 Márta Czakó1,2
Márta Czakó1,2 Ágnes Till1
Ágnes Till1 Judith Zima1Anna Zsigmond1
Judith Zima1Anna Zsigmond1 András Szabó1,2Anita Maász1,2
András Szabó1,2Anita Maász1,2 Béla Melegh1,2Kinga Hadzsiev1,2*
Béla Melegh1,2Kinga Hadzsiev1,2*- 1Department of Medical Genetics, Medical School, University of Pécs, Pécs, Hungary
- 2Szentágothai Research Centre, Pécs, Hungary
Among the diseases with X-linked inheritance and intellectual disability, duplication of the Xp11.23p11.22 region is indeed a rare phenomenon, with less than 90 cases known in the literature. Most of them have been recognized with the routine application of array techniques, as these copy number variations (CNVs) are highly variable in size, occurring in recurrent and non-recurrent forms. Its pathogenic role is not debated anymore, but the information available about the pathomechanism, especially in affected females, is still very limited. It has been observed that the phenotype in females varies from normal to severe, which does not correlate with the size of the duplication or the genes involved, and which makes it very difficult to give an individual prognosis. Among the patients studied by the authors because of intellectual disability, epilepsy, and minor anomalies, overlapping duplications affecting the Xp11.23p11.22 region were detected in three females. Based on our detailed phenotype analysis, we concluded that Xp11.23p11.22 duplication is a neurodevelopmental disorder.
Introduction
The technical possibilities of copy number variation (CNV) detection were significantly improved in the last 15 years, as a result the abnormality causing the pathological phenotype and the mechanism of their development have become known in many diseases (Zhang et al., 1997, 2009; Lupski, 1998; Marshall et al., 2008; Carvalho and Lupski, 2016). Chromosome microarray has become a first-line investigation tool, particularly in patients with intellectual disability (ID), multiple malformations, epilepsy and autism (Miller et al., 2010). In syndromic forms of ID, where morphological abnormalities, behavioral disorders, seizures, and abnormal growth are associated with the disease, the group showing X-linked inheritance is remarkable. This is not surprising considering that based on new data published in the last decade; we know that almost twice as many genes are associated with X-linked mental retardation as thought before (Neri et al., 2018). Among ID patients, the proportion of males is slightly higher (1, 3:1 male to female), 5–10% of the cases shows X-linked inheritance (Froyen et al., 2008). Their studies revealed the role of more than 100 genes located on chromosome X, and a number of CNVs have been described (Froyen et al., 2007; Neri et al., 2018). While among pathogenic CNVs detected on autosomes deletions occur at a higher rate, there are much more duplications occurring on chromosome X (Ropers, 2006; Whibley et al., 2010; Lubs et al., 2012; Vulto-van Silfhout et al., 2013).
The duplication affecting the Xp11.23p11.22 region is unique even among these specific CNVs of X chromosome. It is very rare occurring in both genders (Kokalj Vokac et al., 2002; Bonnet et al., 2006; Froyen et al., 2008, 2012; Monnot et al., 2008; Giorda et al., 2009; Zou and Milunsky, 2009; Holden et al., 2010; Honda et al., 2010; Broli et al., 2011; Chung et al., 2011; Edens et al., 2011; El-Hattab et al., 2011; Flynn et al., 2011; Nizon et al., 2014; Evers et al., 2015; Grams et al., 2015; Moey et al., 2016; Orivoli et al., 2016; Arican et al., 2018; Wang et al., 2020). Inherited and de novo forms are known, all de novo Xp11.23 duplications in which parent of origin has been determined have been paternally inherited (Deng et al., 1990). Generally, duplications of the X chromosome in females are often asymptomatic because X-inactivation process silences the chromosome carrying the duplication (Van den Veyver, 2001). However, in case of Xp11.23p11.22 duplications, the opposite is true: in females with skewed X-inactivation the X chromosome carrying the wild-type allele is silenced, while the abnormal one is active in the majority of the cells. Therefore, females with random X-inactivation develop a milder phenotype.
Based on the cases reported so far, the phenotypic features which develop in males and females affected by duplication of Xp11.23p11.22 are very similar (moderate to severe ID, significant delay of speech development, very specific pattern observed on electroencephalography (Broli et al., 2011) with or without seizures manifestation, and dysmorphic facial features), which is very thought provoking in terms of the pathomechanism. While in boys, the pure increase in gene dose caused by the extra copy may explain the symptoms, the gene dosage assessment in girls is much more complicated due to skewed or random X-inactivation (Bonnet et al., 2006; El-Hattab et al., 2011; Nizon et al., 2014; Orivoli et al., 2016; Wang et al., 2020).
The relationship between the genes affected by the Xp11.23p11.22 duplication and the phenotype is also very complex. Non-recurrent duplications of 0, 3–55 Mb in size have been reported in addition to the recurrent form of about 4.5 Mb, therefore the genes involved in the individual patient’s duplications may be quite different. Patients with this copy number alteration, which have become known so far, show surprisingly similar symptoms despite the variability in CNV size: the resulting symptoms do not correlate with size and gene content of the affected genomic regions (Giorda et al., 2009; Zou and Milunsky, 2009; Flynn et al., 2011; Arican et al., 2018).
In particular, genotype-phenotype analysis of cases with non-recurrent duplication offered opportunity to analyze the relationship between shared symptoms and affected genomic regions. Several studies have found association between certain genes and symptoms (Table 1), however, the individual studies did not examine exactly the same phenotypic traits, which makes the correlation difficult. In a study of six families, Froyen et al. (2008) isolated a minimal overlapping region. Functional analysis of the genes involved identified a causal relationship between elevated gene dosage and intellectual disability only in case of the HUWE1 gene (Froyen et al., 2012). Grams et al. (2015) based on their own analyses and the previously published cases with duplications of Xp11.2 highlighted the importance of two smaller subregions. One of them (Region 1) contains the SHROOM4 and DGKK genes, the other (Region 2) is the same as that was examined by Froyen et al., in which in addition to HUWE1, KDM5C, and IQSEC2 are included as candidate genes (Table 1 and Figure 1). Taken together, based only on the genes with extra copy, the expected phenotype cannot be predicted in individual patients.
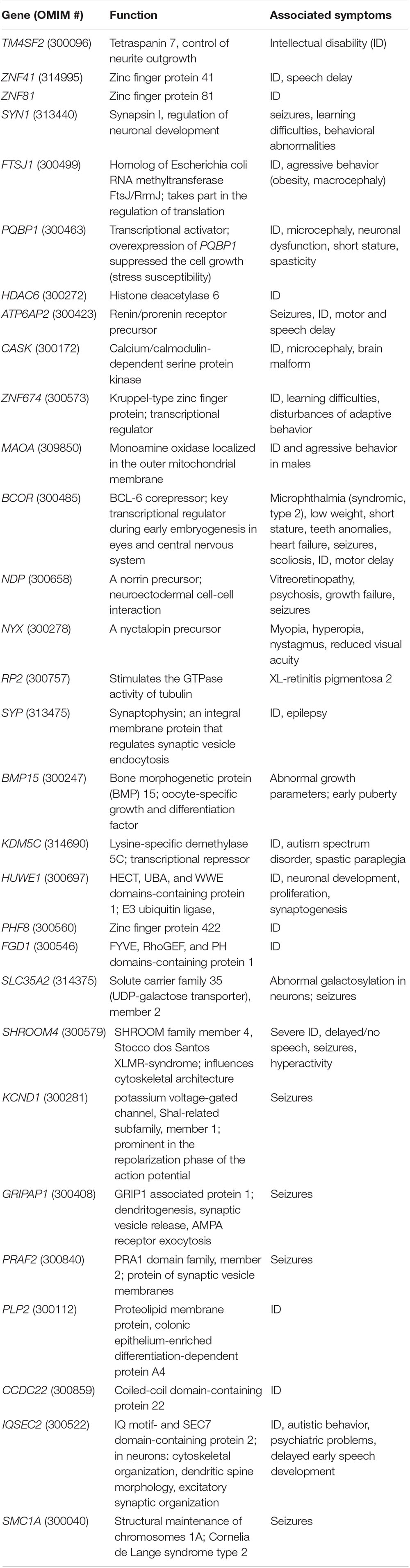
Table 1. Genes involved in Xp11.22p11.23 duplications.
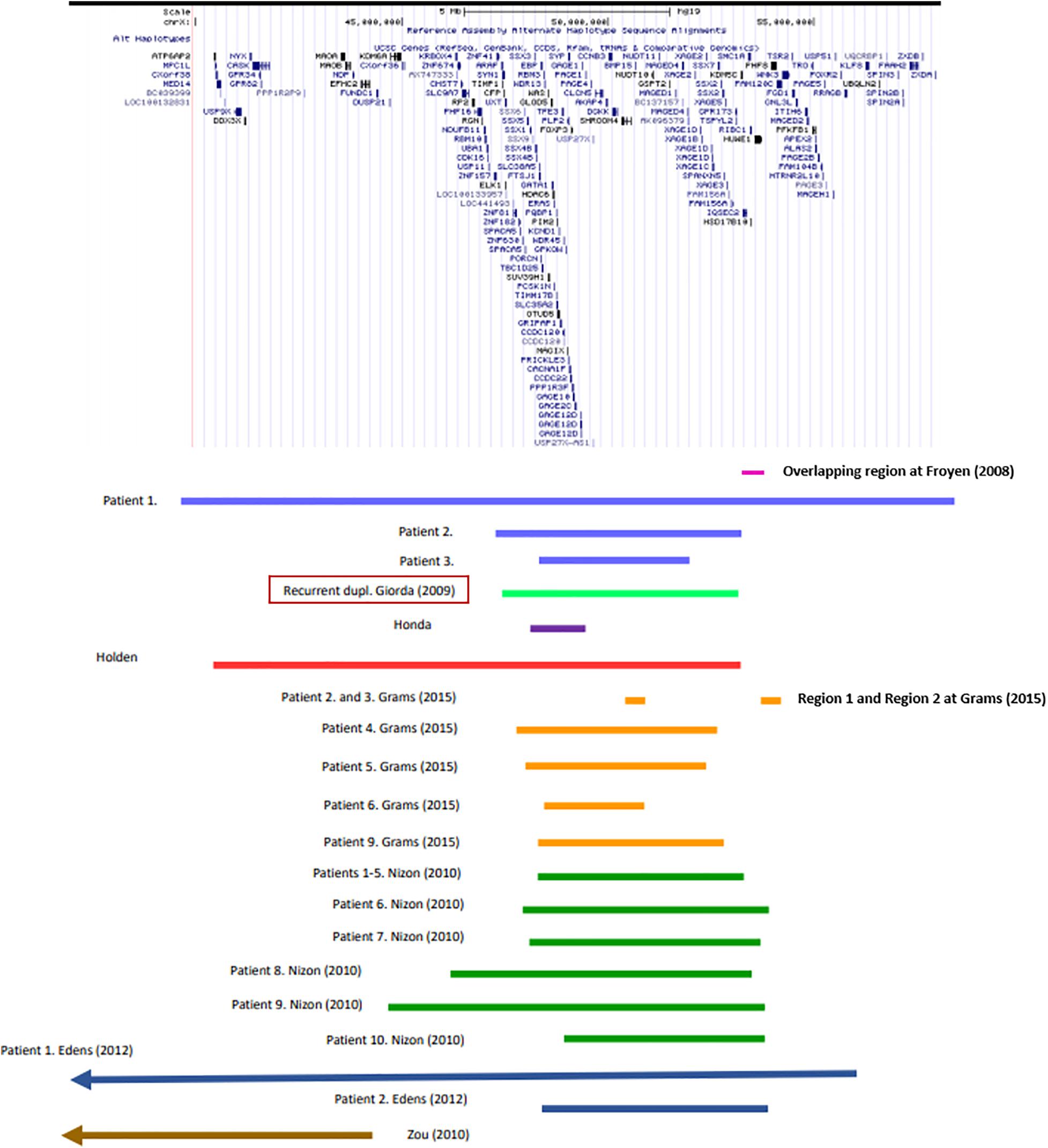
Figure 1. Recurrent and non-recurrent Xp11.22p11.23 duplications of the patients listed in Supplementary Table 1 (UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly).
Within the framework of diagnostic array CGH testing of a cohort of 448 patients presenting ID, epilepsy, and minor anomalies, we detected overlapping duplications affecting the Xp11.23p11.22 region in three female patients. In two girls, we identified the already known recurrent duplication, while in the third patient a large, non-recurrent copy number gain was detected.
Materials and Methods
Subjects
In the Department of Medical Genetics, we collected blood samples from probands with ID, seizures and/or congenital malformations and dysmorphic features, and from their family members. Written informed consent for genetic testing was obtained in genetic counseling from all individuals examined or their guardian, as well as from their healthy relatives. Genomic DNA was isolated from peripheral blood according to standard procedures. This study was performed in accordance with the Hungarian genetic law (XXI/2008).
Patient 1
She is the first child of non-consanguineous parents. The mother has epilepsy and mood disorder; as well the father has mood disorder. Three of the first cousins of the mother are treated with hyperactivity; the maternal grandmother had hearing impairment. One of the father’s first cousins was born from a consanguineous marriage, and had a muscular disorder, without a correct diagnosis. Unfortunately, only the mother was available for genetic testing, the distant relatives were not.
She was born following an uneventful pregnancy in the 39th week of gestation, per vias naturals (birth weight 3,370 g, Apgar scores 9/10). Her early psychomotor development was delayed (determined by Bayley Scales of Infant and Toddler Development); she receives neurohabilitation since her 8 month of age. As a result, she sat at 15 month of age and stood up at the age of 18 months. Delayed language development was detected, at 19 month it is limited to one word. Brain magnetic resonance imaging (MRI) at 16 month of age showed supratentorial abnormalities in the white matter, hypoplasia of corpus callosum, and plexus chorioideus cysts. At 2 years of age, compulsive behavior occurred, with bruxism, hand biting and repetitive hand movements. She does not keep eye contact, and unreasonable laughter can be observed, in addition, her speech is inarticulate. There is a stagnation and slight regression in motor development. Her first epileptic seizure developed at the age of 28 months, appropriate seizure control was achieved by valproate monotherapy (Table 2).
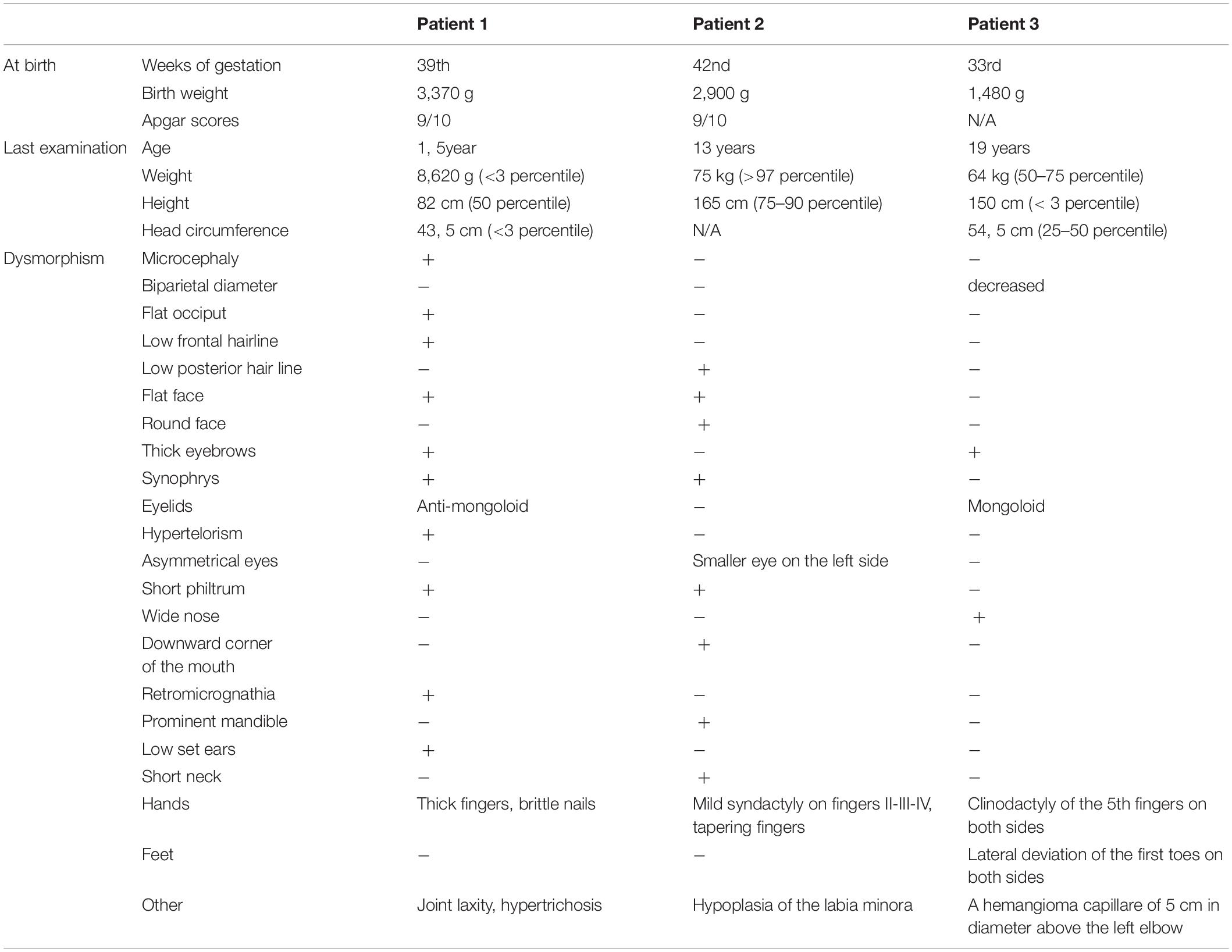
Table 2. Comparison of clinical features in our patients.
She was temporarily cared by an ophthalmologist because of divergent strabismus. She often had upper respiratory infection and inflammation of the eyes. The sequencing of MECP2, FOXG1, CDKL5 genes gave normal results.
Patient 2
She was born from the first, uncomplicated pregnancy of her mother with cesarean section from meconium-stained amniotic fluid. The mother’s sister has short stature, small feet, she is slow moving, with an IQ at the lower limit of normal and similar facial characteristics as our Patient 2 (The sister did not consent to genetic testing).
In the background of feeding difficulties and delayed development of this patient generalized muscle hypotonia was detected at the age of 8 months. As a result of neurohabilitation therapy she walked alone at 28 months of age. Her speech development was severely delayed; she used short sentences from the age of 5–6 years with articulation errors (Budapest-Binet Intelligence Scale). Now she attends a special education school. She has many friends, helps at home and loves to play with a ball. According to the parents, she would be aggressive if not handled well. Brain MRI at age of 1 year showed cerebral atrophy, subdural hygroma, and parietally on the right side a small demyelinated focus was displayed. Three years later the control MRI detected discrete supratentorial, subcortical white matter abnormalities. Weight gain started around the age of 3 years due to compulsive eating, she has regular endocrinological surveillance due to her obesity. Laboratory investigations excluded Prader-Willi syndrome (Table 2).
Patient 3
She was born at the 33rd week of gestation with a weight of 1,480 g after premature rupture of membranes. She was adopted; no family history can be obtained except that her mother has intellectual disability as well. In the perinatal period, she was treated for hypoglycemia, omphalitis, hyperbilirubinemia, and urinary tract infection. Gastroesophageal reflux was confirmed in the background of apnea. Her early psychomotor development was delayed. She was never toilet trained. Her first epileptic seizure developed at the age of 10 years, the EEG (electroencephalography) examination showed fronto-temporal epileptic discharges on the right side. She has been seizure-free with lamotrigine monotherapy for 2 years. The brain MRI detected no abnormalities. Aggression, tantrums have been observed since childhood, risperidone was applied. She attends a special school because of the moderate intellectual disability (tested by Budapest-Binet Intelligence Scale). Regression has been noticed at some developmental areas. Menarche occurred at 10 years of age, the menses is irregular (Table 2).
GTG Banding
Karyotyping from cultured peripheral blood lymphocytes was performed by Giemsa–Trypsin (GTG) banding at 550 bands per haploid set using standard procedures (Caspersson et al., 1970).
Array CGH
Array CGH was performed using Agilent Human Genome Unrestricted G3 ISCA v2 Sureprint 8 × 60K oligo-array (Amadid 021924) (Agilent, Santa Clara, CA) (Kallioniemi et al., 1992). DNA was isolated from peripheral blood leukocytes using the NucleoSpin®Dx Blood DNA Purification Kit (Thermo Fisher Scientific, Waltham, MA) as recommended by the manufacturer. For calculation of the concentration and purity of the isolated DNA NanoDrop spectrophotometer was used. Labeling and hybridization of the samples was made according to the Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis—Enzymatic Labeling Protocol. Washing was performed following the instructions of Agilent Protocol v7.2. The results were obtained by Agilent dual laser scanner G2565CA and processed with Agilent Feature Extraction software (v10.10.1.1.). Agilent Cytogenomics software (v4.0.1.) was used for evaluation of the CNVs. DNA sequence information refers to the public UCSC database (Assembly: Human GRCh37/hg19). The CNVs detected were compared to known aberrations available in public databases like DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources), the Database of Genomic Variants, Clingen Dosage Sensitivity Map, Clinvar, and Ensembl (among others).
X-Inactivation Study
For determination of the X-chromosome inactivation pattern the human androgen receptor gene (AR) assay (HUMARA) was performed on peripheral leukocytes (in case of Patient 1 and her mother, Patient 2 and her parents, and Patient 3 without family members). The assay is based on PCR analysis of the polymorphic CAG repeat containing region of the AR gene, comparing the pattern of DNA samples digested with HpaII methylation sensitive restriction enzyme to undigested samples (Allen et al., 1992).
Results
G-Banding
The karyotype of Patient 1 showed a duplication on the short arm of chromosome X. Chromosome analysis of the mother revealed the presence of the same duplication on one of the X chromosomes. In Patients 2 and 3 the laboratory investigations resulted in a normal female karyotype at 550 bhps resolution.
Array CGH
The breakpoints and sizes of the duplications of chromosome X are the following (according to McGowan-Jordan et al., 2016): Patient 1: arr[GRCh37] Xp11.4p11.21(39969653_58051765) × 3, which means a 18,082 kb duplication; Patient 2: arr[GRCh37] Xp11.23p11.22(46994270_52693966) × 3, with the size of 5,700 kb; and Patient 3.: arr[GRCh37] Xp11.23p11.22(48584351_51956858) × 3, a copy number gain of 3,373 kb, respectively. In addition, common benign variants were detected in all three of the patients. The base pair positions of the genomic imbalances refer to the February 2009 Assembly (GRCh37/hg19). In summary, duplication is of maternal origin for Patient 1 and de novo for Patient 2. In case of Patient 3 none of the parents were available for genetic testing.
X-Inactivation Study
The human androgen receptor assay detected random X-inactivation pattern in Patient 1 and in her mother as well. The results of the test showed similarly random X-inactivation in Patient 2. In contrast, the assay detected non-random X-inactivation pattern in Patient 3 with the same X-chromosome being preferentially inactivated in each of the cells. Unfortunately, in absence of parental samples the origin of the active X chromosome cannot be determined.
Discussion
Studying the literature data on Xp11.22p11.23 duplication, several interesting observations emerge. Although a limited number of such cases have been reported so far, it can be seen that the results of each recent study are inconsistent with one of the previous observations. However, in spite of the size and gene content differing among previously reported patients with Xp11.22p11.23 duplications; it is our opinion that the clinical symptoms are similar. Attempts to link certain symptoms to one or a few genes are remarkably ineffective in this patient group.
Based on a comparison of our three patients and the cases published so far, developmental delay, intellectual disability with varying severity, seizures and different behavioral abnormalities are the most common major symptoms (Supplementary Table 1). This is not surprising, as this genomic region contains a number of genes associated with ID, from which SHROOM4, DGKK, KDM5C, IQSEC2, HSD17B10, and HUWE1 are included in most publications (Table 1). Based on these features, Xp11.22p11.23 duplication could be classified as neurodevelopmental disease. These symptoms are all present in the three patients described here, although the extent of their duplication varies significantly.
To our knowledge, regression has been described rarely in similar patients so far. We observed it in Patients 1 and 3, especially in the field of motor skills. In a male patient with recurrent Xp11.22p11.23 duplication, Flynn et al. (2011) described regression in areas of speech, memory and recognition, furthermore, leading to aggressive behavior. Helm et al. (2017) reported regression of development with co-occurrence of the onset of seizures in a 20 year-old male patient. Until now, we do not know the background of this symptom either.
Behavioral abnormalities are also characteristic of our three patients: compulsive behavior with bruxism, hand biting and repetitive hand movements, no eye contact and unreasonable laughter (Patient 1), aggression (Patients 2 and 3) and tantrums (Patient 3). The authors of studies on Xp11.22p11.23 duplication in the context of autistic behavior and attention deficit and hyperactivity raise the role of KDM5C, SHROOM4 and IQSEC2 genes (Table 1). Our Patients 1 and 2 present such symptoms, in spite of SHROOM4 being present in both of them (and in Patient 3 without similar symptoms), however, KDM5C and IQSEC2 genes are involved only in the duplication of Patient 1. Within the area of Xp11.22p11.23 duplication, in the cases studied so far, two genes have been associated with behavioral abnormalities, SYN1 and ZNF674 (Table 1). Both genes are duplicated in case of Patient 1, only SYN1 is affected in Patient 2. However, all three of our patients struggle with some kind of behavioral disorder.
Epilepsy is an important symptom of the disease; it has been described in about half of the cases reported so far (Giorda et al., 2009; Holden et al., 2010; Edens et al., 2011; Nizon et al., 2014; Grams et al., 2015). Seizures occurred in two of the three patients studied by us (Patient 1 and 3). Table 1 contains 11 genes that may play a role in epilepsy.
It can be seen from the data of Supplementary Table 1 that the somatic parameters of the patients with Xp11.22p11.23 duplication vary widely. Higher than average values for head circumference (OFC) can be seen as typical. Our most noteworthy observation on Patient 1 refers to her OFC (below the 3rd percentile). As far as we know, only two girls has been reported to date with OFC < 3rd percentile (Patient 2 at Edens et al., 2011; Arican et al., 2018). In addition, the mother of Giorda’s Patient 1 should be mentioned with the same OFC value. Examining the role of the underlying genes, we can see that PQBP1 has been associated with microcephaly. One might also speculate that for our Patient 1, it may be explained by the large duplication. However, Patient 1 by Edens et al. (2011) and that of Holden et al. (2010) have large, overlapping duplications also, nevertheless, OFC is at 75th and 90–97th percentile, respectively. The examples listed above demonstrate that studying the size and gene content of Xp11.22p11.23 duplications alone does not provide an explanation for these differences.
Brain MRI showed aspecific abnormalities in a number of reported cases including our cases: Patient 1 and 2 share the supratentorial white matter abnormalities, in addition we observed even corpus callosum hypoplasia and plexus chorioideus cysts in Patient 1., and subdural hygroma in Patient 2 with a small demyelinated focus on the right side.
Duplication of Xp11.22p11.23 is accompanied by dysmorphism in most cases known to date, but the features observed in individual patients are not specific (Table 2). However, it is worth mentioning synophrys and bushy eyebrows, which are present in about half of the patients, including the three girls reported here (Supplementary Table 1 and on the photos by Nizon et al., 2014).
In Patient 1 and 2, eye abnormalities as divergent strabismus (Patient 1), and asymmetrical eyes with smaller eye on the left side (Patient 2) have been also observed. Small eyes are characteristic also for Patient 9 at Grams (2015). In the same study myopia were described at Patient 3 (and in her mother), cataracts, in Patent 5 pseudostrabismus, hyperopia and astigmatism. Early onset myopia and bilateral hypopigmentation of the midperiphery were reported by Zou and Milunsky (2009). Hypermetropic astigmatism occurred in Patient 1 and 2, and recurrent uveitis in Patient 3 described by Giorda et al. (2009). These examples demonstrate that eye disorders are not uncommon in patients with Xp11.22p11.23 duplication, however, only four genes as BCOR, NDP, NYX, and RP2 are known associated with abnormalities of the eyes and these are involved in the duplication of our Patient 1 and the case reported by Zou and Milunsky (2009). The further above mentioned patient’s eye abnormalities cannot be explained by the extra copy of these four genes.
A similar conclusion can be obtained when examining the symptom of early puberty described in about 50% of the patients. As far as we know, BMP15 is the only gene associated with early puberty. BMP15 is affected in all three of our patients, but this symptom is present in two of them only (for similar cases see Supplementary Table 1). Only two patients are listed in Supplementary Table 1 where the duplication does not affect this gene: the 3rd patient reported by Grams et al. (2015) and the case described by Honda et al. (2010). While in the latter case the author did not report data on early puberty, this symptom was described in the patient of Grams et al. (2015). Based on our three patients, age could be an explanation, as Patient 1 not affected by early puberty, is very young. This theory could be applied to 4, 5, and 8th patients of Nizon et al. (2014) (4, 5, and 6 years old girls, respectively) as well. However, Supplementary Table 1 also contains some older patients with BMP15 duplication without early puberty (e.g., 4th patient of Giorda and 9th patient of Grams, both of them with 14 years). Therefore, an extra copy of the BMP15 gene alone may not be a sufficient explanation for the development of this symptom, although reduced penetrance should also be considered.
To examine the causal relationship between Xp11.22p11.23 duplication and phenotype, several factors need to be considered. Which genes and regions may play a role in the development of the symptoms, and which factors may affect the expression of these genes (regulatory elements—primarily cis-acting elements with respect to duplication, the copy number of affected genes, and closely related to this the active functioning copy number influenced by X inactivation).
Among the cases published so far, duplications of various size occur. Therefore, it is difficult to identify a critical region. The most experimental evidence support the role of Region 1 and 2 reported by Grams et al. (2015) (see above). The fact that the known recurrent and non-recurrent Xp11.23 duplications involve these regions is definitely an argument for it (Figure 1). It is an interesting observation that the patient reported by Honda et al. (2010) is the only one shown in Figure 1 whose duplication does not affect these regions. She shows relatively fewer symptoms, mostly intellectual disability that is associated with more genes (see Table 1) being involved in her duplication. Particular attention should be paid to cases where duplication does not overlap with the critical region, but the phenotype does not differ from that of patients with critical region involvement, for example the girl reported by Zou and Milunsky (2009) (see Figure 1). This observation raises the possible role of other factors influencing gene expression, like cis-acting regulatory elements. We still have little information about these for this region now. In any case, studying the ENCODE project data for the genomic region shown in Figure 1, it is striking that H3K27Ac marks are located near the breakpoints of the recurrent duplication which play a role in transcription activation. Based on all this, the genomic region responsible for the phenotype features is most likely to fall into the area of recurrent duplication defined by Giorda et al. (2009).
The occurrence of the Xp11.22p11.23 duplication is well known in both genders, thus, many affected females are described in the literature (e.g., Monnot et al., 2008; Zou and Milunsky, 2009; Holden et al., 2010; Chung et al., 2011; Edens et al., 2011; Evers et al., 2015). According to the authors of Xp11.22p11.23 duplication articles, the cause of the abnormal phenotype is the functional disomy of the genes affected by duplication. This seems to be clear in the case of affected males; however, determination of actually functioning copy number in the female patients is more difficult due to X-inactivation. Not all of the published reports provide data for X-inactivation but in about half of the cases studied, a skewed X-inactivation with preferential inactivation of the normal X chromosome in the majority of the cells was found. In these females, functional disomy is an acceptable causal factor as well. Along this argument, one would expect that females with random X-inactivation show milder clinical features but this is not always the case (Evers et al., 2015; for examples of random X-inactivation see Supplementary Table 1). When interpreting X-inactivation data, it should be taken into account that peripheral blood cells are always examined, and the pattern of X-inactivation may vary from tissue to tissue. An additional challenge for genotype-phenotype analysis is that duplication of Xp11.22p11.23 does not occur exclusively in females with abnormal phenotype. Asymptomatic carrier mothers are reported in some familiar cases, they may have affected offspring of both genders (Giorda et al., 2009; Honda et al., 2010; Grams et al., 2015). In these cases, the normal phenotype does not always involve preferable inactivation of the duplicated X chromosome, i.e., skewed X inactivation does not correct for the effect of duplication. This phenomenon suggests the role of additional regulatory factors also. Among our cases, Patient 1 and her mother who has the same Xp11.22p11.23 duplication have random X-inactivation pattern. Given that the phenotype of Patient 1 is much more severe compared to her mother who has only mild intellectual disability, and the random X inactivation suggests similar active functioning copy number, we must also assume the role of further factors besides the copy number of the duplicated genes. However, the most severe phenotype can be seen in Patient 3 with non-random X-inactivation which reinforces the pathogenic role of elevated copy numbers of the genes involved later on.
Conclusion
The duplication of the p11.22p11.23 region of the short arm of X chromosome, as well as its effect on the phenotype is known from the description of only a limited number of cases to date. The detailed description of the three patients we studied contributes with new observations to the clinical data that have become known so far related to this rare disease. The recognized cases have mostly been examined in diagnostic centers, where the examination possibilities are limited, which in addition to rare occurrence of this abnormality, may also be the reason why very little is currently known about the relationship between genotype and the resulting phenotype. As the comparison of our patients with others reported to date clearly demonstrate, that in addition to the breakpoints of the duplication and the role of the genes involved, a number of other factors influencing gene expression may affect the symptoms that appear.
Deciphering of the secret can be hoped for from systematic analysis of the genomic data, the gene expression, X-inactivation in multiple tissues, as well as other factors involved in the regulation of gene expression. This rare disease with its peculiarities offers an opportunity to gain insight into the functioning of this section of the X chromosome, primarily into the role of regulatory factors and mechanisms.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
JZ and ÁT were responsible for the patient’s clinical genetic examination. AZ contributed to the clinical description. AM, MC, and AS were responsible for methodology and software analysis. KH and ÁT made the conceptualization of the study. ÁT and MC prepared the writing—original draft. KH and BM reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Research on the pathogenesis of rare diseases, developments establishing new diagnostic and therapeutic procedures–GINOP-2.3.2-15-2016-00039; Grant Manager: Ministry of Human Resources, Hungary.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor KK declared a past collaboration with several of the authors MC, T, AS, AM, BM, and KH.
Acknowledgments
We would like to thank the patients and their family members who participated in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.635458/full#supplementary-material
References
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M., and Belmont, J. W. (1992). Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51, 1229–1239.
Arican, P., Cavusoglu, D., Gencpinar, P., Ozyilmaz, B., Ozdemir, T. R., and Dundar, N. O. (2018). A de novo Xp11.23 duplication in a girl with a severe phenotype: expanding the clinical spectrum. J. Pediatr. Genet. 7, 74–77. doi: 10.1055/s-0037-1612598
Bonnet, C., Grégoire, M. J., Brochet, K., Raffo, E., Leheup, B., and Jonveaux, P. (2006). Pure de-novo 5 Mb duplication at Xp11.22-p11.23 in a male: phenotypic and molecular characterization. J. Hum. Genet. 51:815. doi: 10.1007/s10038-006-0023-3
Broli, M., Bisulli, F., Mastrangelo, M., Fontana, E., Fiocchi, I., Zucca, C., et al. (2011). Definition of the neurological phenotype associated with dup (X)(p11.22-p11.23). Epileptic Disord. 13, 240–251. doi: 10.1684/epd.2011.0462
Carvalho, C. M. B., and Lupski, J. R. (2016). Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 17, 224–238. doi: 10.1038/nrg.2015.25
Caspersson, T., Zech, L., Johansson, C., and Modest, E. J. (1970). Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma 30, 215–227.
Chung, B. H., Drmic, I., Marshall, C. R., Grafodatskaya, D., Carter, M., Fernandez, B. A., et al. (2011). Phenotypic spectrum associated with duplication of Xp11.22-p11.23 includes autism spectrum disorder. Eur. J. Med. Genet. 54, e516–e520. doi: 10.1016/j.ejmg.2011.05.008
Deng, H. X., Xia, J. H., Ishikawa, M., and Niikawa, N. (1990). Parental origin and mechanism of formation of X chromosome structural abnormalities: four cases determined with RFLPs. Jinrui Idengaku Zasshi 35, 245–251. doi: 10.1007/BF01876853
Edens, A. C., Lyons, M. J., Duron, R. M., Dupont, B. R., and Holden, K. R. (2011). Autism in two females with duplications involving Xp11.22-p11.23. Dev. Med. Child. Neurol. 53, 463–466. doi: 10.1111/j.1469-8749.2010.03909.x
El-Hattab, A. W., Bournat, J., Eng, P. A., Wu, J. B., Walker, B. A., Stankiewicz, P., et al. (2011). Microduplication of Xp11.23p11.3 with effects on cognition, behavior, and craniofacial development. Clin. Genet. 79, 531–538. doi: 10.1111/j.1399-0004.2010.01496.x
Evers, C., Mitter, D., Strobl-Wildemann, G., Haug, U., Hackmann, K., Maas, B., et al. (2015). Duplication Xp11.22-p14 in females: does X-inactivation help in assessing their significance? Am. J. Med. Genet. A 167A, 553–562. doi: 10.1002/ajmg.a.36897
Flynn, M., Zou, Y. S., and Milunsky, A. (2011). Whole gene duplication of the PQBP1 gene in syndrome resembling renpenning. Am. J. Med. Genet. A 155A, 141–144. doi: 10.1002/ajmg.a.33756
Froyen, G., Belet, S., Martinez, F., Santos-Rebouças, C. B., Declercq, M., Verbeeck, J., et al. (2012). Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am. J. Hum. Genet. 91, 252–264. doi: 10.1016/j.ajhg.2012.06.010
Froyen, G., Corbett, M., Vandewalle, J., Jarvela, I., Lawrence, O., Meldrum, C., et al. (2008). Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 82, 432–443. doi: 10.1016/j.ajhg.2007.11.002
Froyen, G., Van Esch, H., Bauters, M., et al. (2007). Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum. Mutat. 28, 1034–1042. doi: 10.1002/humu.20564
Giorda, R., Bonaglia, M. C., Beri, S., Fichera, M., Novara, F., Magini, P., et al. (2009). Complex segmental duplications mediate a recurrent dup(X)(p11.22-p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females. Am. J. Hum. Genet. 85, 394–400. doi: 10.1016/j.ajhg.2009.08.001
Grams, S. E., Argiropoulos, B., Lines, M., Chakraborty, P., Mcgowan-Jordan, J., Geraghty, M. T., et al. (2016). Genotype-phenotype characterization in 13 individuals with chromosome Xp11.22 duplications. Am. J. Med. Genet. A 170A, 967–977. doi: 10.1002/ajmg.a.37519
Helm, B. M., Powis, Z., Prada, C. E., Casasbuenas-Alarcon, O. L., Balmakund, T., Schaefer, G. B., et al. (2017). The role of IQSEC2 in syndromic intellectual disability: narrowing the diagnostic odyssey. Am. J. Med. Genet. A 173, 2814–2820. doi: 10.1002/ajmg.a.38404
Holden, S. T., Clarkson, A., Thomas, N. S., Abbott, K., James, M. R., and Willatt, L. (2010). A de novo duplication of Xp11.22-p11.4 in a girl with intellectual disability, structural brain anomalies, and preferential inactivation of the normal X chromosome. Am. J. Med. Genet. A 152A, 1735–1740. doi: 10.1002/ajmg.a.33457
Honda, S., Hayashi, S., Imoto, I., Toyama, J., Okazawa, H., Nakagawa, E., et al. (2010). Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis. J. Hum. Genet. 55, 590–599. doi: 10.1038/jhg.2010.74
Kallioniemi, A., Kallioniemi, O. P., Sudar, D., Rutovitz, D., Gray, J. W., Waldman, F., et al. (1992). Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258, 818–821. doi: 10.1126/science.1359641
Kokalj Vokac, N., Seme Ciglenecki, P., Erjavec, A., Zagradisnik, B., and Zagorac, A. (2002). Partial Xp duplication in a girl with dysmorphic features: the change in replication pattern of late-replicating dupX chromosome. Clin. Genet. 61, 54–61. doi: 10.1034/j.1399-0004.2002.610111.x
Lubs, H. A., Stevenson, R. E., and Schwartz, C. E. (2012). Fragile X and X-linked intellectual disability: four decades of discovery. Am. J. Hum. Genet. 90, 579–590. doi: 10.1016/j.ajhg.2012.02.018
Lupski, J. R. (1998). Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends. Genet. 14, 417–422. doi: 10.1016/s0168-9525(98)01555-1558
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
McGowan-Jordan, J., Simons, A., and Schmid, M. (2016). “ISCN 2016,” in. An International System for Human Cytogenomic Nomenclature, 1st Edn, eds J. McGowan-Jordan, A. Simons, and M. Schmid (Basel: Karger).
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86, 749–764. doi: 10.1016/j.ajhg.2010.04.006
Moey, C., Hinze, S. J., Brueton, L., Morton, J., McMullan, D. J., Kamien, B., et al. (2016). Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet. 24, 373–380. doi: 10.1038/ejhg.2015.123
Monnot, S., Giuliano, F., Massol, C., Fossoud, C., Cossée, M., Lambert, J. C., et al. (2008). Partial Xp11.23-p11.4 duplication with random X-inactivation: clinical report and molecular cytogenetic characterization. Am. J. Med. Genet. A. 146A, 1325–1329. doi: 10.1002/ajmg.a.32238
Neri, G., Schwartz, C. E., Lubs, H. A., and Stevenson, R. E. (2018). X-linked intellectual disability update 2017. Am. J. Med. Genet. A. 176, 1375–1388. doi: 10.1002/ajmg.a.38710
Nizon, M., Andrieux, J., Rooryck, C., de Blois, M. C., Bourel-Ponchel, E., Bourgois, B., et al. (2014). Phenotype-genotype correlations in 17 new patients with an Xp11.23p11.22 microduplication and review of the literature. Am. J. Med. Genet. A 167A, 111–122. doi: 10.1002/ajmg.a.36807
Orivoli, S., Pavlidis, E., Cantalupo, G., Pezzella, M., Zara, F., Garavelli, L., et al. (2016). Xp11.22 microduplications including HUWE1: case report and literature review. Neuropediatrics. 47, 51–56. doi: 10.1055/s-0035-1566233
Ropers, H. H. (2006). X-linked mental retardation: many genes for a complex disorder. Curr. Opin. Genet. Dev. 16, 260–269. doi: 10.1016/j.gde.2006.04.017
Van den Veyver, I. B. (2001). Skewed X-inactivation in X-linked disorders. Semin. Reprod. Med. 19, 183–191. doi: 10.1055/s-2001-15398
Vulto-van Silfhout, A. T., Hehir-Kwa, J. Y., van Bon, B. W., Schuurs-Hoeijmakers, J. H., Meader, S., Hellebrekers, C. J., et al. (2013). Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 34, 1679–1687. doi: 10.1002/humu.22442
Wang, Q., Chen, P., Liu, J., Lou, J., Liu, Y., and Yuan, H. (2020). Xp11.22 duplications in four unrelated Chinese families: delineating the genotype-phenotype relationship for HSD17B10 and FGD1. BMC Med. Genom. 13:66. doi: 10.1186/s12920-020-0728-8
Whibley, A. C., Plagnol, V., Tarpey, P. S., Abidi, F., Fullston, T., Choma, M. K., et al. (2010). Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 87, 173–188. doi: 10.1016/j.ajhg.2010.06.017
Zhang, A., Weaver, D. D., and Palmer, C. G. (1997). Molecular cytogenetic identification of four X chromosome duplications. Am. J. Med. Genet. 68, 29–38.
Zhang, F., Gu, W., Hurles, M. E., and Lupski, J. R. (2009). Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 10, 451–481. doi: 10.1146/annurev.genom.9.081307.164217
Keywords: Xp11.23p11.22 duplication, array CGH, X-inactivation, speech and language delay, regression
Citation: Czakó M, Till Á, Zima J, Zsigmond A, Szabó A, Maász A, Melegh B and Hadzsiev K (2021) Xp11.2 Duplication in Females: Unique Features of a Rare Copy Number Variation. Front. Genet. 12:635458. doi: 10.3389/fgene.2021.635458
Received: 30 November 2020; Accepted: 22 March 2021;
Published: 14 April 2021.
Edited by:
Katalin Komlosi, Medical Center University of Freiburg, GermanyReviewed by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyScott Hickey, Nationwide Children’s Hospital, United States
Copyright © 2021 Czakó, Till, Zima, Zsigmond, Szabó, Maász, Melegh and Hadzsiev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kinga Hadzsiev, aGFkenNpZXYua2luZ2FAcHRlLmh1