 Peng Yi
Peng Yi Xiongfeng Xu
Xiongfeng Xu- Department of Orthopedic Surgery, Renmin Hospital of Wuhan University, Wuhan, China
Abstract Objectives Transcriptional changes in cartilage can impact function by causing degradation such as that which occurs during the development of osteoarthritis (OA). Epigenetic regulation may be a key factor leading to transcriptional changes in OA. In this study, we performed a combined analysis of DNA methylation and gene expression microarray datasets and identified key transcription factors (TFs) central to the regulation of gene expression in OA. Methods A DNA methylation profile dataset (GSE63106) and a gene expression profiling dataset (GSE114007) were extracted from the Gene Expression Omnibus (GEO). We used ChAMP methylation analysis and the Limma package to identify differentially methylation genes (DMGs) and differentially expressed genes (DEGs) from normal and human knee cartilage samples in OA. Function enrichment analysis of DMGs was conducted using the DAVID database. A combined analysis of DEGs and DMGs was conducted to identify key TFs in OA. We then validated the mRNA expression of selected TFs in normal and OA cartilage by RT-qPCR. Primary chondrocytes were cultured and treated with the DNA methylation inhibitor 5-Aza-2-deoxycytidine (5-Aza) for functional validation. Results We identified 2,170 differential methylation sites (DMS) containing 1005 genes and 1986 DEGs between normal human and OA cartilage. Functional analysis of DMGs revealed that focal adhesion, extracellular matrix (ECM)-receptor interactions, the PI3K-Akt signaling pathway, and the FoxO signaling pathway were involved in OA. Integrated analysis showed a subset of 17 TFs. Four TFs (ELF3, SOX11, RARA, and FOXD2) were validated. RT-qPCR results showed the mRNA expression of SOX11, RARA, and FOXD2 were consistent with the results from the mRNA expression data. However, the expression of ELF3 could not be validated. Upon 5-Aza-2′-deoxycytidine (5-Aza) treatment, the mRNA levels of ELF3 and SOX11 were down-regulated, whilst RARA was up-regulated, and FOXD2 showed no significant change in expression level. Conclusions the effect of DNA methylation on the transcriptional regulation is related to the distribution of methylated sites across the genome. Epigenetic studies on the positions of DMS in transcriptional units can inform a better understanding of the function of DNA methylation and its transcription regulation.
Introduction
Osteoarthritis (OA) is a common degenerative disease characterized by the destruction of articular cartilage and subchondral bone (Fathollahi et al., 2019). OA is a multifactorial disease with several associated risk factors including sex, age, trauma, obesity, joint injury, joint overuse, and genetic factors such as altered transcriptional control and epigenetic regulation. In OA, transcriptional changes in cartilage can alter its function and cause degradation (Bortoluzzi et al., 2018).
Recent studies have demonstrated that epigenetic regulation may be a key factor leading to transcriptional changes in OA chondrocytes (Barter and Young, 2013; Im and Choi, 2013). DNA methylation is a form of chemical modification in DNA that covalently binds methyl group on the cytosine 5′ carbon of cytosine-phosphate-guanine (CpG) dinucleotides in the genome. Previous studies have found many differentially or aberrantly methylated sites in OA that cause common pathogenesis including degradation of the extracellular matrix (ECM), collagen breakdown, and activation of the inflammatory response (Steinberg et al., 2018). However, the exact mechanisms by which methylation impacts gene transcription and ultimately leads to pathological changes in OA remain to be fully determined.
Previously, it has been reported that the analysis of transcription factors (TFs) is crucial toward understanding biological processes mediated by the epigenetic regulation (Zhu et al., 2016). Recently, methods for genome-wide methylation studies have been developed and applied to demonstrate that the distribution of methylation sites across the genome can regulate gene expression (Harris et al., 2010). Amongst these sites, methylation in gene promoters acts to inhibit transcription. This process has been verified in a range of genes involved in the pathogenesis of OA including genes associated with the ECM(COL9A1), matrix-degradation (MMP13), inflammation (IL-1β), and oxidative stress (SOD2) (Hashimoto et al., 2009; Scott et al., 2010; Bui et al., 2012; Imagawa et al., 2014). These genes showed methylation changes in promoter CpG sites in OA chondrocytes and were negatively correlated at the transcriptional level. It is known that epigenetic changes are essential for tissue development, differentiation, and cell viability (Jones, 2012). However, the functions of methylation sites in enhancers, insulators, and gene body regions, and the detailed mechanisms that affect gene transcription remain to be fully determined.
In this study, we analyzed microarray data (Illumina Infinium Human Methylation 450) from normal and OA human knee cartilage to determine the distribution of CpG sites across the genome. These data were combined with mRNA expression analysis to determine differentially expressed transcription factors (DETFs). Cartilage samples were collected from patients with OA and used to validate changes in mRNA expression levels of the identified TFs. We then performed in vitro experiments to verify the DNA methylation patterns that regulate gene expression.
Materials and Methods
Human Cartilage Samples and Primary Chondrocytes Cell Ccultures
Knee articular cartilage was obtained from three patients who were undergoing knee replacement surgery due to primary knee OA in July 2020. Ethical approval was obtained from the Committee of the Renmin Hospital of Wuhan University and all samples were obtained under informed consent from each patient. All standard biosecurity and institutional safety procedures have been adhered to in all the experiment procedures in this article. Previous studies established a three-region disease progression model system for OA of the knee. The model includes the outer lateral tibial plateau (oLT), the inner lateral tibial plateau (iLT), and the inner medial tibial plateau (iMT) regions of the knee that represent the early, intermediate and late stages of OA (Zhang et al., 2016). Therefore, we obtained normal cartilage and OA cartilage from the oLT and the iMT regions.
Human primary chondrocyte cells (cat. no.CP-H096; Pcocell Life Science and Technology Co., Ltd.) were cultured in DMEM/F12 medium (cat. no. SH30023.01; Hyclone) supplemented with 10% fetal bovine serum (FBS), chondrocyte growth additive, vitamin supplements and 1% double-antibody (penicillin/streptomycin Solution). Cells were incubated at 37°C in 5% CO2. We started the experiment when the chondrocytes were in the logarithmic growth phase. Primary chondrocytes were incubated with or without 10 μM 5-Aza-2′-deoxycytidine (5-Aza) (cat. no. A119533; Aladdin) for 4 days (Iliopoulos et al., 2007; Zhao et al., 2017). Total RNA was extracted from the two groups of cultured chondrocytes to determine the mRNA levels of selected TFs.
DNA Methylation Profiling and Gene Expression Data
The NCBI Gene Expression Omnibus (GEO) database was searched to obtain a DNA methylation profiling dataset (GSE63106) and a gene expression profiling dataset (GSE114007). The inclusion criteria for the selection of samples were; (1) Samples derived from the OA or healthy control cartilage tissue, (2) Sample size was >20, and (3) Samples where the data had been analyzed to assess functional TFs (or transcriptionally functional CpG). GSE63106 was used to collect data from the preserved and affected cartilage samples obtained from 17 knees and 14 hip joints undergoing joint replacement surgery due to primary OA. GSE114007 was used to collect data from 20 OA and 18 normal human knee cartilage tissues.
Analysis of DNA Methylation Profiling Data
We used the ChAMP methylation analysis package in R (v3.6.0) to analyze GSE63106 in all subsequent processes (Morris et al., 2014). Firstly, we loaded intensity data into R (v3.6.0) after filtering unnecessary CpG using the champQC function and generated several plots using the QC.GUI function for data visualization. We then used the champ.norm function with BMIQ to perform a type-II probe normalization of the raw data (Teschendorff et al., 2013). From these analyses, we obtained the adjusted P-values as outputs, log fold change (FC), and the genomic regions between the normal and OA groups. Differentially methylation genes (DMGs) were those including CpG sites with abs (logFC) >0.1 and adjusted P-values < 0.05. The percentage of differential methylation sites (DMS) and the array sites (after filtering) in different genomic regions were statistically analyzed.
Gene Ontology and KEGG Pathway Enrichment Analysis
Gene ontology (GO) and the KEGG signaling pathway enrichment analysis was conducted on DMGs using the DAVID bioinformatics database. Enriched GO terms and KEGG signaling pathways with P-values < 0.05 were deemed as significant.
Identification of Differentially Expressed Genes
As the gene expression profiling datasets extracted from GSE114007 are normalized expression matrices, we did not repeat quantile normalization. To ensure normalization, we used the boxplot function in R to check data normalization. The raw data were analyzed using the R BIOCONDUCTOR packages. Differentially expressed genes (DEGs) were identified through the LIMMA package with the cut-off criteria set as abs (logFC) >1 and adjusted P-value < 0.05.
Identification of Functional Transcription Factors
A list of human TFs and their motifs were downloaded from the “HumanTFs” Website (Lambert et al., 2018). We then compared the DEGs and DMGs with the list of human TFs to identify DETFs whose encoding genes showed simultaneous differential methylation patterns.
Reverse Transcription Quantitative PCR (RT–qPCR)
Total RNA was extracted using Trizol Reagent (cat. no. G3013; Tiangen Biotech, Co., Ltd.) according to the manufacturer’s protocol and transcribed into cDNA using a RevertAid First Strand cDNA Synthesis Kit (cat. no. #K1622, Thermo). The thermocycling conditions were as follows: initial denaturation at 95°C for 30 sec, followed by 40 cycles of 95°C for 15 s, 60°C for 60°s. The melting curve was generated (dissociation, 60°C 95°C at 15 s) to determine normality. FastStart Universal SYBR Green Master (Rox) (Roche) was used for the quantitative analysis. GADPH was used as the internal reference gene. The 2–Δ Δ Cq method was used to analyze the results. The primer sequences of the genes are presented in Table 1.
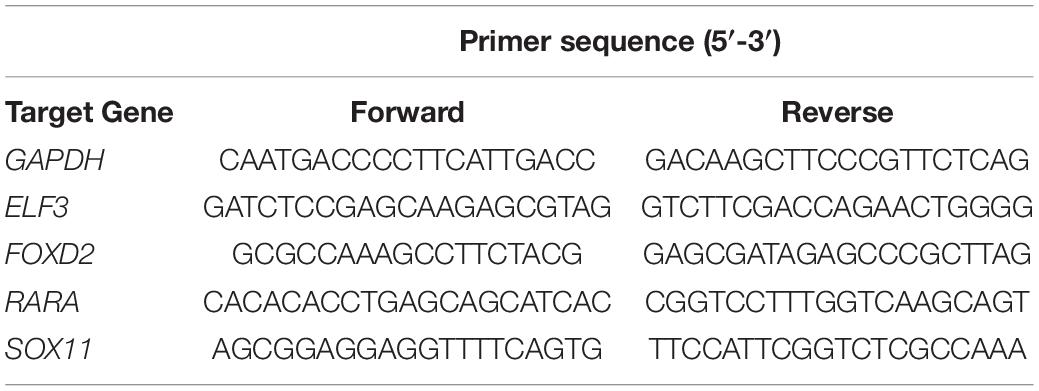
Table 1. Primer sequences of the genes used for RT-qPCR.
Statistical Analysis
All data were expressed as the means ± SEM. Data between the two groups were compared using an unpaired Student’s t-test. Graphs were generated using GraphPad Prism 7 software and P-values of <0.05 considered statistically significant.
Results
Identification of DMGs in Preserved and Affected Cartilage From 17 Knees Joint Samples
We identified 2,170 DMS containing 1005 genes between preserved and affected cartilage. A complete list of the DMS is shown in Supplementary Table 1. Amongst these DMS, 799 sites were hypomethylated and 1,371 site hypermethylated in the OA cartilage. Unsupervised clustering of the 2,170 DMS revealed that samples from normal and OA cartilage could be divided into two main clusters (Figure 1). The first cluster contained eight knee OA samples. The second cluster contained nine knee OA samples and 17 normal samples. Interestingly, the knee OA samples revealed two separate clusters. A previous study reporting the DNA methylome in OA of the knee and hip revealed that OA samples segregated into two groups (Rushton et al., 2014). These results suggest that OA may be divided into two distinct subtypes based on differences in their DNA methylation. The observed differential clustering in the same joint may provide insight toward the development of future diagnostic and therapeutic approaches in OA. However, a critical and challenging question in cluster analysis is whether the identified clusters represent important underlying structure or are artifacts of natural sampling variation (Kimes et al., 2017).
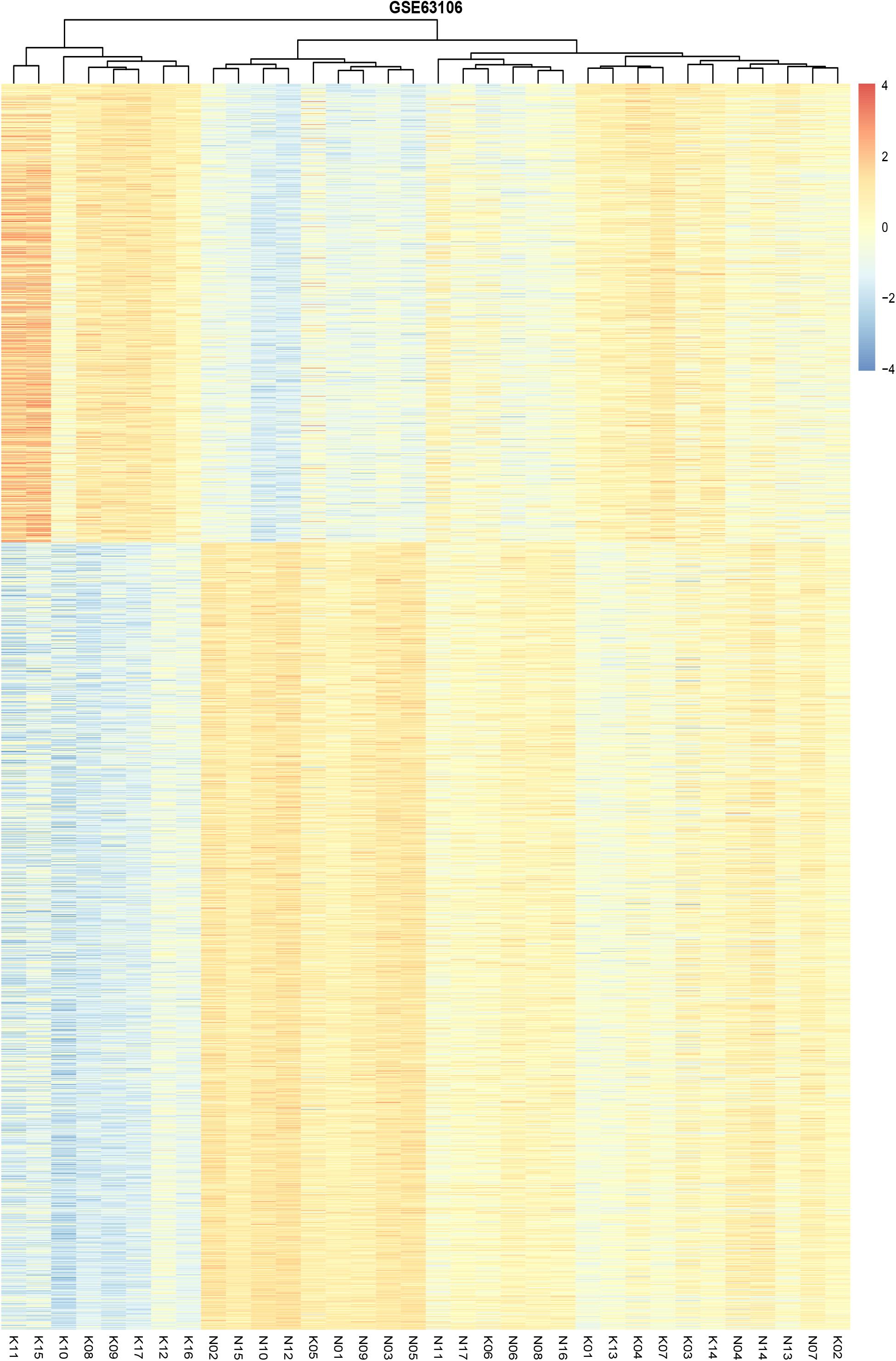
Figure 1. Unsupervised hierarchical clustering of the methylation levels of the DMS from the preserved and affected cartilage, based on the cut-off criteria of abs(logFC) >0.1 and adjusted P-value < 0.05.
Function Enrichment Analysis of DMGs
GO analysis showed the common DMGs were mainly enriched for biological processes including the skeletal system development, osteoblast development, negative regulation of transcription from RNA polymerase II promoter, and ECM organization. For molecular functions, the common DMGs were enriched in phosphatidylinositol-4,5-bisphosphate 3-kinase and 1-phosphatidylinositol-3-kinase activity. For cellular components, the common DMGs were mainly enriched in focal adhesion, the cytoplasm and the actin cytoskeleton (Figure 2A). A complete list of GO analysis is presented in Supplementary Table 2. KEGG pathway analysis demonstrated that common DMGs were mainly enriched in focal adhesion and ECM-receptor interactions, and the PI3K-Akt and FoxO signaling pathways (Figure 2B). A complete list of KEGG pathway analysis is shown in Supplementary Table 3.
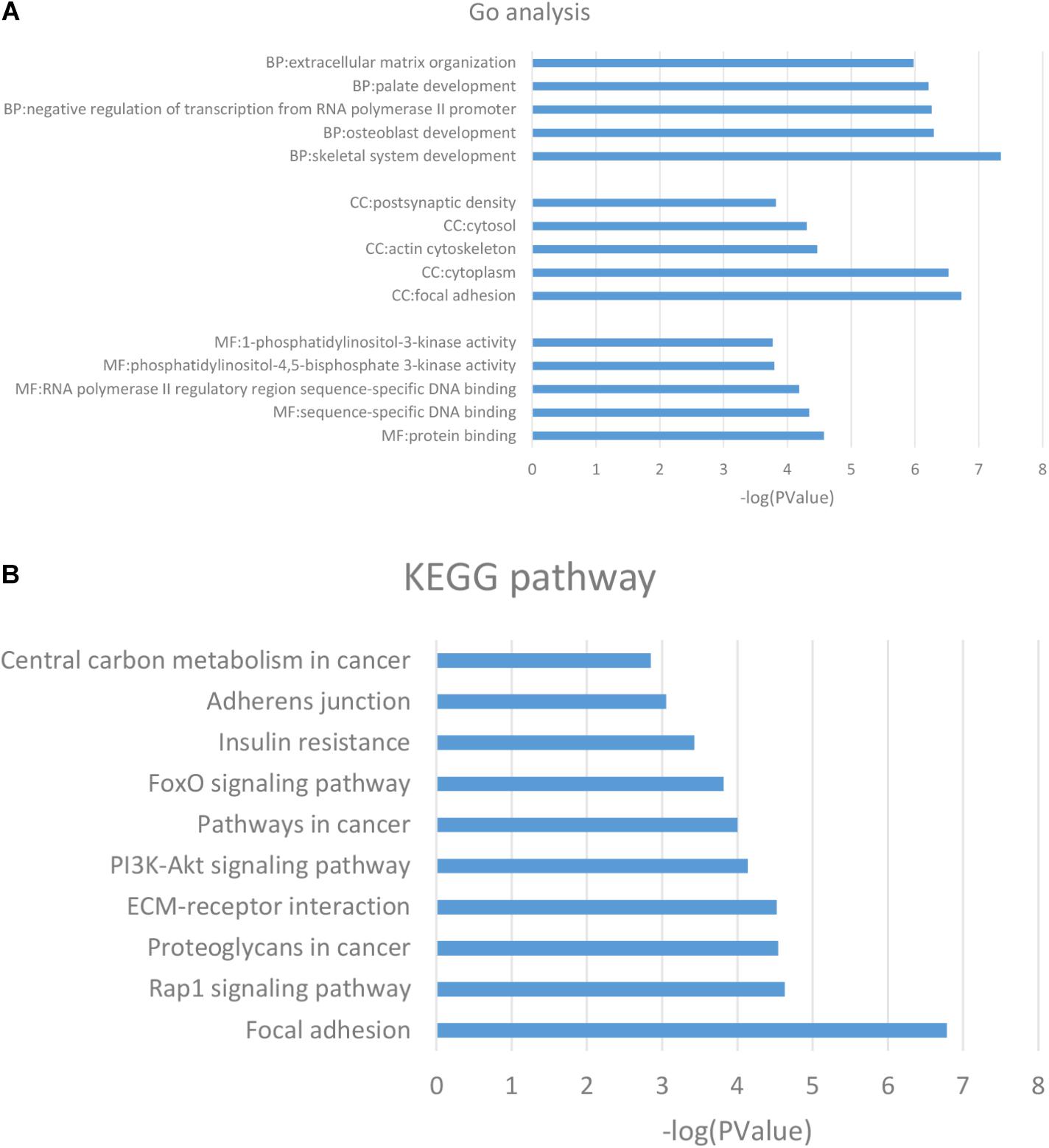
Figure 2. (A) GO analysis of the DMGs. (B) KEGG pathway analysis of the DMGs. The columns are represented as the negative log10 of the P-value. DMGs: differentially methylated genes; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Identification and Characterization of DEGs
A total of 1986 DEGs were identified between the OA and the normal human cartilage. A full list of DEGs is shown in Supplementary Table 4. Amongst these DEGs, 1174 were upregulated and 812 were downregulated in OA. Unsupervised clustering of the 1986 DEGs revealed that samples from normal and OA cartilage could be divided into two main clusters (Figure 3). The first cluster contained 17 normal samples. The second cluster contained 20 knee OA samples and 1 normal sample. Interestingly, the knee OA samples were divided into two different clusters based on their mRNA expression just like the hierarchical clustering of DNA methylation. The specific significance of the differential clustering observed in the same joint deserves further discussion.
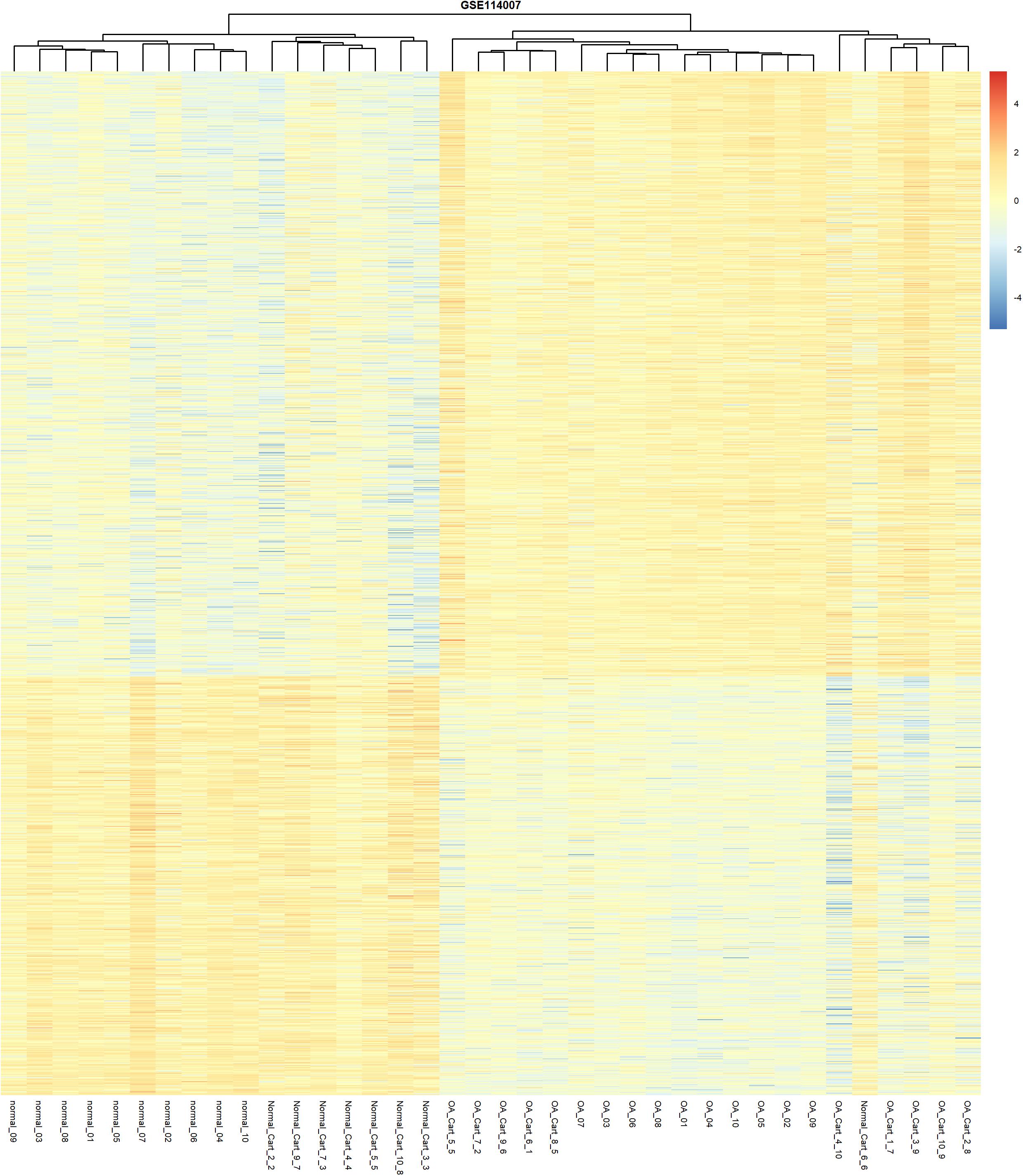
Figure 3. Unsupervised hierarchical clustering of the DEGs, based on their expression levels, from the normal and OA articular cartilage samples using the cut-off criteria of abs (logFC) >1 and adjusted P-value < 0.05.
Statistical Analysis of CpG Sites
Statistical analysis of the number and rate of DMS and the array sites (after screening) was performed in different gene sub-regions. The seven different sub-regions included Transcriptional initiation sites (TSS)200, TSS1500, 1stExon, 3′UTR, 5′UTR, intergenic region (IGR), and gene body. As shown in Table 2, it was observed that 38% of the DMS were enriched in the IGR. In the intragenic regions, 41% of the DMS were significantly enriched in the gene body as opposed to only 17% included in the promoter regions (including TSS200, TSS1500, 5′UTR, and the 1stExon of genes).
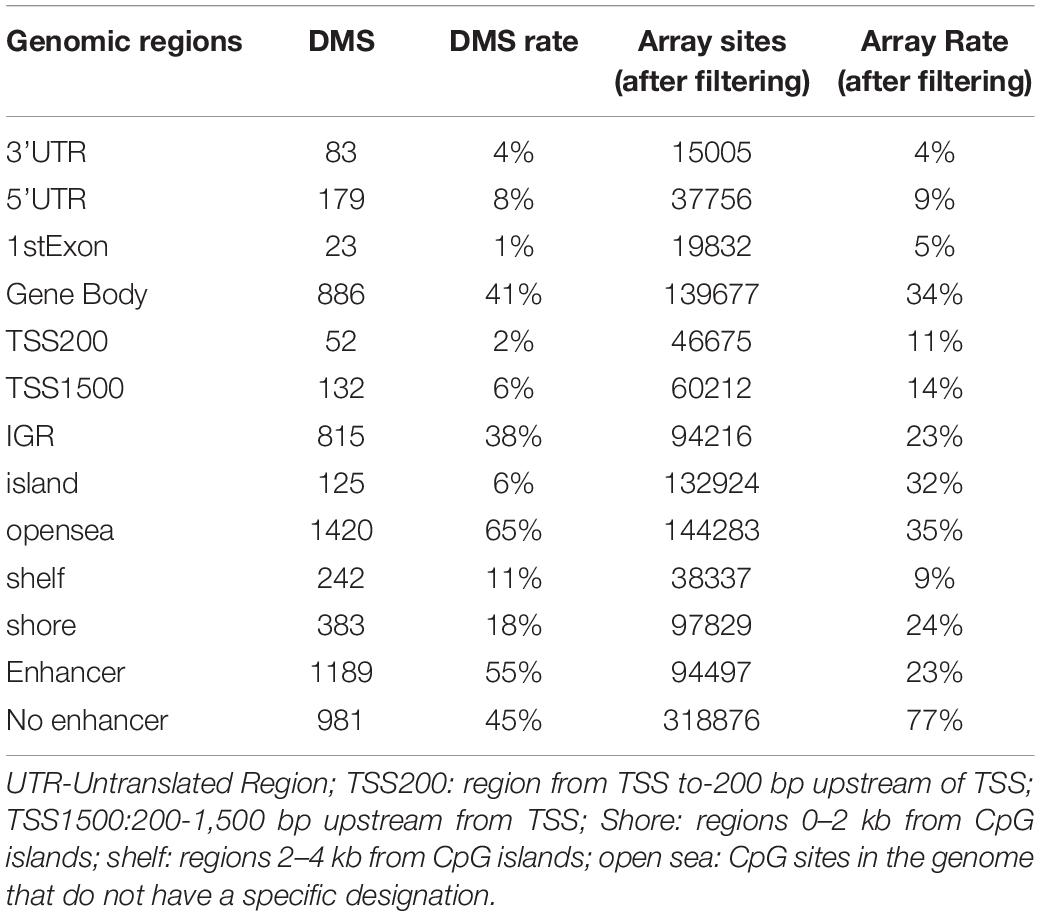
Table 2. DMS and Array sites (after filtering) in different genomic regions.
In addition, we separately analyzed the proportion of DMS and array sites enriched in enhancers. Our results showed that 55% of the DMS were enriched in the enhancer, whilst only 23% were enriched in the array sites. We also analyzed their positions relative to CpG islands (CGI). The positions relative to a CGI included the shore, shelf, and open sea regions. DMS on CGI accounted for 6% whilst the percentage for the array sites was 32%. These data were consistent with previous studies including data from the Human Epigenome Project that showed 80% of the CpG dinucleotides in the human genome are methylated, whereas CGI are usually unmethylated (Brena et al., 2006).
Integrative Analysis of DNA Methylation and Gene Expression
The DEGs and DMGs were compared against the list of human TFs. From the DNA expression profiling dataset, we identified 168 DETFs in 1986 DEGs. Of these 168 DETFs, 21 encoding genes exhibited differential methylation that harbored at least one DMS. Four of these DETFs (HMG2A, ALX4, RORA, and TBX3) were excluded from the study as their encoding genes corresponded with several CpG sites, of which some were hypermethylated and others hypomethylated. The remaining seventeen TFs are shown in Table 3 that included nine hypermethylated genes of which two were up-regulated and seven were down-regulated, and eight hypomethylated genes of which three were up-regulated and five were down-regulated.

Table 3. DETFs harboring DMS in knee articular cartilage from normal and OA patients.
Changes in the Gene Transcription Profiles of Primary Chondrocytes Following Treatment With DNA Methylation Inhibitors
Previous genome-wide methylation studies have shown that promoter methylation negatively correlates with gene expression, whilst methylation in the gene body and 3′UTR is positively correlated with gene expression (Mishra and Guda, 2017). To further explore how the methylation of different genomic regions affects gene transcription, we chose DETFs that conformed to these changes for further investigation including SOX11, RARA,ELF3, and FOXD2. The mRNA expression of selected TFs was then validated. ELF3 and SOX11 were up-regulated in the cartilage of OA patients, whilst RARA and FOXD2 were down-regulated (Figure 4A). Exception for ELF3, the mRNA expression of SOX11, RARA, and FOXD2 were consistent with the results analyzed from the mRNA expression data. Upon treatment with 5-Aza, ELF3, and SOX11 mRNA levels decreased, whilst RARA mRNA levels increased and FOXD2 showed no significant change (Figure 4B). These data indicated that DNA demethylation can affect the mRNA expression levels of these particular TFs.
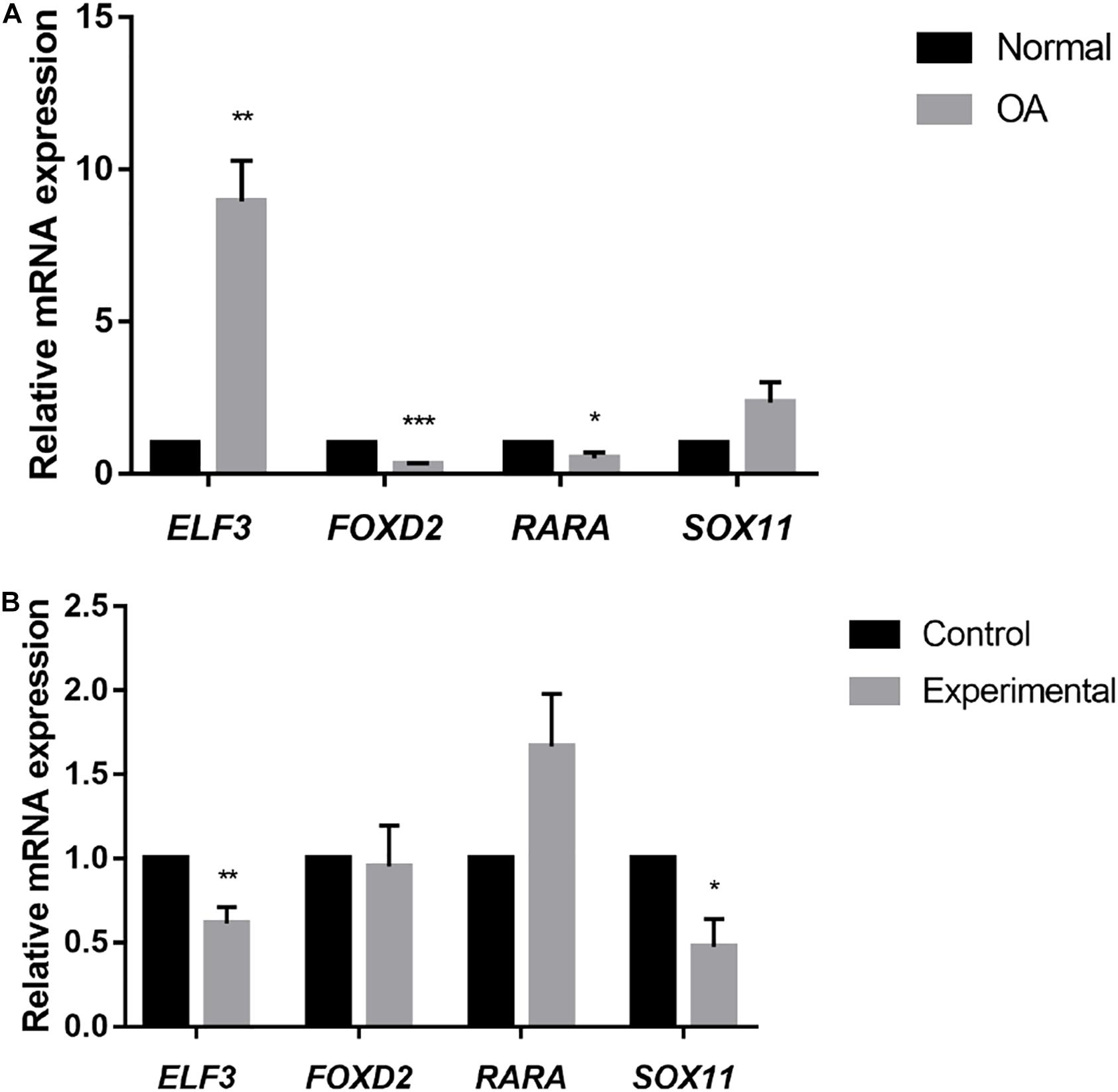
Figure 4. (A) Relative mRNA expression of selected TFs in normal and OA knee articular cartilage, assessed by RT-qPCR. *P < 0.05, **P < 0.01, ***P < 0.001. (B) Relative mRNA expression of selected TFs in cultured primary human chondrocytes (N = 3) treated with 5-Aza (10 μM) or vehicle for 4 days. Values are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Recently, epigenetic regulation has been shown to play an important role in the pathogenesis of OA (den Hollander et al., 2014). In particular, DNA methylation can have a profound impact on the development of OA. In a study by Roach et al. (2005) the abnormal expression of ECM-degrading enzymes including MMP-3, MMP-9, MMP-13, and ADAMTS-4 in OA chondrocytes were shown to be associated with loss of DNA methylation in their promoter region of these genes. However, the mechanisms driving these epigenetic regulatory processes and the observed phenotypes are yet to be determined.
Genome-wide expression profiling analysis has shown that abnormal transcription is essential for the development of OA (Aigner et al., 2006; Karlsson et al., 2010). Previous studies identified key TFs in the pathogenesis of OA such as SOX9, by analyzing DNA methylation in combination with mRNA expression. SOX9 is expressed by mesenchymal stem cells and can promote the expression of matrix proteins in cartilage (Zhang et al., 2019). A previous study showed an increase in SOX9 expression that in OA cartilage was affected by its promoter methylation status. 5-Aza could then reverse the decreased levels of SOX9 gene expression (Kim et al., 2013).
SOX9 can also interact with methylated DNA. Imagawa et al. demonstrated that the DNA methylation status of COL9A1 could influence the binding of SOX9 to DNA. This restrained SOX9-driven promoter activation and down-regulated the expression of COL9A1 (Imagawa et al., 2014). Also, recent progress in epigenetic research has highlighted TFs as a new class of readers and effectors of DNA methylation (Zhu et al., 2016). The analysis of TFs is therefore crucial to explaining the underlying biological processes mediated by epigenetic regulation.
We performed a statistical analysis of CpG sites enriched in different gene sub-regions showing that most DMS were enriched in the IGR (38%) and gene body (41%). Only 17% of DMS were included in the promoter regions. The distance between enhancers and promoters is highly variable. Enhancers are mostly CpG-poor and play an important role in controlling gene expression during development and cell functioning (Lister et al., 2009). The proportion of DMS in enhancer regions (55%) was significantly higher than that in array sites (23% after screening), indicating that epigenetic modifications were more likely to occur in CpG-poor genomic regions (Thurman et al., 2012; Ziller et al., 2013). These data also suggested that the analysis of specific genomic regions during the development of OA may contribute to the understanding of epigenetic functions and their influence on the pathogenesis of OA.
Genome-wide studies have provided detailed information about methylation patterns showing that CpG sites outside TSS have a more complex effect on gene transcription than previously thought. Previous studies focused on the methylation levels of gene promoters. However, gene body methylation remains to be fully characterized. Most studies have focused on CGI at TSS and so DNA methylation is often described as an epigenetic marker for silencing gene expression (Jones, 2012).
In the study of DNA methylation of a single gene in OA, the promoter of COL10A1 was hypomethylation during chondrocyte hypertrophy, and maturation and was associated with increased COL10A1 expression (Zimmermann et al., 2008). Similarly, the methylation levels of CpG sites in the promoters of many metalloproteinases including MMP2, MMP9, MMP13, and ADAMTS4, were reduced and led to up-regulated gene expression in OA cartilage (Roach et al., 2005; Cheung et al., 2009). Methylation in CpG-poor regions of the genome may be more worthy of study than promoter regions.
From the early stages of DNA methylation, it has been known that gene body methylation is characteristic of transcriptional genes (Wolf et al., 1984). Gene bodies are mostly CpG-poor, widely methylated and contain multiple repeating and translocation elements (Jones, 2012). Recently, it was confirmed that there is a widely positive correlation between gene methylation and transcription on the X chromosome (Hellman and Chess, 2007). However, as early as 1999, researchers paradoxically proposed that promoter methylation is inversely correlated with expression, whilst gene body methylation is positively correlated with expression (Jones, 1999).
Gene body methylation is thought to be associated with the regulation of gene splicing. Exons have higher levels of methylation compared to introns, and methylation is more likely to change at exon-intron boundaries (Laurent et al., 2010). However, recent genome-wide studies of methylation have clearly shown that methylation in promoters and 5′UTR are mostly negatively correlated with gene expression, whilst methylation in genes and 3′UTR are positively correlated (Mishra and Guda, 2017).
In this study, we performed a combined analysis of DNA methylation and gene expression microarray datasets. We identified 17 TFs that exhibited simultaneously different methylation and expression patterns between normal and OA cartilage samples. We then validated the mRNA expression of selected TFs and performed functional validation in primary chondrocytes treated with 5-Aza.
Analysis of the mRNA expression data showed that ELF3 was down-regulated in OA cartilage. However, RT-qPCR results showed that ELF3 was up-regulated in OA cartilage. These data suggest that the results obtained from the database analysis are not entirely accurate and require further verification.
We treated cultured human primary chondrocytes with 5-Aza that resulted in the down-regulation of ELF3 mRNA. Although this result was contrary to our expectations, it indicated that methylation in promoters does not always result in gene silencing. Also, previous studies have linked ELF3 dysregulation to epigenetic changes. ELF3 is a member of the ELF subset of the ETS family of TFs. A previous study showed that increased ELF3 levels in OA inhibited COL2A1 by directly binding to ETS sites in the COL2A1 promoter and then binding to the HMG domain of SOX9. This resulted in the repression of CBP/p300 driven HAT activity. The dysregulation of ELF3 was associated with hypermethylation of the ELF3 proximal gene promoter (Otero et al., 2017) which is further supported by our study.
Another study revealed that ELF3 could also activate IL-1β and TNFα-mediated MMP13 transcription by binding to the MMP13 proximal promoter induced by IL-1β (Otero et al., 2012). These studies provide strong evidence that ELF3 is a valuable TF in the pathogenesis of OA. Also, these data revealed that DNA methylation status can not only change the transcription of TFs, but also alter the binding of TFs to DNA target sequences that regulate the expression of genes and cause phenotypic changes.
SOX11 belongs to the group C of the Sox family that is composed of many supergenes with conserved motifs of the HMG box (Kavyanifar et al., 2018). Decreased levels of SOX6 and SOX9 are associated with the phenotypic change of chondrocytes in OA (Sock et al., 2004). Recent studies have also shown that SOX11 is up-regulated in OA tissue and can promote OA by induction of TNF-α (Xu et al., 2019). Our results suggest that the increased level of SOX11 mRNA in OA tissue may be related to methylation in the 3′UTR region of the gene. In addition, the DMS of RARA was located in the 1stExon region, while the DMS of FOXD2 was found in both 1stExon and 3′UTR. It may explain the increased mRNA expression level of RARA after 5-Aza treatment, while the FOXD2 showed no significant change. RARA and FOXD2 do not have an obvious role in OA pathogenesis highlighting this as an important area for further studies.
Our findings on ELF3 indicate that hypermethylation of gene promoters may increase gene transcription. Although methylation in promoters is usually manifested as gene silencing, it is not clear whether methylation is the cause or the result of gene silencing. Studies have found that inactivation of the X chromosome results in methylation of the Hprt gene (Lock et al., 1987). The link between DNA methylation and transcription is likely to be significantly more complex than initially realized. Although DNA methylation cannot always explain changes in transcription, a better understanding of methylation changes in OA cartilage tissue may facilitate the development of novel therapeutic approaches by preventing or reversing epigenetic changes.
There were limitations to our study. Firstly, we did not investigate the normal cartilage from the oLT regions. Undoubtedly, our results would be more representative if we could add qPCR or western-blot to validate the oLT samples. Nevertheless, previous studies of the pathology and transcriptome showed that the oLT regions are very similar to normal. Thus, the oLT regions could serve as a suitable alternative to normal control, which could also reduce the inter-individual variations (Liu et al., 2018). Therefore, the measurement of the cartilage from the oLT regions is considered non-essential. Secondly, the sample size of cartilage tissue we acquired were small. In the future, we will pay more attention to the sample size.
For the first time, we analyzed DNA methylation and mRNA expression data in OA patients according to the distribution of CpG sites across the genome. Our comprehensive analysis identified a subset of DETFs whose gene transcriptional changes may be related to DNA methylation status. These TFs are potential biomarkers and drug targets, and merit further study in the pathogenesis of OA. Also, we found that the distribution of DMS in the genome had obvious characteristics. Moreover, the effect of DNA methylation on transcription is related to the distribution of CpG sites. Further studies on the interaction between epigenetic modification and transcription will provide more effective insights into the pathogenesis of OA.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63106; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114007<.
Ethics Statement
The studies involving human participants were reviewed and approved by the Committee of the Renmin Hospital of Wuhan University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
PY and BQ designed the study. PY, XX, and JY acquired and interpreted the data. PY analyzed the data and was a major contributor in writing the manuscript. XX and JY prepared figures and tables. BQ prepared the manuscript and supervised the study. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant no. 81071494) and Hubei Provincial Science and Technology Support Program (Grant no. 2015BCA316).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.618803/full#supplementary-material
References
Aigner, T., Fundel, K., Saas, J., Gebhard, P. M., Haag, J., Weiss, T., et al. (2006). Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis. Rheum. 54, 3533–3544. doi: 10.1002/art.22174
Barter, M. J., and Young, D. A. (2013). Epigenetic mechanisms and non-coding RNAs in osteoarthritis. Curr. Rheumatol. Rep. 15:353. doi: 10.1007/s11926-013-0353-z
Bortoluzzi, A., Furini, F., and Scire, C. A. (2018). Osteoarthritis and its management - Epidemiology, nutritional aspects and environmental factors. Autoimmun. Rev. 17, 1097–1104. doi: 10.1016/j.autrev.2018.06.002
Brena, R. M., Huang, T. H., and Plass, C. (2006). Toward a human epigenome. Nat. Genet. 38, 1359–1360. doi: 10.1038/ng1206-1359
Bui, C., Barter, M. J., Scott, J. L., Xu, Y., Galler, M., Reynard, L. N., et al. (2012). cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 26, 3000–3011. doi: 10.1096/fj.12-206367
Cheung, K. S., Hashimoto, K., Yamada, N., and Roach, H. I. (2009). Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol. Int. 29, 525–534. doi: 10.1007/s00296-008-0744-z
den Hollander, W., Ramos, Y. F., Bos, S. D., Bomer, N., van der Breggen, R., Lakenberg, N., et al. (2014). Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann. Rheum. Dis. 73, 2208–2212. doi: 10.1136/annrheumdis-2014-205980
Fathollahi, A., Aslani, S., Jamshidi, A., and Mahmoudi, M. (2019). Epigenetics in osteoarthritis: novel spotlight. J. Cell. Physiol. 234, 12309–12324. doi: 10.1002/jcp.28020
Harris, R. A., Wang, T., Coarfa, C., Nagarajan, R. P., Hong, C., Downey, S. L., et al. (2010). Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 28, 1097–1105. doi: 10.1038/nbt.1682
Hashimoto, K., Oreffo, R. O., Gibson, M. B., Goldring, M. B., and Roach, H. I. (2009). DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis. Rheum. 60, 3303–3313. doi: 10.1002/art.24882
Hellman, A., and Chess, A. (2007). Gene body-specific methylation on the active X chromosome. Science 315, 1141–1143. doi: 10.1126/science.1136352
Iliopoulos, D., Malizos, K. N., and Tsezou, A. (2007). Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann. Rheum. Dis. 66, 1616–1621. doi: 10.1136/ard.2007.069377
Im, G. I., and Choi, Y. J. (2013). Epigenetics in osteoarthritis and its implication for future therapeutics. Expert. Opin. Biol. Ther. 13, 713–721. doi: 10.1517/14712598.2013.764410
Imagawa, K., de Andres, M. C., Hashimoto, K., Itoi, E., Otero, M., Roach, H. I., et al. (2014). Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis. Rheumatol. 66, 3040–3051. doi: 10.1002/art.38774
Jones, P. A. (1999). The DNA methylation paradox. Trends. Genet. 15, 34–37. doi: 10.1016/s0168-9525(98)01636-9
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
Karlsson, C., Dehne, T., Lindahl, A., Brittberg, M., Pruss, A., Sittinger, M., et al. (2010). Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis. Cartilage. 18, 581–592. doi: 10.1016/j.joca.2009.12.002
Kavyanifar, A., Turan, S., and Lie, D. C. (2018). SoxC transcription factors: multifunctional regulators of neurodevelopment. Cell. Tissue. Res. 371, 91–103. doi: 10.1007/s00441-017-2708-7
Kim, K. I., Park, Y. S., and Im, G. I. (2013). Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J. Bone. Miner. Res. 28, 1050–1060. doi: 10.1002/jbmr.1843
Kimes, P. K., Liu, Y. F., Hayes, D. N., and Marron, J. S. (2017). Statistical significance for hierarchical clustering. Biometrics 73, 811–821. doi: 10.1111/biom.12647
Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y., and Albu, M. (2018). The human transcription factors. Cell 172, 650–665. doi: 10.1016/j.cell.2018.01.029
Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C. T., et al. (2010). Dynamic changes in the human methylome during differentiation. Genome. Res. 20, 320–331. doi: 10.1101/gr.101907.109
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Liu, Y., Chang, J. C., Hon, C. C., Fukui, N., Tanaka, N., Zhang, Z., et al. (2018). Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 8:15499. doi: 10.1038/s41598-018-33779-z
Lock, L. F., Takagi, N., and Martin, G. R. (1987). Methylation of the Hprt gene on the inactive X occurs after chromosome inactivation. Cell 48, 39–46. doi: 10.1016/0092-8674(87)90353-9
Mishra, N. K., and Guda, C. (2017). Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 8, 28990–29012. doi: 10.18632/oncotarget.15993
Morris, T. J., Butcher, L. M., Feber, A., Teschendorff, A. E., Chakravarthy, A. R., Wojdacz, T. K., et al. (2014). ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 30, 428–430. doi: 10.1093/bioinformatics/btt684
Otero, M., Peng, H., Hachem, K. E., Culley, K. L., Wondimu, E. B., Quinn, J., et al. (2017). ELF3 modulates type II collagen gene (COL2A1) transcription in chondrocytes by inhibiting SOX9-CBP/p300-driven histone acetyltransferase activity. Connect. Tissue. Res. 58, 15–26. doi: 10.1080/03008207.2016.1200566
Otero, M., Plumb, D. A., Tsuchimochi, K., Dragomir, C. L., Hashimoto, K., Peng, H., et al. (2012). E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem. 287, 3559–3572. doi: 10.1074/jbc.M111.265744
Roach, H. I., Yamada, N., Cheung, K. S., Tilley, S., Clarke, N. M., Oreffo, R. O., et al. (2005). Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis. Rheum. 52, 3110–3124. doi: 10.1002/art.21300
Rushton, M. D., Reynard, L. N., Barter, M. J., Refaie, R., Rankin, K. S., Young, D. A., et al. (2014). Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis. Rheumatol. 66, 2450–2460. doi: 10.1002/art.38713
Scott, J. L., Gabrielides, C., Davidson, R. K., Swingler, T. E., Clark, I. M., Wallis, G. A., et al. (2010). Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 69, 1502–1510. doi: 10.1136/ard.2009.119966
Sock, E., Rettig, S. D., Enderich, J., Bosl, M. R., Tamm, E. R., and Wegner, M. (2004). Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell. Biol. 24, 6635–6644. doi: 10.1128/MCB.24.15.6635-6644.2004
Steinberg, J., Brooks, R. A., Southam, L., Bhatnagar, S., Roumeliotis, T. I., Hatzikotoulas, K., et al. (2018). Widespread epigenomic, transcriptomic and proteomic differences between hip osteophytic and articular chondrocytes in osteoarthritis. Rheumatology (Oxford) 57, 1481–1489. doi: 10.1093/rheumatology/key101
Teschendorff, A. E., Marabita, F., Lechner, M., Bartlett, T., Tegner, J., Gomez-Cabrero, D., et al. (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196. doi: 10.1093/bioinformatics/bts680
Thurman, R. E., Rynes, E., Humbert, R., Vierstra, J., Maurano, M. T., Haugen, E., et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. doi: 10.1038/nature11232
Wolf, S. F., Jolly, D. J., Lunnen, K. D., Friedmann, T., and Migeon, B. R. (1984). Methylation of the hypoxanthine phosphoribosyltransferase locus on the human X chromosome: implications for X-chromosome inactivation. Proc. Natl. Acad. Sci. U.S.A. 81, 2806–2810. doi: 10.1073/pnas.81.9.2806
Xu, S., Yu, J., Wang, Z., Ni, C., Xia, L., and Tang, T. (2019). SOX11 promotes osteoarthritis through induction of TNF-alpha. Pathol. Res. Pract. 215:152442. doi: 10.1016/j.prp.2019.152442
Zhang, M., Theleman, J. L., Lygrisse, K. A., and Wang, J. (2019). Epigenetic mechanisms underlying the aging of articular cartilage and osteoarthritis. Gerontology 65, 387–396. doi: 10.1159/000496688
Zhang, Y., Fukui, N., Yahata, M., Katsuragawa, Y., Tashiro, T., Ikegawa, S., et al. (2016). Genome-wide DNA methylation profile implicates potential cartilage regeneration at the late stage of knee osteoarthritis. Osteoarthritis. Cartilage. 24, 835–843. doi: 10.1016/j.joca.2015.12.013
Zhao, L., Wang, Q., Zhang, C., and Huang, C. (2017). Genome-wide DNA methylation analysis of articular chondrocytes identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in osteoarthritis. Clin. Rheumatol. 36, 2335–2342. doi: 10.1007/s10067-017-3667-9
Zhu, H., Wang, G., and Qian, J. (2016). Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 17, 551–565. doi: 10.1038/nrg.2016.83
Ziller, M. J., Gu, H., Muller, F., Donaghey, J., Tsai, L. T., Kohlbacher, O., et al. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. doi: 10.1038/nature12433
Keywords: DNA methylation, gene transcription, genomic regions, chondrocytes, osteoarthritis
Citation: Yi P, Xu X, Yao J and Qiu B (2021) Analysis of mRNA Expression and DNA Methylation Datasets According to the Genomic Distribution of CpG Sites in Osteoarthritis. Front. Genet. 12:618803. doi: 10.3389/fgene.2021.618803
Received: 18 October 2020; Accepted: 29 March 2021;
Published: 15 April 2021.
Edited by:
Jorg Tost, Commissariat à l’Energie Atomique et aux Energies Alternatives, FranceReviewed by:
Shirin Kadler, Technical University of Berlin, GermanyMaría Del Carmen De Andrés Gonzalez, Instituto de Investigación Biomedica de A Coruña Hospital Teresa Herrera, Spain
Copyright © 2021 Yi, Xu, Yao and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Qiu, cWJ0ZzE2M0AxNjMuY29t