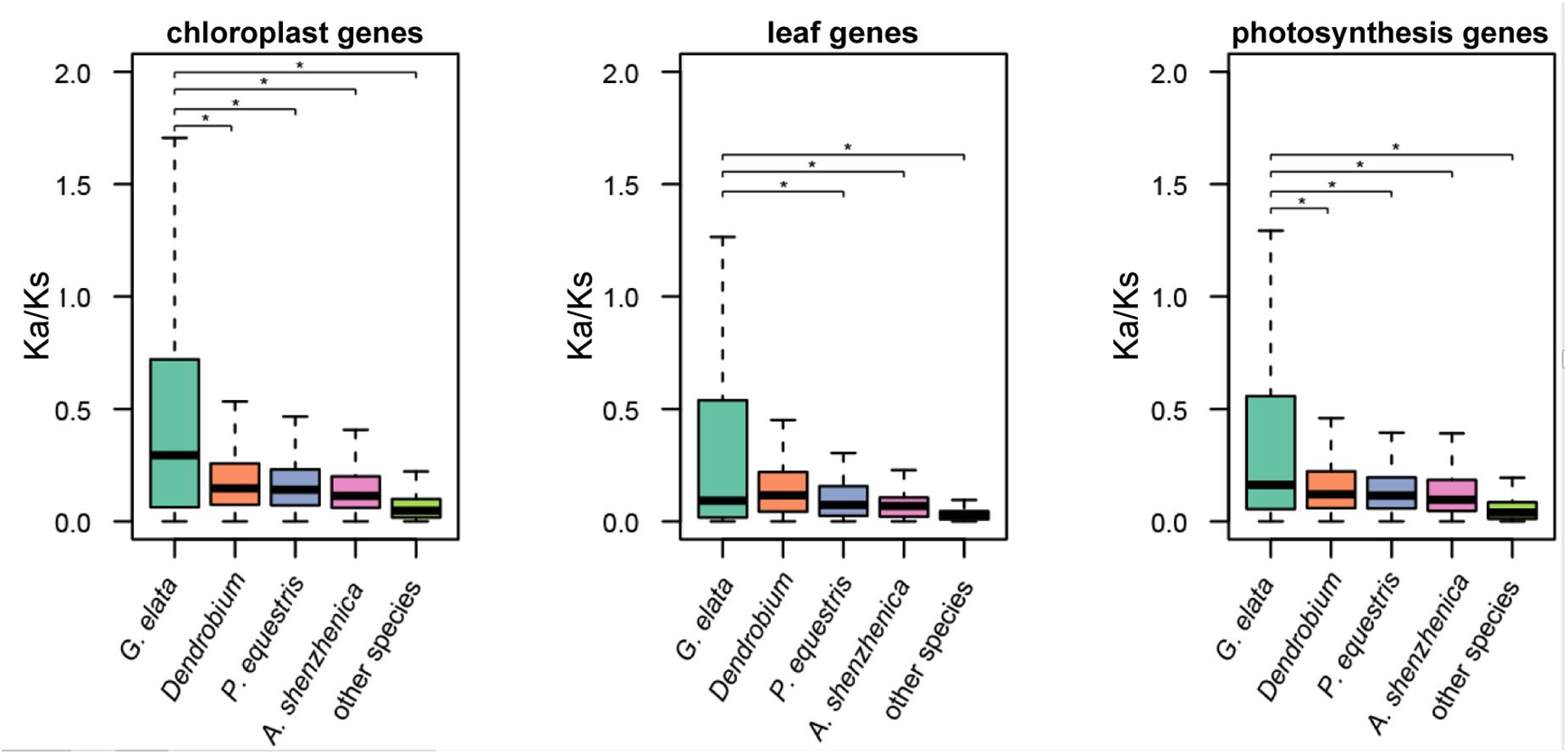
Figure 3. Relaxation of selective constraints on nuclear-encoded genes. Ka/Ks ratios of nuclear-encoded plastid genes and genes involved in leaf development in G. elata, other orchids (including D. officinale, P. equestris, D. catenatum, and A. shenzhenica) and other green plants (including A. thaliana and O. sativa). *P < 0.05.
Analysis of Symbiotic Genes in G. elata Genome
Since G. elata cannot perform photosynthesis, it relies on symbiotic genes for nutrition, so symbiotic genes are also attracting attention in the Analysis of Symbiotic Genes in Gastrodia Genome. G. elata exists underground without leaves and chloroplasts, with the nutrients necessary for growth being supplied by the symbiotic fungus Armillaria mellea (Sa et al., 2003) fungus. Gastrodia antifungal protein (GAFP, also known as gastrodianin) was first purified from the cortex of the terminal corm of G. elata (Hu et al., 1988). It is mainly distributed throughout the epidermis and cortex layer of G. elata (Liu et al., 1993; Hu and Huang, 1994) and shows a strong fungistatic activity against a broad spectrum of fungi, including A. mellea (Hu and Huang, 1994), Rhizoctonia solani, Valsa ambiens, Gibberella zeae, Ganoderma lucidum, and Botrytis cinerea in vitro to reach plants symbiosis with microorganisms (Wang et al., 2001). In the G. elata genome, we identified 20 GAFP genes, 10 GAFP genes in A. shenzhenica, and 0 in A. thaliana, which indicates that GAFP has expanded in G. elata (Supplementary Table 16). It is known that strigolactone can stimulate hyphal branching and development of arbuscular mycorrhizal fungi, which increases the chances of an encounter with a host plant (Kretzschmar et al., 2012). Strigolactone is an important signal for the establishment of the symbiosis relationship between G. elata and A. mellea. Its mechanism of action is similar to that of promoting symbiosis between plants and mycorrhiza. In this article, we have identified the key genes for the biosynthesis of strigolactone CCDs (Delaux et al., 2012). In the G. elata genome, 12 CCDs genes were identified, whereas ten and nine CCDs genes were identified in A. shenzhenica and A. thaliana, respectively. In order to verify whether the CCDs gene has positive selection in G. elata, we further performed the Ka/Ks analysis. The Ka/Ks values of these genes in G. elata were significantly higher than those in D. catenatum and P. equestris. It showed that, compared with some orchid and non-orchid plant species, CCDs have expanded in the G. elata genome.
Genomic Basis for the Achlorophyllous Phenotype in G. elata
We next investigated nuclear-encoded genes involved in the plant pigment synthesis in the G. elata genome. We detected no copies of four core genes in the chlorophyll biosynthetic pathway (UROS, CAO, LPOR, and VDE; Supplementary Figure 5a). This is in line with the result that no signals of major photosynthetic pigments could be detected via the HPLC in the G. elata plant tissue (compared to the other two orchids D. officinale and P. equestris) (Supplementary Figure 5b). Moreover, three essential genes (PDV1, PDV2, and ARC3) involved in the chloroplast division were also not detected (Supplementary Figure 6). These results clearly showed the genomic basis for the achlorophyllous phenotype in G. elata.
Conclusion
The G. elata genome provides an illuminating model for probing evolutionary molecular changes associated with leaf loss, plastid degeneration, and plant-fungal symbioses. Both plastid-encoded and nuclear-encoded photosynthesis-related genes in G. elata showed evidence of the relaxation of selective constraints. Besides the profound changes in the photosynthetic system, it is possible that future research could identify the underlying mechanisms for the rewiring of basic metabolic pathways, the evolution of TEs, gene death, and generation of new genes, since an improved G. elata genome is now available. Thus, the genome sequence of G. elata would be a valuable resource for future investigations of the evolution of orchids and non-photosynthetic plants at both the genomic and the whole-organism levels.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI database under BioProject PRJNA394702.
Author Contributions
SSC, GHZ, and WLS collected the samples and performed the experiments. XW, YZW, and XD completed the data analysis. YD, WC, WW, MA, and JHM edited and modified the manuscript. All authors read and approved the manuscript.
Funding
This study was supported by “The Thousand Talents Program” (Class B) of the government of China, National Key R&D Program of China (2019YFC1711100), Yunnan Provincial Key Programs of Yunnan Eco-friendly Food International Cooperation Research Center Project (2019ZG00908), and the Guangxi Innovation-Driven Development Project (GuiKe AA18242040).
Conflict of Interest
XW was employed by the company Jiaxing Synbiolab Biotechnology Co., Ltd. and she declared that Jiaxing Synbiolab Biotechnology Co., Ltd. plays no role in the funding, design, analysis, and publication of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.580568/full#supplementary-material
Footnotes
- ^ http://sourceforge.net/projects/soapdenovo2/files/GapCloser/
- ^ https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.3.4.3
- ^ http://ccb.jhu.edu/software/tophat/index.shtml
- ^ https://github.com/trinityrnaseq/trinityrnaseq/releases
- ^ http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/index.html
- ^ ftp://ftp.ebi.ac.uk/pub/software/unix/iprscan/5/5.32-71.0/interproscan-5.32-71.0-64-bit.tar.gz
- ^ http://www.drive5.com/muscle/downloads.htm
- ^ http://nbisweden.github.io/MrBayes/download.html
- ^ http://evomics.org/resources/software/molecular-evolution-software/paml/
- ^ http://dogma.ccbb.utexas.edu
- ^ https://chlorobox.mpimp-golm.mpg.de/OGDraw-Downloads.html
References
Barrett, C. F., and Davis, J. I. (2012). The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am. J. Bot. 99, 1513–1523. doi: 10.3732/ajb.1200256
Bidartondo, M. I. (2005). The evolutionary ecology of myco-heterotrophy. New Phytol. 167, 335–352. doi: 10.1111/j.1469-8137.2005.01429.x
Cai, J., Liu, X., Vanneste, K., Proost, S., Tsai, W. C., Liu, K. W., et al. (2015). The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 47, 65–72. doi: 10.1038/ng.3149
Cribb, P., and Killmann, E. F. A. D. (2010). A revision of Gastrodia (Orchidaceae: Epidendroideae, Gastrodieae) in tropical Africa. Kew Bull. 65, 315–321. doi: 10.1007/s12225-010-9193-4
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Delaux, P. M., Xie, X., Timme, R. E., Puech-Pages, V., Dunand, C., Lecompte, E., et al. (2012). Origin of strigolactones in the green lineage. New Phytol. 195, 857–871. doi: 10.1111/j.1469-8137.2012.04209.x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hu, Z., and Huang, Q. Z. (1994). Induction and accumulation of the antifungal protein in Gastrodia elata. Acta Bot. Yunnan 16, 169–177.
Hu, Z., Yang, Z., and Wang, J. (1988). Isolation and partial characterization of an antifungal protein from Gastrodia elata corm. Acta Bot. Yunnanica 10, 373–380.
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jheng, C. F., Chen, T. C., Lin, J. Y., Chen, T. C., Wu, W. L., and Chang, C. C. (2012). The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant Sci. 190, 62–73. doi: 10.1016/j.plantsci.2012.04.001
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Krause, K. (2008). From chloroplasts to “cryptic” plastids: evolution of plastid genomes in parasitic plants. Curr. Genet. 54, 111–121. doi: 10.1007/s00294-008-0208-8
Kretzschmar, T., Kohlen, W., Sasse, J., Borghi, L., Schlegel, M., Bachelier, J. B., et al. (2012). A petunia ABC protein controls strigolactone-dependent symbiotic signalling and branching. Nature 483, 341–344. doi: 10.1038/nature10873
Lahti, D. C., Johnson, N. A., Ajie, B. C., Otto, S. P., Hendry, A. P., Blumstein, D. T., et al. (2009). Relaxed selection in the wild. Trends Ecol. Evol. 24, 487–496. doi: 10.1016/j.tree.2009.03.010
Lan, J., Xu, J. T., and Li, J. S. (1994). Study on symbiotic relation between Gastrodia elata and Armillariella mellea by autoradiography. Mycosystema 13, 219–222.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leake, J. R. (1994). The biology of myco-heterotrophic (‘saprophytic’) plants. New Phytol. 127, 171–216. doi: 10.1111/j.1469-8137.1994.tb04272.x
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liang, Y., Xiao, W., Hui, L., Yang, T., Lian, J., Yang, R., et al. (2015). The genome of dendrobium officinale illuminates the biology of the important traditional chinese orchid herb. Mol. Plant 008, 922–934. doi: 10.1016/j.molp.2014.12.011
Liu, J., Jin-tang, X., He, W., Hong-xiang, L., and Yong-ru, S. (1993). Detection and immunofiuorescence localization of antifungal protein of gastrodiaelata. J. Integr. Plant Biol. 35, 593–599.
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Merckx, V., Bidartondo, M. I., and Hynson, N. A. (2009). Myco-heterotrophy: when fungi host plants. Ann. Bot. 104, 1255–1261. doi: 10.1093/aob/mcp235
Merckx, V., and Freudenstein, J. V. (2010). Evolution of mycoheterotrophy in plants: a phylogenetic perspective. New Phytol. 185, 605–609. doi: 10.1111/j.1469-8137.2009.03155.x
Ning, Z., Bai, X. F., Jian-Tao, L. V., Yang, J. F., and Gui-Ping, X. U. (2010). Study on symbiotic mechanism between gastrodia elata blume and armillaria mellea in tissue culture system. Med. Plant 34, 95–96.
Palmer, J. D., and Thompson, W. F. (1982). Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 29, 537–550. doi: 10.1016/0092-8674(82)90170-2
Read, D. J. (1991). Mycorrhizas in ecosystems. Cell. Mol. Life Sci. 47, 376–391. doi: 10.1007/BF01972080
Sa, Q., Wang, Y., Li, W., Zhang, L., and Sun, Y. (2003). The promoter of an antifungal protein gene from Gastrodia elata confers tissue -specific and fungus-inducible expression patterns and responds to both salicylic acid and jasmonic acid. Plant Cell Rep. 22, 79–84. doi: 10.1007/s00299-003-0664-z
Sandelius, A. S., and Aronsson, H. (2009). “The chloroplast: interactions with the environment,” in Plant Cell Monographs, ed. P. Nick (Cham: Springer).
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Wang, X., Bauw, G., Van Damme, E. J., Peumans, W. J., Chen, Z. L., Van Montagu, M., et al. (2001). Gastrodianin-like mannose-binding proteins: a novel class of plant proteins with antifungal properties. Plant J. 25, 651–661. doi: 10.1046/j.1365-313x.2001.00999.x
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2017). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Wicke, S., Muller, K. F., De Pamphilis, C. W., Quandt, D., Wickett, N. J., Zhang, Y., et al. (2013). Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell 25, 3711–3725. doi: 10.1105/tpc.113.113373
Wicke, S., Schaferhoff, B., DePamphilis, C. W., and Muller, K. F. (2014). Disproportional plastome-wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Mol. Biol. Evol. 31, 529–545. doi: 10.1093/molbev/mst261
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yan, L., Wang, X., Liu, H., Tian, Y., Lian, J., Yang, R., et al. (2015). The genome of dendrobium officinale illuminates the biology of the important traditional chinese orchid herb. Mol. Plant 8, 922–934. doi: 10.1016/j.molp.2014.12.011
Yang, P., Zhou, H., Qian, J., Xu, H., Shao, Q., Li, Y., et al. (2016). The complete chloroplast genome sequence of Dendrobium officinale. Mitochondrial DNA DNA Mapp. Seq. Anal. 27, 1262–1264. doi: 10.3109/19401736.2014.945547
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9:1615. doi: 10.1038/s41467-018-03423-5
Zhang, G. Q., Liu, K. W., Li, Z., Lohaus, R., Hsiao, Y. Y., Niu, S. C., et al. (2017). The Apostasia genome and the evolution of orchids. Nature 549, 379–383. doi: 10.1038/nature23897
Keywords: Gastrodia elata, genome, achlorophyllous, relaxed selection, non-photosynthetic
Citation: Chen S, Wang X, Wang Y, Zhang G, Song W, Dong X, Arnold ML, Wang W, Miao J, Chen W and Dong Y (2020) Improved de novo Assembly of the Achlorophyllous Orchid Gastrodia elata. Front. Genet. 11:580568. doi: 10.3389/fgene.2020.580568
Received: 06 July 2020; Accepted: 19 October 2020;
Published: 19 November 2020.
Edited by:
Wei Chen, North China University of Science and Technology, ChinaReviewed by:
Liangsheng Zhang, Zhejiang University, ChinaXiaohua Jin, Institute of Botany (CAS), China
Copyright © 2020 Chen, Wang, Wang, Zhang, Song, Dong, Arnold, Wang, Miao, Chen and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhua Miao, bWpoMTk2MkB2aXAuMTYzLmNvbQ==; Wei Chen, d2NoZW5udHJAZ21haWwuY29t; Yang Dong, bG95YWx5YW5nQDE2My5jb20=
†These authors have contributed equally to this work