 Nan Zhang
Nan Zhang Brittani Bewick
Brittani Bewick Guangbin Xia2
Guangbin Xia2 Denis Furling
Denis Furling Tetsuo Ashizawa
Tetsuo Ashizawa- 1Department of Neurology, Houston Methodist Research Institute, Houston, TX, United States
- 2Indiana University School of Medicine, Fort Wayne, IN, United States
- 3Institut National de la Sante et de la Recherche Medicale (INSERM), Centre de Recherche en Myologie (CRM), Association Institut de Myologie, Sorbonne Université, Paris, France
Cas13a, an effector of type VI CRISPR-Cas systems, is an RNA guided RNase with multiplexing and therapeutic potential. This study employs the Leptotrichia shahii (Lsh) Cas13a and a repeat-based CRISPR RNA (crRNA) to track and eliminate toxic RNA aggregates in myotonic dystrophy type 1 (DM1) – a neuromuscular disease caused by CTG expansion in the DMPK gene. We demonstrate that LshCas13a cleaves CUG repeat RNA in biochemical assays and reduces toxic RNA load in patient-derived myoblasts. As a result, LshCas13a reverses the characteristic adult-to-embryonic missplicing events in several key genes that contribute to DM1 phenotype. The deactivated LshCas13a can further be repurposed to track RNA-rich organelles within cells. Our data highlights the reprogrammability of LshCas13a and the possible use of Cas13a to target expanded repeat sequences in microsatellite expansion diseases.
Introduction
Recent advances in genome editing have become increasingly important for addressing monogenic disorders. Microsatellite expansion diseases are a group of more than 30 incurable neurological and neuromuscular disorders that are caused by the expansion of 3–10 nucleotide repeats in the residing gene. In affected individuals, repeat expansions can occur in both coding and non-coding regions, resulting in toxic protein gain-of-function and RNA gain-of-function, respectively. Large repeat size often correlates positively with intergenerational instability and disease severity. When repeat expansions are located in non-coding regions, diseases such as myotonic dystrophy types 1 and 2 (DM1/2), C9orf72-amyotrophic lateral sclerosis/frontotemporal dementia (C9orf72-ALS/FTD), fragile-X tremor ataxia syndrome (FXTAS), spinocerebellar ataxias types 8, 10, 12, 31, and 36 (SCA 8/10/12/31/36), Huntington’s disease-like 2 (HDL2), and Fuch’s endothelial corneal dystrophy (FECD) occur (Mohan et al., 2014; Rohilla and Gagnon, 2017; Zhang and Ashizawa, 2017).
DM1 is the most prevalent adult-onset muscular dystrophy for which there is currently no treatment. This multisystemic disease affects approximately 1 in 8,000 individuals and is caused by a CTG repeat expansion within the 3′-untranslated region (3′-UTR) of the DMPK gene (Brook et al., 1992; Mahadevan et al., 1992; Thornton, 2014). The widely accepted pathogenic mechanism suggests that the expanded CUG repeat is retained in the nucleus and forms RNA foci, sequestering the MBNL (Muscleblind) family of regulators of alternative splicing and inducing upregulation of CELF1 (CUG-BP and Elav-like family member 1, another regulator of alternative splicing) through PKC (protein kinase C)-mediated phosphorylation, and together leading to a cascade of downstream missplicing events (Jiang et al., 2004; Kuyumcu-Martinez et al., 2007; Chau and Kalsotra, 2015). Other than spliceopathy, pathomechanisms involving transcription, mRNA transport and localization, polyadenylation, conventional and repeat-associated non-AUG (RAN) translation, and microRNA expression may also contribute to disease etiology to different extents (Ebralidze et al., 2004; Krol et al., 2007; Du et al., 2010; Huichalaf et al., 2010; Rau et al., 2011; Fernandez-Costa et al., 2013; Timchenko, 2013; Batra et al., 2014; Kim et al., 2014; Goodwin et al., 2015; Gourdon and Meola, 2017; Zhang and Ashizawa, 2017; Cappella et al., 2018; Cerro-Herreros et al., 2018).
Several therapeutic approaches have been designed to treat DM1 and can be broadly grouped as (Nelson et al., 2017): (1) targeting DNA repeats [e.g., CRISPR-Cas9 with CTG-flanking guide sgRNAs in DM1 myoblasts, induced pluripotent stem cells and myogenic cells (Provenzano et al., 2017; van Agtmaal et al., 2017; Dastidar et al., 2018; Wang et al., 2018) and TALEN in yeast (Richard et al., 2014)], (2) degrading RNA repeats [e.g., synthetic siRNAs (Sobczak et al., 2013), chemically modified antisense oligonucleotide (ASO) gapmers (Wheeler et al., 2012; Jauvin et al., 2017; Manning et al., 2017), bleomycin-cleavage modules (Rzuczek et al., 2017), deactivated Cas9 (dCas9) conjugated to a PIN domain (Batra et al., 2017), engineered human U7-snRNA-CUG (Francois et al., 2011), hammerhead ribozymes (Langlois et al., 2003), and small RNase mimics (Nguyen et al., 2015)], (3) dissociating CUG-MBNL interactions [e.g., small molecules (Gareiss et al., 2008; Warf et al., 2009; Childs-Disney et al., 2013; Nguyen et al., 2015; Rzuczek et al., 2015), miniPEG-γ peptides with terminal pyrenees (Hsieh et al., 2018), and steric-blocking morpholino ASOs (Wheeler et al., 2009)], and (4) small molecules that target cellular pathways independent of CUG expansions (Lopez-Morato et al., 2018). Furthermore, dCas9 has been used to impede CUG repeat transcription through steric blockage (Pinto et al., 2017), and the RIBOTAC technology coupled with RNase L could potentially be adopted to target repeat expansions in DM1 (Costales et al., 2018). Given that DNA editing using CRISPR-Cas9 is hindered by large deletions and rearrangements (Kosicki et al., 2018), p53-mediated DNA damage response (Haapaniemi et al., 2018; Ihry et al., 2018), and RNA polymerase movement (Clarke et al., 2018), an RNA-based platform might serve as an alternative therapeutic strategy. Synthetic siRNAs typically do not target nuclear repeat RNA efficiently, possibly due to the preferential distribution of the RISC (RNA-induced silencing complex) in the cytoplasm. ASOs, when administered peripherally for muscle degeneration, are often not taken up by local tissues. For instance, the IONIS-DMPKRX human trial for DM1 patients did not reach the desired endpoints1. In addition, chemically modified ASOs with high-affinity nucleic analogs [e.g., locked nucleic acid (LNA) or constrained ethyl (cEt) nucleic acid] can induce RNase H1-dependent hepatotoxicity and apoptosis in both mice and cell models (Kakiuchi-Kiyota et al., 2014; Burel et al., 2016; Kasuya et al., 2016). Thus, an alternative reprogrammable RNA-based strategy may provide important therapeutic value in treating DM1, and by extension other microsatellite expansion diseases. However, that is not to say that the alternative reprogrammable RNA-based strategy does not face its own mechanistic and delivery challenges.
The recently discovered CRISPR-Cas13a protein belongs to the type VI-A CRISPR-Cas system and is an RNA-guided dual-functional RNase (East-Seletsky et al., 2016; Liu et al., 2017; Shmakov et al., 2017; Smargon et al., 2017; Konermann et al., 2018; Yan et al., 2018). Cas13a from Leptotrichia shahii (Lsh) utilizes a guide CRISPR RNA (crRNA) containing a 28-nucleotide (nt) spacer sequence to cleave its target RNA. The design of crRNA requires the 3′-protospacer flanking sequence (PFS) to end with A, U, or C (Abudayyeh et al., 2016). The N-terminal domain (NTD) and Helical 1 of LshCas13a are responsible for crRNA maturation, while the two C-terminal HEPN domains are responsible for target RNA cleavage preferentially at uracils (Abudayyeh et al., 2016; Liu et al., 2017). This dual-functional nature enables multiplexing of several spacers in a single precursor crRNA (Gootenberg et al., 2018). The CRISPR-Cas13a system holds great potential for scalable RNA editing applications with reprogrammable crRNA. Evidently since its discovery, the system has been used for attomolar RNA detection by taking advantage of its collateral RNA cleavage (East-Seletsky et al., 2017; Gootenberg et al., 2018; Myhrvold et al., 2018), RNA knockdown in Escherichia coli and mammalian and plant cells (Abudayyeh et al., 2017; Adams et al., 2017; Zhao et al., 2018), and base editing with a deactivated Cas13a conjugated to ADAR2 (adenosine deaminase acting on RNA type 2) (Cox et al., 2017; Jing et al., 2018). Other sub-classes of Cas13 proteins (Cas13b, c and d) have been identified (Smargon et al., 2017; Konermann et al., 2018; Yan et al., 2018).
In this study, we repurposed the human codon optimized LshCas13a protein to track and degrade toxic RNA with a repeat-based crRNA. We observed CUG repeat cleavage biochemically and in a DM1 patient-derived myoblast cell line through lentiviral delivery. In the transduced DM1 myoblast cell line, we observed CUG RNA foci reduction, DMPK RNA knockdown and near-complete reversal of DM1-associated splicing defects in the tested genes.
Results
Engineering a Deactivated LshCas13a Tracker to Visualize CUG Repeat RNA Foci in the Nucleus
The R1278A mutant of LshCas13a was previously reported to have lost RNA cleavage activity but retained high RNA binding affinity [>6-fold tighter than wildtype protein (Abudayyeh et al., 2016)]. One application of this mutant is to be engineered as an RNA tracker in cells. The formation of nuclear CUG RNA foci is a hallmark of DM1 cells; currently the means to detect such foci are limited. We hypothesize that the deactivated LshCas13aR1278A mutant tethered to an eGFP (enhanced green fluorescent protein, hereafter the conjugate is named dLshCas13a) could serve as a tool to visualize RNA foci that cause DM1. Under such premises, the LshCas13aR1278A-eGFP sequence was first cloned into the pX330 backbone along with two terminal nuclear localization signals (NLSs), the pU6-driven direct repeat (DR) hairpin, and two inverted type II restriction sites for spacer insertion (Figures 1A,B). Dual NLS-tags have been extensively used in RNA targeting as well as DNA editing Cas systems (Cong et al., 2013; Nelles et al., 2016; Konermann et al., 2018). Given the primary goal of this study is to track and eliminate nuclear CUG foci, we decided to adopt the dual NLS labeling system. Without the presence of crRNA, the dual NLS-labeled dLshCas13a largely resides in the nucleus of COS-M6 cells (Figure 1C). Subsequently to track DM1 repeats, a 28 nt spacer complementary to CUG RNA was designed with a 3′-PFS of uracil (hereafter named crDM1, Figure 1B). To create the expanded CUG RNA foci, the DT960 plasmid encoding human DMPK exons 11–15 with 960 interrupted CTG repeats was co-transfected with the eGFP-dLshCas13a-crDM1 tracker plasmid into COS-M6 cells (Figure 1D). Fluorescence in situ hybridization (FISH) detected punctate colocalization between CUG RNA foci and the CUG-targeting dLshCas13a (Figure 1D). In contrast, the non-targeting dLshCas13a with a 28nt spacer against a lambda phage sequence (crλNT) showed significantly less colocalization as calculated by Pearson’s correlation (p = 0.0009, Supplementary Figure 1). The use of Cos-M6 cells overexpresses expanded CUG RNA to mimic and encourage RNA foci formation as seen in patient cells, however, Cas13a proteins are also overexpressed that could potentially form macromolecular aggregates. To date, most of the published Cas-based RNA tracking systems have been established in either HEK293FT or U2OS cells (Nelles et al., 2016; Abudayyeh et al., 2017; Konermann et al., 2018) – both are overexpressing cell lines. Such Cas protein aggregates have not been reported or had impacts on cell proliferation, global RNA size distribution and RNA integrity. We conclude that the dLshCas13a tracker enables direct visualization of the expanded CUG RNA foci in COS-M6 cells.
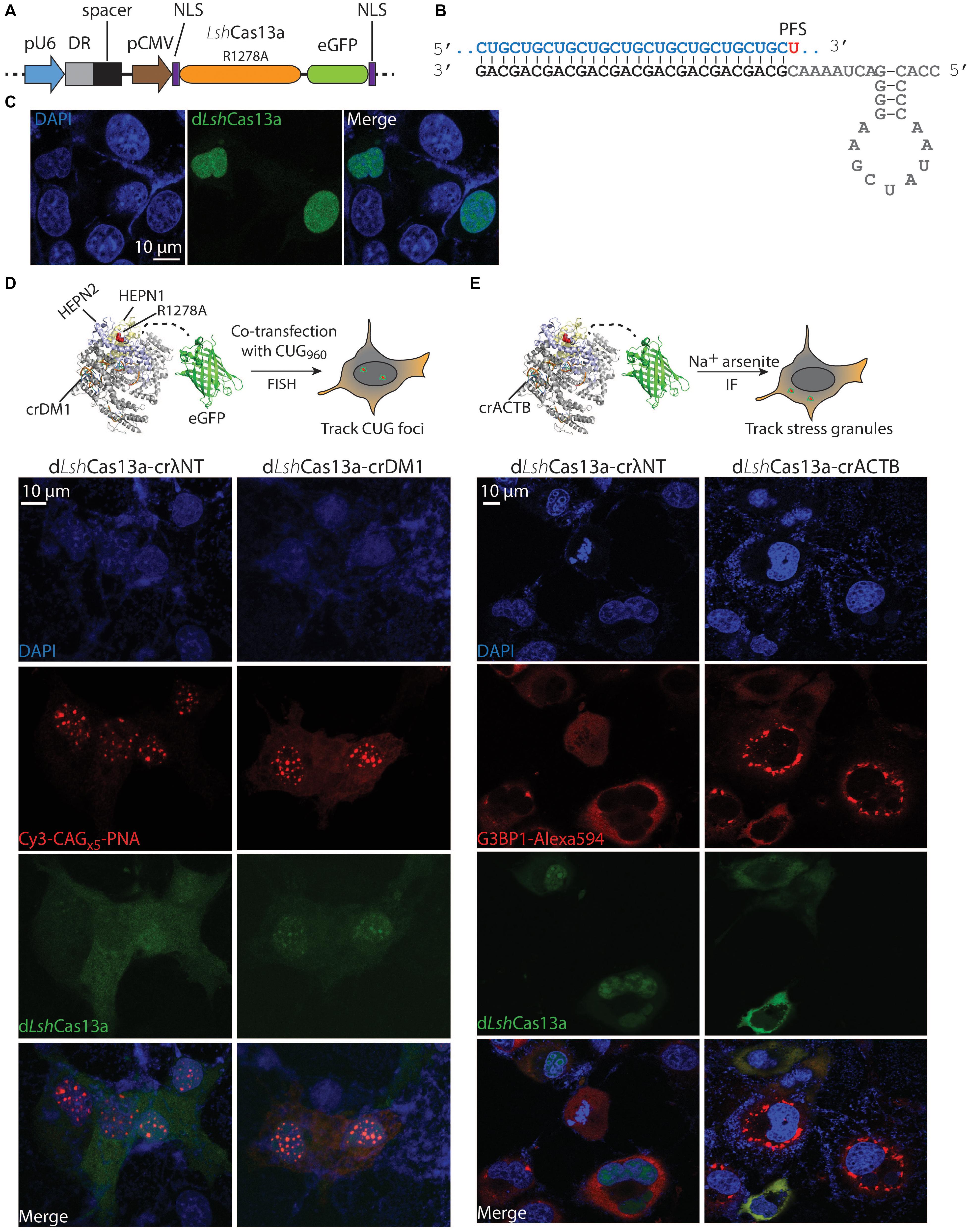
Figure 1. Tracking the expanded CUG RNA foci and stress granules in COS-M6 cells. (A) Mammalian expression vector pX330 encoding crRNA under a pU6 promoter and dLshCas13a (LshCas13aR1278A-eGFP) under a pCMV promoter. The crRNA contains the direct repeat hairpin (DR, gray box) and the spacer (black box). The dLshCas13a is flanked by an N-terminal nucleoplasmin NLS and a C-terminal c-Myc NLS (Ray et al., 2015). (B) Structure of the crDM1 with DR sequence highlighted in gray, spacer in black and target CUG repeats in blue. The 3′-PFS uracil is highlighted in red. (C) Without the expression of crRNA, dLshCas13a primarily localizes in the nucleus. (D) Comparison of non-targeting (crλNT) and CUG-targeting (crDM1) dLshCas13a tracking nuclear CUG foci in COS-M6 cells. The expression of the interrupted CUGx960 repeats is driven by a CMV promoter in DT960 plasmid. (E) Comparison of non-targeting (crλNT) and ACTB-targeting (crACTB) dLshCas13a tracking cytoplasmic stress granules in COS-M6 cells. The stress condition was induced by adding 200 μM sodium arsenite for 1 h 30 min before fixation. Stress granules are labeled with anti-G3BP1 Alexa 594 antibodies by immunofluorescence (IF).
dLshCas13a Can Be Reprogrammed to Visualize Stress Granules in the Cytoplasm
Stress granules are cytoplasmic, translationally silent RNA-protein aggregates that resemble the expanded CUG RNA foci; their role in neurodegeneration has been increasingly recognized (Li et al., 2013; Zhang and Ashizawa, 2017; Benarroch, 2018). Many polyadenylated mRNAs (e.g., ACTB mRNA) and protein markers (e.g., Ras GTPase-activating protein-binding protein 1 – G3BP1) are known to localize to stress granules under stress conditions (Tourriere et al., 2003; Unsworth et al., 2010; Matsuki et al., 2013; Bley et al., 2015). To test the reprogrammability of dLshCas13a to track stress granules in COS-M6 cells, we generated a crRNA targeting the 3′-UTR of the ACTB gene (crACTB, Figure 1E) into the tracker plasmid. After treating COS-M6 cells with sodium arsenite to induce stress (Nelles et al., 2016; Wheeler et al., 2016; Abudayyeh et al., 2017), we observed significant overlap between the ACTB-targeting tracker and the antibody-labeled G3BP1 stress granule marker, in comparison to the non-targeting dLshCas13a (p = 0.0069, Figure 1E and Supplementary Figure 2). Our data are in line with existing literature where crRNA may be a primary drive of target recognition and Cas protein complex localization, even though the dLshCas13a protein is dual NLS-tagged. Consistently, another Cas13a protein that is predominantly nucleus-localized translocates to the cytoplasm when complexed with a crRNA targeting ACTB mRNA in the stress granule; while dual NLS-tagged RCas9 translocates to the cytoplasm when complexed with a GAPDH-targeting sgRNA (Nelles et al., 2016; Abudayyeh et al., 2017). Overall, the above data demonstrate the versatility of dLshCas13a-eGFP in tracking nuclear and cytoplasmic RNA targets. By inference, we suggest that dLshCas13a equipped with appropriate crRNA can be used to track other RNA-rich organelles in the cell.
LshCas13a Promotes CUG Repeat Cleavage in Biochemical Assays and in Patient-Derived Myoblasts
To investigate if LshCas13a can cleave CUG repeats biochemically, we expressed and purified the protein in E. coli (Figure 2A and Supplementary Figures 3A,B). To obtain the target CUG repeats, 30 CTG repeats were generated by self-priming PCR, cloned into the pcDNA3.1/hygro(+) vector, linearized and in vitro transcribed under a T7 promoter. In the presence of crDM1, we observed ≈58% total target cleavage in 16 h (Figure 2B). This is in marked contrast to the background cleavage as observed in the reaction without crDM1 (≈30% of total RNA target cleaved, Figure 2B). Interestingly, multiple target-guide RNA duplexes were observed under denaturing conditions (7 M Urea and 30% formamide at 95°C, Supplementary Figure 3C). LshCas13a seems to be able to overcome the stability of these CUG-CAG duplexes and achieve efficient cleavage. Overall, the above data suggest that LshCas13a promotes CUG repeat RNA cleavage in biochemical assays.
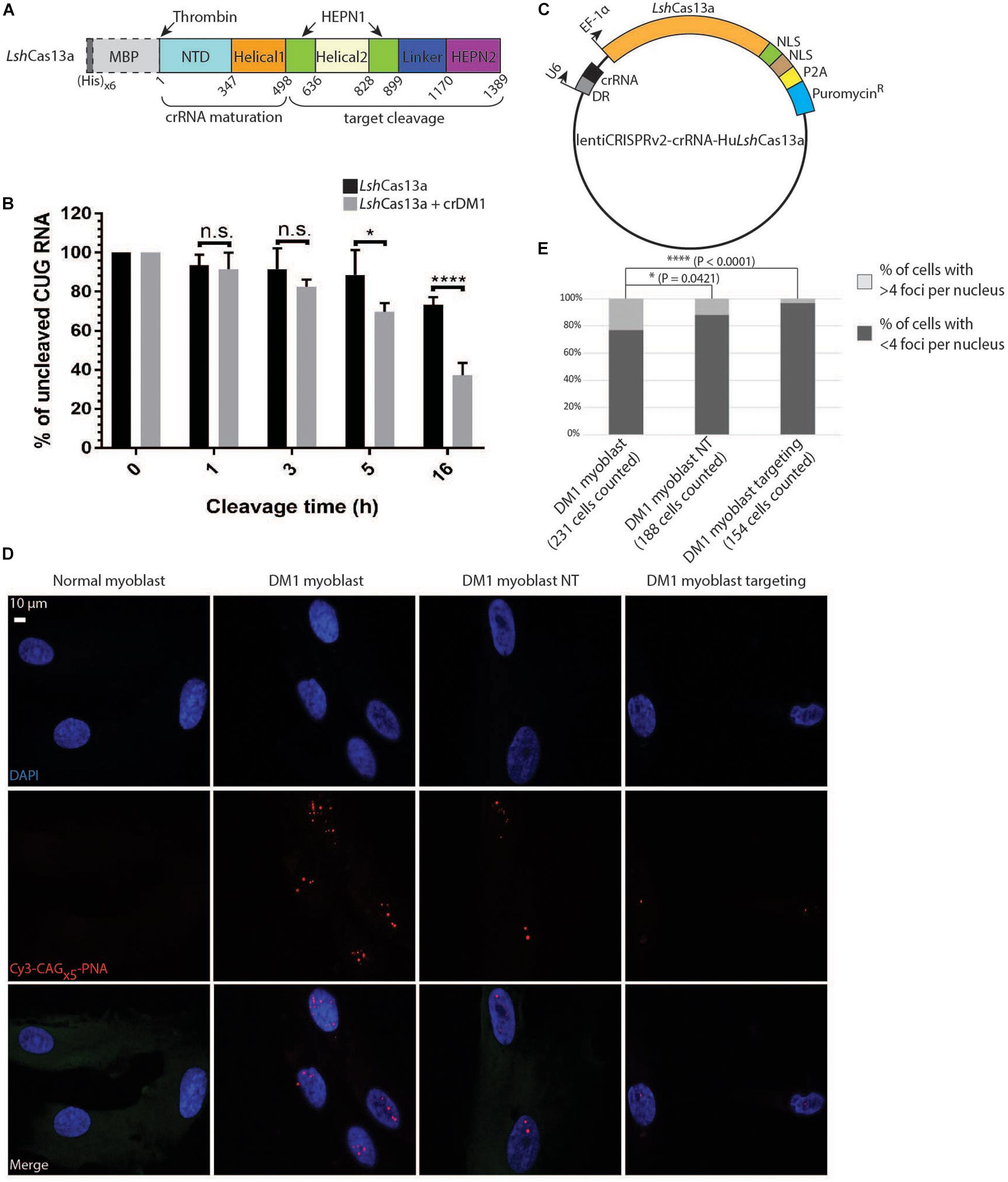
Figure 2. Degradation of CUG repeats in biochemical assays and in DM1 patient-derived myoblasts. (A) Domain organization of LshCas13a purified in E. coli (not drawn to scale). NTD stands for N-terminal domain. The (His)x6 and maltose binding protein (MBP) tags were removed after purification by thrombin cleavage (dotted box). (B) Cleavage assay of CUGx30 RNA targets using purified LshCas13a with or without in vitro transcribed crDM1. The RNA cleavage products were separated on a 10% TBE-Urea gel and quantified by Fiji ImageJ. The amount of uncut RNA at each time point was expressed as a percentage of the quantity at 0 h (100%). Error bars were calculated from four independent experiments using the same protein and RNA preparations. (C) Lentiviral construct for LshCas13a-crRNA delivery. The EF-1α-driven LshCas13a was conjugated to a nucleoplasmin NLS, an SV40 NLS, a P2A cleavage signal and an in-frame puromycin-resistant (puromycinR) gene. (D) Comparison of CUG RNA foci in normal myoblasts, untreated DM1 myoblasts, non-targeted DM1 myoblasts (DM1 myoblast NT), and CUG-targeted DM1 myoblasts (DM1 myoblast targeting) by FISH. (E) Quantification of foci counts of cell lines created in panel (D). The number of cells used for quantification was gathered from two experiments each with two technical replicates.
We next examined the ability of LshCas13a to degrade endogenous CUG repeats in a DM1 patient derived myoblast cell line (>176 CTG repeats). To ensure that all tested DM1 myoblasts express LshCas13a and crRNA, we constructed the lentiCRISPR v2-crRNA-LshCas13a vector with type II restriction sites for spacer insertion, and subsequently used the constructs for lentiviral transduction into the cell line (Figure 2C). Puromycin treatment of the transduced cell lines (5 days at 2 μg/ml) selected for cells expressing the puromycin-resistant gene in frame with LshCas13a and a P2A cleavage signal (Figure 2C). By performing FISH, we detected a significant reduction of RNA foci post transduction of the CUG-targeting LshCas13a (Figures 2D,E). Approximately 3% of DM1 myoblasts transduced with CUG-targeting LshCas13a contained >4 foci per cell, as opposed to 23% of the non-treated cells and 12% of cells transduced with non-targeting LshCas13a (Figure 2E). Taken together, the above data indicate that LshCas13a can indeed cleave the expanded CUG RNA foci in DM1 patient derived myoblasts.
LshCas13a Rescues Splicing Defects in DM1 Patient-Derived Myoblasts
Splicing misregulation is a cardinal feature of DM1, which is characteristic of an adult-to-embryonic switch in splicing patterns. The misspliced embryonic mRNA isoforms are thought unable to fulfill the adult tissue function, resulting in gradual neurodegeneration (Chau and Kalsotra, 2015). As RNA foci can be indicative of RNA binding protein sequestration and missplicing, we hypothesize that the observed reduction of RNA foci post LshCas13a treatment could potentially translate to a reversal of splicing defects. We tested splicing patterns of six genes that are known to be regulated by MBNL1 and contribute to DM1 pathogenesis [troponin T2 – TNNT2, insulin receptor – INSR, sarcoplasmic/endoplasmic reticulum calcium ATPase 1 – ATP2A1 or SERCA1, dystonin – DST, and Muscleblind 1/2 – MBNL 1/2 (Savkur et al., 2001; Kuyumcu-Martinez and Cooper, 2006; Hino et al., 2007; Childs-Disney et al., 2013; Santoro et al., 2013; Arandel et al., 2017; Batra et al., 2017; Renna et al., 2017, 2019)]. We observed a missplicing rescue in all six tested genes in DM1 myoblasts transduced with CUG-targeting LshCas13a (Figure 3A and Supplementary Figure 4). However, we also observed a partial rescue with the non-targeting LshCas13a treatment (Figure 3A). We further performed immunofluorescence (IF) against MBNL1 in normal, untreated DM1, non-targeting and CUG-targeting DM1 myoblast cells (Supplementary Figure 5) and observed a more dispersed MBNL1 distribution in LshCas13a treated cells (Supplementary Figure 5), which may account for the splicing rescue.
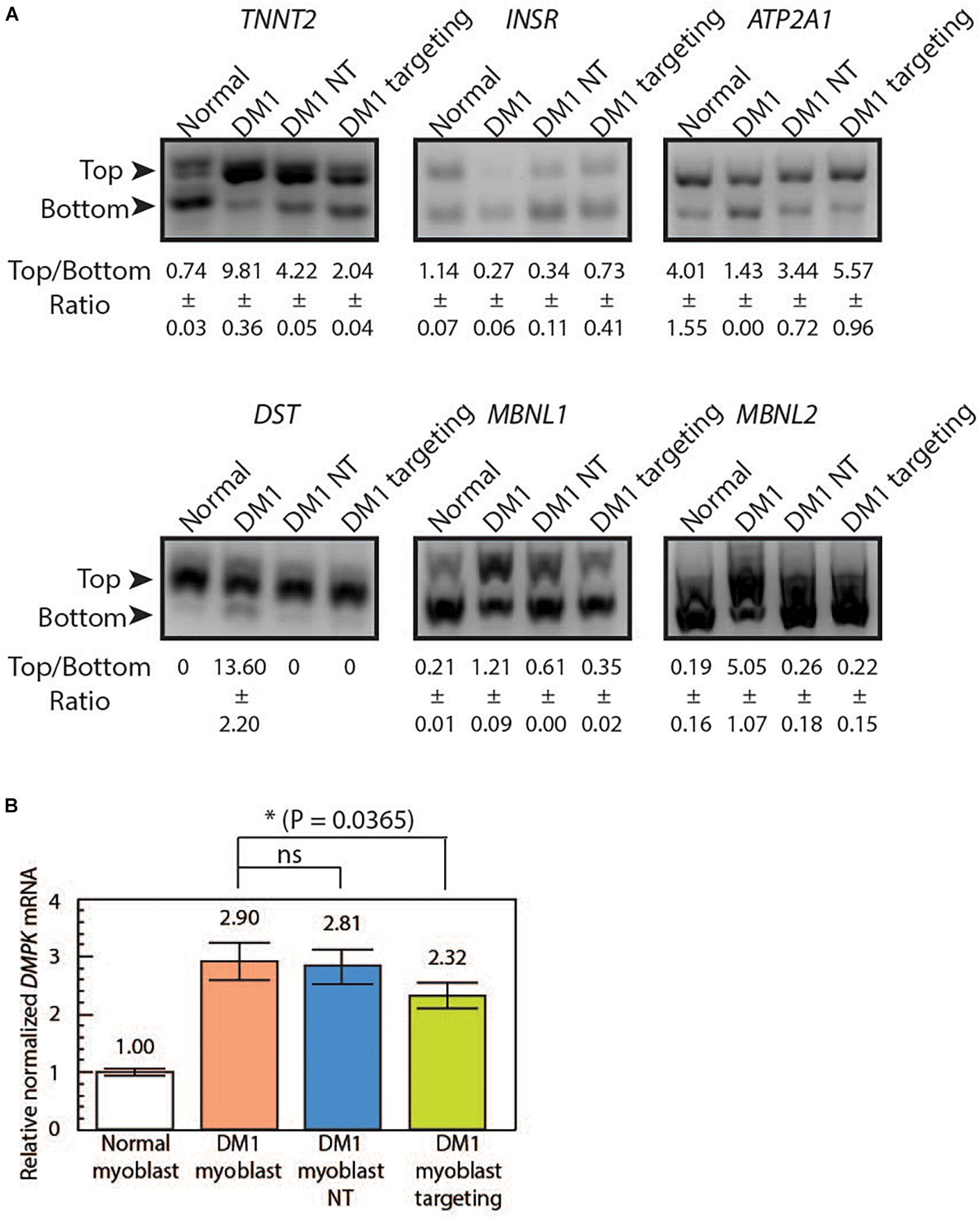
Figure 3. LshCas13a reverses downstream missplicing events and knocks down DMPK mRNA quantity in transduced DM1 myoblasts. (A) Splicing patterns from six MBNL1-regulated genes were analyzed in normal myoblasts, untreated DM1 myoblasts, DM1 myoblasts NT and DM1 myoblasts targeting. The ratios of top and bottom bands (as indicated by arrows) were quantified with standard deviation (±SD) in Fiji ImagJ from two separate experiments. (B) The DMPK mRNA level was measured using qRT-PCR from the four cell lines. The relative DMPK mRNA level from each cell line was normalized to that of the normal myoblast (white bar). Three biological replicates each with three technical replicates were used in this assay.
LshCas13a Marginally Reduces DMPK mRNA Level in Treated Cells
To further test if the mRNA level of the repeat-residing DMPK gene was affected by the CUG-targeting LshCas13a, we extracted the total RNA from normal myoblasts, DM1 myoblasts, non-targeted DM1 myoblasts, and CUG-targeting DM1 myoblasts. Quantitative RT-PCR was subsequently performed using DMPK primers specific to exon 9. By setting the DMPK mRNA level from normal myoblasts as one (Figure 3B white bar), we calculated the relative normalized DMPK mRNA level from each cell line. We observed a 2.9-fold increase in DMPK mRNA level in DM1 myoblasts (Figure 3B orange bar vs. white bar). The non-targeted DM1 myoblasts showed a 3% reduction in DMPK mRNA level compared to the non-treated DM1 myoblasts (Figure 3B blue bar vs. orange bar, 2.81 vs. 2.9). In contrast, the CUG-targeting DM1 myoblasts showed a 20% reduction in DMPK mRNA level (Figure 3B green bar vs. orange bar, 2.32 vs. 2.9). The above data suggest that the LshCas13a treatment significantly reduced CUG RNA foci and rescued several gene missplicing defects without compromising the DMPK mRNA level. We have designed qRT-PCR primers to check potential off-target effects on two human RNAs that contain short CUG repeats, namely MAP3K4 and CASK. Cas13a knocks down about 50% of MAP3K4 mRNA and 30% of CASK mRNA in DM1 myoblasts (either non-targeting or CUG-targeting, Supplementary Figures 6A,B). Despite the off-target knockdown, the residual MAP3K4 and CASK mRNAs remain within or higher than the normal range and may be sufficient to sustain normal cellular activity without yielding detrimental effects.
Conclusion
Expansions of unstable short repeats in the human genome cause an array of neurological and neuromuscular diseases. Although symptomatically and pathologically diverse, these diseases do share an overlapping molecular mechanism, that is, the accumulation of expanded RNA. The expanded RNA can induce cellular toxicity by sequestering various RNA binding proteins as an RNA gain-of-function, or by translation into expanded proteins as a protein gain-of-function. In light of recent findings on RAN translation and the presence of RNA nuclear foci in Huntington’s disease (Zu et al., 2011; Banez-Coronel et al., 2015; Rue et al., 2016; Cleary and Ranum, 2017; Urbanek et al., 2017), the distinction between the two types of toxicity has blurred. Thus, a therapeutic strategy that can target the repeat RNA upstream of the pathogenic cascade may help transition treatment from palliative care to prevention or reversal of disease progression.
In this study, we utilized a recently discovered CRISPR-Cas13a family member LshCas13a to track and degrade the expanded CUG repeats that cause DM1. The expanded CUG repeats are known to form RNA foci with sequestered proteins in the nucleus. Such RNA-protein rich complexes can be an ideal candidate to test the validity of using the eGFP-conjugated dLshCas13a as an RNA tracker. However, the tendency of CUG expansion to form complex gel-like structures may pose access barriers for crRNA (Jain and Vale, 2017). Inferring from the fact that ASOs composed of 3–25 CAG repeats can bind and degrade CUG RNA foci (Mulders et al., 2009; Evers et al., 2011; Lee et al., 2012), we speculate that a repeat-based crRNA with a spacer of 28nt (annealing to approximately 9 CUG repeats) should be sufficient to guide the dLshCas13a tracker to RNA foci. Indeed, we observed statistically significant colocalization between the tracker and CUGx960 repeat foci in COS-M6 cells. By designing a crRNA targeting the 3′-UTR of the ACTB mRNA, we were able to image stress granules in the cytoplasm. These results suggest that dLshCas13a-eGFP is a flexible and reprogrammable RNA tracker that can reach different cellular compartments with the appropriate crRNA. A negative-feedback system using zinc finger self-targeting and KRAB domain repression on dLshCas13a may help further reduce background noise (Abudayyeh et al., 2017).
We showed biochemically that the purified LshCas13a cleaved more than 50% of its CUG target within 16 h in the presence of the repeat-based crRNA (Figure 2B). We attribute the relatively slow cleavage rate to the formation of multiple denaturation-resistant RNA duplexes (Supplementary Figure 3C). These RNA duplex conformations can survive 7 M urea and 30% formamide and may pose a significant energy barrier for target loading into LshCas13a. We also observed non-specific background CUG cleavage by LshCas13a in the absence of a crRNA (Figure 2B), which is different from the collateral RNA degradation commonly seen in the Cas13a family where a non-targeted RNA is cleaved by the activated Cas13a (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Konermann et al., 2018; Myhrvold et al., 2018; Yan et al., 2018). We suggest three explanations for the non-specific RNA cleavage without a properly matched crRNA: (1) The digestion time required for CUG repeats (16 h) is much longer than that for non-repetitive targets [typically 1–2 h (Abudayyeh et al., 2016)], possibly due to repeat RNA’s complex three-dimensional folding [hairpin and gel-like nature (Tian et al., 2000; Jain and Vale, 2017)]. (2) It is evident that in the apo state LshCas13a crystal structure a portion of the target loading channel and the two HEPN cleavage modules are located on the surface of the protein (Liu et al., 2017). The observed non-specific CUG cleavage without crRNA in this study could be due to stochastic loading of target into the apo LshCas13a over the course of digestion. (3) Given the large size of LshCas13a, the protein might not be stable (Supplementary Figures 3A,B); partial degradation may give rise to peptides without proper catalytic constraints. However, it is also possible that Cas13a may exhibit an intrinsic RNA cleavage activity even in the absence of crRNA-target base pairing. In parallel, dCas9 conjugated to a GFP cleaves CUG repeats as efficiently as does dCas9 conjugated to a PIN RNase domain (Batra et al., 2017), suggesting that target promiscuity might be common amongst Cas proteins. In addition, the background cleavage we observed is unlikely due to the CUG imperfect hairpin structure that may mimic the bulged 5′-crRNA handle (Napierala and Krzyzosiak, 1997; Nalavade et al., 2013), because substitutions of key residues in the 5′-crRNA hairpin result in complete abolishment of Cas13a activity (Abudayyeh et al., 2016; Liu et al., 2017).
The ability of LshCas13a to cleave the expanded CUG repeats was further demonstrated by lentiviral delivery into patient-derived DM1 myoblast cells. By FISH we showed that > 20% of these cells contain >4 RNA foci per cell (Figure 2E). The CUG-targeting LshCas13a reduced the population of DM1 myoblasts with >4 RNA foci per cell by approximately 20% (Figure 2E). The incomplete removal of foci was different from those observed with ASOs and PIN-conjugated dCas9 (PIN-dCas9) (Wheeler et al., 2012; Batra et al., 2017), largely due to different enzymatic mechanisms at work. ASO gapmers that form DNA-RNA hybrids elicit endogenous RNase H activity to degrade their target RNA. The PIN-dCas9 with repeat-based sgRNA (without a PAMer sequence) may operate under a complex mechanism, for: (1) The dCas9-sgRNA complex is known to bind more tightly to double-stranded DNA and DNA-RNA heteroduplex than to single-stranded RNA without a PAMer sequence (O’Connell et al., 2014). (2) The PIN domain derived from human SMG6 has high similarity of overall fold to RNase H, and the catalytic triad of acidic residues in the PIN domain are also crucial for RNase H activity (Glavan et al., 2006). Despite the incomplete foci clearance by LshCas13a, the CUG-targeting LshCas13a was sufficient to yield a reversal of splicing defects in six MBNL1-regulated genes that are known to contribute to DM1 phenotype (Figure 3A). The above observation raises the question as to whether a complete RNA foci elimination is required to see phenotypic improvement in cells. We are currently establishing DM1 myoblasts differentiated from induced pluripotent stem cells (iPSCs) with different repeat lengths to further validate LshCas13a’s capacity to track and eliminate CUG foci and to rescue missplicing under varying RNA toxicity load.
Off-target effects and delivery are two major concerns of using CRISPR-Cas systems. Although crRNA could be designed to target unique sequences flanking DMPK CUG repeats to reduce off-target effects, it is possible that such cleavage may not extend to the repeat region and the liberated “pure” CUG repeats may still exert toxicity as shown to form gel-like structures that trap MBNL (Jain and Vale, 2017). By designing repeat-based crRNA, we initially speculate that the accumulation of CUG repeat expansions would provide more target sites for LshCas13a to cleave, which may dissipate potential off-target effects. In line with our speculation, a small molecule with a bleomycin-cleavage module binding to every 2 CUG repeats preferentially cleaves CUG500 over CUG5 and other short CUG-containing RNA (Rzuczek et al., 2017). Furthermore, ASOs with CAG5, CAG7, or CAG10 silence long CUG transcripts more efficiently than short CUG transcripts in DM1 myoblasts (Gonzalez-Barriga et al., 2013). Our experiments show that the levels of DMPK and other CUG-containing mRNAs are affected by CUG-targeting LshCas13a, but the residual mRNA levels are comparable to or higher than the wildtype levels. Further analyses with different DM1-relevant cell lines (e.g., cardiomyocytes and neurons), and animal models will provide additional information on phenotypic correction and off-target effects. Finally, the LshCas13a-crRNA locus is approximately 7.8 kb in size and poses a challenge for adeno-associated viral (AAV) delivery (a packaging capacity of around 4.5 kb). In light of recent success of using a dual-vector AAV delivery system for gene editing (Bengtsson et al., 2017), it is possible to encode LshCas13a (≈4.1 kb) and crRNA on two separate AAV vectors. Overall, our findings highlight the potential use of Cas13a to modulate the repeat RNA level. With our growing understanding of crRNA design and prediction and the choice of different Cas13 proteins at hand, the CRISPR-Cas13a strategy could potentially be extended to treating other microsatellite expansion diseases.
Materials and Methods
Ethics
The studies involving human participants were reviewed and approved by the Institutional Review Board of the University of Florida. The patients/participants provided their written informed consent to participate in this study.
Plasmid Constructs
The pcDNA3.1hygro-CTGx30 plasmids used in in vitro transcription were cloned into the vector backbone (Thermo Fisher) with NheI and HindIII. Wildtype LshCas13a was cloned from pC001 (Addgene #79150, gift from Feng Zhang) into pAcGFP-C1 (Clonetech) and subsequently underwent site-directed mutagenesis to create the pAcGFP-C1-LshC2c2R1278A plasmid. To construct the tracker vector, the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid (Addgene #42230, gift from Feng Zhang) was modified through the following steps: (1) Replacing the pU6-Chimeric gRNA scaffold with pU6-DR-crRNA containing two inverted BsaI sites (IDT). The BsaI sites were designed for spacer insertion. (2) Replacing the CBh promoter with pCMV from pAcGFP-C1. (3) Primer walk to concatenate nucleoplasmin NLS, LshCas13aR1278A, linker, eGFP (from the pC015-dLwCas13a-NF plasmid, Addgene #91905, gift from Feng Zhang) and c-Myc NLS. To construct the lentiviral expression vector, the pU6-sgRNA scaffold sequence was replaced with pU6-crRNA (IDT) in the lentiCRISPR v2 plasmid (Addgene #52961, gift from Feng Zhang). Subsequently, the SpCas9 sequence was replaced with the concatenated LshCas13a-nucleoplasmin NLS-SV40 NLS-P2A-pyromycin resistant gene.
Cell Culture and Transfection
COS-M6 cells (an African green monkey kidney cell line) were commercially obtained and cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (GIBCO) and passaged at 80–90% confluency. HEK293FT cells were cultured in GlutaMAX supplemented with 10% FBS and 1% penicillin/streptomycin and passaged at 80–90% confluency. Myoblasts were cultured in skeletal muscle growth medium (Cell Applications) and passaged at 70–80% confluency. COS-M6 cells were seeded at 5x105 cells per well of a 6-well plate the day before transfection. For FISH studies, 2 μg of DT960 and 2 μg of the tracker plasmid were mixed with 10 μl of Lipofectamine 2000 (Thermo Fisher) in OptiMem during transfection. For FISH-IF studies, 2 μg of the ACTB tracker vector at a 1:5 ratio to Lipofectamine 2,000 were used.
Lentiviral Production and Transduction
LentiCRISPR v2-derived constructs encoding dLshCas13a and crRNA were co-transfected with psPAX2 (Addgene #12260, packaging vector, gift from Didier Trono) and pMD2.G (Addgene #12259, envelop vector, gift from Didier Trono) into HEK293FT cells at a ratio of 3.3:2.5:1 (a total of 17 μg DNA) on a 10 cm plate with Fugene 6 (Promega). Viruses produced at 48 and 72 h were filtered with 0.45 μm PVDF filter units (Millex-HV), checked for titer with Lenti-X GoStix (Takara), and used for transduction on myoblasts in the presence of 8 μg/ml polybrene per well of a 6-well plate. Cells were selected in 2 μg/ml puromycin for 5 days.
RNA Fluorescence in situ Hybridization
Experiments were conducted as described in Manning et al. (2017). Twenty-four hours post transfection, cells on round coverslips were fixed in 4% PFA in PBS for 15 min, permeabilized in 0.2% Triton X-100 in PBS for 10 min, and incubated in 40% formamide/2x SSC for 10 min. Cells were then incubated with 200 nM Cy3-labeled PNA probes (PNA Bio) in the hybridization buffer (40% formamide/2x SSC/0.02% BSA) at 37°C for 1 h and washed with 40% formamide/1x SSC at 37°C for 1 h, twice with 1x SSC for 30 min, and four times with PBS (5 min each) before mounted in mounting medium with DAPI (Vectorlabs). Images were taken at x60 objective on a Nikon A1 Confocal Imaging System with filters specifically for Cy3 and eGFP under a sequential stimulation mode. For quantification of RNA foci in each cell line, data were collected from two independent experiments each with two technical replicates. All images within each experiment were taken under identical exposure and transmittance.
Immunofluorescence
To induce stress granule formation, 200 μM sodium arsenite were added per well of a 6-well plate at 37°C for 1 h 30 min. After 1x PBS wash, cells were fixed in 4% PFA in PBS for 15 min, permeabilized in 0.2% Triton X-100 in PBS for 10 min, and blocked in 5% BSA in PBST for 1 h. Mouse monoclonal anti-G3BP1-Alexa 594 (H-10, Santa Cruz, CA, United States) antibody was added to cells at 4 μg/ml in 5% BSA-PBST at 4°C overnight. After four PBS washes (5 min each), cells were mounted in mounting medium containing DAPI for imaging. Two biological samples each with two technical replicates were imaged. The mouse monoclonal anti-MBNL1 antibody (Sigma M3320, clone number 3A4-1E9, immunogen – recombinant MBNL1) was used for IF.
LshCas13a Protein Expression and Purification
LshCas13a was expressed from the pC001-huLshC2c2-MBP vector (gift from Feng Zhang) in E. coli BL21 (DE3) cells. E. coli cells were grown in 1 L LB broth (Sigma) at 37°C to reach OD600 = 0.4–0.7 before induction with 0.5 mM IPTG at 18°C overnight. Cells were harvested by centrifugation at 5,000 rpm, resuspended in lysis buffer (20 mM Tris–HCl pH7.5/500 mM NaCl/5% glycerol), and frozen at −80°C overnight.
Frozen cells were thawed, sonicated on ice for two 10 min cycles of 40% amplitude 2 s on-off, and centrifuged at 14,000 rpm to obtain the soluble fraction. The soluble lysate was incubated with NiNTA agarose beads (Qiagen) of a 5 mL bed volume, washed with 7 volumes of wash buffer (50 mM Tris–HCl pH7.5/2 M NaCl/5 mM MgCl2/5% glycerol), eluted under a linear gradient of elution buffer (50 mM Tris–HCl pH7.5/2 M NaCl/5 mM MgCl2/1 M imidazole/5% glycerol), and dialyzed in 50 mM Tris–HCl pH7.5/500 mM NaCl/5 mM MgCl2/1 mM DTT/5% glycerol. The dialyzed LshCas13a was further purified with amylose agarose (NEB) of a 3 ml bed volume with wash buffer (20 mM Tris–HCl pH7.5/1 M NaCl/5% glycerol) and elution buffer (20 mM Tris–HCl pH7.5/1 M NaCl/10 mM Maltose/5% glycerol). The (His)x6-MBP tag was removed by thrombin (EMD Millipore) at 4°C overnight. The tag, uncleaved LshCas13a and biotinylated thrombin were captured by a mixture of streptavidin and NiNTA agarose beads in column. The flow-through that contains the purified protein was dialyzed in 50 mM Tris–HCl pH7.5/500 mM NaCl/1 mM DTT/5% glycerol.
Generation of RNA
The pcDNA3.1 hygro-CTGx30 plasmids were linearized and gel purified. Approximately 500 ng of DNA template were used to synthesize RNA repeats at 30°C overnight using the HiScribe T7 High Yield Kit (NEB) according to the manufacturer’s protocol. For crRNA synthesis, a synthetic short T7 primer was annealed to the synthetic crRNA (both oligos were ordered from IDT, Oligonucleotide list, Supplementary Table 1) in the annealing buffer (30 mM HEPES pH7.5/100 mM potassium acetate/2 mM magnesium acetate) at 95°C for 5 min, followed by a gradual cool-down to room temperature. All the transcribed RNA underwent Turbo DNase I (Thermo Fisher) digestion and purified with RNA Clean & Concentrator (Zymo Research).
LshCas13a in vitro Cleavage Assay
The cleavage assays were performed in 10 μl volumes, containing: nuclease buffer (40 mM Tris–HCl pH7.5/60 mM NaCl/6 mM MgCl2), 450 nM HuLshCas13a, 100 nM target RNA and 100 nM crRNA. The crRNA was pre-loaded with LshCas13a at 37°C for 15 min before the target RNA was added. Reactions were allowed to proceed over a period of time (0–16 h), quenched with 2x TBE-Urea loading dye (BioRad) mixed with 3x Formamide dye (100% formamide/0.5 M EDTA), boiled at 95°C for 5 min, and loaded on a 10% TBE-Urea gel. Gels were stained with SYBR Gold (Thermo Fisher) at a 1:10,000 dilution for 20 min before imaged using an Azure c400 scanner (Azure Biosystems).
qRT-PCR and Missplicing Assays
Total RNA extractions on 10 cm plates were performed with RNeasy Mini Kit (Qiagen), followed by Turbo DNase I digestion and column purification. PCR reactions were performed on purified RNA with β-actin primers to ensure the absence of genomic DNA contamination for each sample. A 1 μg input of total RNA was converted to cDNA using the iScript cDNA Synthesis Kit (BioRad) according to the manufacturer’s protocol. Typically, 2.5 μl of the diluted cDNA (1:3 dilution) were used for qRT-PCR on a CFX96 Real-time System (BioRad). Three biological replicates, each with three technical replicates, were performed.
For the missplicing assay, 2.5 μl of the diluted cDNA were incubated with Amplitaq Gold 360 Master Mix (Thermo Fisher) with 95°C 5 min, 35x (95°C 30 s, 61°C 30 s and 72°C 1 min), 72°C 5 min and 4°C hold. Reactions were resolved on a 2% agarose gel, scanned using Azure c400, and quantified using Fiji ImagJ. Two independent experiments were performed for each sample.
Quantification and Statistical Analysis
RNA foci counts were performed in Fiji ImageJ with the “Find Maxima” function at a threshold level of 80. Significance levels were calculated using Chi-square with 4 foci per cell as a cut-off.
Colocalization between stress granules and the dLshCas13a tracker was analyzed in Fiji ImageJ. Images were processed with “Split Channels.” Each cell was applied with an ROI mask before analyzed by “Coloc 2” to get the Pearson’s R value. R values were tabulated and two-tailed t-tests were performed in GraphPad Prism 7. Statistical values used: ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
In vitro RNA Cleavage and Splicing Were Quantified Using Fiji ImageJ
All qRT-PCR data were analyzed in BioRad CFX Manager. Two-tailed t-tests were performed in GraphPad Prism 7. Statistical values used: ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
NZ and BB carried out cloning. NZ expressed and purified LshCas13a and performed biochemical cleavage assays, performed FISH, FISH/IF, missplicing assays and qRT-PCR, and devised the experiments. GX and DF provided critical experimental cell lines. NZ and TA performed data analysis and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the AFM-Telethon Trampoline Grant (#21601) awarded to NZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anjana Tiwari and Vera Hesham for critical comments of the manuscript. We thank Chantel Potvin and Mona Bisase for helping with the logistics of the project. We thank Thomas Cooper M. D., Kassie Manning, Arseniy Kolonin, and Ashish Rao for providing critical cell lines and protocols.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.594576/full#supplementary-material
Footnotes
References
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. doi: 10.1038/nature24049
Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D. B., et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353:aaf5573. doi: 10.1126/science.aaf5573
Adams, B. D., Parsons, C., Walker, L., Zhang, W. C., and Slack, F. J. (2017). Targeting noncoding RNAs in disease. J. Clin. Invest. 127, 761–771.
Arandel, L., Polay Espinoza, M., Matloka, M., Bazinet, A., De Dea Diniz, D., Naouar, N., et al. (2017). Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech. 10, 487–497. doi: 10.1242/dmm.027367
Banez-Coronel, M., Ayhan, F., Tarabochia, A. D., Zu, T., Perez, B. A., Tusi, S. K., et al. (2015). RAN translation in huntington disease. Neuron 88, 667–677. doi: 10.1016/j.neuron.2015.10.038
Batra, R., Charizanis, K., Manchanda, M., Mohan, A., Li, M., Finn, D. J., et al. (2014). Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 56, 311–322. doi: 10.1016/j.molcel.2014.08.027
Batra, R., Nelles, D. A., Pirie, E., Blue, S. M., Marina, R. J., Wang, H., et al. (2017). Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell 170, 899–912.e10. doi: 10.1016/j.cell.2017.07.010
Benarroch, E. E. (2018). Cytoplasmic RNA granules, ribostasis, and neurodegeneration. Neurology 90, 553–562. doi: 10.1212/wnl.0000000000005172
Bengtsson, N. E., Hall, J. K., Odom, G. L., Phelps, M. P., Andrus, C. R., Hawkins, R. D., et al. (2017). Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 8:14454.
Bley, N., Lederer, M., Pfalz, B., Reinke, C., Fuchs, T., Glass, M., et al. (2015). Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res. 43:e26. doi: 10.1093/nar/gku1275
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 69:385.
Burel, S. A., Hart, C. E., Cauntay, P., Hsiao, J., Machemer, T., Katz, M., et al. (2016). Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 44, 2093–2109. doi: 10.1093/nar/gkv1210
Cappella, M., Perfetti, A., Cardinali, B., Garcia-Manteiga, J. M., Carrara, M., Provenzano, C., et al. (2018). High-throughput analysis of the RNA-induced silencing complex in myotonic dystrophy type 1 patients identifies the dysregulation of miR-29c and its target ASB2. Cell Death Dis. 9:729.
Cerro-Herreros, E., Sabater-Arcis, M., Fernandez-Costa, J. M., Moreno, N., Perez-Alonso, M., Llamusi, B., et al. (2018). miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 9:2482.
Chau, A., and Kalsotra, A. (2015). Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev. Dyn. 244, 377–390. doi: 10.1002/dvdy.24240
Childs-Disney, J. L., Stepniak-Konieczna, E., Tran, T., Yildirim, I., Park, H., Chen, C. Z., et al. (2013). Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat. Commun. 4:2044.
Clarke, R., Heler, R., MacDougall, M. S., Yeo, N. C., Chavez, A., Regan, M., et al. (2018). Enhanced bacterial immunity and mammalian genome editing via RNA-polymerase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol. Cell 71, 42–55.e8.
Cleary, J. D., and Ranum, L. P. (2017). New developments in RAN translation: insights from multiple diseases. Curr. Opin. Genet. Dev. 44, 125–134. doi: 10.1016/j.gde.2017.03.006
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823.
Costales, M. G., Matsumoto, Y., Velagapudi, S. P., and Disney, M. D. (2018). Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc. 140, 6741–6744. doi: 10.1021/jacs.8b01233
Cox, D. B. T., Gootenberg, J. S., Abudayyeh, O. O., Franklin, B., Kellner, M. J., Joung, J., et al. (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. doi: 10.1126/science.aaq0180
Dastidar, S., Ardui, S., Singh, K., Majumdar, D., Nair, N., Fu, Y., et al. (2018). Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 46, 8275–8298. doi: 10.1093/nar/gky548
Du, H., Cline, M. S., Osborne, R. J., Tuttle, D. L., Clark, T. A., Donohue, J. P., et al. (2010). Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 17, 187–193. doi: 10.1038/nsmb.1720
East-Seletsky, A., O’Connell, M. R., Burstein, D., Knott, G. J., and Doudna, J. A. (2017). RNA targeting by functionally orthogonal type VI-A CRISPR-Cas enzymes. Mol. Cell 66, 373–383.e3.
East-Seletsky, A., O’Connell, M. R., Knight, S. C., Burstein, D., Cate, J. H., Tjian, R., et al. (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. doi: 10.1038/nature19802
Ebralidze, A., Wang, Y., Petkova, V., Ebralidse, K., and Junghans, R. P. (2004). RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science 303, 383–387. doi: 10.1126/science.1088679
Evers, M. M., Pepers, B. A., van Deutekom, J. C., Mulders, S. A., den Dunnen, J. T., Aartsma-Rus, A., et al. (2011). Targeting several CAG expansion diseases by a single antisense oligonucleotide. PLoS One 6:e24308. doi: 10.1371/journal.pone.0024308
Fernandez-Costa, J. M., Garcia-Lopez, A., Zuniga, S., Fernandez-Pedrosa, V., Felipo-Benavent, A., Mata, M., et al. (2013). Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 22, 704–716. doi: 10.1093/hmg/dds478
Francois, V., Klein, A. F., Beley, C., Jollet, A., Lemercier, C., Garcia, L., et al. (2011). Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat. Struct. Mol. Biol. 18, 85–87. doi: 10.1038/nsmb.1958
Gareiss, P. C., Sobczak, K., McNaughton, B. R., Palde, P. B., Thornton, C. A., and Miller, B. L. (2008). Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: discovery of lead compounds targeting myotonic dystrophy (DM1). J. Am. Chem. Soc. 130, 16254–16261. doi: 10.1021/ja804398y
Glavan, F., Behm-Ansmant, I., Izaurralde, E., and Conti, E. (2006). Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J. 25, 5117–5125. doi: 10.1038/sj.emboj.7601377
Gonzalez-Barriga, A., Mulders, S. A., van de Giessen, J., Hooijer, J. D., Bijl, S., van Kessel, I. D., et al. (2013). Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids 2:e81. doi: 10.1038/mtna.2013.9
Goodwin, M., Mohan, A., Batra, R., Lee, K. Y., Charizanis, K., Fernandez Gomez, F. J., et al. (2015). MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. 12, 1159–1168. doi: 10.1016/j.celrep.2015.07.029
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., and Zhang, F. (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. doi: 10.1126/science.aaq0179
Gourdon, G., and Meola, G. (2017). Myotonic dystrophies: state of the art of new therapeutic developments for the CNS. Front. Cell Neurosci. 11:101. doi: 10.3389/fncel.2017.00101
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B., and Taipale, J. (2018). CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930. doi: 10.1038/s41591-018-0049-z
Hino, S., Kondo, S., Sekiya, H., Saito, A., Kanemoto, S., Murakami, T., et al. (2007). Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum. Mol. Genet. 16, 2834–2843. doi: 10.1093/hmg/ddm239
Hsieh, W. C., Bahal, R., Thadke, S. A., Bhatt, K., Sobczak, K., Thornton, C., et al. (2018). Design of a “Mini” nucleic acid probe for cooperative binding of an RNA-repeated transcript associated with myotonic dystrophy type 1. Biochemistry 57, 907–911. doi: 10.1021/acs.biochem.7b01239
Huichalaf, C., Sakai, K., Jin, B., Jones, K., Wang, G. L., Schoser, B., et al. (2010). Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 24, 3706–3719. doi: 10.1096/fj.09-151159
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946.
Jain, A., and Vale, R. D. (2017). RNA phase transitions in repeat expansion disorders. Nature 546, 243–247. doi: 10.1038/nature22386
Jauvin, D., Chretien, J., Pandey, S. K., Martineau, L., Revillod, L., Bassez, G., et al. (2017). Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids 7, 465–474. doi: 10.1016/j.omtn.2017.05.007
Jiang, H., Mankodi, A., Swanson, M. S., Moxley, R. T., and Thornton, C. A. (2004). Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079–3088. doi: 10.1093/hmg/ddh327
Jing, X., Xie, B., Chen, L., Zhang, N., Jiang, Y., Qin, H., et al. (2018). Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 46:e90. doi: 10.1093/nar/gky433
Kakiuchi-Kiyota, S., Koza-Taylor, P. H., Mantena, S. R., Nelms, L. F., Enayetallah, A. E., Hollingshead, B. D., et al. (2014). Comparison of hepatic transcription profiles of locked ribonucleic acid antisense oligonucleotides: evidence of distinct pathways contributing to non-target mediated toxicity in mice. Toxicol. Sci. 138, 234–248. doi: 10.1093/toxsci/kft278
Kasuya, T., Hori, S., Watanabe, A., Nakajima, M., Gahara, Y., Rokushima, M., et al. (2016). Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci. Rep. 6:30377. doi: 10.1038/srep30377
Kim, Y. K., Mandal, M., Yadava, R. S., Paillard, L., and Mahadevan, M. S. (2014). Evaluating the effects of CELF1 deficiency in a mouse model of RNA toxicity. Hum. Mol. Genet. 23, 293–302. doi: 10.1093/hmg/ddt419
Konermann, S., Lotfy, P., Brideau, N. J., Oki, J., Shokhirev, M. N., and Hsu, P. D. (2018). Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676e14.
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771. doi: 10.1038/nbt.4192
Krol, J., Fiszer, A., Mykowska, A., Sobczak, K., de Mezer, M., and Krzyzosiak, W. J. (2007). Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 25, 575–586. doi: 10.1016/j.molcel.2007.01.031
Kuyumcu-Martinez, N. M., and Cooper, T. A. (2006). Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog. Mol. Subcell Biol. 44, 133–159. doi: 10.1007/978-3-540-34449-0_7
Kuyumcu-Martinez, N. M., Wang, G. S., and Cooper, T. A. (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78. doi: 10.1016/j.molcel.2007.07.027
Langlois, M. A., Lee, N. S., Rossi, J. J., and Puymirat, J. (2003). Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol. Ther. 7, 670–680. doi: 10.1016/s1525-0016(03)00068-6
Lee, J. E., Bennett, C. F., and Cooper, T. A. (2012). RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U.S.A. 109, 4221–4226. doi: 10.1073/pnas.1117019109
Li, Y. R., King, O. D., Shorter, J., and Gitler, A. D. (2013). Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372. doi: 10.1083/jcb.201302044
Liu, L., Li, X., Wang, J., Wang, M., Chen, P., Yin, M., et al. (2017). Two distant catalytic sites are responsible for C2c2 RNase activities. Cell 168, 121–134.e12. doi: 10.1016/j.cell.2016.12.031
Lopez-Morato, M., Brook, J. D., and Wojciechowska, M. (2018). Small molecules which improve pathogenesis of myotonic dystrophy type 1. Front. Neurol. 9:349. doi: 10.3389/fneur.2018.00349
Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., et al. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 255, 1253–1255. doi: 10.1126/science.1546325
Manning, K. S., Rao, A. N., Castro, M., and Cooper, T. A. (2017). BNA(NC) gapmers revert splicing and reduce RNA foci with low toxicity in myotonic dystrophy cells. ACS Chem. Biol. 12, 2503–2509. doi: 10.1021/acschembio.7b00416
Matsuki, H., Takahashi, M., Higuchi, M., Makokha, G. N., Oie, M., and Fujii, M. (2013). Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 18, 135–146. doi: 10.1111/gtc.12023
Mohan, A., Goodwin, M., and Swanson, M. S. (2014). RNA-protein interactions in unstable microsatellite diseases. Brain Res. 1584, 3–14. doi: 10.1016/j.brainres.2014.03.039
Mulders, S. A., van den Broek, W. J., Wheeler, T. M., Croes, H. J., van Kuik-Romeijn, P., de Kimpe, S. J., et al. (2009). Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 13915–13920. doi: 10.1073/pnas.0905780106
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. doi: 10.1126/science.aas8836
Nalavade, R., Griesche, N., Ryan, D. P., Hildebrand, S., and Krauss, S. (2013). Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 4:e752. doi: 10.1038/cddis.2013.276
Napierala, M., and Krzyzosiak, W. J. (1997). CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 272, 31079–31085. doi: 10.1074/jbc.272.49.31079
Nelles, D. A., Fang, M. Y., O’Connell, M. R., Xu, J. L., Markmiller, S. J., Doudna, J. A., et al. (2016). Programmable RNA tracking in live cells with CRISPR/Cas9. Cell 165, 488–496. doi: 10.1016/j.cell.2016.02.054
Nelson, C. E., Robinson-Hamm, J. N., and Gersbach, C. A. (2017). Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 13, 647–661. doi: 10.1038/nrneurol.2017.126
Nguyen, L., Luu, L. M., Peng, S., Serrano, J. F., Chan, H. Y., and Zimmerman, S. C. (2015). Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J. Am. Chem. Soc. 137, 14180–14189. doi: 10.1021/jacs.5b09266
O’Connell, M. R., Oakes, B. L., Sternberg, S. H., East-Seletsky, A., Kaplan, M., and Doudna, J. A. (2014). Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266. doi: 10.1038/nature13769
Pinto, B. S., Saxena, T., Oliveira, R., Mendez-Gomez, H. R., Cleary, J. D., Denes, L. T., et al. (2017). Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479–490e5.
Provenzano, C., Cappella, M., Valaperta, R., Cardani, R., Meola, G., Martelli, F., et al. (2017). CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol. Ther. Nucleic Acids 9, 337–348. doi: 10.1016/j.omtn.2017.10.006
Rau, F., Freyermuth, F., Fugier, C., Villemin, J. P., Fischer, M. C., Jost, B., et al. (2011). Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18, 840–845. doi: 10.1038/nsmb.2067
Ray, M., Tang, R., Jiang, Z., and Rotello, V. M. (2015). Quantitative tracking of protein trafficking to the nucleus using cytosolic protein delivery by nanoparticle-stabilized nanocapsules. Bioconjug. Chem. 26, 1004–1007. doi: 10.1021/acs.bioconjchem.5b00141
Renna, L. V., Bose, F., Brigonzi, E., Fossati, B., Meola, G., and Cardani, R. (2019). Aberrant insulin receptor expression is associated with insulin resistance and skeletal muscle atrophy in myotonic dystrophies. PLoS One 14:e0214254. doi: 10.1371/journal.pone.0214254
Renna, L. V., Bose, F., Iachettini, S., Fossati, B., Saraceno, L., Milani, V., et al. (2017). Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS One 12:e0184987. doi: 10.1371/journal.pone.0184987
Richard, G. F., Viterbo, D., Khanna, V., Mosbach, V., Castelain, L., and Dujon, B. (2014). Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS One 9:e95611. doi: 10.1371/journal.pone.0095611
Rohilla, K. J., and Gagnon, K. T. (2017). RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol. Commun. 5:63.
Rue, L., Banez-Coronel, M., Creus-Muncunill, J., Giralt, A., Alcala-Vida, R., Mentxaka, G., et al. (2016). Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J. Clin. Invest. 126, 4319–4330. doi: 10.1172/jci83185
Rzuczek, S. G., Colgan, L. A., Nakai, Y., Cameron, M. D., Furling, D., Yasuda, R., et al. (2017). Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 13, 188–193. doi: 10.1038/nchembio.2251
Rzuczek, S. G., Southern, M. R., and Disney, M. D. (2015). Studying a drug-like, RNA-focused small molecule library identifies compounds that inhibit RNA toxicity in myotonic dystrophy. ACS Chem. Biol. 10, 2706–2715. doi: 10.1021/acschembio.5b00430
Santoro, M., Masciullo, M., Bonvissuto, D., Bianchi, M. L., Michetti, F., and Silvestri, G. (2013). Alternative splicing of human insulin receptor gene (INSR) in type I and type II skeletal muscle fibers of patients with myotonic dystrophy type 1 and type 2. Mol. Cell Biochem. 380, 259–265. doi: 10.1007/s11010-013-1681-z
Savkur, R. S., Philips, A. V., and Cooper, T. A. (2001). Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29, 40–47. doi: 10.1038/ng704
Shmakov, S., Smargon, A., Scott, D., Cox, D., Pyzocha, N., Yan, W., et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 15, 169–182.
Smargon, A. A., Cox, D. B. T., Pyzocha, N. K., Zheng, K., Slaymaker, I. M., Gootenberg, J. S., et al. (2017). Cas13b Is a Type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol. Cell 65, 618–630e7.
Sobczak, K., Wheeler, T. M., Wang, W., and Thornton, C. A. (2013). RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 21, 380–387. doi: 10.1038/mt.2012.222
Tian, B., White, R. J., Xia, T., Welle, S., Turner, D. H., Mathews, M. B., et al. (2000). Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 6, 79–87. doi: 10.1017/s1355838200991544
Timchenko, L. (2013). Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int. J. Biochem. Cell Biol. 45, 2280–2287. doi: 10.1016/j.biocel.2013.06.010
Tourriere, H., Chebli, K., Zekri, L., Courselaud, B., Blanchard, J. M., Bertrand, E., et al. (2003). The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160, 823–831. doi: 10.1083/jcb.200212128
Unsworth, H., Raguz, S., Edwards, H. J., Higgins, C. F., and Yague, E. (2010). mRNA escape from stress granule sequestration is dictated by localization to the endoplasmic reticulum. FASEB J. 24, 3370–3380. doi: 10.1096/fj.09-151142
Urbanek, M. O., Fiszer, A., and Krzyzosiak, W. J. (2017). Reduction of Huntington’s disease RNA foci by CAG repeat-targeting reagents. Front. Cell Neurosci. 11:82. doi: 10.3389/fncel.2017.00082
van Agtmaal, E. L., Andre, L. M., Willemse, M., Cumming, S. A., van Kessel, I. D. G., van den Broek, W., et al. (2017). CRISPR/Cas9-induced (CTGCAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 25, 24–43. doi: 10.1016/j.ymthe.2016.10.014
Wang, Y., Hao, L., Wang, H., Santostefano, K., Thapa, A., Cleary, J., et al. (2018). Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol Ther. 26, 2617–2630. doi: 10.1016/j.ymthe.2018.09.003
Warf, M. B., Nakamori, M., Matthys, C. M., Thornton, C. A., and Berglund, J. A. (2009). Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 18551–18556. doi: 10.1073/pnas.0903234106
Wheeler, J. R., Matheny, T., Jain, S., Abrisch, R., and Parker, R. (2016). Distinct stages in stress granule assembly and disassembly. eLife 5:e18413.
Wheeler, T. M., Leger, A. J., Pandey, S. K., MacLeod, A. R., Nakamori, M., Cheng, S. H., et al. (2012). Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115. doi: 10.1038/nature11362
Wheeler, T. M., Sobczak, K., Lueck, J. D., Osborne, R. J., Lin, X., Dirksen, R. T., et al. (2009). Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325, 336–339. doi: 10.1126/science.1173110
Yan, W. X., Chong, S., Zhang, H., Makarova, K. S., Koonin, E. V., Cheng, D. R., et al. (2018). Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol. Cell 70, 327–339.e5.
Zhang, N., and Ashizawa, T. (2017). RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 44, 17–29. doi: 10.1016/j.gde.2017.01.005
Zhao, X., Liu, L., Lang, J., Cheng, K., Wang, Y., Li, X., et al. (2018). A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 431, 171–181. doi: 10.1016/j.canlet.2018.05.042
Keywords: myotonic dystrophy, neurodegeneration, CRISPR-Cas13a, RNA targeting, stress granule
Citation: Zhang N, Bewick B, Xia G, Furling D and Ashizawa T (2020) A CRISPR-Cas13a Based Strategy That Tracks and Degrades Toxic RNA in Myotonic Dystrophy Type 1. Front. Genet. 11:594576. doi: 10.3389/fgene.2020.594576
Received: 04 September 2020; Accepted: 23 November 2020;
Published: 10 December 2020.
Edited by:
Philipp Kapranov, Huaqiao University, ChinaReviewed by:
Giovanni Meola, University of Milan, ItalyPartha Sarathi Sarkar, University of Texas Medical Branch at Galveston, United States
Copyright © 2020 Zhang, Bewick, Xia, Furling and Ashizawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Zhang, bnpoYW5nQGhvdXN0b25tZXRob2Rpc3Qub3Jn; Tetsuo Ashizawa, dGFzaGl6YXdhQGhvdXN0b25tZXRob2Rpc3Qub3Jn