 Chia-Hsiang Chen
Chia-Hsiang Chen Min-Chih Cheng
Min-Chih Cheng Tsung-Ming Hu
Tsung-Ming Hu Lieh-Yung Ping
Lieh-Yung Ping- 1Department of Psychiatry, Chang Gung Memorial Hospital, Taoyuan, Taiwan
- 2Department and Graduate Institute of Biomedical Sciences, Chang Gung University, Taoyuan, Taiwan
- 3Department of Psychiatry, Yuli Branch, Taipei Veterans General Hospital, Hualien, Taiwan
Schizophrenia is a chronic, devastating mental disorder with complex genetic components. Given the advancements in the molecular genetic research of schizophrenia in recent years, there is still a lack of genetic tests that can be used in clinical settings. Chromosomal microarray analysis (CMA) has been used as first-tier genetic testing for congenital abnormalities, developmental delay, and autism spectrum disorders. This study attempted to gain some experience in applying chromosomal microarray analysis as a first-tier genetic test for patients with schizophrenia. We consecutively enrolled patients with schizophrenia spectrum disorder from a clinical setting and conducted genome-wide copy number variation (CNV) analysis using a chromosomal microarray platform. We followed the 2020 “Technical Standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)” to interpret the clinical significance of CNVs detected from patients. We recruited a total of 60 patients (36 females and 24 males) into this study. We detected three pathogenic CNVs and one likely pathogenic CNV in four patients, respectively. The detection rate was 6.7% (4/60, 95% CI: 0.004–0.13), comparable with previous studies in the literature. Also, we detected thirteen CNVs classified as uncertain clinical significance in nine patients. Detecting these CNVs can help establish the molecular genetic diagnosis of schizophrenia patients and provide helpful information for genetic counseling and clinical management. Also, it can increase our understanding of the pathogenesis of schizophrenia. Hence, we suggest CMA is a valuable genetic tool and considered first-tier genetic testing for schizophrenia spectrum disorders in clinical settings.
Introduction
Schizophrenia is a chronic debilitating mental disorder marked by delusions, hallucinations, erratic emotions, bizarre behaviors, and cognitive deficits. Approximately 1% of the general population is affected by the disease. It is a complex disorder involving genetic and environmental factors, and genetic factors play a significant role in the genesis of schizophrenia. Recent advances in genetic and genomic technology have significantly increased our understanding of the genetic architecture of schizophrenia (Avramopoulos, 2018). Genetic variants associated with schizophrenia are heterogeneous, including hundreds of common variants with modest clinical effects and multiple rare mutations with high penetrance and varied clinical expressivity (Foley et al., 2017; Henriksen et al., 2017).
Copy number variations (CNVs) are deleted and duplicated genomic DNA segments resulting from aberrant chromosomal rearrangement. They are associated with human health and diseases (Harel and Lupski, 2018; Hu et al., 2018). CNVs are divided into common and rare CNVs according to their populational frequencies. Accumulating evidence indicates that rare CNVs contribute significantly to the genetic basis of neuropsychiatric disorders, such as intellectual disability, autism spectrums disorder (ASD), and schizophrenia (Rutkowski et al., 2017; Lowther et al., 2017a). Identifying disease-associated CNVs can help establish cell and animal models of neuropsychiatric disorders to elucidate the pathogenesis and facilitate new therapeutic regimens (Nomura and Takumi, 2012; Flaherty and Brennand, 2017; Rutkowski et al., 2017; Takumi and Tamada, 2018).
The Psychiatric Genomics Consortium CNV Analysis Group performed a genome-wide CNV analysis in 21,094 patients with schizophrenia and 20,227 controls. They confirmed a global enrichment of CNV burden in patients with schizophrenia compared to controls. Additionally, they reported rare recurrent CNVs at eight genetic loci reaching a genome-wide significance. The odds ratios of CNVs at these eight loci range from 3.8 to infinity (Marshall et al., 2017), supporting a substantial role of rare CNVs in the genesis of schizophrenia.
Chromosomal microarray analysis (CMA) is a molecular genetic tool that can identify the location and the size of CNV with high resolution and precision (Rosenfeld and Patel, 2017; Shaikh, 2017). It has been recommended as the first-tier genetic test for patients with developmental disabilities or congenital abnormalities, as it significantly increases the diagnostic yield of children affected with global developmental delay, autism spectrum disorders, or multiple congenital abnormalities (Manning et al., 2010; Miller et al., 2010; Michelson et al., 2011). Costain and colleagues conducted a prospective community-based chromosomal microarray analysis of 459 patients with schizophrenia in a catchment area. They found that 8.1% of their study subjects carried rare large (>500 kb) CNVs of clinical significance. Hence, they suggested using CMA as a genetic test for schizophrenia (Costain et al., 2013). Lowther and colleagues reported a CNV survey of 546 patients with schizophrenia from six community clinics; they found 39/546 (7.1%) patients had at least one pathogenic CNV (Lowther et al., 2017b). Baker and colleagues also proposed CMA as a routine clinical genetic test for schizophrenia (Baker et al., 2014), in view that identification of pathogenic CNVs can help establish the genetic diagnosis and improve the medical and mental health care of patients with schizophrenia.
We previously reported three novel rare CNVs associated with schizophrenia in three families, respectively (Liao et al., 2012), indicating the potential clinical utility of chromosomal microarray as a genetic test for schizophrenia in our population. This study attempted to gain some experiences of applying CMA as a first-tier genetic test for schizophrenia spectrum disorders in a clinical setting. We consecutively recruited patients with schizophrenia in our clinical practice and conducted a genome-wide CNV analysis for them. Here, we report our findings in a sample of 60 patients recruited from the outpatient unit of our hospital.
Materials and Methods
Subjects
All the patients were residents of Taiwan. We recruited patients fulfilling the diagnostic criteria of schizophrenia (295.9) or schizoaffective disorder (295.70) according to the DSM-5 (Diagnostic and Statistical Manual of Mental Disorder-5th edition) into this study. After interviewing and reviewing their medical records, patients with a history of substance abuse, intellectual disability, childhood-onset psychiatric disorders, head injury, or medical conditions associated with psychiatric disorders were excluded. The Review Board of Chang Gung Memorial Hospital-Linkou approved the study with the approved number of 104-6522A3 (201506522A3). We obtained informed consent from each participant after a full explanation of this study.
Genome-Wide Copy Number Variation Analysis
We extracted genomic DNA from venous blood from each participant using the Smart Genomic DNA Extraction kit (Intelligent Biomedicine, Taipei, Taiwan) according to the manufacturer’s instructions. DNA was subjected to genome-wide copy number variation analysis using CytoScan HD Array (Affymetrix Inc., Santa Clara, CA, United States) following the manufacturer’s protocol. The Genomic Medicine Core Laboratory of Chang Gung Memorial Hospital-Linkou (Taoyuan, Taiwan) did the experiment, and the raw data were analyzed using the software Chromosomal Analysis Suite Version 3.3.0.139 (r10838) (Affymetrix Inc., Santa Clara, CA, United States). Gain and loss of CNVs were analyzed at the resolution of 50 probes and 100 kb. The genomic coordinates of CNVs followed the human genome sequences version GRCh37/hg19.
Real-Time Quantitative PCR
The real-time quantitative PCR (RT-qPCR) method was used as a complementary method to verify the authenticity of CNVs detected by the CytoScan HD array. Genes encompassed by the CNV of interest were selected. A comparative ddCt method was used for the analysis of RT-qPCR data. The Ct of the target gene was subtracted by the Ct of a reference gene (dCt), then normalized to a control subject (ddCt). The relative fold change to a normal subject was determined as 2−ddCt. We performed experiments using the StepOnePlus machine (Applied Biosystems, Forster City, CA) with the SYBR green method according to the manufacturer’s instructions, the experiments. Target genes, primer sequences, optimal annealing temperature, and the amplicon sizes are listed in Supplementary Table S1.
Assessment of the Pathogenicity of Copy Number Variation
To assess the clinical significance of CNVs detected from patients, we followed the “Technical Standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)” (Riggs et al., 2020). This report recommended using a semiquantitative, point-based scoring system to classify CNVs into five categories used in sequence variation classification (Richards et al., 2015), including pathogenic, likely pathogenic, uncertain significance, likely benign, and benign. We used the recommended web-based ClinGen CNV Pathogenicity Calculator (http://cnvcalc.clinicalgenome.org/cnvcalc/) to record and calculate the final score of CNVs detected in our sample. CNVs with a score of equal or greater than 0.99 were classified pathogenic, while scores between 0.90 and 0.98 were likely pathogenic. CNVs with scores between −0.89 and 0.89 belonged to the uncertain significance (VUS) category, while scores between −0.90 and −0.98 belonged to the likely benign category. CNVs with scores less than −0.99 were classified as benign. We defined pathogenic and likely pathogenic CNVs as of clinical relevance in this study.
Results
Identification of Pathogenic and Likely Pathogenic Copy Number Variations
We recruited 60 patients into this study from May 2016 to April 2019, including 36 females and 24 males. The age ranged from 20 to 70 years old. The mean age was 41 years old, with a standard deviation of 12 years old. We detected three pathogenic CNVs and one likely pathogenic CNV in four respective patients from this sample. The three pathogenic CNVs included a 443 kb microduplication at 15q13.3, a 1,489 kb microdeletion at 22q11.21, and a 912 kb microduplication at 2q23.1. The likely pathogenic CNV was a 188 kb microduplication at 12q24.11. The readouts of three pathogenic CNVs and one likely pathogenic CNV in the Chromosomal Analysis Suite are shown in Figure 1. The authenticity of these CNVs was further verified using real-time quantitative PCR. The detection rate of pathogenic and likely pathogenic CNVs together in this sample was 6.7% (4/60, 95% CI: 0.004–0.13). The genetic information of these four clinically relevant CNVs, including their location, size, and encompassed genes, is listed in Table 1.
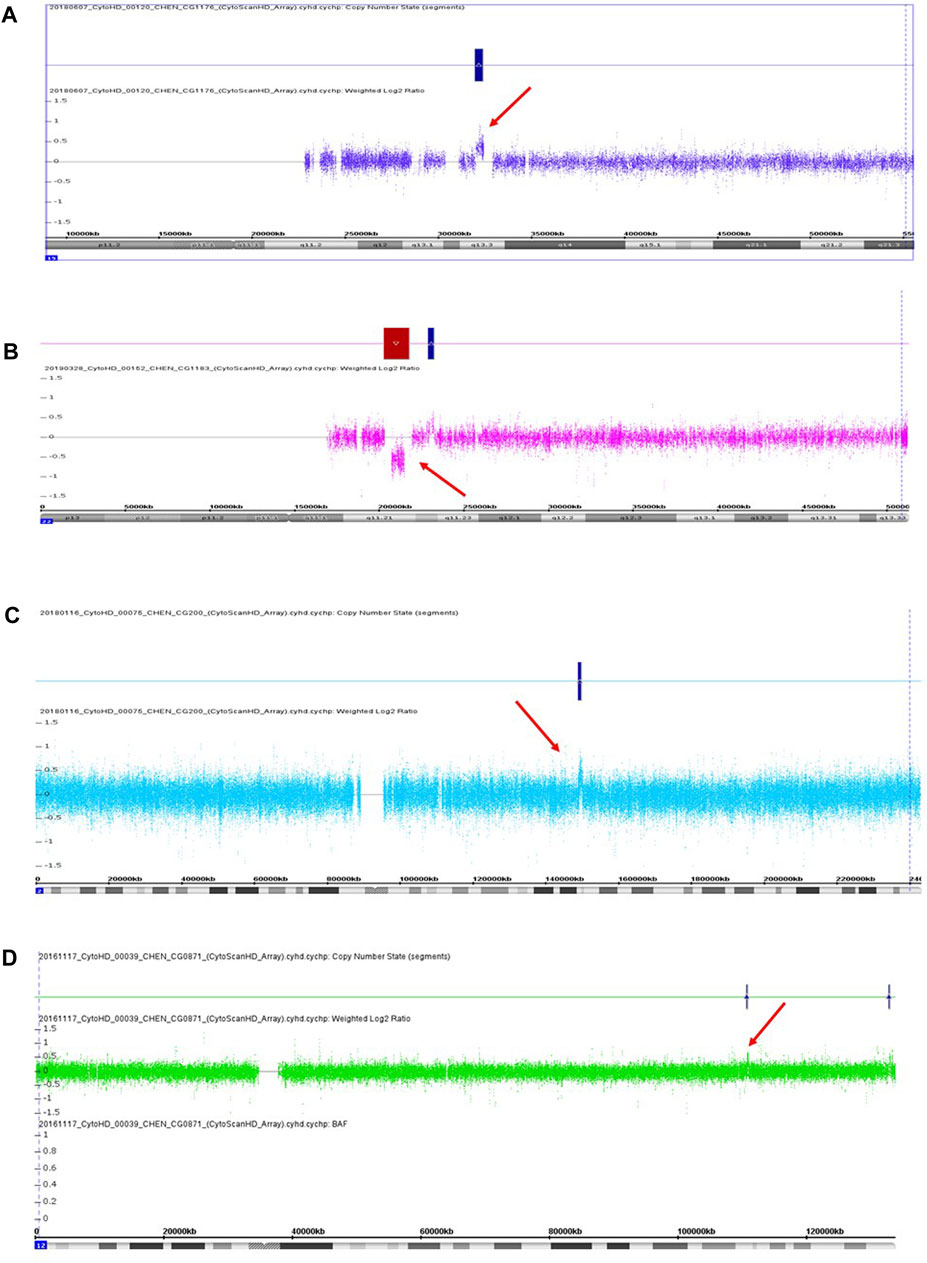
FIGURE 1. Three pathogenic CNVs (A–C) and one likely pathogenic CNV (D) were detected in this study. (A) A 433 kb microduplication at 15q13.3 in the patient CG1176. (B) A 1,489 kb microdeletion at 22q11.21 in the patient CG1183. (C) A 912 kb microduplication at 2q13.1 in the patient CG0200. (D) A 188 kb microduplication at 12q24.11 in the patient CG0871. Red arrow indicates the location of CNV.
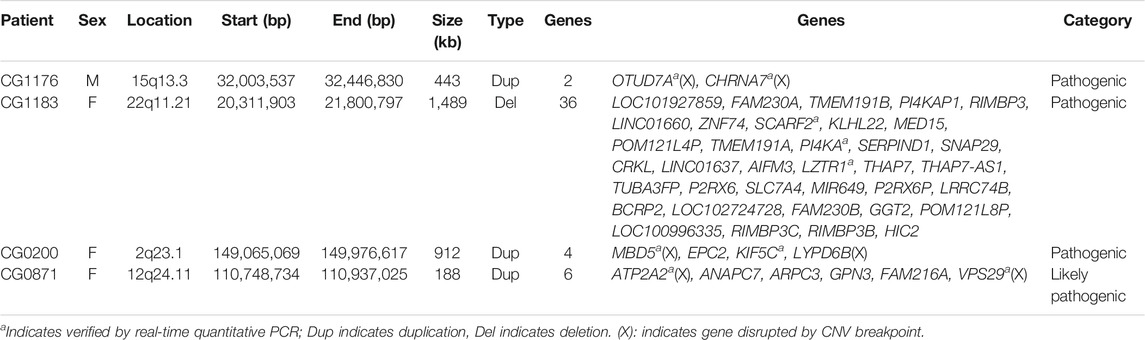
TABLE 1. Pathogenic and likely pathogenic CNVs detected in this study.
The first pathogenic CNV was a 433 kb microduplication at 15q13.3, which was detected in a male patient (CG1176) without a family history of mental illness. He suffered from several acute psychotic episodes in his twenties and was subsequently diagnosed with schizophrenia. His psychotic symptoms included auditory hallucination, the idea of reference, a delusion of persecution, irritability, and agitation. He had an unremarkable birth history and normal psychosocial development before the onset of his mental illness. Also, he did not have physical abnormalities, an intellectual disability, or neurological symptoms. His psychotic symptoms were under control after antipsychotics treatment, but he still suffered from residual auditory hallucinations. Currently, he keeps a job and has good compliance with long-term antipsychotic medication.
The second pathogenic CNV was a 1,489 kb microdeletion at 22q11.21, present in a female patient without a family history. Her birth history and psychosocial development were unremarkable before the onset of her mental illness. She did not have physical abnormalities, an intellectual disability, or neurological symptoms. In her twenties, she started to express psychotic symptoms of loosening of association, irrelevant speech, unstable emotions, and auditory hallucinations and was subsequently diagnosed with schizoaffective disorder. She received antipsychotics and mood stabilizer treatment and had a good response, but she did not comply with medication well due to the lack of insight. Her mental symptoms recurred several times after discontinuation of her medication, which led to her social function impairment.
The third pathogenic CNV was a 912 kb microduplication at 2q23.1, occurring in a female patient without a family history of mental illness. In her twenties, she was diagnosed with schizophrenia due to psychotic symptoms, such as auditory hallucinations, the idea of reference, delusions of being controlled and possessed. She did not have physical abnormalities, an intellectual disability, or neurological symptoms. Also, her birth history and normal psychosocial development were unremarkable before the onset of her mental illness. She attempted suicide once due to commanding hallucinations. Her social function deteriorated despite continuous antipsychotic treatment.
The likely pathogenic CNV was a 188 kb microduplication at 12q24.11, identified in a female patient without a family history of mental illness. She was diagnosed with schizoaffective disorder in her twenties. Her mental symptoms included auditory hallucination, loose association, and irrelevant speech. Also, she had depressive episodes in her course of illness. She had normal psychosocial development before the onset of her mental illness. Also, she did not have physical abnormalities, an intellectual disability, or neurological symptoms. She had a reasonable response to antipsychotic and antidepressant treatment.
Identification of Copy Number Variations With Uncertain Significance
Furthermore, we detected thirteen CNVs classified as uncertain significance in nine patients. The authenticity of these thirteen CNVs was also verified using real-time quantitative PCR. The genetic information of these thirteen CNVs with uncertain significance, including their location, size, and encompassed genes, is listed in Table 2. The patient CG0200 carried one CNV with uncertain significance (a microduplication at 4q13.1) and one pathogenic CNV (a microduplication at 2q23.1). The patient CG0027 carried two CNVs with uncertain significance, while the patient CG0314 carried four CNVs with uncertain significance. The other six patients carried only one CNV with uncertain significance.
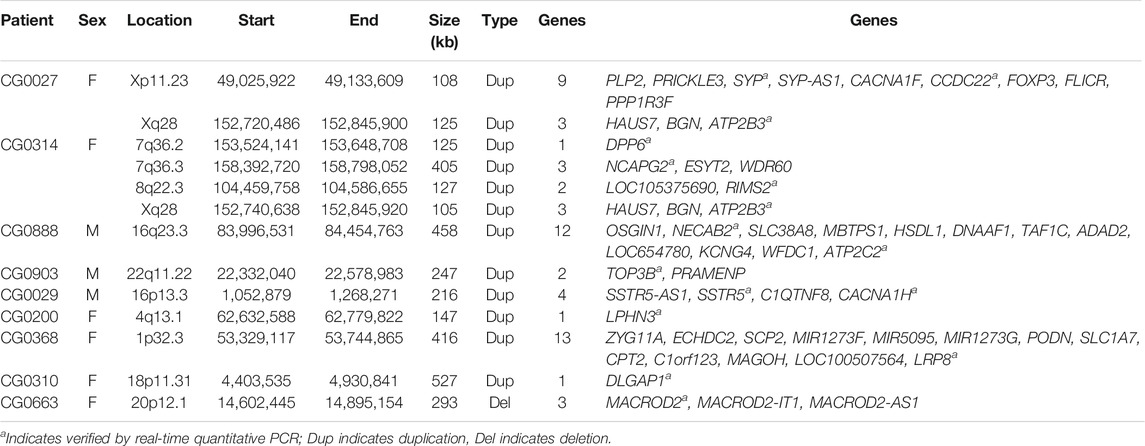
TABLE 2. CNVs with uncertain significance detected in this study.
Discussion
CMA has been used as a first-tier genetic test in patients with intellectual disability, congenital abnormalities, and autism spectrum disorders (Miller et al., 2010), and several studies suggested the clinical utility of CMA for schizophrenia and other psychiatric disorders (Costain et al., 2013; Baker et al., 2014; Lowther et al., 2017a; Lowther et al., 2017b). We conducted CMA in a sample of patients with schizophrenia or schizoaffective recruited consecutively from a clinical setting to test the utility of CMA in searching for the genetic etiology for schizophrenia patients. We selected patients without comorbidity of other diagnoses, such as substance abuse, head injury, medical conditions associated with brain dysfunction, and neurological disorders. We also excluded patients with the comorbid diagnosis with intellectual disability, as one study reported that schizophrenia with a low intelligence quotient (IQ) had a higher pathogenic CNV rate than those without low IQ (Lowther et al., 2017b).
We detected four clinically relevant CNVs in four respective patients. The detection rate was 6.7%, comparable to the prevalence rate of 8.1% in a community-based sample of 595 unrelated patients (Costain et al., 2013), and the 7.1% in a study of 546 community-based patients (Lowther et al., 2017b). Together, these studies support that CMA is an instrumental tool in detecting the genetic etiology in a portion of schizophrenia patients. Identifying clinically relevant CNVs in schizophrenia can provide helpful information for genetic counseling and guide clinical management for patients in clinical settings. (Baker et al., 2014). Furthermore, the identification of clinically relevant CNVs in schizophrenia can increase our understanding of the pathogenesis of schizophrenia.
The first pathogenic CN = V reported in this study was a 443 kb microduplication at 15q13.3 in the male patient CG1176. The breakpoints of the microduplication intersected with the OTUD7A and CHRNA7, respectively, resulting in the disruption of these two genes. CNVs at 15q13.3, including microdeletion and microduplication, are associated with multiple neuropsychiatric disorders, such as epilepsy, intellectual disability, autism spectrum disorders, and schizophrenia (Gillentine and Schaaf, 2015). There are seven genes at the 15q13.3 CNV region, including FAN1, MTMR10, TRPM1, MIR211, KLF13, OTUD7A, and CHRNA7. Several lines of study have suggested that CHRNA7 is a candidate gene for the neuropsychiatric phenotypes in patients with 15q13.3 CNVs (Gillentine and Schaaf, 2015; Gillentine et al., 2017). CHRNA7 encodes the subunit protein to form the alpha-7 nicotinic receptor, a homopentameric ligand-gated ion channel regulating inhibitory and excitatory neurotransmission in the brain. Alpha-7 nicotinic receptor is involved in the molecular mechanism of attention, learning, and memory (Bouzat et al., 2018; Natarajan et al., 2020). Postmortem studies showed reduced immunoreactivity of alpha-7 nicotinic receptors in the brains of schizophrenia (Freedman et al., 2000). Hence, the alpha-7 nicotinic receptor’s reduced function was related to the sensory gating deficits and impaired cognitive function in schizophrenia. Alpha-7 nicotinic receptor has been considered the drug target for cognitive enhancement in schizophrenia patients (Freedman, 2014). Hence, disruption of the CHRNA7 by the 443 kb microduplication may contribute to the schizophrenia pathogenesis of our patient.
The OTUD7A encodes a deubiquitinating enzyme highly expressed in the brain. Uddin and colleagues combined whole-genome sequencing, human brain gene expression analysis, and study of a mouse model with a syntenic heterozygous deletion of 15q13; they found reduced expression of OTUD7A in the dendritic spine of the cortical neurons that contributed to the dendrite outgrowth abnormalities in the heterozygous 15q13 deletion mice. They proposed that the OTUD7A was the major regulatory gene for 15q13.3 microdeletion syndrome phenotypes (Uddin et al., 2018). Similarly, Yin and colleagues studied Otud7a-knockout mice; they also found that Otud7a-null mice had decreased dendritic spine density and recapitulated many clinical features of 15q13.3 syndrome. They suggested that decreased OTUD7A dosage was the major contributor to the neurodevelopmental phenotypes associated with 15q13.3 microdeletion syndrome due to the aberrant dendritic spine density and activity (Yin et al., 2018).
Further, mutations of the OTUD7A were associated with neurodevelopment disorders. Garret and colleagues reported a homozygous missense mutation of OTUD7A in a male patient with severe global developmental delay, language impairment, and epileptic encephalopathy. His parents and a younger brother who manifested learning disability were heterozygous carriers of the missense mutation (Garret et al., 2020). Suzuki and colleagues also reported the biallelic loss of function of OTUD7A in a male patient with hypotonia, intellectual disability, and seizures (Suzuki et al., 2021). These findings suggest the OTUD7A plays a critical role in brain development, and haploinsufficiency of the OTUD7A may contribute to the pathogenesis of our patient.
The second pathogenic CNV identified in this study was a 1,489 kb microdeletion at 22q11.21 in the female patient CG1183. 22q11.2 is a hot region with recurrent CNVs in humans associated with various neurodevelopmental disorders such as intellectual disability, autism spectrum disorders, and schizophrenia. Researchers classified CNVs at 22q11.2 as proximal, central, and distal according to their locations (Burnside, 2015; Woodward et al., 2019). The 1,489 kb microdeletion in this patient belonged to the central 22q11.21 deletion, encompassing 36 genes (Table 1). SNAP29 is one of the genes deleted in our patient. SNAP29 is one of the SNARE (Soluble NSF Attachment REceptor) proteins that regulate a wide range of cellular functions. Studies showed that SNAP29 was involved in autophagy, synaptic transmission, cell division, and infection (Mastrodonato et al., 2018). A polymorphism at the promoter of the SNAP29 was reported to be associated with schizophrenia (Saito et al., 2001; Wonodi et al., 2005). Using a functional genomic approach, Forsyth and colleagues discovered that genes in the 22q11.2 locus formed an extensive protein network and regulated the developmental modules associated with schizophrenia and autism spectrum disorders. They also suggested that SEPT5, PI4KA, and SNAP29 were candidate drivers of 22q11.2 synaptic pathology relevant to schizophrenia and autism spectrum disorder (Forsyth et al., 2020). Both PI4KA and SNAP29 were located at the deleted region in our patient. The 22q11.2 deletion syndrome working groups of the Enhancing Neuroimaging Genetics through Meta-Analysis (ENIGMA) recently reported that patients with 22q11.2 deletion syndrome had reduced cortical surface area and increased cortical thickness. They found that AIFM3 was associated with the cortical surface area deviance, while P2RX6, was associated with cortical thickness deviance (Forsyth et al., 2021). These two genes were also included in the deleted region in our patient. Thus, the microdeletion in this patient contains several genes that may work together and leads to the pathogenesis of her clinical phenotypes.
The third pathogenic CNV detected in this study was a 912 kb microduplication at 2q23.1 in the female patient CG200. Four genes were present in this region, including two duplicated genes, EPC2 and KIF5C, and two breakpoints-disrupted genes, MBD5 and LYPD6B. KIF5C is a member of kinesin superfamily proteins that are molecular motors involved in cargo transport in the central nervous system (Duquesne et al., 2020). Pathogenic mutations KIF5C are rare but cause abnormal cortical development and result in severe developmental delay, absent language, seizures, and abnormal behaviors in affected patients (Michels et al., 2017; Duquesne et al., 2020), indicating the critical role of KIF5C in the normal development of the cortex. The duplication of KIF5C in our patient may lead to increased expression of KIF5C in the brain and contribute to the pathogenesis of schizophrenia, which is distinct from the clinical symptoms caused by loss-of-function KIF5C pathogenic variants.
The other important gene that may contribute to the pathogenesis of schizophrenia in this patient is the MBD5. MBD5 encodes a member of the methyl-CpG-binding domain (MBD) family proteins which bind specifically to methylated DNA and regulate gene expression. Haploinsufficiency of MBD5 causes neurodevelopmental disorders such as intellectual disability, autism spectrum disorders, and seizures. Furthermore, patients with haploinsufficiency of MBD5 may present with psychiatric disorders, including anxiety, bipolar disorder, and schizophrenia (Hodge et al., 2014; Kushima et al., 2017), suggesting the variable expressivity of MBD5 haploinsufficiency. The cause of MBD5 haploinsufficiency can be due to 2q23.1 deletions encompassing the MBD5, intragenic deletion of MBD5, and pathogenic mutations of MBD5 (Mullegama et al., 1993). The breakpoint of the 912 kb microduplication in the patient CG0200 intersected with the MBD5, leading to disruption of the MBD5. Our patient presented schizophrenia symptoms only. She did not present other MBD5-related neurodevelopmental symptoms such as developmental delay, speech impairment, seizure, and abnormal behaviors (Mullegama et al., 2015; Mullegama and Elsea, 2016), suggesting other factors may modify the clinical presentations of the MBD5-haploinsufficiency.
This study detected a 188 kb microduplication at 12q24.11, classified as likely pathogenic CNV in the female patient CG0871. This microduplication encompassed six genes, including ATP2A2, ANAPC7, ARPC3, GPN3, FAM216A, and VPS29. Among these genes, the ATP2A2 and VPS29 were interrupted by the breakpoints of the microduplication, respectively. ATP2A2 encodes an ATPase, which helps pump the calcium ion from cytosol to sarcoplasmic reticulum lumen. Haploinsufficient mutations of the ATP2A2 cause a rare autosomal dominant form of skin disorder, Darier disease (Ruiz-Perez et al., 1999). Patients with Darier disease have an increased risk for bipolar disorder and schizophrenia (Cederlöf et al., 2015; Gordon-Smith et al., 2018), indicating that mutations of ATP2A2 have pleiotropic effects on the brain. Notably, our patient manifested schizophrenia symptoms only. She did not present any skin disorder, suggesting psychiatric manifestations might be independent of skin pathology in patients with ATP2A2 haploinsufficiency. A large-scale genome-wide association study discovered that ATP2A2 was significantly associated with schizophrenia, indicating that the ATP2A2 was a risk gene for schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
Another gene disrupted by the microduplication in this patient was the VPS29 which encodes a subunit protein of the retromer complex. Retromer complex is involved in the recycling of proteins from endosomes to the plasma membrane. Dysfunction of the retromer complex is associated with some neurodegenerative disorders (Reitz, 2018). A recent study reported that a VPS29 variant associated with decreased VPS29 expression in the brain significantly increased the risk for schizophrenia, suggesting reduced VPS29 expression in the postmortem brains of schizophrenia is a biological mechanism underlying schizophrenia (Byrne et al., 2020). Hence, the disruption of the VPS29 in our patient might contribute to the pathogenesis of her schizophrenia. Further study is needed to address this issue.
In this patient, four genes, ANAPC7, ARPC3, GPN3, and FAM216A, were duplicated in the microduplication. In these four genes, ARPC3 might implicate the schizophrenia pathogenesis of this patient. ARPC3 encodes one of the seven subunits of the Arp2/3 protein complex, which is involved in the actin polymerization in cells (Welch et al., 1997). Several studies showed that disruption of ARPC3 was associated with aberrant synaptic plasticity in the brain and behavioral abnormalities in mice (Kim et al., 2013; Spence et al., 2016; Kim et al., 2020). Nevertheless, whether overexpression of the ARPC3 is associated with brain pathology and behavioral problems remains to be elucidated.
Besides four clinically relevant CNVs, we detected thirteen CNVs classified as uncertain significance in nine patients in this study. The patient CG0200 carried a microduplication of 147 kb at 4p13.1, classified as uncertain significance (Table 2), and a pathogenic microdeletion at 2q23.1 (Table 1). The microduplication at 4p13.1 intersected with the LPHN3. Variants of LPHN3 were associated with attention deficit hyperactivity disorder, autism spectrum disorder, and substance use disorder (Martinez et al., 2016; Arcos-Burgos et al., 2019; Kappel et al., 2019). Hence, this CNV might contribute to the patient’s symptoms.
The female patient CG0027 carried two CNVs with uncertain significance, including a 108 kb microduplication at Xp11.23 and a 125 kb microduplication at Xq28 (Table 2). The 108 kb microduplication at Xp11,23 encompassed nine genes (Table 2). Among these genes, PLP2, CCDC22, and SYP mutations were associated with X-linked intellectual disability (Zhang et al., 2007; Tarpey et al., 2009; Kolanczyk et al., 2015). Some schizophrenia patients were found to have rare SYP mutations (Shen et al., 2012). Furthermore, the 125 kb microduplication at Xq28 encompassed the ATP2B3 associated with X-linked spinocerebellar ataxia (Zanni et al., 2012; Feyma et al., 2016). This patient did not have an intellectual disability and other neurological symptoms. Hence, this patient’s psychiatric manifestations might be related to the random inactivation of these genes at the X chromosome and varied clinical symptoms.
The patient CG0314 carried four CNVs classified as uncertain significance. They were a 125 kb microduplication at 7q36.2, a 405 kb microduplication at 7q36.3, a 127 kb microduplication at 8q22.3, and a 105 kb microduplication at Xq28 (Table 2). The 105 kb microduplication at Xq28 also included the ATP2B3, whose mutations were associated with X-linked spinocerebellar ataxia (Zanni et al., 2012; Feyma et al., 2016). The 125 kb microduplication at 7q36.2 disrupted the DPP6, whose mutations were associated with autism (Marshall et al., 2008; Egger et al., 2014), autosomal dominant microcephaly, intellectual disability (Liao et al., 2013), and neurodegenerative dementia (Cacace et al., 2019). The 405 kb microduplication at 7q36.3 covered the NCAGP2, whose mutations were associated with microcephaly and intellectual disability (Perche et al., 2013; Khan et al., 2019). Finally, the 127 kb microduplication at 8q22.3 contained the RIMS2, which was proposed as a risk gene for autism (Fan et al., 2018a). Taken together, these four CNVs might work independently or synergistically to contribute to schizophrenia’s pathogenesis. Several studies reported that multiple CNVs might interact and result in varied clinical phenotypes of developmental and neuropsychiatric disorders, including intellectual disability, congenital abnormalities (Girirajan et al., 2012), autism spectrum disorder (Gau et al., 2012), and schizophrenia (Williams et al., 2013; Rudd et al., 2014; Kushima et al., 2017). Our findings of multiple CNVs in three patients in this study agreed with these studies and supported the multiple-hits hypothesis of schizophrenia. Notably, additional hits are not limited to CNV. They may include rare or common genetic variants with various effect sizes and non-genetic factors (Williams et al., 2013; Feigenson et al., 2014; Rudd et al., 2014).
Six patients (CG0888, CG0903, CG0029, CG0368, CG0310, and CG0663) carried one CNV with uncertain significance, respectively (Table 2). The 458 kb microduplication at 16q23.3 in the patient CG0888 encompassed the NECAB2, which encodes a neuronal calcium-binding protein that interacts with and modulates adenosine A2A receptor (Canela et al., 2007) and metabotropic glutamate receptor type 5 (Canela et al., 2009). The 247 kb microduplication at 22q11.22 in the patient CG0903 interrupted the TOP3B, whose mutations were associated with several neurodevelopmental disorders including schizophrenia, autism, and epilepsy (Stoll et al., 2013; Ahmad et al., 2017; Daghsni et al., 2018). The 216 kb microduplication at 16p13.3 in the patient CG0029 spanned the SSTR5. CNVs encompassing the SSTR5 were reported in patients with schizophrenia before (Rodríguez-Santiago et al., 2010; Saus et al., 2010). The 416 kb microduplication at 1p32.3 in the patient CG0368 intersected with the LRP8, which encodes the low-density lipoprotein receptor-related protein 8. The LRP8 was considered a susceptibility gene for schizophrenia and bipolar disorder (Li et al., 2016; Xiao et al., 2020). The 527 kb microduplication at 18p11.3 in the patient CG0310 interrupted the DLGAP1, which encodes the DLG associated protein 1. Rare missense mutations of the DLGAP1 were detected in patients with schizophrenia before (Li et al., 2013; Xing et al., 2016), while common polymorphisms of the DLGAP1 influenced the executive function in attention deficit hyperactivity disorder (Fan et al., 2018b). Finally, the 293 kb microdeletion at 20p12.1 in the patient CG0663 covered the MACROD2 gene. CNVs involving the MACROD2 gene were detected in patients with attention deficit hyperactivity disorder, autism (Lionel et al., 2011; Bacchelli et al., 2020), and major depression with suicide attempts (Perlis et al., 2012). Although these CNVs were classified as uncertain significance according to the guidelines used in this study, their relationship with schizophrenia needs further evaluation as more studies come along in the future.
In this study, we feedback our CNV fidnings to the patients and their families. We discussed the clinical significance of their CNV findings and answered their questions regarding the origin of their CNV findings and the possible treatment in the future, given the limited treatment based on the CNV findings at this moment. We also attempted to conduct a family study to trace the origins of three pathogenic CNVs and one likely pathogenic CNV identified in this study. However, we could not collect DNA from both parents because some parents passed away, divorced, separated, or were reluctant to donate their DNA. Hence, we were not able to report the origins of these four CNVs in this study.
In conclusion, given a small sample size of sixty patients recruited consecutively from a clinical setting, we identified three pathogenic CNVs, one likely pathogenic, and thirteen CNVs with uncertain significance in this study. Further, we found three patients carrying multiple CNVs. The detection of these CNVs enriches our understanding of the underlying genetic deficits associated with schizophrenia spectrum disorders. Hence, we suggest that CMA is a valuable tool for a first-tier genetic test for schizophrenia in clinical settings. It can help establish the patients’ molecular genetic diagnosis, provides helpful information for genetic counseling, and increases our understanding of the pathogenesis of schizophrenia.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by The Review Board of Chang Gung Memorial Hospital-Linkou, Taoyuan City, Taiwan. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
C-HC designed the study and obtained research funding. C-HC, T-MH, and L-YP diagnosed and recruited the study samples. M-CC conducted the experiments. C-HC and M-CC analyzed and interpreted the data. C-HC wrote the first draft of the article; all authors contributed to and have approved the final article.
Funding
The study was supported by the Grants CMRPG3F1021, CMRPG3F1022, and CMRPG3F1023 from Chang Gung Memorial Hospital, Taoyuan, Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Genomic Medicine Core Laboratory of Chang Gung Memorial Hospital-Linkou, Taoyuan, Taiwan, for performing the genome-wide CNV analysis for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.620496/full#supplementary-material
References
Ahmad, M., Shen, W., Li, W., Xue, Y., Zou, S., Xu, D., et al. (2017). Topoisomerase 3β Is the Major Topoisomerase for mRNAs and Linked to Neurodevelopment and Mental Dysfunction. Nucleic Acids Res. 45 (5), 2704–2713. doi:10.1093/nar/gkw1293
Arcos-Burgos, M., Velez, J. I., Vélez, J. I., Martinez, A. F., Ribasés, M., Ramos-Quiroga, J. A., et al. (2019). ADGRL3 (LPHN3) Variants Predict Substance Use Disorder. Transl. Psychiatry 9 (1), 42. doi:10.1038/s41398-019-0396-7
Avramopoulos, D. (2018). Recent Advances in the Genetics of Schizophrenia. Mol. Neuropsychiatry 4 (1), 35–51. doi:10.1159/000488679
Bacchelli, E., Cameli, C., Viggiano, M., Igliozzi, R., Mancini, A., Tancredi, R., et al. (2020). An Integrated Analysis of Rare CNV and Exome Variation in Autism Spectrum Disorder Using the Infinium PsychArray. Sci. Rep. 10 (1), 3198. doi:10.1038/s41598-020-59922-3
Baker, K., Costain, G., Fung, W. L. A., and Bassett, A. S. (2014). Chromosomal Microarray Analysis-A Routine Clinical Genetic Test for Patients with Schizophrenia. Lancet Psychiatry 1 (5), 329–331. doi:10.1016/s2215-0366(14)70308-6
Bouzat, C., Lasala, M., Nielsen, B. E., Corradi, J., and Esandi, M. d. C. (2018). Molecular Function of α7 Nicotinic Receptors as Drug Targets. J. Physiol. 596 (10), 1847–1861. doi:10.1113/jp275101
Burnside, R. D. (2015). 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet. Genome Res. 146 (2), 89–99. doi:10.1159/000438708
Byrne, E. M., Zhu, Z., Qi, T., Skene, N. G., Bryois, J., Pardinas, A. F., et al. (2020). Conditional GWAS Analysis to Identify Disorder-specific SNPs for Psychiatric Disorders. Mol. Psychiatry 26, 2070. doi:10.1038/s41380-020-0705-9
Cacace, R., Heeman, B., Heeman, B., Van Mossevelde, S., De Roeck, A., Hoogmartens, J., et al. (2019). Loss of DPP6 in Neurodegenerative Dementia: a Genetic Player in the Dysfunction of Neuronal Excitability. Acta Neuropathol. 137 (6), 901–918. doi:10.1007/s00401-019-01976-3
Canela, L., Fernández-Dueñas, V., Albergaria, C., Watanabe, M., Lluís, C., Mallol, J., et al. (2009). The Association of Metabotropic Glutamate Receptor Type 5 with the Neuronal Ca2+-Binding Protein 2 Modulates Receptor Function. J. Neurochem. 111 (2), 555–567. doi:10.1111/j.1471-4159.2009.06348.x
Canela, L., Luján, R., Lluís, C., Burgueño, J., Mallol, J., Canela, E. I., et al. (2007). The Neuronal Ca2+-Binding Protein 2 (NECAB2) Interacts with the Adenosine A2A Receptor and Modulates the Cell Surface Expression and Function of the Receptor. Mol. Cell Neurosci. 36 (1), 1–12. doi:10.1016/j.mcn.2007.05.007
Cederlöf, M., Bergen, S. E., Långström, N., Larsson, H., Boman, M., Craddock, N., et al. (2015). The Association between Darier Disease, Bipolar Disorder, and Schizophrenia Revisited: a Population-Based Family Study. Bipolar Disord. 17 (3), 340–344. doi:10.1111/bdi.12257
Costain, G., Lionel, A. C., Merico, D., Forsythe, P., Russell, K., Lowther, C., et al. (2013). Pathogenic Rare Copy Number Variants in Community-Based Schizophrenia Suggest a Potential Role for Clinical Microarrays. Hum. Mol. Genet. 22 (22), 4485–4501. doi:10.1093/hmg/ddt297
Daghsni, M., Lahbib, S., Fradj, M., Sayeb, M., Kelmemi, W., Kraoua, L., et al. (2018). TOP3B: A Novel Candidate Gene in Juvenile Myoclonic Epilepsy? Cytogenet. Genome Res. 154 (1), 1–5. doi:10.1159/000486945
Duquesne, S., Nassogne, M.-C., Clapuyt, P., Stouffs, K., and Sznajer, Y. (2020). Phenotype Description in KIF5C Gene Hot-Spot Mutations Responsible for Malformations of Cortical Development (MCD). Eur. J. Med. Genet. 63 (9), 103991. doi:10.1016/j.ejmg.2020.103991
Egger, G., Roetzer, K. M., Noor, A., Lionel, A. C., Mahmood, H., Schwarzbraun, T., et al. (2014). Identification of Risk Genes for Autism Spectrum Disorder through Copy Number Variation Analysis in Austrian Families. Neurogenetics 15 (2), 117–127. doi:10.1007/s10048-014-0394-0
Fan, Y., Du, X., Liu, X., Wang, L., Li, F., and Yu, Y. (2018). Rare Copy Number Variations in a Chinese Cohort of Autism Spectrum Disorder. Front. Genet. 9, 665. doi:10.3389/fgene.2018.00665
Fan, Z., Qian, Y., Lu, Q., Wang, Y., Chang, S., and Yang, L. (2018). DLGAP1 and NMDA Receptor-Associated Postsynaptic Density Protein Genes Influence Executive Function in Attention Deficit Hyperactivity Disorder. Brain Behav. 8 (2), e00914. doi:10.1002/brb3.914
Feigenson, K. A., Kusnecov, A. W., and Silverstein, S. M. (2014). Inflammation and the Two-Hit Hypothesis of Schizophrenia. Neurosci. Biobehav. Rev. 38, 72–93. doi:10.1016/j.neubiorev.2013.11.006
Feyma, T., Ramsey, K., Huentelman, M. J., Craig, D. W., Padilla-Lopez, S., Narayanan, V., et al. (2016). Dystonia in ATP2B3 -associated X-Linked Spinocerebellar Ataxia. Mov. Disord. 31 (11), 1752–1753. doi:10.1002/mds.26800
Flaherty, E. K., and Brennand, K. J. (2017). Using hiPSCs to Model Neuropsychiatric Copy Number Variations (CNVs) Has Potential to Reveal Underlying Disease Mechanisms. Brain Res. 1655, 283–293. doi:10.1016/j.brainres.2015.11.009
Foley, C., Corvin, A., and Nakagome, S. (2017). Genetics of Schizophrenia: Ready to Translate?. Curr. Psychiatry Rep. 19 (9), 61. doi:10.1007/s11920-017-0807-5
Forsyth, J. K., Mennigen, E., Lin, A., Sun, D., Vajdi, A., Kushan-Wells, L., et al. (2021). Prioritizing Genetic Contributors to Cortical Alterations in 22q11.2 Deletion Syndrome Using Imaging Transcriptomics. Cereb. Cortex 31 (7), 3285–3298. doi:10.1093/cercor/bhab008
Forsyth, J. K., Nachun, D., Gandal, M. J., Geschwind, D. H., Anderson, A. E., Coppola, G., et al. (2020). Synaptic and Gene Regulatory Mechanisms in Schizophrenia, Autism, and 22q11.2 Copy Number Variant-Mediated Risk for Neuropsychiatric Disorders. Biol. Psychiatry 87 (2), 150–163. doi:10.1016/j.biopsych.2019.06.029
Freedman, R., Adams, C. E., and Leonard, S. (2000). The Alpha7-Nicotinic Acetylcholine Receptor and the Pathology of Hippocampal Interneurons in Schizophrenia. J. Chem. Neuroanat. 20 (3-4), 299–306. doi:10.1016/s0891-0618(00)00109-5
Freedman, R. (2014). α7-Nicotinic Acetylcholine Receptor Agonists for Cognitive Enhancement in Schizophrenia. Annu. Rev. Med. 65, 245–261. doi:10.1146/annurev-med-092112-142937
Garret, P., Ebstein, F., Delplancq, G., Dozieres‐Puyravel, B., Boughalem, A., Auvin, S., et al. (2020). Report of the First Patient with a Homozygous OTUD7A Variant Responsible for Epileptic Encephalopathy and Related Proteasome Dysfunction. Clin. Genet. 97 (4), 567–575. doi:10.1111/cge.13709
Gau, S. S.-F., Liao, H.-M., Hong, C.-C., Chien, W.-H., and Chen, C.-H. (2012). Identification of Two Inherited Copy Number Variants in a Male with Autism Supports Two-Hit and Compound Heterozygosity Models of Autism. Am. J. Med. Genet. 159B (6), 710–717. doi:10.1002/ajmg.b.32074
Gillentine, M. A., Berry, L. N., Goin-Kochel, R. P., Ali, M. A., Ge, J., Guffey, D., et al. (2017). The Cognitive and Behavioral Phenotypes of Individuals with CHRNA7 Duplications. J. Autism Dev. Disord. 47 (3), 549–562. doi:10.1007/s10803-016-2961-8
Gillentine, M. A., and Schaaf, C. P. (2015). The Human Clinical Phenotypes of Altered CHRNA7 Copy Number. Biochem. Pharmacol. 97 (4), 352–362. doi:10.1016/j.bcp.2015.06.012
Girirajan, S., Rosenfeld, J. A., Coe, B. P., Parikh, S., Friedman, N., Goldstein, A., et al. (2012). Phenotypic Heterogeneity of Genomic Disorders and Rare Copy-Number Variants. N. Engl. J. Med. 367 (14), 1321–1331. doi:10.1056/nejmoa1200395
Gordon-Smith, K., Green, E., Grozeva, D., Tavadia, S., Craddock, N., and Jones, L. (2018). Genotype-phenotype Correlations in Darier Disease: A Focus on the Neuropsychiatric Phenotype. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177 (8), 717–726. doi:10.1002/ajmg.b.32679
Harel, T., and Lupski, J. R. (2018). Genomic Disorders 20 Years On-Mechanisms for Clinical Manifestations. Clin. Genet. 93 (3), 439–449. doi:10.1111/cge.13146
Henriksen, M. G., Nordgaard, J., and Jansson, L. B. (2017). Genetics of Schizophrenia: Overview of Methods, Findings and Limitations. Front. Hum. Neurosci. 11, 322. doi:10.3389/fnhum.2017.00322
Hodge, J. C., Mitchell, E., Pillalamarri, V., Toler, T. L., Bartel, F., Kearney, H. M., et al. (2014). Disruption of MBD5 Contributes to a Spectrum of Psychopathology and Neurodevelopmental Abnormalities. Mol. Psychiatry 19 (3), 368–379. doi:10.1038/mp.2013.42
Hu, L., Yao, X., Huang, H., Guo, Z., Cheng, X., Xu, Y., et al. (2018). Clinical Significance of Germline Copy Number Variation in Susceptibility of Human Diseases. J. Genet. Genomics 45 (1), 3–12. doi:10.1016/j.jgg.2018.01.001
Kappel, D. B., Schuch, J. B., Rovaris, D. L., da Silva, B. S., Müller, D., Breda, V., et al. (2019). ADGRL3 Rs6551665 as a Common Vulnerability Factor Underlying Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder. Neuromol. Med. 21 (1), 60–67. doi:10.1007/s12017-019-08525-x
Khan, T. N., Khan, K., Sadeghpour, A., Reynolds, H., Perilla, Y., McDonald, M. T., et al. (2019). Mutations in NCAPG2 Cause a Severe Neurodevelopmental Syndrome that Expands the Phenotypic Spectrum of Condensinopathies. Am. J. Hum. Genet. 104 (1), 94–111. doi:10.1016/j.ajhg.2018.11.017
Kim, I. H., Kim, N., Kim, S., Toda, K., Catavero, C. M., Courtland, J. L., et al. (2020). Dysregulation of the Synaptic Cytoskeleton in the PFC Drives Neural Circuit Pathology, Leading to Social Dysfunction. Cel Rep. 32 (4), 107965. doi:10.1016/j.celrep.2020.107965
Kim, I. H., Racz, B., Wang, H., Burianek, L., Weinberg, R., Yasuda, R., et al. (2013). Disruption of Arp2/3 Results in Asymmetric Structural Plasticity of Dendritic Spines and Progressive Synaptic and Behavioral Abnormalities. J. Neurosci. 33 (14), 6081–6092. doi:10.1523/jneurosci.0035-13.2013
Kolanczyk, M., Krawitz, P., Hecht, J., Hupalowska, A., Miaczynska, M., Marschner, K., et al. (2015). Missense Variant in CCDC22 Causes X-Linked Recessive Intellectual Disability with Features of Ritscher-Schinzel/3C Syndrome. Eur. J. Hum. Genet. 23 (5), 633–638. doi:10.1038/ejhg.2014.109
Kushima, I., Aleksic, B., Nakatochi, M., Shimamura, T., Shiino, T., Yoshimi, A., et al. (2017). High-resolution Copy Number Variation Analysis of Schizophrenia in Japan. Mol. Psychiatry 22 (3), 430–440. doi:10.1038/mp.2016.88
Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., and Chen, C. H. (2013). Genetic Analysis of the DLGAP1 Gene as a Candidate Gene for Schizophrenia. Psychiatry Res. 205 (1-2), 13–17. doi:10.1016/j.psychres.2012.08.014
Li, M., Huang, L., Huang, L., Grigoroiu-Serbanescu, M., Bergen, S. E., Landén, M., et al. (2016). Convergent Lines of Evidence Support LRP8 as a Susceptibility Gene for Psychosis. Mol. Neurobiol. 53 (10), 6608–6619. doi:10.1007/s12035-015-9559-6
Liao, C., Fu, F., Li, R., Yang, W.-q., Liao, H.-y., Yan, J.-r., et al. (2013). Loss-of-function Variation in the DPP6 Gene Is Associated with Autosomal Dominant Microcephaly and Mental Retardation. Eur. J. Med. Genet. 56 (9), 484–489. doi:10.1016/j.ejmg.2013.06.008
Liao, H. M., Chao, Y. L., Huang, A. L., Cheng, M. C., Chen, Y. J., Lee, K. F., et al. (2012). Identification and Characterization of Three Inherited Genomic Copy Number Variations Associated with Familial Schizophrenia. Schizophr Res. 139 (1-3), 229–236. doi:10.1016/j.schres.2012.05.015
Lionel, A. C., Crosbie, J., Barbosa, N., Goodale, T., Thiruvahindrapuram, B., Rickaby, J., et al. (2011). Rare Copy Number Variation Discovery and Cross-Disorder Comparisons Identify Risk Genes for ADHD. Sci. Transl. Med. 3 (95), 95ra75. doi:10.1126/scitranslmed.3002464
Lowther, C., Costain, G., Baribeau, D. A., and Bassett, A. S. (2017). Genomic Disorders in Psychiatry-What Does the Clinician Need to Know? Curr. Psychiatry Rep. 19 (11), 82. doi:10.1007/s11920-017-0831-5
Lowther, C., Merico, D., Costain, G., Waserman, J., Boyd, K., Noor, A., et al. (2017). Impact of IQ on the Diagnostic Yield of Chromosomal Microarray in a Community Sample of Adults with Schizophrenia. Genome Med. 9 (1), 105. doi:10.1186/s13073-017-0488-z
Manning, M., Hudgins, L., Professional, P., and Guidelines, C. (2010). Array-based Technology and Recommendations for Utilization in Medical Genetics Practice for Detection of Chromosomal Abnormalities. Genet. Med. 12 (11), 742–745. doi:10.1097/gim.0b013e3181f8baad
Marshall, C. R., Howrigan, D. P., Merico, D., Thiruvahindrapuram, B., Wu, W., Greer, D. S., et al. (2017). Contribution of Copy Number Variants to Schizophrenia from a Genome-wide Study of 41,321 Subjects. Nat. Genet. 49 (1), 27–35. doi:10.1038/ng.3725
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 82 (2), 477–488. doi:10.1016/j.ajhg.2007.12.009
Martinez, A. F., Abe, Y., Hong, S., Molyneux, K., Yarnell, D., Löhr, H., et al. (2016). An Ultraconserved Brain-Specific Enhancer within ADGRL3 (LPHN3) Underpins Attention-Deficit/Hyperactivity Disorder Susceptibility. Biol. Psychiatry 80 (12), 943–954. doi:10.1016/j.biopsych.2016.06.026
Mastrodonato, V., Morelli, E., and Vaccari, T. (2018). How to Use a Multipurpose SNARE: The Emerging Role of Snap29 in Cellular Health. CST 2 (4), 72–81. doi:10.15698/cst2018.04.130
Michels, S., Foss, K., Park, K., Golden-Grant, K., Saneto, R., Lopez, J., et al. (2017). Mutations of KIF5C Cause a Neurodevelopmental Disorder of Infantile-Onset Epilepsy, Absent Language, and Distinctive Malformations of Cortical Development. Am. J. Med. Genet. 173 (12), 3127–3131. doi:10.1002/ajmg.a.38496
Michelson, D. J., Shevell, M. I., Sherr, E. H., Moeschler, J. B., Gropman, A. L., and Ashwal, S. (2011). Evidence Report: Genetic and Metabolic Testing on Children with Global Developmental Delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 77 (17), 1629–1635. doi:10.1212/wnl.0b013e3182345896
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 86 (5), 749–764. doi:10.1016/j.ajhg.2010.04.006
Mullegama, S. V., Alaimo, J. T., Chen, L., and Elsea, S. H. (2015). Phenotypic and Molecular Convergence of 2q23.1 Deletion Syndrome with Other Neurodevelopmental Syndromes Associated with Autism Spectrum Disorder. Int. J. Mol. Sci. 16 (4), 7627–7643. doi:10.3390/ijms16047627
Mullegama, S. V., Mendoza-Londono, R., and Elsea, S. H. (1993). “MBD5 Haploinsufficiency,” in GeneReviews(R). Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaaet al. (Seattle (WA)).
Mullegama, S. V., and Elsea, S. H. (2016). Clinical and Molecular Aspects of MBD5-Associated Neurodevelopmental Disorder (MAND). Eur. J. Hum. Genet. 24 (9), 1235–1243. doi:10.1038/ejhg.2016.35
Natarajan, K., Mukhtasimova, N., Corradi, J., Lasala, M., Bouzat, C., and Sine, S. M. (2020). Mechanism of Calcium Potentiation of the α7 Nicotinic Acetylcholine Receptor. J. Gen. Physiol. 152 (9), e202012606. doi:10.1085/jgp.202012606
Nomura, J., and Takumi, T. (2012). Animal Models of Psychiatric Disorders that Reflect Human Copy Number Variation. Neural Plast. 2012, 589524. doi:10.1155/2012/589524
Perche, O., Menuet, A., Marcos, M., Liu, L., Pâris, A., Utami, K. H., et al. (2013). Combined Deletion of Two Condensin II System Genes (NCAPG2 and MCPH1) in a Case of Severe Microcephaly and Mental Deficiency. Eur. J. Med. Genet. 56 (11), 635–641. doi:10.1016/j.ejmg.2013.07.007
Perlis, R. H., Ruderfer, D., Hamilton, S. P., and Ernst, C. (2012). Copy Number Variation in Subjects with Major Depressive Disorder Who Attempted Suicide. PLoS One 7 (9), e46315. doi:10.1371/journal.pone.0046315
Reitz, C. (2018). Retromer Dysfunction and Neurodegenerative Disease. CG 19 (4), 279–288. doi:10.2174/1389202919666171024122809
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–423. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Rodríguez-Santiago, B., Brunet, A., Sobrino, B., Serra-Juhé, C., Flores, R., Armengol, L., et al. (2010). Association of Common Copy Number Variants at the Glutathione S-Transferase Genes and Rare Novel Genomic Changes with Schizophrenia. Mol. Psychiatry 15 (10), 1023–1033. doi:10.1038/mp.2009.53
Rosenfeld, J. A., and Patel, A. (2017). Chromosomal Microarrays: Understanding Genetics of Neurodevelopmental Disorders and Congenital Anomalies. J. Pediatr. Genet. 6 (1), 42–50. doi:10.1055/s-0036-1584306
Rudd, D. S., Axelsen, M., Epping, E. A., Andreasen, N. C., and Wassink, T. H. (2014). A Genome-wide CNV Analysis of Schizophrenia Reveals a Potential Role for a Multiple-Hit Model. Am. J. Med. Genet. 165 (8), 619–626. doi:10.1002/ajmg.b.32266
Ruiz-Perez, V. L., Carter, S. A., Healy, E., Todd, C., Rees, J. L., Steijlen, P. M., et al. (1999). ATP2A2 Mutations in Darier's Disease: Variant Cutaneous Phenotypes Are Associated with Missense Mutations, but Neuropsychiatry Features Are Independent of Mutation Class. Hum. Mol. Genet. 8 (9), 1621–1630. doi:10.1093/hmg/8.9.1621
Rutkowski, T. P., Schroeder, J. P., Gafford, G. M., Warren, S. T., Weinshenker, D., Caspary, T., et al. (2017). Unraveling the Genetic Architecture of Copy Number Variants Associated with Schizophrenia and Other Neuropsychiatric Disorders. J. Neurosci. Res. 95 (5), 1144–1160. doi:10.1002/jnr.23970
Saito, T., Guan, F., Papolos, D. F., Rajouria, N., Fann, C. S. J., and Lachman, H. M. (2001). Polymorphism in SNAP29 Gene Promoter Region Associated with Schizophrenia. Mol. Psychiatry 6 (2), 193–201. doi:10.1038/sj.mp.4000825
Saus, E., Brunet, A., Armengol, L., Alonso, P., Crespo, J. M., Fernández-Aranda, F., et al. (2010). Comprehensive Copy Number Variant (CNV) Analysis of Neuronal Pathways Genes in Psychiatric Disorders Identifies Rare Variants within Patients. J. Psychiatr. Res. 44 (14), 971–978. doi:10.1016/j.jpsychires.2010.03.007
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological Insights from 108 Schizophrenia-Associated Genetic Loci. Nature 511 (7510), 421–427. doi:10.1038/nature13595
Shaikh, T. H. (2017). Copy Number Variation Disorders. Curr. Genet. Med. Rep. 5 (4), 183–190. doi:10.1007/s40142-017-0129-2
Shen, Y. C., Tsai, H. M., Ruan, J. W., Liao, Y. C., Chen, S. F., and Chen, C. H. (2012). Genetic and Functional Analyses of the Gene Encoding Synaptophysin in Schizophrenia. Schizophr Res. 137 (1-3), 14–19. doi:10.1016/j.schres.2012.01.028
Spence, E. F., Kanak, D. J., Carlson, B. R., and Soderling, S. H. (2016). The Arp2/3 Complex Is Essential for Distinct Stages of Spine Synapse Maturation, Including Synapse Unsilencing. J. Neurosci. 36 (37), 9696–9709. doi:10.1523/jneurosci.0876-16.2016
Stoll, G., Pietiläinen, O. P. H., Linder, B., Suvisaari, J., Brosi, C., Hennah, W., et al. (2013). Deletion of TOP3β, a Component of FMRP-Containing mRNPs, Contributes to Neurodevelopmental Disorders. Nat. Neurosci. 16 (9), 1228–1237. doi:10.1038/nn.3484
Suzuki, H., Inaba, M., Yamada, M., Uehara, T., Takenouchi, T., Mizuno, S., et al. (2021). Biallelic Loss of OTUD7A Causes Severe Muscular Hypotonia, Intellectual Disability, and Seizures. Am. J. Med. Genet. 185 (4), 1182–1186. doi:10.1002/ajmg.a.62054
Takumi, T., and Tamada, K. (2018). CNV Biology in Neurodevelopmental Disorders. Curr. Opin. Neurobiol. 48, 183–192. doi:10.1016/j.conb.2017.12.004
Tarpey, P. S., Smith, R., Pleasance, E., Whibley, A., Edkins, S., Hardy, C., et al. (2009). A Systematic, Large-Scale Resequencing Screen of X-Chromosome Coding Exons in Mental Retardation. Nat. Genet. 41 (5), 535–543. doi:10.1038/ng.367
Uddin, M., Unda, B. K., Kwan, V., Holzapfel, N. T., White, S. H., Chalil, L., et al. (2018). OTUD7A Regulates Neurodevelopmental Phenotypes in the 15q13.3 Microdeletion Syndrome. Am. J. Hum. Genet. 102 (2), 278–295. doi:10.1016/j.ajhg.2018.01.006
Welch, M. D., DePace, A. H., Verma, S., Iwamatsu, A., and Mitchison, T. J. (1997). The Human Arp2/3 Complex Is Composed of Evolutionarily Conserved Subunits and Is Localized to Cellular Regions of Dynamic Actin Filament Assembly. J. Cel Biol. 138 (2), 375–384. doi:10.1083/jcb.138.2.375
Williams, H. J., Monks, S., Murphy, K. C., Kirov, G., O'Donovan, M. C., and Owen, M. J. (2013). Schizophrenia Two-Hit Hypothesis in Velo-Cardio Facial Syndrome. Am. J. Med. Genet. 162 (2), 177–182. doi:10.1002/ajmg.b.32129
Wonodi, I., Hong, L. E., Avila, M. T., Buchanan, R. W., Carpenter, W. T., Stine, O. C., et al. (2005). Association between Polymorphism of the SNAP29 Gene Promoter Region and Schizophrenia. Schizophr Res. 78 (2-3), 339–341. doi:10.1016/j.schres.2005.03.023
Woodward, K. J., Stampalia, J., Vanyai, H., Rijhumal, H., Potts, K., Taylor, F., et al. (2019). Atypical Nested 22q11.2 Duplications between LCR22B and LCR22D Are Associated with Neurodevelopmental Phenotypes Including Autism Spectrum Disorder with Incomplete Penetrance. Mol. Genet. Genomic Med. 7 (2), e00507. doi:10.1002/mgg3.507
Xiao, X., Yu, H., Li, J., Wang, L., Li, L., Chang, H., et al. (2020). Further Evidence for the Association between LRP8 and Schizophrenia. Schizophrenia Res. 215, 499–505. doi:10.1016/j.schres.2017.05.002
Xing, J., Kimura, H., Wang, C., Ishizuka, K., Kushima, I., Arioka, Y., et al. (2016). Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci. Rep. 6, 27491. doi:10.1038/srep27491
Yin, J., Chen, W., Chao, E. S., Soriano, S., Wang, L., Wang, W., et al. (2018). Otud7a Knockout Mice Recapitulate Many Neurological Features of 15q13.3 Microdeletion Syndrome. Am. J. Hum. Genet. 102 (2), 296–308. doi:10.1016/j.ajhg.2018.01.005
Zanni, G., Cali, T., Kalscheuer, V. M., Ottolini, D., Barresi, S., Lebrun, N., et al. (2012). Mutation of Plasma Membrane Ca2+ ATPase Isoform 3 in a Family with X-Linked Congenital Cerebellar Ataxia Impairs Ca2+ Homeostasis. Proc. Natl. Acad. Sci. 109 (36), 14514–14519. doi:10.1073/pnas.1207488109
Keywords: copy number variation, schizophrenia, chromosome microarray analysis, genetic test, rare mutation
Citation: Chen C-H, Cheng M-C, Hu T-M and Ping L-Y (2021) Chromosomal Microarray Analysis as First-Tier Genetic Test for Schizophrenia. Front. Genet. 12:620496. doi: 10.3389/fgene.2021.620496
Received: 23 October 2020; Accepted: 20 September 2021;
Published: 01 October 2021.
Edited by:
Francis J. McMahon, National Institutes of Health (NIH), United StatesReviewed by:
Mandy Johnstone, University of Edinburgh, United KingdomKuanjun He, Inner Mongolia University for Nationalities, China
Copyright © 2021 Chen, Cheng, Hu and Ping. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chia-Hsiang Chen, Y2NoZW4zODAxQGdtYWlsLmNvbQ==