 Bo Zhang
Bo Zhang Yanlin Gu
Yanlin Gu Guoqin Jiang
Guoqin Jiang- General Surgery Department, The Second Affiliated Hospital of Soochow University, Suzhou, China
Purpose: N6-methyladenosine (m6A) is the most prevalent modification in mRNA methylation which has a wide effect on biological functions. This study aims to figure out the efficacy of m6A RNA methylation regulator-based biomarkers with prognostic significance in breast cancer.
Patients and Methods: The 23 RNA methylation regulators were firstly analyzed through ONCOMINE, then relative RNA-seq transcriptome and clinical data of 1,096 breast cancer samples and 112 normal tissue samples were acquired from The Cancer Gene Atlas (TCGA) database. The expressive distinction was also showed by the Gene Expression Omnibus (GEO) database. The gene expression data of m6A RNA regulators in human tissues were acquired from the Genotype-Tissue Expression (GTEx) database. The R v3.5.1 and other online tools such as STRING, bc-GeneExminer v4.5, Kaplan-Meier Plotter were applied for bioinformatics analysis.
Results: Results from ONCOMINE, TCGA, and GEO databases showed distinctive expression and clinical correlations of m6A RNA methylation regulators in breast cancer patients. The high expression of YTHDF3, ZC3H13, LRPPRC, and METTL16 indicated poor survival rate in patients with breast cancer, while high expression of RBM15B pointed to a better survival rate. Both univariate and multivariate Cox regression analyses revealed that age and risk scores were related to overall survival (OS). Univariate analysis also delineated that stage, tumor (T) status, lymph node (N) status, and metastasis (M) status were associated with OS. From another perspective, Kaplan-Meier Plotter platform showed that the relatively high expression of YTHDF3 and LRPPRC and the relatively low expression of RBM15B, ZC3H13, and METTL16 in breast cancer patients had worse Relapse-Free Survival (RFS). Breast Cancer Gene-Expression Miner v4.5 showed that LRPPRC level was negatively associated with ER and PR expression, while METTL16, RBM15B, ZC3H13 level was positively linked with ER and PR expression. In HER-2 (+) breast cancer patients, the expression of LRPPRC, METTL16, RBM15B, and ZC3H13 were all lower than the HER-2 (−) group.
Conclusion: The significant difference in expression levels and prognostic value of m6A RNA methylation regulators were analyzed and validated in this study. This signature revealed the potential therapeutic value of m6A RNA methylation regulators in breast cancer.
Introduction
According to the data GLOBALCAN 2018, breast cancer is the leading cause of cancer-related deaths and the most frequently diagnosed cancer in females with different phenotypes due to genetic and epigenetic diversity (Bray et al., 2018). Benefiting from the early stage diagnosis and treatment, the 5-year relative survival rate of breast cancer patients primarily diagnosed at stage I reaches to nearly 100%, whereas the survival rate of patients first diagnosed at stage IV is only 26% (Miller et al., 2019). Accurate diagnosis, detection and treatment are always effective approaches to improve the prognosis of breast cancer patients. In breast cancer research, epitranscriptome has been focused for its important role in biological functions. The post-transcriptional modifications are widely located in messenger RNA (mRNA) and non-coding RNA (Shi et al., 2019). Until now, over 170 kinds of RNA modifications have been identified (Boccaletto et al., 2018). The RNA methylated modifications occurring on adenosine present in different forms, such as N6-methyladenosine (m6A), N6-2′-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A). In the 1970s, N6-methyladenosine (m6A) was first identified as the most prevalent modification in mRNA. It has been proved to be a reversible and widespread internal adenosine modification particularly enriched in 3′ UTRs codon of mammalian mRNA (Meyer et al., 2012). m6A also has a cooperative effect on RNA functions, such as stability, metabolism, transcriptional regulation and intracellular signaling (Roundtree et al., 2017). The biological importance of m6A can be embodied in embryonic self-renewal capability (Wang et al., 2014), hematopoietic stem/progenitor cells (HSPCs) emergence (Zhang et al., 2017), circadian control (Fustin et al., 2018), heat shock response (Zhou et al., 2015), neuronal functions (Lence et al., 2016), and tumorigenesis (Lan et al., 2019).
RNA modification is mediated by a series of interplays including “writers” (methyltransferases), “readers” (binding proteins), and “erasers” (demethylases) which regulate a complicated biological regulation process. The “writers” include methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), methyltransferase-like 16 (METTL16), William tumor 1 associated protein (WTAP), vir like m6A methyltransferase associated (VIRMA, also named KIAA1429), zinc finger CCCH domain-containing protein 13 (ZC3H13), RNA binding motif protein 15 (RBM15), and RNA binding motif protein 15B (RBM15B). The “readers” are composed of the YTH family: YTH domain containing 1 (YTHDC1), YTH domain containing 2 (YTHDC2), YTH domain-containing family protein 1 (YTHDF1), YTH domain-containing family protein 2 (YTHDF2), YTH domain-containing family protein 3 (YTHDF3), heterogeneous nuclear ribonucleoprotein C (HNRNPC), FMRP translational regulator 1 (FMR1), leucine-rich pentatricopeptide-repeat containing (LRPPRC), heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), insulin like growth factor binding proteins (IGFBPs): IGFBP1, IGFBP2, IGFBP3 and heterogeneous nuclear ribonucleoprotein G (HNRNPG, also named RBMX). The “erasers” are fat mass and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) (Warda et al., 2017; Yang et al., 2018). Each m6A RNA methylation regulator serves a critical role in the RNA methylation process. Although in the past few years, researches have been done to decipher the interactions coupled with m6A RNA methylation regulators, the nature of RNA modifications and their biological functions in breast cancer remain unclear.
To further clarify the expression of various RNA regulators and their impacts on the prognosis of breast cancer, we obtained the expression of 23 m6A RNA regulators from ONCOMINE. We downloaded breast cancer datasets from TCGA, which included 1096 breast cancer samples and 112 normal tissue samples to analyze the distinct expressions of 23 RNA methylation regulators and their clinical characteristics in breast cancer. The expressive results were also confirmed by the GEO database. Through the COX regression analysis and the least absolute shrinkage and selection operator (LASSO) regression, we found five m6A methylation regulators which were significant to clinical prognosis. Kaplan-Meier Plotter and bc-GenExMiner v4.5 were used to further explore the clinicopathological and prognostic value of the five m6A methylation regulators. The Gene Ontology (GO) analysis of 23 regulators and the Gene Set Enrichment Analysis (GSEA) may help to shed light on their biological research value.
Materials and Methods
Datasets and Study Cohort
ONCOMINE Database
ONCOMINE database1 is a systematic and comprehensive aggregation of microarray datasets. The 23 m6A RNA methylation regulators were initially analyzed through ONCOMINE by evaluating their expression in various cancer types compared with the normal. We searched each regulator for the Cancer vs. Normal Analysis and typed with the threshold of p < 0.05 and with the gene ranking at the top 10%.
TCGA Database
The RNA-seq transcriptome data of 1,096 breast cancer samples and 112 normal tissue samples were downloaded from the TCGA database2. Clinical data of breast cancer patients was also downloaded from TCGA. All data were normalized by the expectation-maximization method and converted into Sample IDs by Perl. Patients without survival information were excluded.
GEO Database
The differential expression of m6A RNA methylation regulators between breast cancer tissues and adjacent normal tissues was verified by GSE70905. The data was downloaded from the GEO database3. The GSE70905 included 47 breast adenocarcinoma and 47 paired adjacent normal breast tissues.
Kaplan-Meier Plotter
The Kaplan-Meier curves were displayed via Kaplan-Meier Plotter4 with the log-rank test. It was performed to provide survival information of m6A RNA methylation regulators in breast cancer patients.
Breast Cancer Gene-Expression Miner v4.5
A public statistical mining tool, the Breast Cancer Gene-Expression Miner v4.55, was used to evaluate the correlations between mRNA expression and clinicopathological characteristics, such as ER, PR, and HER-2 status, as well as the Nottingham Prognostic Index (NPI) and the Scarff-Bloom-Richardson (SBR) grading.
Other Online Databases
The Protein-Protein Interaction (PPI) network was used to exhibit the comprehensive functional annotation of related proteins (version11.0)6. Besides, the gene expression of m6A RNA methylation regulators in human tissues was acquired from the GTEx database7. The data was acquired in a way where strict ethical guidelines were followed.
Selection of m6A Methylation Regulators
A total of 23 regulators were selected according to the previous study, including ALKBH5, FTO, HNRNPC, HNRNPA2B1, KIAA1429, METTL14, METTL3, METTL16, RBM15, RBMX, RBM15B, WTAP, YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, ZC3H13, IGF2BP1, IGF2BP2, IGF2BP3, FMR1, and LRPPRC. Then their expressive correlations and clinicopathological characteristics were analyzed.
Bioinformatic Analysis
The expressive distinction and correlation of 23 regulators were analyzed through “Limma” package by using R v3.5.18 with the cut-off criteria of p < 0.05. A heatmap diagram was drawn to compare the expression of m6A RNA methylation regulators between tumors and normal tissues. Vioplot diagrams were constructed to visualize the expression of 23 regulators of breast cancer and normal tissues. Besides, GO analysis of 17 regulators was performed through “GO PLOT” and “Digest” package. Univariate Cox regression analysis and LASSO Cox regression model were used to correlate prognostic m6A regulators, and the corresponding results showed that five regulators significantly correlated with the prognosis of breast cancer according to the statistical analysis of log-rank p-value (p < 0.05) and the hazard ratio (HR) with 95% confidence intervals. If the HR > 1, the gene expression shows a positive correlation with OS, while HR < 1 indicates a negative correlation with OS. Additionally, the receiver operating characteristic (ROC) curves were used to evaluate sensitivity and specificity. Next, all the samples selected by LASSO analysis were divided into two groups judged by the risk score (RS). Patients with RS above the mean were assigned to the high-risk group while the rest patients with RS below the mean were assigned to the low-risk groups. The GSEA was performed to study the functions m6A RNA methylation regulators by using “kegg.v7.1 symbol.gmt” package.
Statistical Analysis
The expression of m6A RNA methylation regulators between breast cancer and normal tissues were compared through the Wilcoxon test. The high-risk group and the low-risk group were classified according to the median risk score. The chi-square test was used to compare the relationship between the clinicopathological variables and risk score. Cox univariate and multivariate analyses were performed to compare the relationship between clinicopathological variables and risk score. The t-test was used to compare the difference between the two groups classified by risk scores. The value p < 0.05 was considered to be statistically significant.
Results
Expression of m6A RNA Methylation Regulators in Breast Cancer
The ONCOMINE analysis revealed the gene expression of the 23 RNA methylation regulators in different types of cancer compared with normal tissues (Figure 1). From the ONCOMINE database, we had a brief view of the expression of the genes. METTL3, METTL16, ZC3H13, YTHDC1 and FTO were expressed at a low level in breast cancer, while KIAA1429, RBM15, and YTHDF1 were expressed at a high level. Then the m6A RNA methylation regulators were analyzed to compare the expression level through the data from the TCGA database. The expression levels in each regulator were compared by the average level of all tissue samples. YTHDF1 (p < 0.001), HNRNPA2B1 (p < 0.001), HNRNPC (p < 0.001), LRPPRC (p < 0.001), KIAA1429 (p < 0.001), RBM15 (p < 0.001), FMR1 (p < 0.01), IGF2BP1 (p < 0.01), YTHDF2 (p < 0.05), and IGF2BP3 (p < 0.05) were over-expressed at the mean level in breast cancer tissues compared with normal tissues, while ZC3H13 (p < 0.001), METTL14 (p < 0.001), YTHDC1 (p < 0.001), WTAP (p < 0.001), IGF2BP2 (p < 0.001), FTO (p < 0.001), and METTL16 (p < 0.001) were found to be under-expressed in breast cancer tissues in comparison with normal tissues (Figures 2A,B). In GSE70905, YTHDC1 (p < 0.05), IGFBP1 (p < 0.001), ALKBH5 (p < 0.001), WTAP (p < 0.001), YTHDF3 (p < 0.001) and HNRNPA2B1 (p < 0.001) were down-regulated in breast cancer to the adjacent. While YTHDC2 (p < 0.05), HNRNPC (p < 0.001), METTL16 (p < 0.001), RBMX (p < 0.05), FMR1 (p < 0.05), RBM15B (p < 0.05), KIAA1429 (p < 0.001), IGFBP3 (p < 0.05), LRPPRC (p < 0.05) and ZC3H13 (p = 0.001) were up-regulated (Figure 2C). The correlations between m6A RNA methylation regulators and clinicopathological features, such as N status, M status, T status, pathological stage, age, and survival state were investigated. The T status (p < 0.001), stage (p < 0.01), and age (p < 0.01) were found to be relevant to the expression level of m6A RNA methylation regulators (Figure 2D). Spearman correlation analysis was performed to compare the relationship between each other. YTHDC1 and METTL14 showed the most significant positive correlation (Figure 2E). The PPI network showed from a novel perspective that the regulators were strongly linked with each other and the line sickness intuitively disclosed the strength of interactions (Figure 2F).
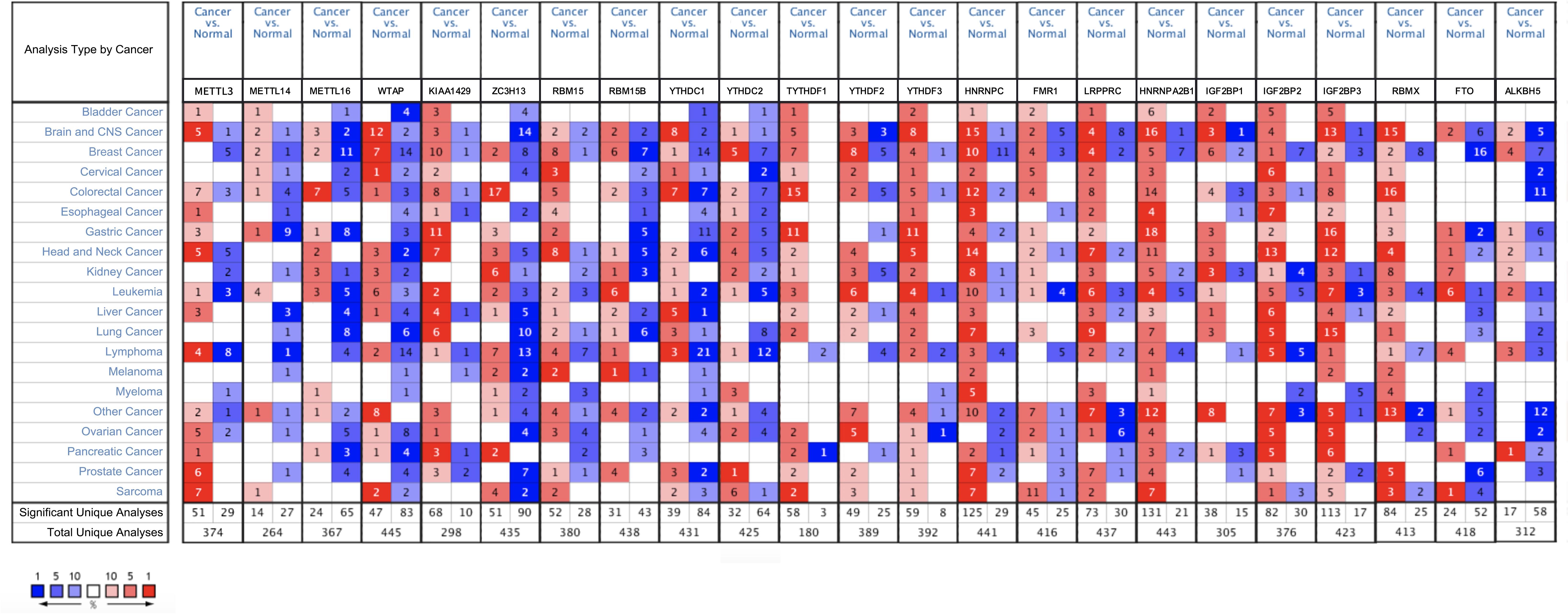
Figure 1. The overall mRNA expression of 23 RNA methylation regulators in various cancer types. The number in the small check signifies the number of datasets which meet the criteria. The red means high expression while the blue means low expression of relative genes in different analyses. And the shade in the box indicates the gene ranks. Statistically significant values are demarcated with the threshold p < 0.05.
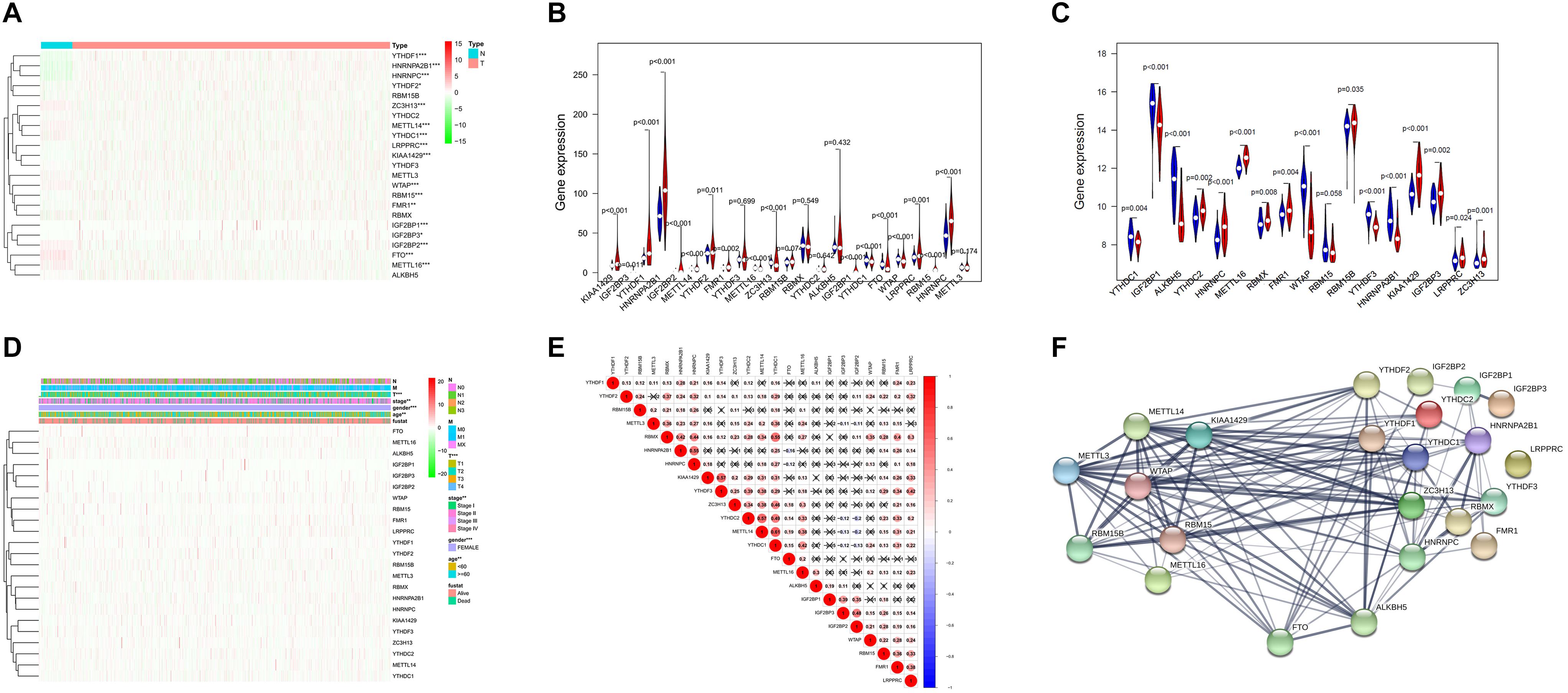
Figure 2. Expression and correlation of m6A RNA methylation regulation factors in breast cancer. (A) Expression heatmap of 23 m6A RNA regulators in breast cancer tissues and normal tissues. Vioplot demonstrates the expression of the regulation factors with the comparison between breast cancer tissues and normal tissues through the data from The Cancer Gene Atlas (TCGA) (B) and Gene Expression Omnibus (GEO) (C). The white spot in every “violin” presents the median expression level. (D) The correlations between m6A RNA methylation regulators and clinicopathological features. (E) Spearman correlation analysis of m6A RNA methylation regulators. (F) The PPI network of m6A RNA methylation regulators. *p < 0.05, **p < 0.01, ***p < 0.001.
Expression and Biological Function Annotation of m6A RNA Methylation Regulators
In order to figure out the expression of RNA methylation regulators in normal human tissues, the GTEx database was used as a supplementary data to verify their research value. On a broad range of tissue types, HNRNPA2B1, HNRNPC, LRPPRC, and YTHDF2 had correspondingly high expression in breast tissues. The regulators such as IGFBP1, IGFBP3 were relatively low by contrast (Figure 3A). The GO analysis showed that these m6A RNA methylation regulators participated in the regulation of mRNA metabolic process, the RNA catabolic process and the regulation of mRNA stability (Figures 3B,C).
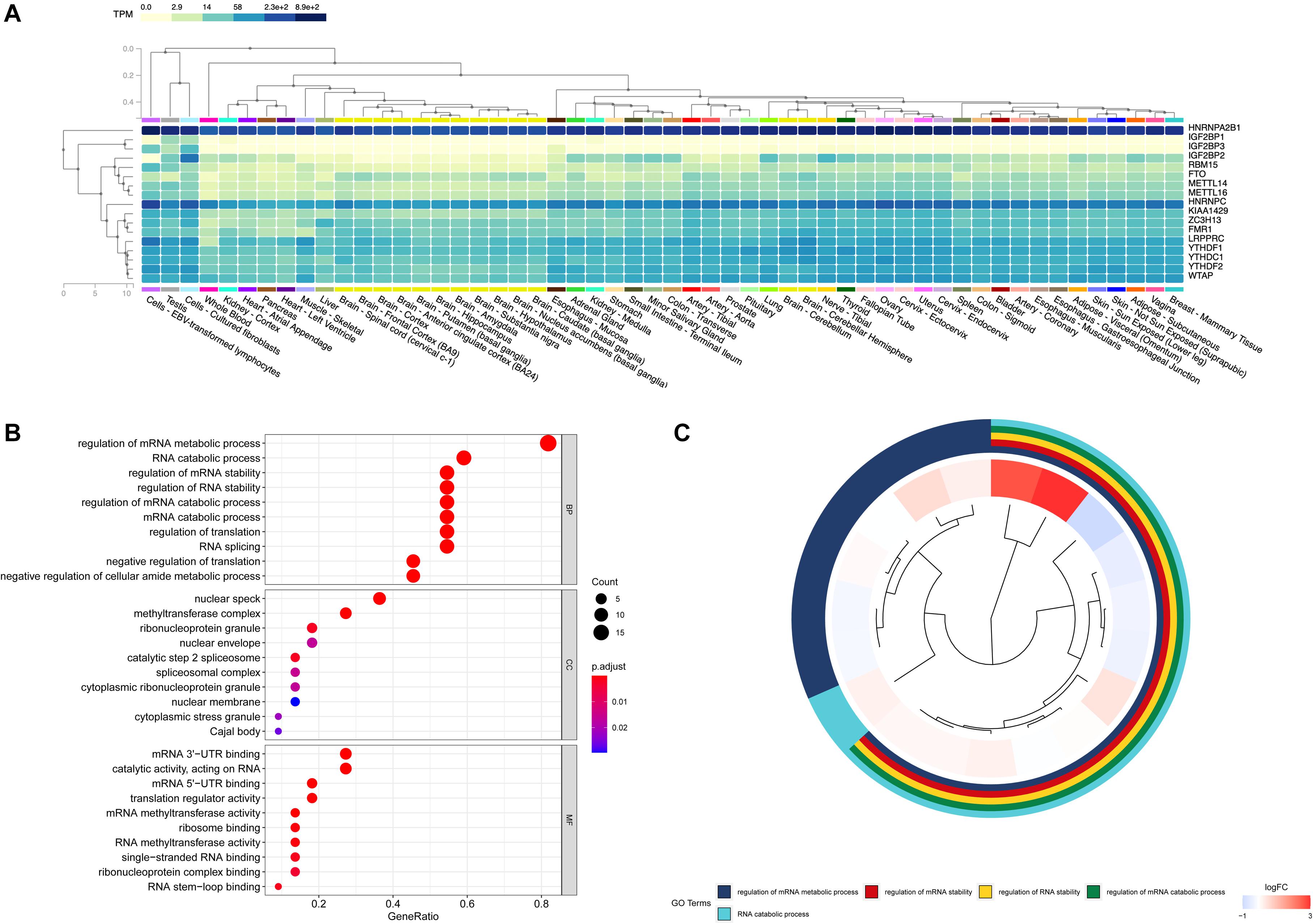
Figure 3. Expression and biological functional annotation of m6A RNA methylation regulators. (A) The expression levels of 17 m6A RNA regulators in normal tissues from the Genotype-Tissue Expression (GTEx) database. (B,C) The Gene Ontology (GO) terms of the genes, including molecular function, biological process, and cellular components.
Prognostic Value and Risk Signature of m6A RNA Methylation Regulators
Cox univariate analysis was performed to select the m6A RNA methylation regulators that were associated with prognosis. The results showed that 5 out of 23 regulators had a close correlation with overall survival (OS). High expression of YTHDF3 (HR: 1.025, 95% CI: 1.009–1.042), ZC3H13 (HR: 1.039, 95% CI: 1.006–1.074), LRPPRC (HR: 1.016, 95% CI: 1.001–1.031) and METTL16 (HR: 1.101, 95% CI: 1.003–1.207) indicated poor survival in patients with breast cancer, while high expression of RBM15B (HR: 0.931, 95% CI: 0.888–0.977) corresponds with better survival (Figure 4A). To figure out the most significant prognostic regulators, RBM15B, YTHDF3, ZC3H13, LRPPRC, and METTL16 were selected with the criteria of p < 0.05 to perform LASSO Cox regression algorithm. Results showed that these five regulators were all strong prognostic factors. Then RBM15B, YTHDF3, ZC3H13, LRPPRC, and METTL16 were selected to evaluate risk characteristics (Figures 4B,C). We divided patients into the low-risk group and the high-risk group according to the median risk score based on the coefficients from LASSO analysis. The survival analysis of the five-gene risk signature demonstrated that the high-risk group patients had a significantly poorer prognosis than the low-risk patients (p < 0.001) (Figure 4D).
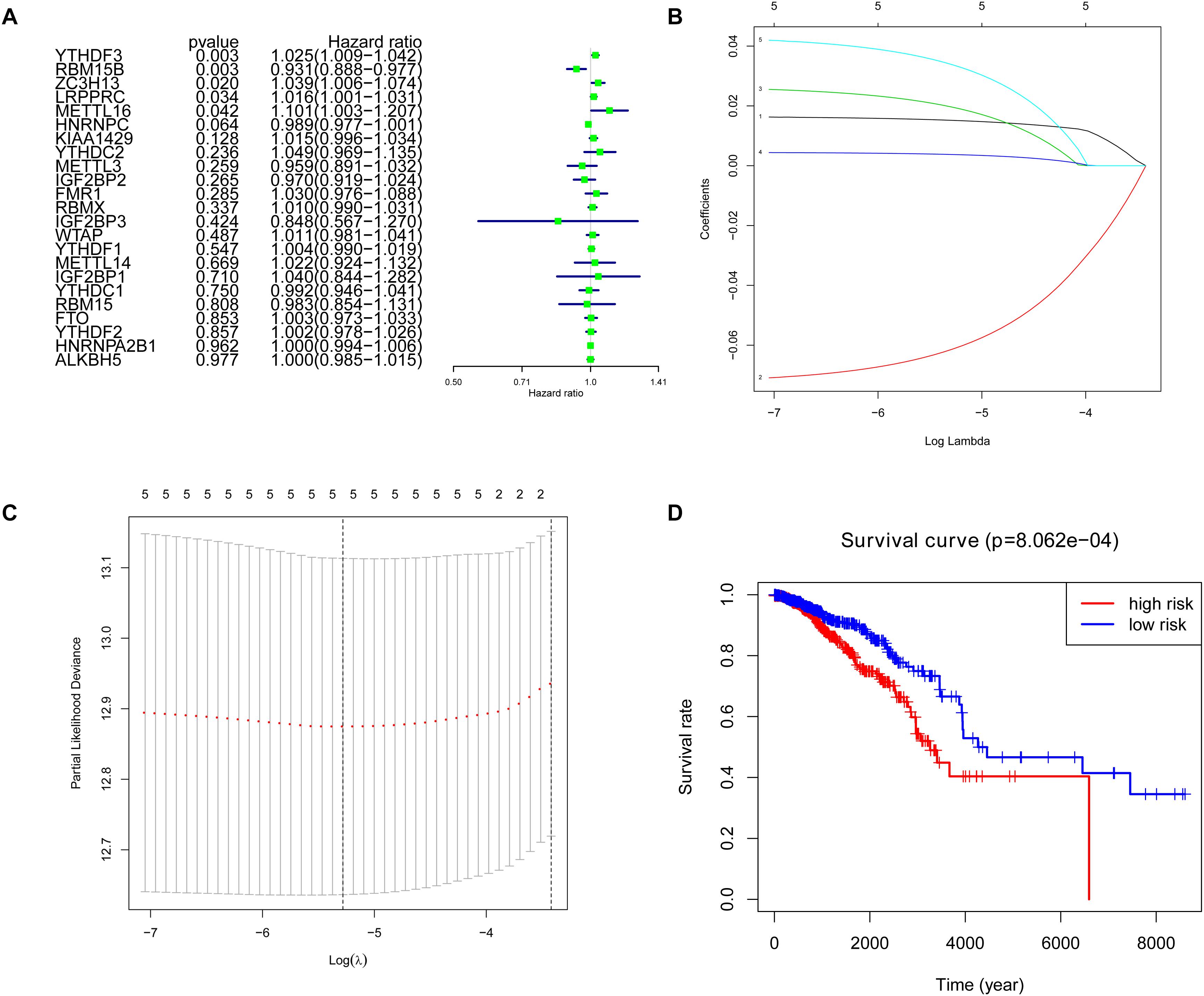
Figure 4. Selection of clinical-pathological m6A RNA regulators with prognostic value. (A) Univariate Cox regression analysis showed the p-value, hazard ratio (HR), and 95% confidence interval (CI) of 23 m6A RNA methylation regulators. (B,C) Five regulators were selected for risk coefficient calculation through the least absolute shrinkage and selection operator (LASSO) Cox regression. (D) Kaplan-Meier Curve showed the overall survival of high and low-risk groups according to the risk score.
Relationship Between Five m6A RNA Methylation Regulators and Clinicopathological Features of Breast Cancer
The five regulators of m6A RNA methylation regulators vary differently among clinicopathological characteristics (Figure 5A). When the data was divided into high-risk and low-risk groups, it can be observed that RBM15B was in low expression, while YTHDF3, LRPPRC, ZC3H13, and METTL16 were in high expression in the high-risk group. We further performed the receiver operating characteristic (ROC) curve to evaluate the predictive specificity and sensitivity, and the area under the curve (AUC) was determined to be 0.643 (Figure 5B). Both univariate and multivariate Cox regression analyses indicated that age and risk scores were related to OS. Univariate analyses also revealed that stage, T status, N status, and M status were associated with OS (Figures 5C,D). After retrieving the RNA-seq data including the TCGA and SCAN-B databases, we got the comparison outcomes with ER, PR, HER-2 and PAM50 subtypes (Figure 6). LRPPRC was negatively associated with ER and PR expression, while METTL16, RBM15B, and ZC3H13 were positively associated with ER and PR expression. In HER-2 (+) breast cancer patients, LRPPRC, METTL16, RBM15B, and ZC3H13 expressed lower than the HER-2 (−). LRPPRC showed the highest expression in basal-like breast cancer subtype. METTL16 expressed at a lower level in HER-E and basal-like subtypes. RBM15B expressed higher in Luminal A and normal breast-like subtypes of breast cancer. ZC3H13 expressed lower in basal-like, HER-E and Luminal B subtypes than in Luminal A and basal-like. However, no obvious difference among these five subtypes was observed with regards to YTHDF3 expression. These results were also verified by METABRIC database in bc-GenExMiner v4.5 (Supplementary Figure 1). LRPPRC and YTHDF3 were negatively associated with ER and PR expression, while METTL16 and RBM15B were positively associated with ER and PR expression. In HER-2 (+) breast cancer patients, METTL16 expressed lower than the HER-2 (−), but YTHDF3 expressed higher than the HER-2 (−). And we got similar results of PAM50 subtypes from METABRIC database. As a prognostic factor in breast cancer, the NPI and SBR histological grade is widely applied to predict tumor prognosis. Patients with high NPI and SBR grade tend to have a poor prognosis. Results from bc-GenExMiner v4.5 showed that patients with higher NPI and SBR grade tended to have higher expression of LRPPRC and lower expression of METTL16, RBM15B, and ZC3H13 (Figure 7). These results were also confirmed by METABRIC database in bc-GenExMiner v4.5 (Supplementary Figure 2). Patients with higher NPI and SBR grade tend to have higher expression of LRPPRC and YTHDF3 and lower expression of METTL16 and RBM15B. The prognostic value of m6A RNA methylation regulators on RFS in breast cancer patients was analyzed via the Kaplan-Meier Plotter platform. The results in breast cancer patients showed that the relatively high expression of YTHDF3 and LRPPRC were remarkably associated with worse RFS, whereas relatively high expression of RBM15B, ZC3H13, and METTL16 had better RFS (Figure 8).
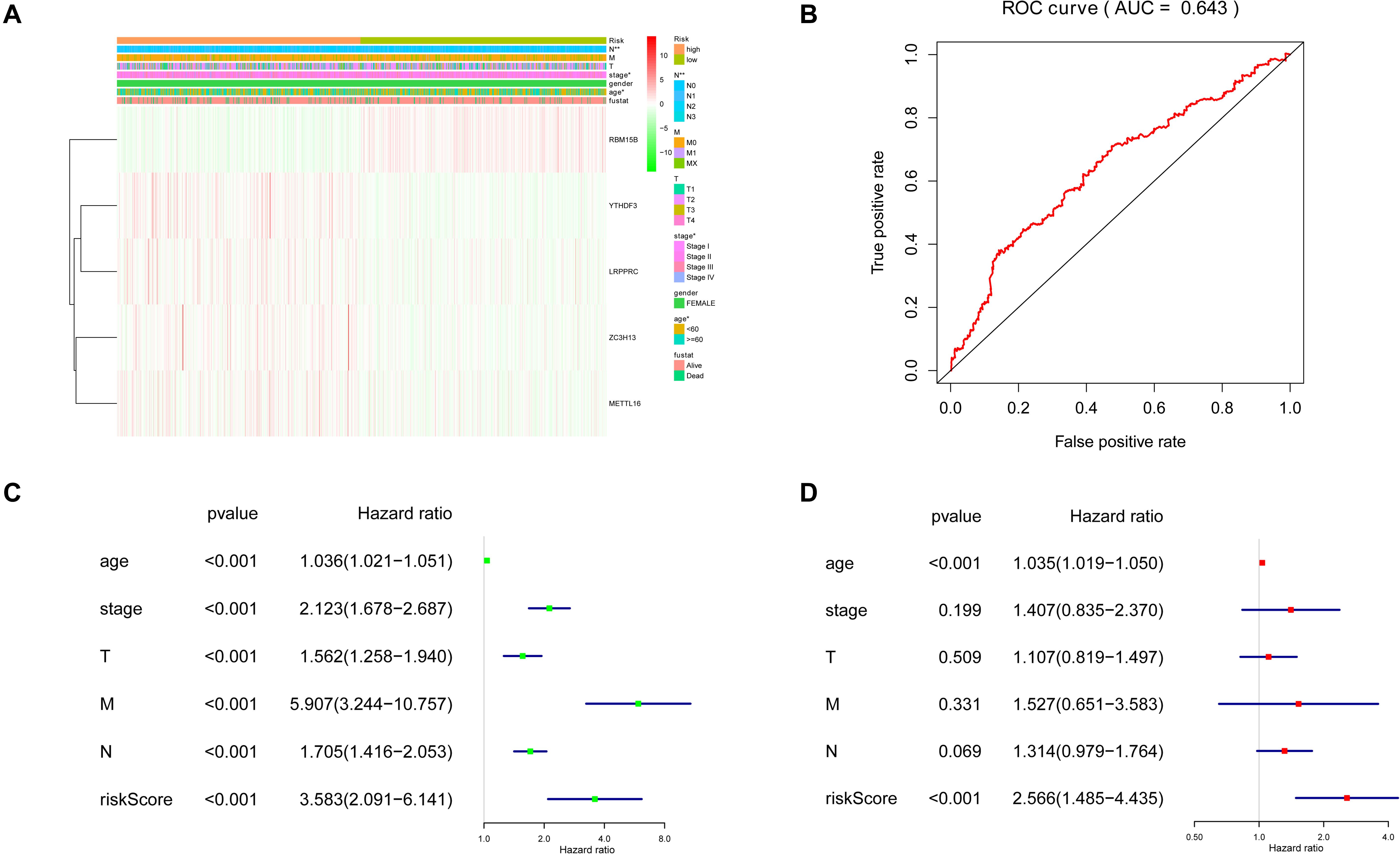
Figure 5. Clinicopathological features of the two risk subgroups based on five selected regulators. (A) The heatmap shows the expression levels of five m6A RNA regulators in high and low-risk groups. (B) The receiver operating characteristic (ROC) curves were used to validate the predictive specificity and sensitivity. The association between clinicopathological factors and overall survival of breast cancer patients through univariate (C) and multivariate (D) Cox regression analyses.
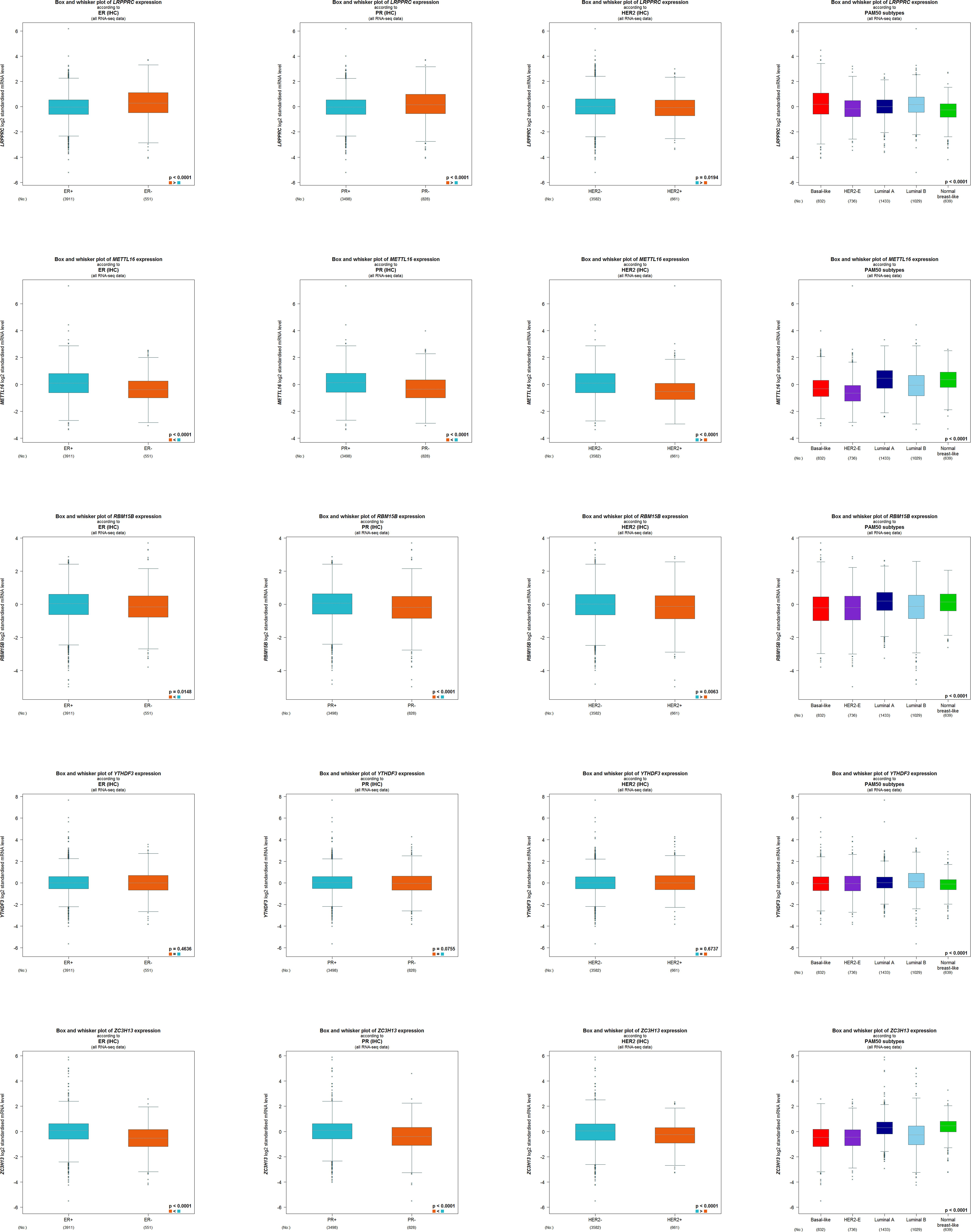
Figure 6. RNA-seq data from bc-GenExMiner v4.5 showing the comparison outcomes with ER, PR, HER-2, and PAM50 subtypes on the expression level of YTHDF3, RBM15B, ZC3H13, LRPPRC, and METTL16.
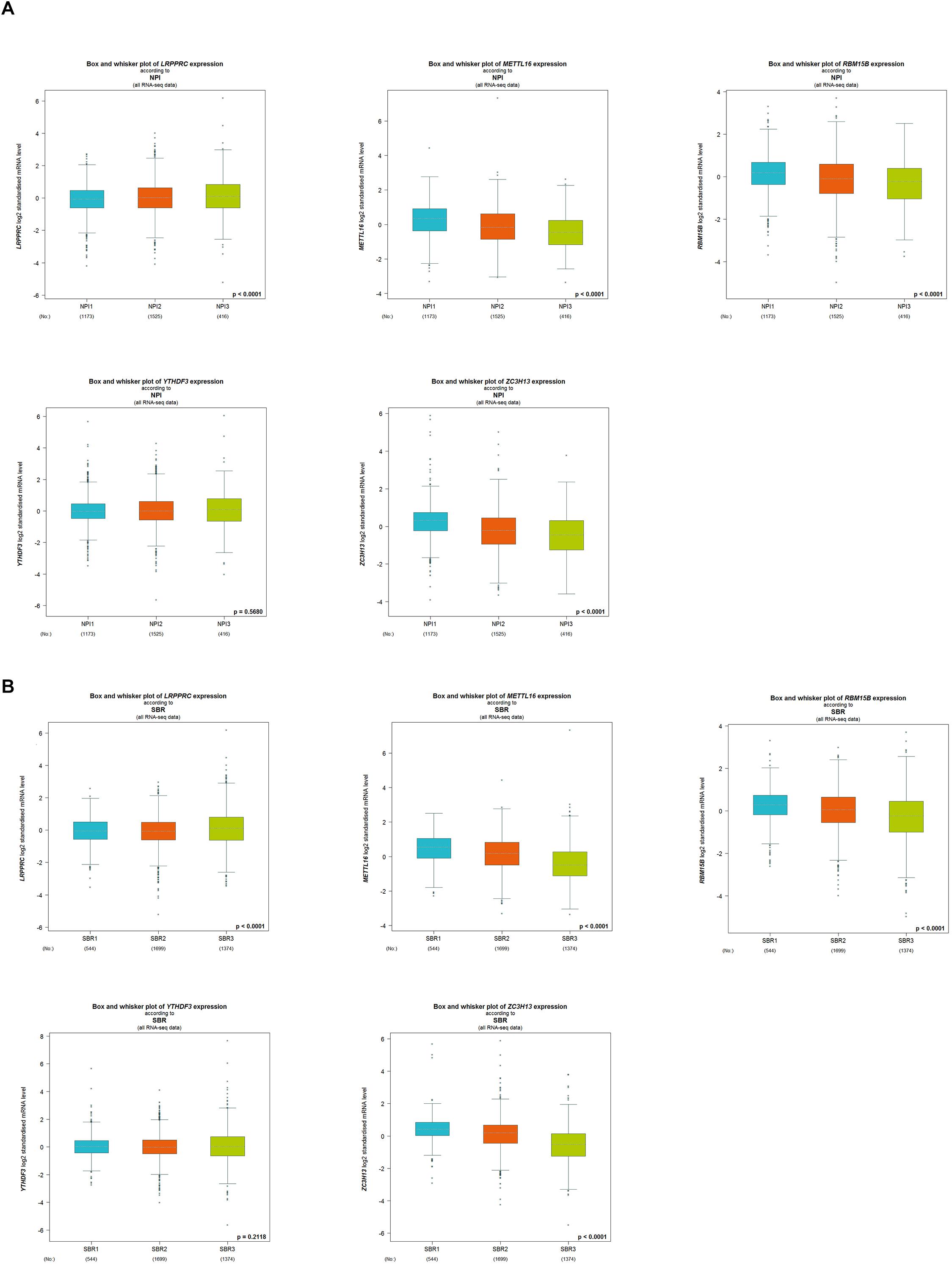
Figure 7. The correlation between the expression level of LRPPRC, METTL16, RBM15B, YTHDF3, and ZC3H13 and the Nottingham prognostic index (NPI) (A) and the Scarff-Bloom-Richardson (SBR) (B) grading. Patients with higher NPI and SBR grade tend to have higher expression of LRPPRC and lower expression of METTL16, RBM15B, and ZC3H13 (p < 0.0001).
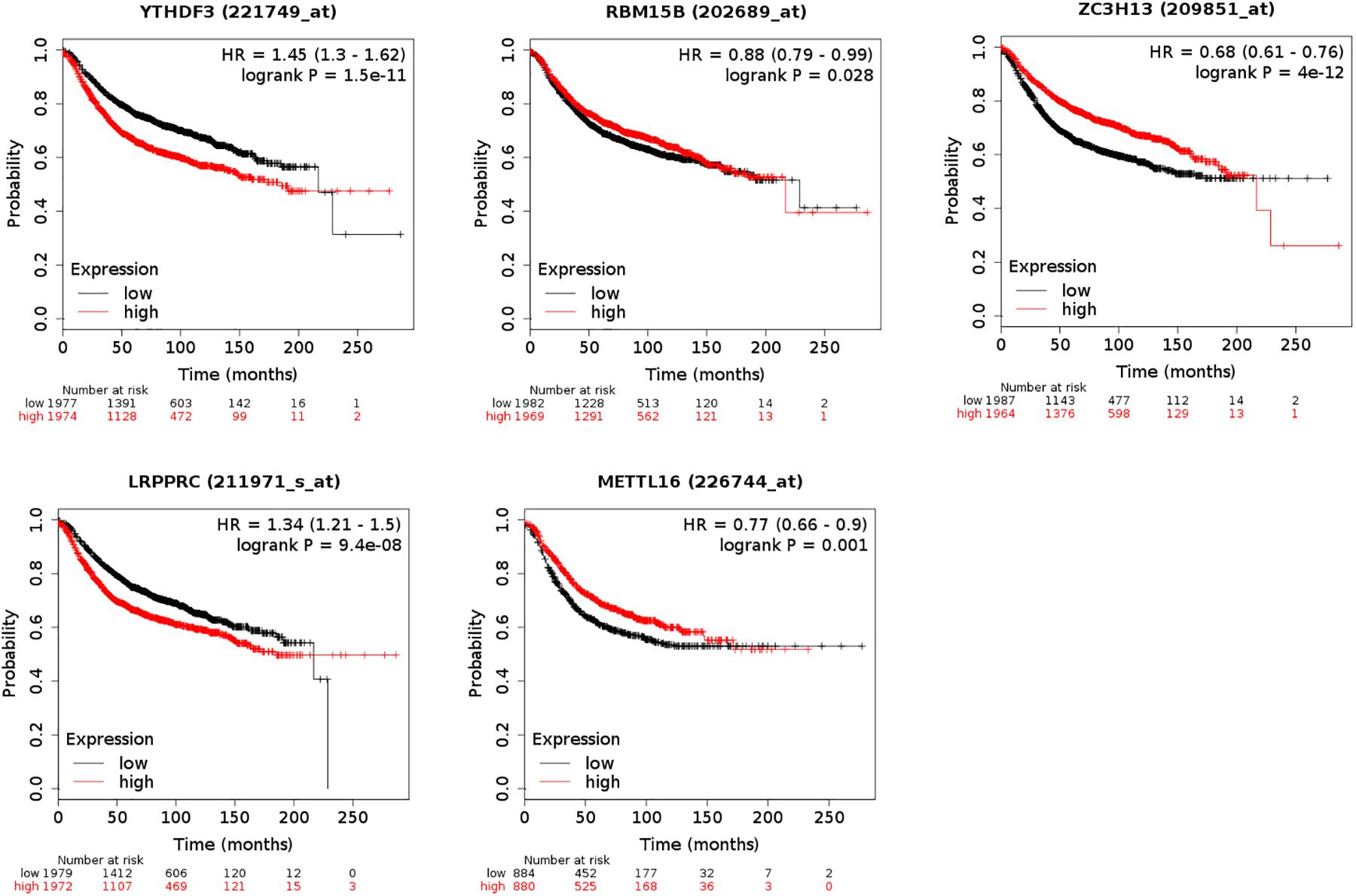
Figure 8. Kaplan-Meier Plotter showing the prognostic value of five m6A RNA methylation regulators on Relapse-Free Survival (RFS) in breast cancer patients. Breast cancer patients with relatively high expression of YTHDF3 and LRPPRC as well as relatively low expression of ZC3H13, METTL16, and RBM15B presented with worse RFS.
Associated Biological Pathways of Five m6A RNA Methylation Regulators
The GSEA analysis was performed to identify associated pathways. We selected significantly enriched signaling pathways based on their normalized enrichment score (NES) and normalized p-value (Figure 9). YTHDF3 was enriched in mTOR signaling pathway (NES = 1.98, p = 0.021), neurotrophin signaling pathway (NES = 1.98, p = 0.023), Notch signaling pathway (NES = 2.10, p = 0.012), pathways in cancer (NES = 1.84, p = 0.047) and Wnt signaling pathway (NES = 1.86, p = 0.042). RBM15B was enriched in aminoacyl tRNA biosynthesis (NES = 2.12, p = 0.011), mTOR signaling pathway (NES = 1.94, p = 0.023), Notch signaling pathway (NES = 2.10, p = 0.008), pathways in cancer (NES = 1.82, p = 0.049), and Wnt signaling pathway (NES = 1.86, p = 0.044). ZC3H13 was enriched in aminoacyl tRNA biosynthesis (NES = 2.13, p = 0.005), lysine degradation (NES = 2.39, p < 0.001), mTOR signaling pathway (NES = 1.95, p = 0.024), Notch signaling pathway (NES = 2.15, p = 0.005), and Wnt signaling pathway (NES = 1.87, p = 0.038). LRPPRC was enriched in cell cycle (NES = 2.28, p = 0.002), homologous recombination pathway (NES = 1.95, p = 0.017), RNA degradation (NES = 2.64 p < 0.001), ubiquitin-mediated proteolysis (NES = 2.63, p < 0.001), and Wnt signaling pathway (NES = 2.01, p = 0.015). LRPPRC was down-regulated in arachidonic acid metabolism (NES = −2.31, p = 0.002). METTL16 was enriched in ErbB signaling pathway (NES = 1.96, p = 0.010), mismatch repair (NES = 2.01, p = 0.008), RNA degradation (NES = 2.59, p < 0.001), and Wnt signaling pathway (NES = 2.39, p = 0.004). METTL16 was down-regulated in arachidonic acid metabolism (NES = −2.26, p = 0.004).
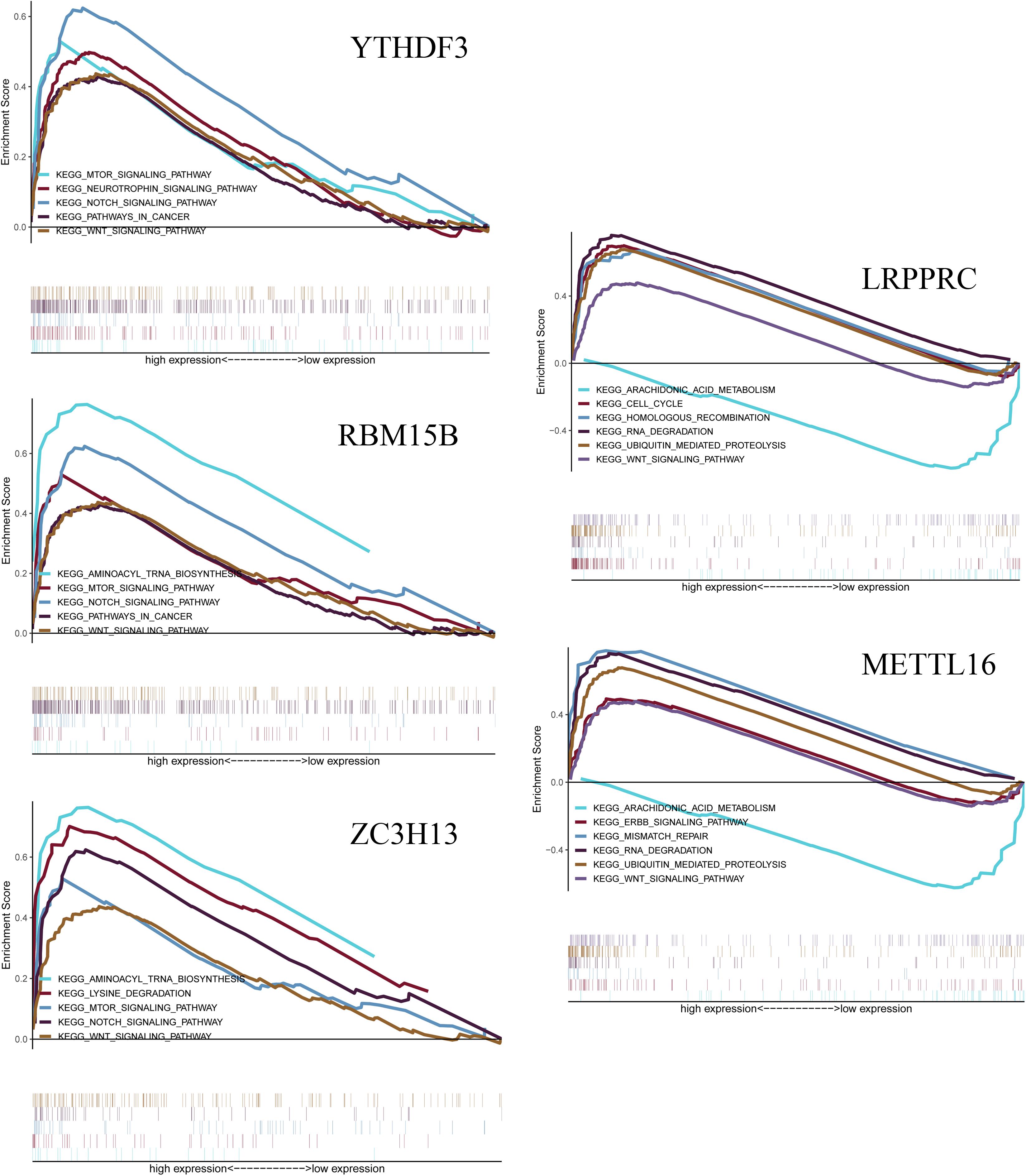
Figure 9. Gene Set Enrichment Analysis (GSEA) shows that the five m6A RNA methylation regulators, including YTHDF3, RBM15B, ZC3H13, LRPPRC, METTL16. The results are based on their normalized enrichment score (NES) and normalized p-value. Pathways with positive NES are positively correlated with the genes, and pathways with negative NES are negatively correlated with the genes.
Discussion
The 5-year relative survival rate of breast cancer has been improved significantly due to the improvement of earlier diagnosis and treatment. However, there is still a long way to go for new therapeutic targets for breast cancer treatment. The m6A RNA methylation is an important post-transcriptional factor that affects tumor occurrence and development. Previous studies show a close relationship between RNA methylation and breast cancer. Hypoxia-inducible factor (HIF)-1α-and HIF-2α-dependent ALKBH5 in breast cancer cells can be stimulated under hypoxia condition, leading to demethylation of NANOG mRNA. Furthermore, the high expression level of NANOG leads to the enrichment of breast cancer stem cells (BCSCs) (Zhang et al., 2016). Cai et al. (2018) reported that the mTORC1 activator mammalian hepatitis B X-interacting protein (HBXIP) can active METTL3 by prohibiting the expression of oncogenes-related miRNA let-7g. While the up-regulated METTL3 can simultaneously promote the HBXIP expression. Thereby, HBXIP, METTL3 and let-7g form a feedback loop accelerating proliferation and invasion of breast cancer. The “writer” KIAA1429 is confirmed to be up-regulated in breast cancer, and 5′-fluorouracil is effective for exhibiting the expression of KIAA1429 and its methylated target cyclin-dependent kinase 1 (CDK1) (Qian et al., 2019). These studies mostly concentrate on few methylation regulators, such as METTL3, METTL14, KIAA1429, FTO, and ALKBH5. With the continuing discovery of novel m6A regulators, there is an ever-increasing demand for in-depth research into RNA methylation about breast cancer.
In our present study, we analyzed the expression of the 23 RNA methylation regulators in the round from ONCOMINE database. Based on 1,096 tumor tissues and 112 normal tissues from the TCGA database, the analytic outcomes showed that 17 out of 23 regulators had statistical significance (p < 0.05). Surprisingly, some researches have verified that the repression of HNRNPC can inhibit two breast cancer cell lines (MCF7 and T47D) proliferation through the accumulation of double-stranded RNA (dsRNA), the binding ligand of RIG-I, and anti-proliferation activity induced by the stimulated IFN cytokine (Wu et al., 2018). The silencing of the m6A methyltransferase has an effect on the stability of gene expression, such as the interference of the TP53 signaling pathway which is a vital tumor suppressor gene (Dominissini et al., 2012). The p53 protein binds closely with HNRNPC and causes destabilization of HNRNPC. Under the treatment of doxorubicin, lncRNA SNHG1 and HNRNPC can reach a balance on the mediation of p53 (Shen et al., 2017). In line with the TCGA outcomes, up-regulated HNRNPA2B1 is concluded to promote breast cancer tumorigenic potential by activating extracellular-signal-regulated kinase 1/2 (ERK1/2), and signal transducer and activator of transcription 3 (STAT3). Knockdown of HNRNPA2B1 can prohibit proliferation, accelerate apoptosis and prolong S phase in vitro, while restraining tumorigenicity in vivo (Hu et al., 2017).
The m6A RNA methylation regulators are of great significance in the occurrence, development and prognosis of various types of cancers. Recently, by using bioinformatic databases, researchers have discovered its importance in tumor therapies. The variations and/or mutations of m6A RNA methylation regulators occurring in the acute myeloid leukemia (AML) patients present a close relation with the mutations of TP53 may disclose the poor prognosis in AML (Kwok et al., 2017). Patients with high levels of mRNA methylation resulting from overexpression of reader proteins and methyltransferase complexes showed poor survival benefits in prostate cancer (Ji et al., 2020). In the subgroup construction signature of TCGA and GEO database, the m6A regulators showed a great value in malignant progression and prognosis of uveal melanoma (UM) (Tang et al., 2020).
According to the clinical database of breast cancer of TCGA, RBM15B, YTHDF3, ZC3H13, METTL16, and LRPPRC were picked up indicating strong associations with clinical characteristics. We used a variety of databases to verify their impact on the survival and prognosis of breast cancer patients and its potential research significance. The outcome revealed a clear direction for further study in vivo and in vitro. RBM15B is the paralog of RBM15, and they are confirmed to bind METTL3 relying on the meditation of WTAP (Patil et al., 2016). BRCA1-associated protein-1 (BAP1) is found to be expressed at a low level in breast cancer patients, while RBM15B has a positive correlation with BAP1 in invasive breast cancer (Shahriyari et al., 2019). YTH domain protein family contains several m6A-binding proteins playing parts as readers in m6A methylation but with different functions. YTHDF1 and YTHDF3 work cooperatively to promote ribosome loading, while YTHDF2 accelerates the decay of mRNA and YTHDC1 acts as a recruiter for mRNA splicing (Xiao et al., 2016; Hsu et al., 2017; Li et al., 2017). YTHDF3 has been verified to correlate with several kinds of carcinoma, such as colorectal cancer (CRC) (Liu et al., 2019), gastric cancer (Zhang et al., 2019), bladder cancer (Jin et al., 2019), breast cancer (Liu et al., 2019), and so on. In the ONCOMINE and TCGA databases, we didn’t notice a considerable difference in the expression of RBM15B and YTHDF3 in breast cancer. Whether it has an impact on the occurrence, development and prognosis of breast cancer needs to be further confirmed by research. ZC3H13 is a novel “writer” mediating m6A methylation. The ablation of ZC3H13 can lead to an obvious decrease of m6A enrichment, especially at the 3′end of the mRNA. It is also speculated that ZC3H13 is a component of the RBM15/ZC3H13/WTAP/VIRMA/HAIKAI complex, also refers to the m6A-METTL-associated complex, which regulates m6A methylation either on its own or interacts with METTL3/METTL14 complex (Knuckles et al., 2018). In the triple-negative subtype of inflammatory breast cancer (TN-IBC), ZC3H13 is down-regulated in contrast to TN-non-IBC (Funakoshi et al., 2019). The outcome of this research is consistent with the results of our bioinformatics analysis. METTL16 was first found to have a close correlation with a cancer-promoting long non-coding RNA: metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) (Brown et al., 2016). As a cancer-promoting factor, MALAT1 is closely related to certain biological characteristics such as vascular epithelialization, migration and distant metastasis, postoperative fever, and systemic inflammatory response in breast cancer (Huang et al., 2018; Li et al., 2018; Gomes et al., 2019). METTL16 is also reported in U6 small nuclear (snRNA), pre-mRNAs and other lncRNA (Warda et al., 2017). METTL16 binds the UACAGAGAA sequence on the 3′ UTR of MAT2A mRNA sequence for MAT2A splicing, the procedure of which can encode the SAM synthetase expression and form a methylation pattern regulated according to SAM level which is quite different from the way that METTL3-METTL14-WTAP complex acts (Pendleton et al., 2017). Former research has pointed out that the biochemical synthesis of SAM is a unique metabolic “soft spot” of triple-negative BCSCs whose drug-resistance can be eradicated by combining methionine depletion with MAT2A suppression (Strekalova et al., 2019). Although these regulators failed to present desired and consistent outcomes in the analysis of expression levels and prognostic values, it’s worth further exploring their biological functions and interactions in breast cancer. Besides, the subtypes of breast cancer may interfere in RNA methylation which indicates new research interests in exploring different subtypes. LRPPRC is an RNA-binding protein regulating mitochondrial DNA-encoded mRNA and also transactivating nuclear DNAs. The immunohistochemical detection of several types of cancers, including lung adenocarcinoma, breast cancer, oesophageal squamous cell cancer, gastric adenocarcinoma, etc., shows that LRPPRC is abundantly expressed in cancer tissues in contrast to the adjacent normal tissues. It can promote apoptosis resistance and invasive activity of cancer cells (Tian et al., 2012). Research on BCSCs shows that the over-expression of microRNA-1 (miR-1) can target the 3′ UTRs of mitochondrial inner membrane organizing system 1 (MINOS1) and glycerol-3-phosphate dehydrogenase 2 (GPD2) under the participation of LRPPRC, which results in mitochondrial damage (Zhang et al., 2019). However, similar effects upon m6A RNA methylation need to be proved.
m6A RNA methylation is a complex regulative system that acts through methylation and demethylation on different targets. Its effect is also reflected in “promoter” or “suppressor” in tumorigenesis and development. With the improvement of RNA methylation detection technology, an increasing number of regulators are found, which reveals a more complicated regulation system. Further studies are needed to be done to verify these findings and their interactive mechanisms. Besides, the way to control specificity and to strengthen monitoring of RNA methylation regulators in the dynamic post-transcriptional balance is also a great challenge. But we believe, as a promising research area in epigenetics, RNA methylation would bring a new dawn of hope for combating breast cancer.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: http://www.cancergenome.nih.gov/ and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70905.
Author Contributions
GJ supervised and conceptualized the study. BZ searched the articles and designed the study. BZ and YG made the figures and wrote this manuscript. All authors read and revised the final manuscript.
Funding
This study was supported by the Suzhou Health Planning Commission’s Key Clinical Diagnosis, Treatment Program (LCZX201606), the National Natural Science Foundation of China (81873730), and the Jiangsu Women and Children Health Key Discipline Program (FXK201758).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the reviewers for their constructive comments. We thank Sancy Mary Joyson for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.604597/full#supplementary-material
Footnotes
- ^ https://www.oncomine.org/
- ^ http://www.cancergenome.nih.gov/
- ^ https://www.ncbi.nlm.nih.gov/geo/
- ^ http://kmplot.com/analysis/
- ^ http://bcgenex.centregauducheau.fr/
- ^ http://string-db.org
- ^ https://www.gtexportal.org/
- ^ http://www.r-project.org/
References
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Brown, J. A., Kinzig, C. G., Degregorio, S. J., and Steitz, J. A. (2016). Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 113, 14013–14018. doi: 10.1073/pnas.1614759113
Cai, X., Wang, X., Cao, C., Gao, Y., Zhang, S., Yang, Z., et al. (2018). HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 415, 11–19. doi: 10.1016/j.canlet.2017.11.018
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Funakoshi, Y., Wang, Y., Semba, T., Masuda, H., Hout, D., Ueno, N. T., et al. (2019). Comparison of molecular profile in triple-negative inflammatory and non-inflammatory breast cancer not of mesenchymal stem-like subtype. PLoS One 14:e0222336. doi: 10.1371/journal.pone.0222336
Fustin, J. M., Kojima, R., Itoh, K., Chang, H. Y., Ye, S., Zhuang, B., et al. (2018). Two Ck1δ transcripts regu lated by m6A methylation code for two antagonistic kinases in the control of the circadian clock. Proc. Natl. Acad. Sci. USA 115, 5980–5985. doi: 10.1073/pnas.1721371115
Gomes, C. P., Nobrega-Pereira, S., Domingues-Silva, B., Rebelo, K., Alves-Vale, C., Marinho, S. P., et al. (2019). An antisense transcript mediates MALAT1 response in human breast cancer. BMC Cancer 19:771. doi: 10.1186/s12885-019-5962-0
Hsu, P. J., Zhu, Y., Ma, H., Guo, Y., Shi, X., Liu, Y., et al. (2017). Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27:99. doi: 10.1038/cr.2017.99
Hu, Y., Sun, Z., Deng, J., Hu, B., Yan, W., Wei, H., et al. (2017). Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol. 39:1010428317694318. doi: 10.1177/1010428317694318
Huang, X. J., Xia, Y., He, G. F., Zheng, L. L., Cai, Y. P., Yin, Y., et al. (2018). MALAT1 promotes angiogenesis of breast cancer. Oncol. Rep. 40, 2683–2689. doi: 10.3892/or.2018.6705
Ji, G., Huang, C., He, S., Gong, Y., Song, G., Li, X., et al. (2020). Comprehensive analysis of m6A regulators prognostic value in prostate cancer. Aging 12, 14863–14884. doi: 10.18632/aging.103549
Jin, H., Ying, X., Que, B., Wang, X., Chao, Y., Zhang, H., et al. (2019). N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBio Med. 47, 195–207. doi: 10.1016/j.ebiom.2019.07.068
Knuckles, P., Lence, T., Haussmann, I. U., Jacob, D., Kreim, N., Carl, S. H., et al. (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 32, 415–429. doi: 10.1101/gad.309146.117
Kwok, C. T., Marshall, A. D., Rasko, J. E., and Wong, J. J. (2017). Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J. Hematol. Oncol. 10:39. doi: 10.1186/s13045-017-0410-6
Lan, Q., Liu, P. Y., Haase, J., Bell, J. L., Huttelmaier, S., and Liu, T. (2019). The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 79, 1285–1292. doi: 10.1158/0008-5472.Can-18-2965
Lence, T., Akhtar, J., Bayer, M., Schmid, K., Spindler, L., Ho, C. H., et al. (2016). m(6)A modulates neuronal functions and sex determination in Drosophila. Nature 540, 242–247. doi: 10.1038/nature20568
Li, A., Chen, Y. S., Ping, X. L., Yang, X., Xiao, W., Yang, Y., et al. (2017). Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 27, 444–447. doi: 10.1038/cr.2017.10
Li, Z., Xu, L., Liu, Y., Fu, S., Tu, J., Hu, Y., et al. (2018). LncRNA MALAT1 promotes relapse of breast cancer patients with postoperative fever. Am. J. Transl. Res. 10, 3186–3197.
Liu, L., Liu, X., Dong, Z., Li, J., Yu, Y., Chen, X., et al. (2019). N6-methyladenosine-related Genomic Targets are Altered in Breast Cancer Tissue and Associated with Poor Survival. J. Cancer 10:35053. doi: 10.7150/jca.35053
Liu, X., Liu, L., Dong, Z., Li, J., Yu, Y., Chen, X., et al. (2019). Expression patterns and prognostic value of m(6)A-related genes in colorectal cancer. Am. J. Transl. Res. 11, 3972–3991.
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Miller, K. D., Nogueira, L., Mariotto, A. B., Rowland, J. H., Yabroff, K. R., Alfano, C. M., et al. (2019). Cancer treatment and survivorship statistics. CA A Cancer J. Clin. 69, 363–385. doi: 10.3322/caac.21565
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. doi: 10.1038/nature19342
Pendleton, K. E., Chen, B., Liu, K., Hunter, O. V., Xie, Y., Tu, B. P., et al. (2017). The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835.e814. doi: 10.1016/j.cell.2017.05.003
Qian, J. Y., Gao, J., Sun, X., Cao, M. D., Shi, L., Xia, T. S., et al. (2019). KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene 38, 6123–6141. doi: 10.1038/s41388-019-0861-z
Roundtree, I.A., Evans, M.E., Pan, T., and He, C. (2017). Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187-1200. doi: 10.1016/j.cell.2017.05.045
Shahriyari, L., Abdel-Rahman, M., and Cebulla, C. (2019). BAP1 expression is prognostic in breast and uveal melanoma but not colon cancer and is highly positively correlated with RBM15B and USP19. PLoS One 14:e0211507. doi: 10.1371/journal.pone.0211507
Shen, Y., Liu, S., Fan, J., Jin, Y., Tian, B., Zheng, X., et al. (2017). Nuclear retention of the lncRNA SNHG1 by doxorubicin attenuates hnRNPC-p53 protein interactions. EMBO Rep. 18, 536–548. doi: 10.15252/embr.201643139
Shi, H., Wei, J., and He, C. (2019). Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 74, 640–650. doi: 10.1016/j.molcel.2019.04.025
Strekalova, E., Malin, D., Weisenhorn, E. M. M., Russell, J. D., Hoelper, D., Jain, A., et al. (2019). S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat. 175, 39–50. doi: 10.1007/s10549-019-05146-7
Tang, J., Wan, Q., and Lu, J. (2020). The prognostic values of m6A RNA methylation regulators in uveal melanoma. BMC Cancer 20:674. doi: 10.1186/s12885-020-07159-8
Tian, T., Ikeda, J., Wang, Y., Mamat, S., Luo, W., Aozasa, K., et al. (2012). Role of leucine-rich pentatricopeptide repeat motif-containing protein (LRPPRC) for anti-apoptosis and tumourigenesis in cancers. Eur. J. Cancer 48, 2462–2473. doi: 10.1016/j.ejca.2012.01.018
Wang, Y., Li, Y., Toth, J. I, Petroski, M. D., Zhang, Z., and Zhao, J. C. (2014). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198. doi: 10.1038/ncb2902
Warda, A. S., Kretschmer, J., Hackert, P., Lenz, C., Urlaub, H., Hobartner, C., et al. (2017). Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014. doi: 10.15252/embr.201744940
Wu, Y., Zhao, W., Liu, Y., Tan, X., Li, X., Zou, Q., et al. (2018). Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 37:e99017. doi: 10.15252/embj.201899017
Xiao, W., Adhikari, S., Dahal, U., Chen, Y. S., Hao, Y. J., Sun, B. F., et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 61, 507–519. doi: 10.1016/j.molcel.2016.01.012
Yang, Y., Hsu, P. J., Chen, Y. S., and Yang, Y. G. (2018). Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 28, 616–624. doi: 10.1038/s41422-018-0040-8
Zhang, C., Chen, Y., Sun, B., Wang, L., Yang, Y., Ma, D., et al. (2017). m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276.
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., et al. (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 113, E2047–E2056. doi: 10.1073/pnas.1602883113
Zhang, C., Zhang, M., Ge, S., Huang, W., Lin, X., Gao, J., et al. (2019). Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 8, 4766–4781. doi: 10.1002/cam4.2360
Zhang, S., Liu, C., and Zhang, X. (2019). Mitochondrial Damage Mediated by miR-1 Overexpression in Cancer Stem Cells. Mol. Ther. Nucleic Acids 18, 938–953. doi: 10.1016/j.omtn.2019.10.016
Keywords: N6-methyladenosine (m6A) RNA methylation, breast carcinoma, post-transcriptional modification, bioinformatics analysis, prognosis
Citation: Zhang B, Gu YL and Jiang GQ (2020) Expression and Prognostic Characteristics of m6 A RNA Methylation Regulators in Breast Cancer. Front. Genet. 11:604597. doi: 10.3389/fgene.2020.604597
Received: 09 September 2020; Accepted: 19 November 2020;
Published: 10 December 2020.
Edited by:
Jia Meng, Xi’an Jiaotong-Liverpool University, ChinaReviewed by:
Jinhua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaJustin Jong-Leong Wong, Royal Prince Alfred Hospital, Australia
Copyright © 2020 Zhang, Gu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoqin Jiang, amlhbmdfZ3VvcWluQDE2My5jb20=
†These authors have contributed equally to this work