 Gloria Kafui Esi Zodanu1
Gloria Kafui Esi Zodanu1 Mónika Oszlánczi2Kálmán Havasi2Anita Kalapos2Gergely Rácz2Márta Katona3
Mónika Oszlánczi2Kálmán Havasi2Anita Kalapos2Gergely Rácz2Márta Katona3 Anikó Ujfalusi4
Anikó Ujfalusi4 Orsolya Nagy4Márta Széll1
Orsolya Nagy4Márta Széll1 Dóra Nagy1*
Dóra Nagy1*- 1Department of Medical Genetics, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 2Second Department of Internal Medicine and Cardiology Centre, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 3Department of Pediatrics, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 4Division of Clinical Genetics, Department of Laboratory Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hungary
Congenital heart defects (CHD) are the most common developmental abnormalities, affecting approximately 0.9% of livebirths. Genetic factors, including copy number variations (CNVs), play an important role in their development. The most common CNVs are found on chromosome 22q11.2. The genomic instability of this region, caused by the eight low copy repeats (LCR A-H), may result in several recurrent and/or rare microdeletions and duplications, including the most common, ∼3 Mb large LCR A-D deletion (classical 22q.11.2 deletion syndrome). We aimed to screen 22q11.2 CNVs in a large Hungarian pediatric and adult CHD cohort, regardless of the type of their CHDs. All the enrolled participants were cardiologically diagnosed with non-syndromic CHDs. A combination of multiplex ligation-dependent probe amplification (MLPA), chromosomal microarray analysis and droplet digital PCR methods were used to comprehensively assess the detected 22q11.2 CNVs in 212 CHD-patients. Additionally, capillary sequencing was performed to detect variants in the TBX1 gene, a cardinal gene located in 22q11.2. Pathogenic CNVs were detected in 5.2% (11/212), VUS in 0.9% and benign CNVs in 1.8% of the overall CHD cohort. In patients with tetralogy of Fallot the rate of pathogenic CNVs was 17% (5/30). Fifty-four percent of all CNVs were typical proximal deletions (LCR A-D). However, nested (LCR A-B) and central deletions (LCR C-D), proximal (LCR A-D) and distal duplications (LCR D-E, LCR D-H, LCR E-H, LCR F-H) and rare combinations of deletions and duplications were also identified. Segregation analysis detected familial occurrence in 18% (2/11) of the pathogenic variants. Based on in-depth clinical information, a detailed phenotype–genotype comparison was performed. No pathogenic variant was identified in the TBX1 gene. Our findings confirmed the previously described large phenotypic diversity in the 22q11.2 CNVs. MLPA proved to be a highly efficient genetic screening method for our CHD-cohort. Our results highlight the necessity for large-scale genetic screening of CHD-patients and the importance of early genetic diagnosis in their clinical management.
Introduction
Congenital heart defects (CHDs) are the most common congenital developmental defects and affect approximately 0.9% of livebirths (van der Linde et al., 2011). Thirty to forty percent of CHDs are syndrome-associated and are caused by copy number variants (CNVs) or a mutation in a single gene. The most common human CNVs affect chromosomal region 22q11.2 (Fahed et al., 2013; Digilio and Marino, 2016). Proximal microdeletions of 1.5–3 Mb in this chromosomal region typically include the sequence between low copy repeat regions A and D (LCR A-D, LCR A-B) and may lead to the classical phenotype of 22q11.2 deletion syndrome, also known as DiGeorge syndrome. Central (LCR B-D, LCR C-D) or distal deletions (LCR C-H) may cause other, variable phenotypes (Burnside, 2015; Kruszka et al., 2017). Duplications have also been identified in this chromosomal region and are associated with even more significant phenotypic variability than deletions (Wentzel et al., 2008).
DiGeorge syndrome (also known as velocardiofacial syndrome and conotruncal anomaly face syndrome) is mostly characterized by CHD, thymus hypoplasia, immunodeficiency and skeletal, gastrointestinal and urogenital defects as well as by developmental delay, learning difficulties, susceptibility to neuropsychiatric disorders and, in some cases, by mild to moderate intellectual disability. 22q11.2 CNVs have reduced penetrance and incomplete expression and may be detected in asymptomatic or mildly affected individuals; approximately 7–10% of the cases are familial (McDonald-McGinn et al., 1993-2020; Campbell et al., 2018).
The T-box transcription factor 1 (TBX1) gene is located within the proximal 22q11.2 region, encodes a transcription factor that plays an important role in early embryonic development and is hypothesized to contribute to 22q11.2 deletion phenotype as well as to non-syndromic CHDs. (Griffin et al., 2010; Heike et al., 2010; Guo et al., 2011).
Clinical diagnosis may be challenging and significantly delayed due to the large phenotypic spectrum resulting from 22q11.2 CNVs (from asymptomatic appearance to multiple defects) (van Engelen et al., 2010). Previous studies have drawn attention to the importance of routine screening for 22q11.2 CNVs in patients with congenital heart defects, especially with conotruncal anomalies (Wozniak et al., 2010; Huber et al., 2014; Goldmuntz, 2020).
Based on the low number of patients referred for 22q11.2 CNV analysis at our genetic department over the last decade, we hypothesized that some patients with 22q11.2 CNVs—especially in the adult population—may have remained undiagnosed. The aim of our study was therefore to test for 22q11.2 CNVs and TBX1 gene variants for the pediatric and adult patients of the Southern-Hungarian CHD Registry, cardiologically diagnosed with non-syndromic CHDs, and to carry out genotype–phenotype comparison in positive cases based on in-depth clinical data.
Materials and Methods
Overall, 212 unrelated patients (110 females, 112 males; mean age: 26.9 years; age range: 2 weeks to 74 years) previously cardiologically diagnosed with non-syndromic congenital heart defects were enrolled in the study at the University of Szeged between 2016 and 2019. The distribution of the patients with the different CHD types are presented in Table 1.
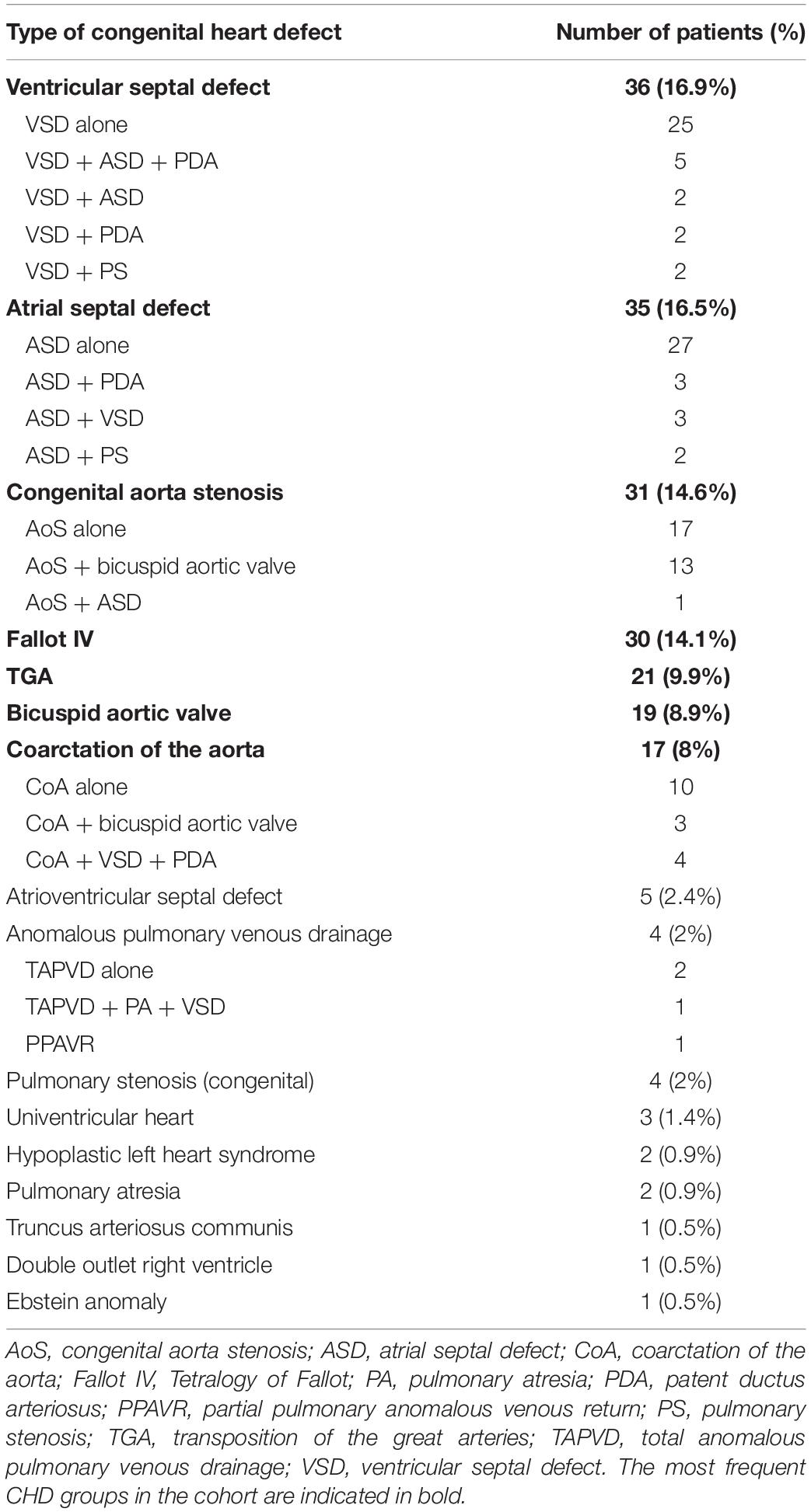
Table 1. The distribution of different types of congenital heart defects among patients (N = 212).
The DNA of 211 Hungarian individuals with no CHDs (confirmed with cardiological examination), and with no family history of CHD (144 females, 67 males, mean age: 37 years, age range: 8–73 years) was used as controls for the comparative analyses.
In positive cases, genetic testing was offered to all first-degree family members.
All investigations were performed according to the Helsinki Declaration 2008 and approved by the National Medical Research Council (No CHD-01/2016—IF-6299-8/2016) and the Local Ethical Committee of the University of Szeged (No 105/2016-SZTE). Participants/legal guardians/parents gave their informed consent to the study.
Sample Preparation and Multiplex Ligation-Dependent Probe Amplification (MLPA)
DNA was extracted from peripheral blood with the QIAamp DNA Blood Mini Kit (QIAGEN, Gödöllõ, Hungary).
To detect CNVs in the 22q11.2 locus, all patient samples were processed using the P250-B2 DiGeorge SALSA MLPA Probemix (IVD, MRC-Holland, Amsterdam) according to the manufacturer’s instructions. The MLPA probe mix contained 48 probes, 29 of which are located in the 22q11.2 region (24 in the LCR A to H region and 5 in the Cat-Eye syndrome region) and 19 in regions 4q35, 8p23, and 9q34 (Kleefstra syndrome), 10p14 (DiGeorge syndrome 2) and 17p13 and 22q13 (Phelan-McDermid syndrome), deletions in the latter may result in phenotypical similarity to DGS. Amplicon fragment length analysis was performed on an ABI 3500 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA) and analyzed by Coffalyser.net software (MRC-Holland, Amsterdam).
Validation of Positive Cases: FISH, Chromosomal Microarray Analysis, ddPCR
MLPA was repeated for all samples in which CNVs were found. Deletions and duplications were confirmed with an independent method, including FISH (Vysis DiGeorge Region LSI N25 SO/ARSA SGn Probes, Abbott Molecular Inc., Des Plaines, IL, United States, and SureFISH 22q11.21 CRKL, Agilent Technologies, Cedar Creek, TX, United States), a supplementary MLPA kit (P372-SALSA MLPA Microdeletions 6, MRC-Holland, Amsterdam, Netherlands) or chromosomal microarray analysis (CMA, Affymetrix, CytoScan 750 K, Thermo Fisher Scientific, Waltham, MA, United States). CMA was performed as described by Nagy et al. (2019). In cases where one probe was deleted, the probe region was sequenced with bidirectional capillary sequencing to exclude MLPA-interfering SNPs in the sample DNA. These validation methods confirmed all positive MLPA results (i.e., no false positives).
A droplet digital PCR (ddPCR) method was designed for the confirmation of recurrent single-probe CNVs in the TOP3B gene from CHD patient samples. This method was also used to determine the frequency of TOP3B CNVs in the control cohort as well. The analysis was performed on the QX100 Droplet Digital PCR system (Bio-Rad Laboratories, Hercules, CA, United States), according to the manufacturer’s instruction. Primers and probes were designed for TOP3B exon 7 and for the PRDM15 gene as reference region on chromosome 21 (Supplementary Material). TOP3B CNVs found in the controls with ddPCR were confirmed with MLPA.
Sequencing of the TBX1 Gene
Bidirectional capillary sequencing of TBX1 coding regions was performed for all patient samples with an ABI 3500 Genetic Analyzer. The primers used are listed in Supplementary Material. The non-synonymous variants (all located in exon 9 of TBX1 gene) were tested in the control cohort as well.
CNV and Variant Interpretation
Identified CNVs and single nucleotide variants (SNVs) were classified according to the standards and guidelines of the American College of Medical Genetics (Richards et al., 2015; Riggs et al., 2020). The following websites and databases were used for CNV interpretation: Database of Chromosomal Imbalance, Phenotype of Humans using Ensemble Resources (DECIPHER, Firth et al., 2009), Database of Genomic Variation (DGV, MacDonald et al., 2013), PubMed and GeneReviews (McDonald-McGinn et al., 1993-2020). For SNV interpretation, VarSome (Kopanos et al., 2011), ClinVar (Landrum et al., 2020), and Genome Aggregation Database (GnomAD, Karczewski et al., 2020) databases were used.
Statistical Analysis
GraphPad Prism (GraphPad Software, San Diego, California, United States), version 4.00 for Windows, was used for statistical analysis. The frequency of TOP3B CNVs and TBX1 variants in the patient cohort was compared with the frequency in the control cohort and also with the frequency in the global dataset of GnomAD using the Fisher exact test and χ2-test. P < 0.05 was considered to be statistically significant.
Results
Distribution of CHD Types in Patients
In the CHD cohort, the four most common CHD types were ventricular septal defect (VSD), atrial septal defect (ASD), congenital aorta stenosis (AoS) and tetralogy of Fallot (TOF) (Table 1). In 81% of the patients, only one cardiac entity was diagnosed; whereas, in 19% of the cases, two or more CHDs occurred together. The distribution of the different CHDs among the South-Hungarian Registry patients corresponded well with the frequency described in the literature (van der Linde et al., 2011).
Distribution of Positive MLPA Results and Classification of the Detected CNVs
Overall, 17 cases of 212 patients (8%) diagnosed prior with non-syndromic CHD were yielded positive MLPA result, and after evaluation 11 of these copy number changes (5.2%) were interpreted as pathogenic variant, two as variant of unknown significance (VUS, 0.9%) and four as benign (1.8%) (Figure 1 and Supplementary Material). The most frequent CNVs of the positive MLPA results were microdeletions (8/17); however, microduplications (7/17) and a combination of deletions and duplications (2/17) were also observed.

Figure 1. Pathogenic variants, variants of uncertain significance and benign 22q11.2 copy number variations in the CHD cohort. Cen, centromere; CES, Cat eye syndrome region; A-H yellow boxes: low copy repeat regions in locus 22q11.2. Genes indicated between the LCR regions, are the ones that have corresponding probes in the P250-B2 DiGeorge SALSA MLPA Probemix; LCR: low copy repeat region. Dark red: pathogenic microdeletion; Dark blue: pathogenic microduplication; Middle red: microdeletion of uncertain significance; Middle blue: microduplication of uncertain significance; Light red: benign microdeletion; Light blue: benign microduplication. LCR A-D: the typical ∼2.5–3 Mb microdeletion in region 22q11.21; LCR A-B: ∼1.5 Mb proximal microdeletion in region 22q11.21; LCR C-D: ∼0.5 Mb central microdeletion in region 22q11.21; LCR D-E: ∼1.2 Mb central microduplication in region 22q11.21q11.22; LCR D-H: ∼3.1–3.5 Mb central-distal microduplication in region 22q11.21q23; LCR E-H: ∼1.55–2 Mb distal microduplication in region 22q11.22q11.23; LCR F-H: ∼1–1.2 Mb distal microduplication in region 22q11.23; Asterix denotes familial CNVs. AoS, congenital aorta stenosis; AVSD, atrioventricular septal defect; bicuspid AV: bicuspid aortic valve; CoA, coarctation of the aorta; Fallot IV, tetralogy of Fallot; PDA, patent ductus arteriosus; TAPVD, total anomalous pulmonary venous drainage; VSD, ventricular septal defect.
Among pathogenic CNVs 7 microdeletions, 2 duplications and 1 combination of a deletion and a duplication was detected, while among the VUS one duplication and one combined CNV and among the benign variants one deletion and three duplications.
Pathogenic results were observed most frequently in the TOF group: in 17% of all TOF patients, followed by the group of bicuspid aortic valve with 10% (Figure 1 and Table 2).

Table 2. The distribution of the pathogenic and VUS 22q11 CNVs in the different CHD groups.
Based on the interpretation guidelines (Riggs et al., 2020), 11 CNVs were interpreted as pathogenic:
6 typical deletions of LCR A-D, one proximal nested deletion of LCR A-B, two duplications of LCR A-D, one combination of the proximal deletion of LCR A-D with a duplication of LCR E-H and one duplication of LCR D-H. Two further CNVs (one combination of a central deletion of LCR C-D with duplication of LCR D-E and one duplication of LCR F-H) were classified as VUS (Figure 1 and Table 3). Four CNVs (three 268 kbp duplications and one 278 kbp deletion) were detected in the TOP3B gene and resulted from one probe alteration in the MLPA reaction. These CNVs were confirmed with chromosomal microarray analysis (Supplementary Material). Considering the relatively high proportion of TOP3B CNVs in our patient cohort (overall 4/212, 1.9%: deletion in 1/212, 0.5% and duplication in 3/212, 1.4%), we decided to perform an independent analysis with ddPCR to determine the frequency of TOP3B CNVs in the healthy controls. The TOP3B deletion was detected in one control sample (0.5%) and a duplication in four control samples (1.9%); i.e., CNVs were identified in 2.4% of the controls (5/211). The difference in the CNV frequency between patients and controls was not significant (p = 0.751). Thus, we ultimately classified TOP3B CNVs as rare benign variants, which are more frequent in the Hungarian population than in the global database (frequency in DECIPHER: 0.36%).
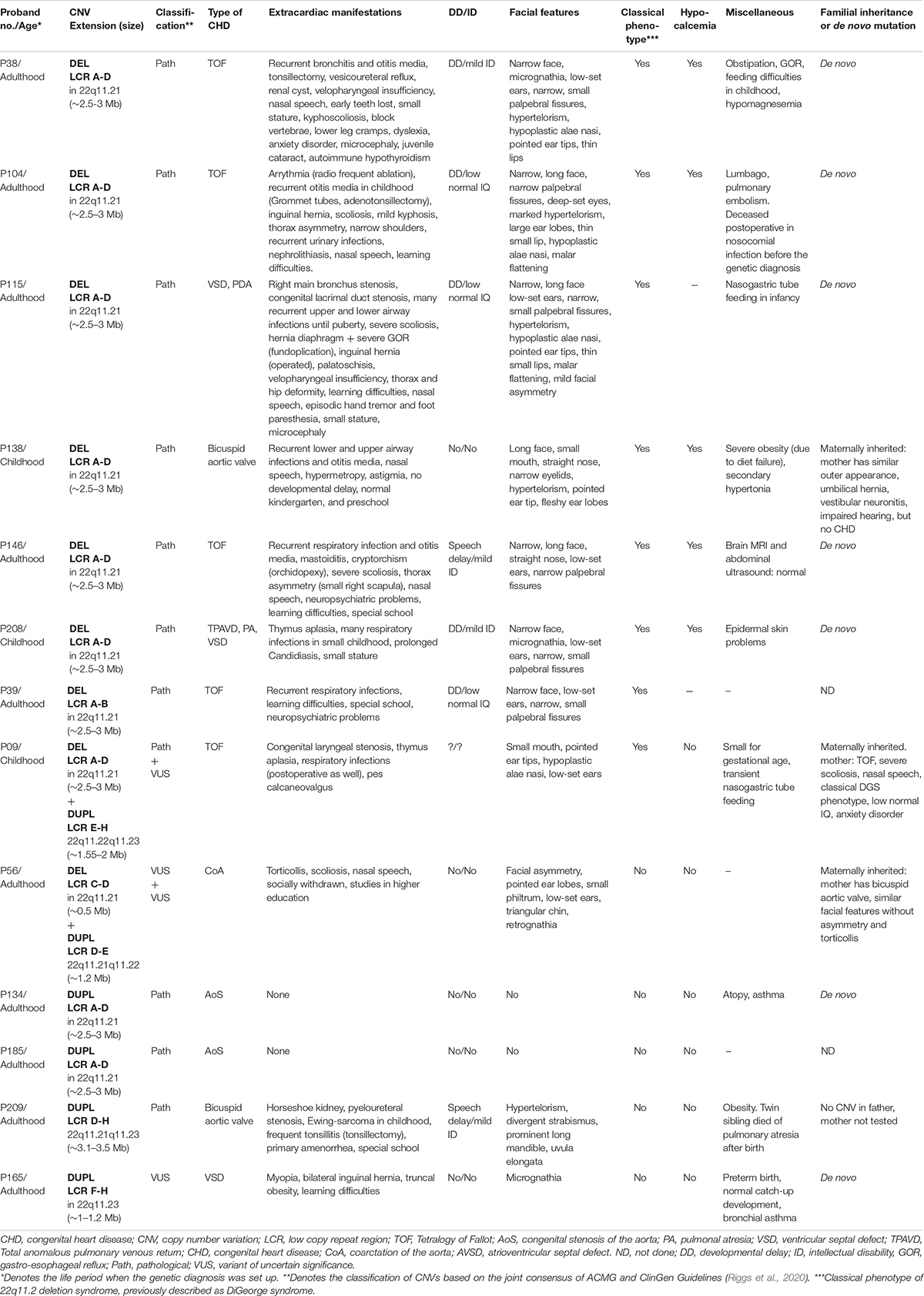
Table 3. Clinical features of probands and parents with pathogenic and VUS MLPA results.
Familial Segregation
It was possible to perform segregation analysis for 14 of the 17 positive cases. Six cases proved to be familial (Figure 1), two of these were for patients with pathogenic CNVs, one for a patient with VUS and three for patients with benign TOP3B variants. In addition to the TOP3B microduplication, proband P32 also had a 201-bp microduplication on chromosome 17p13.3, which included the YWHAE gene (Supplementary Material). Both chromosome imbalances were inherited from an asymptomatic parent. Two asymptomatic siblings also carried the YWHAE microduplication but without the TOP3B CNV. Therefore, the YWHAE CNV was interpreted as a rare benign variant. In case of the other TOP3B microduplications (P51 and P69) one healthy parent carried also the variant. In the patient with the TOP3B deletion, segregation analysis was not performed, the proband had two healthy children. The individuals with TOP3B CNVs were excluded from the genotype–phenotype comparison based on these results, the high frequency of TOP3B CNV in controls and the fact that these patients displayed no other malformations or comorbidities in addition to CHD.
The segregation analysis detected familial occurrence for 18% (2/11) of the pathogenic CNVs and 50% (1/2) of VUS. In these three familial cases (P138, P09 and P56), the proband’s mother carried the same chromosome imbalance. The phenotypes of the mothers were the same severity (P138) or milder (P09, P56) than the probands’. The suspicion of an underlying 22q11.2 CNV prior to the genetic testing was not raised at any of the affected family members.
For proband P09 and for the proband’s mother, the typical ∼2.5–3 Mb 22q11.2 microdeletion was combined with a distal ∼1.5–2 Mb 22q11.2 microduplication. The segregation analysis of the family showed that the maternal grandmother and one sibling of the mother carried only the duplication with no cardiological symptom or developmental malformation, although they had been diagnosed with mild anxiety disorder and depression. The deletion occurred most probably de novo in the mother and was transferred to the child.
The mother of proband P56 had only bicuspid aortic valve (clinically diagnosed only after the genetic diagnosis) and similar facial features as the proband without torticollis or severe scoliosis.
The clinical features of patients and family members are shown in Table 3.
Genotype-Phenotype Comparison
The probands’ age at the genetic diagnosis with pathogenic or VUS 22q11.2 CNVs ranged from 2 months to 52 years (median age: 21 years). Three patients out of 13 were diagnosed in childhood, one child in the first year of life. The two oldest patients and the affected family members were born before the molecular diagnostic era. No correlation could be observed between the severity of the phenotype and the age at the diagnosis.
The prevalence of common clinical features for different CNVs is comparable to previously reported prevalence data in the literature (Table 4). Patients presented with more marked phenotypic features for 22q11.2 microdeletions than with microduplications in the same region. In addition to the CHDs, the typical microdeletions of LCR A-D—with or without accompanying CNVs—resulted in the classical phenotype of 22q11.2 deletion syndrome. The co-occurring duplication in proband P09 and mother has not modified their phenotype significantly compared to other LCR A-D microdeletion phenotypes. In proband P56 and his mother with the combination of central deletion and distal duplication, the phenotype differed completely from that of 22q11.2 deletion syndrome (except the CHD) and the impact of the duplication could not be determined precisely (Table 3).
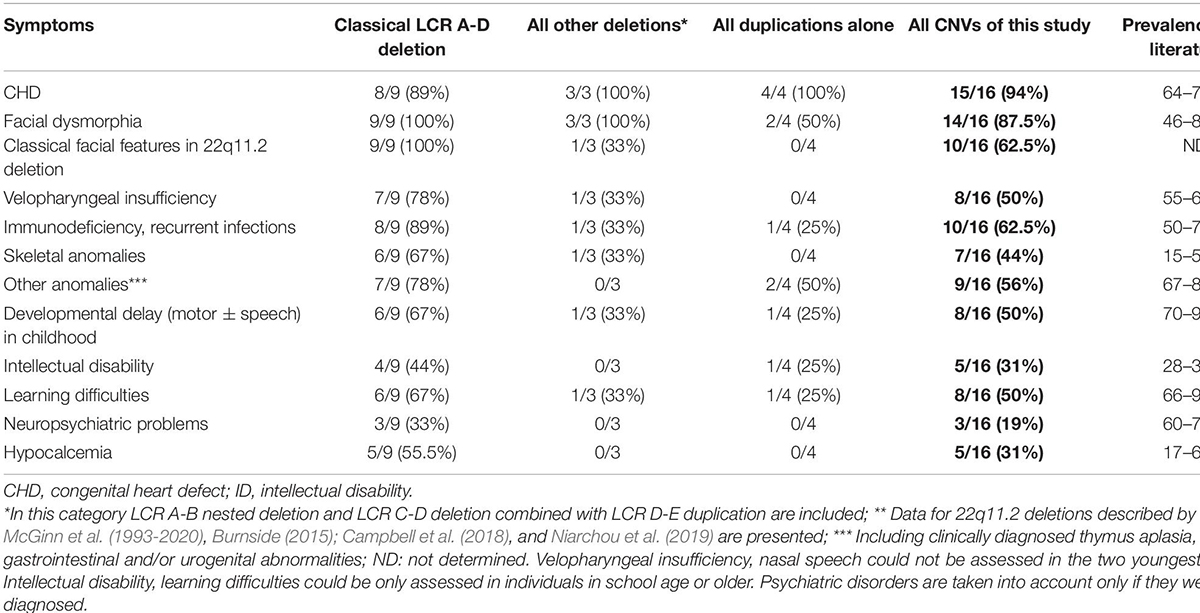
Table 4. The prevalence of the common clinical features in all probands and family members carrying pathogenic CNVs and VUS in the 22q11.2 region compared to the prevalence in the literature.
Among patients with deletions, Fallot tetralogy was the most common CHD. Among patients with duplications, congenital aorta stenosis, coarctation of the aorta and bicuspid aortic valve were the most common CHD types (Table 2). These three entities may be considered on the spectrum for one disease.
CHDs were overrepresented in our 22q11.2 CNV patients and their affected family members compared to data in the literature (94% vs. 74%), which may be the result of the patient enrollment criteria. Neuropsychiatric disorders were underrepresented among our patients (19% vs. 60%). Other characteristics (facial features, velopharyngeal insufficiency, immunodeficiency, hypocalcemia, skeletal anomalies, developmental delay, and learning difficulties) had a distribution in our cohort similar to that described in the literature (Table 4). The presence of immunodeficiency was deduced from the recurrence of respiratory and ear infections occurring mostly in childhood. Based on regular laboratory check-ups, the average absolute lymphocyte count was in the lower normal range (2.13 G/L, normal range: 1.5–3.2 G/L); whereas the average relative lymphocyte count was below normal (23.8%, normal range: 27–34%). Flow-cytometry and serum immunoglobulin levels were not measured regularly. The immune status and infections of 22q11.2 CNV patients were not strictly controlled before the genetic diagnosis. Before genetic diagnosis, proband P104 died of a fulminant postoperative infection (Table 3).
Hypocalcemia (average serum calcium level: 1.84 mmol/l, normal range: 2.2–2.55 mmol/l) was often present in patients with the typical 22q11.2 microdeletions—with or without clinical symptoms. However, hypocalcemia was not considered relevant for therapy before genetic diagnosis. Severe hypomagnesemia was also detected in one 22q11.2 microdeletion patient. The thrombocyte count was in the low normal range with an average of 156 × 109/l. Thyroid and parathyroid hormone levels and vitamin D levels were not measured in these patients before genetic diagnosis.
These laboratory abnormalities could not be consistently identified for patients with CNVs other than the typical microdeletion.
Results of the TBX1 Gene Sequencing
No apparently pathogenic variant was detected in the TBX1 gene. For CHD patients, three missense variants were found in exon 9: c.1189A>A; p.Asn397His with a 21% minor allele frequency (MAF), c.1049G>A; p.Gly350Asp with 0.48% MAF and c.1341_1342insCCGCACGCGCAT; p.Ala450_His453dup with 0.24% MAF (Table 5). The frequency of the p.Asn397His variant was also 21% for the controls. The two less frequent variants were also detected in one of the proband’s healthy parents and are listed in the Hungarian or the global database with very low frequencies (Table 5). Of the 10 probands and mothers with proximal 22q11.2 microdeletions encompassing the TBX1 gene, two (20%) carried the common p.Asn397His variant in hemizygous form (P09 and P138). Proband P09 exhibited a severe phenotype; whereas proband P138 presented only milder symptoms. The two rare variants were not detected in any of the microdeletion patients. Based on the allele frequencies, ACMG criteria and segregation analyses, all three variants were ultimately classified as benign.
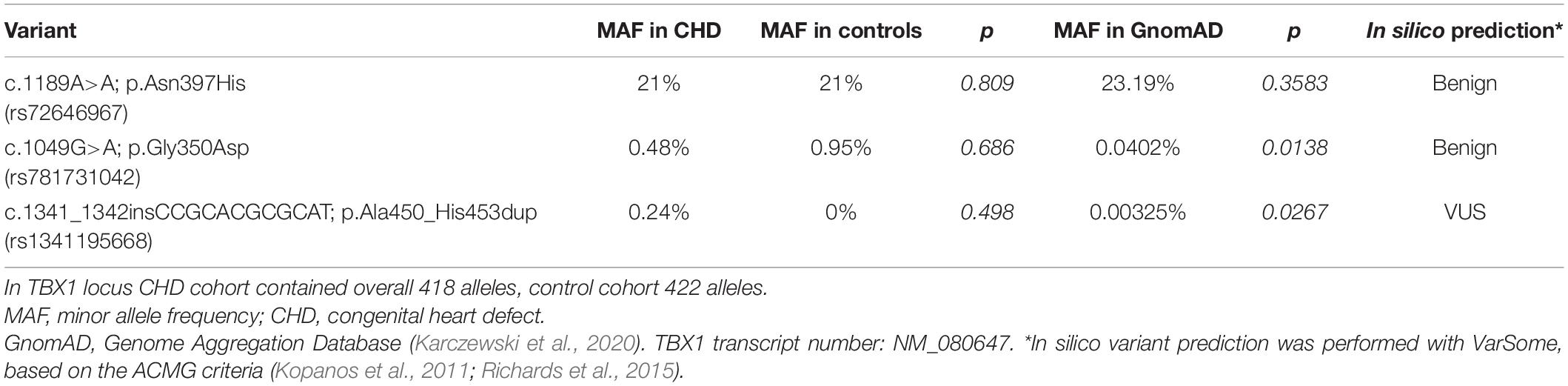
Table 5. Minor allele frequencies of TBX1 variants in the CHD cohort and control cohort compared to the allele frequency in the global database.
Discussion
This was the first systemic, large-scale genetic screening study of Hungarian CHD patients. All patients with cardiologically verified CHDs were enrolled in the study without further selection. Although the enrolled patients were cardiologically diagnosed with non-syndromic CHDs prior to this study, 13 were found to be syndromic after the genetic screening.
We observed a higher median age (21 years) and a similar or wider age range (0.17–52 years) at the genetic diagnosis in our cohort as compared to previously described cohorts (median age: 17.3 years, range: 0.1–59.4 years in Canadian patients; median age: 2.9 years, range: 0–17.6 years in American patients) (Palmer et al., 2018). This difference may partly be explained by the fact, that the 22q11.2 duplication patients with more variable phenotypes were also included in the present study, whereas only 22q11.2 deletions were analyzed by Palmer and colleagues.
The frequency of CHDs was representative and corresponded to the frequency described in large epidemiological studies (van der Linde et al., 2011).
All types of CNVs in the 22q11.2 chromosomal region were present in 8% of the CHD cohort, while pathogenic CNVs in 5.2%, VUS in 0.9% and benign CNVs in 1.8%. Our patients presented pathogenic 22q11.2 CNVs more often compared to other CHD cohorts, such as 1.27% in Brazilian, 2.8% in Cameroonian and 2.9% in Chinese population (Huber et al., 2014; Wonkam et al., 2017; Li et al., 2019). However, this difference may be also explained by the fact that most of these studies focused on the detection of 22q11.2 deletion but not on duplications.
For tetralogy of Fallot, the proportion of pathogenic CNVs was significantly higher, 17% in our cohort, which is in agreement with the fact that 22q11.2 CNVs are common in conotruncal heart defects (McDonald-McGinn et al., 1993-2020). 22q11.2 deletion can be detected in approximately 20% of all conotruncal heart defects (Wozniak et al., 2010), within this category its prevalence can be as high as ∼50% in interrupted aortic arch type B, ∼35% in truncus arteriosus or 10–25% in tetralogy of Fallot (Goldmuntz, 2020). All these suggest an absolute indication for 22q11.2 CNV analysis in these CHD groups, especially when co-occurring with at least one extracardiac manifestation or dysmorphic traits (Wozniak et al., 2010).
Congenital bicuspid aortic valve is common (0.5–2%) and, without complication of stenosis, regurgitation or dissection, considered a largely benign congenital heart defect (Li et al., 2017). However, based on our results, it should not be ignored in genetic testing.
The most common (64%) pathogenic CNV among our patients was the typical microdeletion of the LCR A-D region on chromosome 22q11.2, which is in agreement with the literature (Du et al., 2020). The frequency of deletions decreased toward the LCR F-H region, which was reflected in our results as well, since nested and central deletions were rare, and distal deletions were not detected. Proximal and distal duplication as well as two combined CNVs were also identified. Although most patients with the typical LCR A-D deletion showed the majority of the characteristic features of 22q11.2 deletion syndrome (velopharyngeal insufficiency, skeletal malformation, gastrointestinal and nephrological anomalies, hypocalcemia, frequent infections due to immunodeficiency and common facial features), these symptoms were present less frequently (Table 4) with the non-typical deletions and the duplications, as expected (Burnside, 2015; Du et al., 2020). In addition to the presence of CHDs, no typical common characteristics could be found for these patients. This may be due to the low number of patients with single CNVs in our cohort or to the even wider phenotypic spectrum of these CNVs, e.g., in the case of 22q11.2 duplications (Yu et al., 2019). Therefore, several individuals with only mild symptoms or no detectable malformation or dysmorphia may remain undetected.
Hypocalcemia, hypomagnesemia, lymphocytopenia, thrombocytopenia, and abnormalities of the thyroid, parathyroid hormone or vitamin D levels may remain undiscovered in 22q11.2 deletion patients, especially without genetic diagnosis. However, these conditions may significantly contribute to co-morbidities, such as increased susceptibility to infections, bleeding diathesis and heightened prevalence of autoimmune disorders, and, thus, should be considered for treatment (Lambert et al., 2018; Goldmuntz, 2020; Legitimo et al., 2020).
Neuropsychiatric disorders (attention deficit hyperactivity disorder, autism spectrum disorder, schizophrenia, anxiety symptoms and sleep disturbances) are frequent in patients with 22q11.2 CNVs (Brzustowicz and Bassett, 2012; Moulding et al., 2020). Complex presentation of three or more psychiatric traits may occur in 73% of patients with 22q11.2 CNVs (Niarchou et al., 2014; Chawner et al., 2019; Niarchou et al., 2019). However, these were markedly underrepresented (19%) in our patient cohort (Table 4), and this is most probably due to the lack of awareness and screening rather than to their absence. This result further emphasizes the importance of multidisciplinary management of patients with 22q11.2 CNVs.
The large phenotypic variability of 22q11.2 microdeletions has recently been the focus of much research but is still not yet fully understood. The haploinsufficiency of the coding genes, including TBX1, DGCR8, CRKL among others, alone does not seem to account for the highly variable phenotypes and incomplete penetrance of affected individuals (Du et al., 2020). Some recent studies have investigated the role of possible genetic and epigenetic factors which contribute to the diversity of phenotypes associated with 22q11.2 deletions (Brzustowicz and Bassett, 2012; Bertini et al., 2017; Du et al., 2020). Breakpoint analysis of the LCR A-D region showed that small variations in the deletion size within this region have no significant role on phenotypic variability (Bertini et al., 2017). Pathogenic sequential variations in the remaining single copy of the genes (with an emphasis on TBX1 gene) encompassed in the deleted region, were not yet revealed by previous investigation (Brzustowicz and Bassett, 2012). And this was further supported by our TBX1 sequencing results, since no pathogenic TBX1 variant was detected in our patients with 22q11.2 CNVs or in the overall CHD-cohort. Thus, pathogenic TBX1 mutations may be causal most probably only in a small fraction of CHD patients and 22q11.2 CNV patients, if at all. It was also hypothesized that sequential variations elsewhere in the genome (for example, de novo mutations in histone modifying genes) may collectively contribute to this diversity (Zaidi et al., 2013). Bertini and colleagues have investigated additional rare and common CNVs in typical 22q11.2 patients (2017). According to their results, these additional CNVs often contain miRNA genes or mitochondrial genes, which may interact with 22q11.2 deletion and lead to metabolic and energetic problems rather than a decreased dosage of morphogenetic genes. The DGCR8 gene is located within the typical 22q11.2 region and plays a crucial role in miRNA biosynthesis and, in combination with other CNV-miRNAs, may orchestrate highly variable phenotypic outcomes (Bertini et al., 2017). Previously, these additional CNVs, miRNAs have been investigated exclusively for 22q11.2 microdeletion patients, but not for duplication patients. Studying these duplication patients may further refine the diversity.
The family segregation study proved to be beneficial in cases with pathogenic CNVs, since in 18% further affected family members were identified. The number of familial cases was higher in our cohort than the previously described 6–10% (McDonald-McGinn et al., 1993-2020; Goldmuntz, 2020). The affected family members in our cohort exhibited similar or milder symptoms than the probands. This phenomenon has already been observed (Wozniak et al., 2010; Goldmuntz, 2020).
22q11.2 is considered one of the most unstable regions of the human genome, due to the low-copy repeat regions on chromosome 22. This instability predisposes the region to deletions and duplications through non-allelic homologous recombination events. Hence, the presence of a parental CNV may trigger the development of another CNV in the same or nearby chromosomal region in the offspring, as seen in the family P09 and as described by Capra et al. (2013).
In conclusion, based on the present results and on those described in the literature (Wozniak et al., 2010; Li et al., 2019; Goldmuntz, 2020), we suggest the implementation of the genetic screening of CNVs in the postnatal management of CHD patients, regardless of the type of CHDs. For this purpose, MLPA is a cost-effective, fast and specific method suitable for the screening of a large number of samples. Patients and families benefit greatly from early diagnosis, through the regular cardiological, orthopedic, endocrinological, immunological, neurodevelopmental, and psychiatric follow-ups, the more aggressive infection control and the possibility of positive family planning.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the National Medical Research Council (No. CHD-01/2016—IF-6299-8/2016) and by the Local Ethical Committee of the University of Szeged (No. 105/2016-SZTE). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
DN and MS: conceptualization, review, editing, and supervision. DN, GZ, MO, AK, KH, GR, MK, AU and ON: methodology, investigation, and validation. DN and MO: data curation. GZ and DN: writing and original draft preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Hungarian Scientific Research Fund (Grant No. 5S441-A202) and GINOP-2.3.2-15-2016-00039 grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Zsuzsanna Horváth-Gárgyán, Blanka Godza, Dóra Isaszegi, and Anikó Gárgyán for their skilled technical assistance and Dr. Shannon Frances for providing language help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.635480/full#supplementary-material
References
Bertini, V., Azzarà, A., Legitimo, A., Milone, R., Battini, R., Consolini, R., et al. (2017). Deletion extents are not the cause of clinical variability in 22q11.2 deletion syndrome: does the interaction between DGCR8 and miRNA-CNVs play a major role? Front. Genet. 8:47. doi: 10.3389/fgene.2017.00047
Brzustowicz, L. M., and Bassett, A. S. (2012). MiRNA-mediated risk for schizophrenia in 22q11.2 deletion syndrome. Front. Genet. 3:291. doi: 10.3389/fgene.2012.00291
Burnside, R. D. (2015). 22q11.21 deletion syndromes: a review of proximal, central, and distal deletions and their associated features. Cytogenet. Genome Res. 146, 89–99. doi: 10.1159/000438708
Campbell, I. M., Sheppard, S. E., Crowley, T. B., McGinn, D. E., Bailey, A., McGinn, M. J., et al. (2018). What is new with 22q? An update from the 22q and you center at the children’s hospital of Philadelphia. Am. J. Med. Genet. A 176, 2058–2069. doi: 10.1002/ajmg.a.40637
Capra, V., Mascelli, S., Garrè, M. L., Nozza, P., Vaccari, C., Bricco, L., et al. (2013). Parental imbalances involving chromosomes 15q and 22q may predispose to the formation of de novo pathogenic microdeletions and microduplications in the offspring. PLoS One 8:e57910. doi: 10.1371/journal.pone.0057910
Chawner, S. J. R. A., Owen, M. J., Holmans, P., Raymond, F. L., Skuse, D., Hall, J., et al. (2019). Genotype-phenotype associations in children with copy number variants associated with high neuropsychiatric risk in the UK (IMAGINE-ID): a case-control cohort study. Lancet Psychiatry 6, 493–505. doi: 10.1016/S2215-0366(19)30123-3
Digilio, M. C., and Marino, B. (2016). What is new in genetics of congenital heart defects? Front. Pediatr. 4:120. doi: 10.3389/fped.2016.00120
Du, Q., de la Morena, M. T., and van Oers, N. S. C. (2020). The genetics and epigenetics of 22q11.2 deletion syndrome. Front. Genet. 10:1365. doi: 10.3389/fgene.2019.01365
Fahed, A. C., Gelb, B. D., Seidman, J. G., and Seidman, C. E. (2013). Genetics of congenital heart disease: the glass half empty. Circ. Res. 112, 707–720. doi: 10.1161/CIRCRESAHA.112.300853
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D., et al. (2009). DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am. J. Hum. Genet. 84, 524–533. doi: 10.1016/j.ajhg.2009.03.010
Goldmuntz, E. (2020). 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. 184C, 64–72. doi: 10.1002/ajmg.c.31774
Griffin, H. R., Töpf, A., Glen, E., Zweier, C., Stuart, A. G., Parsons, J., et al. (2010). Systematic survey of variants in TBX1 in non-syndromic tetralogy of Fallot identifies a novel 57 base pair deletion that reduces transcriptional activity but finds no evidence for association with common variants. Heart 96, 1651–1655. doi: 10.1136/hrt.2010.200121
Guo, T., McDonald-McGinn, D., Blonska, A., Shanske, A., Bassett, A. S., Chow, E., et al. (2011). Genotype and cardiovascular phenotype correlations with TBX1 in 1,022 velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Hum. Mutat. 32, 1278–1289. doi: 10.1002/humu.21568
Heike, C. L., Starr, J. R., Rieder, M. J., Cunningham, M. L., Edwards, K. L., Stanaway, I. B., et al. (2010). Single nucleotide polymorphism discovery in TBX1 in individuals with and without 22q11.2 deletion syndrome. Birth Defects Res. A Clin. Mol. Teratol. 88, 54–63. doi: 10.1002/bdra.20604
Huber, J., Peres, V. C., de Castro, A. L., dos Santos, T. J., da Fontoura Beltrão, L., de Baumont, A. C., et al. (2014). Molecular screening for 22Q11.2 deletion syndrome in patients with congenital heart disease. Pediatr. Cardiol. 35, 1356–1362. doi: 10.1007/s00246-014-0936-0
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2011). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Kruszka, P., Addissie, Y. A., McGinn, D. E., Porras, A. R., Biggs, E., Share, M., et al. (2017). 22q11.2 deletion syndrome in diverse populations. Am. J. Med. Genet. A 173, 879–888. doi: 10.1002/ajmg.a.38199
Lambert, M. P., Arulselvan, A., Schott, A., Markham, S. J., Crowley, T. B., Zackai, E. H., et al. (2018). The 22q11.2 deletion syndrome: cancer predisposition, platelet abnormalities and cytopenias. Am. J. Med. Genet. A 176, 2121–2127. doi: 10.1002/ajmg.a.38474
Landrum, M. J., Chitipiralla, S., Brown, G. R., Chen, C., Gu, B., Hart, J., et al. (2020). ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844. doi: 10.1093/nar/gkz972
Legitimo, A., Bertini, V., Costagliola, G., Baroncelli, G. I., Morganti, R., Valetto, A., et al. (2020). Vitamin D status and the immune assessment in 22q11.2 deletion syndrome. Clin. Exp. Immunol. 200, 272–286. doi: 10.1111/cei.13429
Li, Y., Wei, X., Zhao, Z., Liao, Y., He, J., Xiong, T., et al. (2017). Prevalence and complications of bicuspid aortic valve in chinese according to echocardiographic database. Am. J. Cardiol. 120, 287–291. doi: 10.1016/j.amjcard.2017.04.025
Li, Z., Huang, J., Liang, B., Zeng, D., Luo, S., Yan, T., et al. (2019). Copy number variations in the GATA4, NKX2-5, TBX5, BMP4 CRELD1, and 22q11.2 gene regions in Chinese children with sporadic congenital heart disease. J. Clin. Lab. Anal. 33:e22660. doi: 10.1002/jcla.22660
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L., and Scherer, S. W. (2013). The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992. doi: 10.1093/nar/gkt958
McDonald-McGinn, D. M., Hain, H. S., Emanuel, B. S., and Zackai, E. H. (1993-2020). “22q11.2 Deletion Syndrome,” in GeneReviews® [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Moulding, H. A., Bartsch, U., Hall, J., Jones, M. W., Linden, D. E., Owen, M. J., et al. (2020). Sleep problems and associations with psychopathology and cognition in young people with 22q11.2 deletion syndrome (22q11.2DS). Psychol. Med. 50, 1191–1202. doi: 10.1017/S0033291719001119
Nagy, O., Szakszon, K., Biró, B. O., Mogyorósy, G., Nagy, D., Nagy, B., et al. (2019). Copy number variants detection by microarray and multiplex ligation-dependent probe amplification in congenital heart diseases. J. Biotechnol. 299, 86–95. doi: 10.1016/j.jbiotec.2019.04.025
Niarchou, M., Chawner, S. J. R. A., Fiksinski, A., Vorstman, J. A. S., Maeder, J., Schneider, M., et al. (2019). Attention deficit hyperactivity disorder symptoms as antecedents of later psychotic outcomes in 22q11.2 deletion syndrome. Schizophr. Res. 204, 320–325. doi: 10.1016/j.schres.2018.07.044
Niarchou, M., Zammit, S., van Goozen, S. H., Thapar, A., Tierling, H. M., Owen, M. J., et al. (2014). Psychopathology and cognition in children with 22q11.2 deletion syndrome. Br. J. Psychiatry 204, 46–54. doi: 10.1192/bjp.bp.113.132324
Palmer, L. D., Butcher, N. J., Boot, E., Hodgkinson, K. A., Heung, T., Chow, E. W. C., et al. (2018). Elucidating the diagnostic odyssey of 22q11.2 deletion syndrome. Am. J. Med. Genet. A 176, 936–944. doi: 10.1002/ajmg.a.38645
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. doi: 10.1038/s41436-019-0686-8
van der Linde, D., Konings, E. E., Slager, M. A., Witsenburg, M., Helbing, W. A., Takkenberg, J. J., et al. (2011). Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247. doi: 10.1016/j.jacc.2011.08.025
van Engelen, K., Topf, A., Keavney, B. D., Goodship, J. A., van der Velde, E. T., Baars, M. J., et al. (2010). 22q11.2 Deletion Syndrome is under-recognised in adult patients with tetralogy of Fallot and pulmonary atresia. Heart 96, 621–624. doi: 10.1136/hrt.2009.182642
Wentzel, C., Fernström, M., Ohrner, Y., Annerén, G., and Thuresson, A. C. (2008). Clinical variability of the 22q11.2 duplication syndrome. Eur. J. Med. Genet. 51, 501–510. doi: 10.1016/j.ejmg.2008.07.005
Wonkam, A., Toko, R., Chelo, D., Tekendo-Ngongang, C., Kingue, S., Dahoun, S., et al. (2017). The 22q11.2 deletion syndrome in congenital heart defects: prevalence of microdeletion syndrome in cameroon. Glob. Heart 12, 115–120. doi: 10.1016/j.gheart.2017.01.003
Wozniak, A., Wolnik-Brzozowska, D., Wisniewska, M., Glazar, R., Materna-Kiryluk, A., Moszura, T., et al. (2010). Frequency of 22q11.2 microdeletion in children with congenital heart defects in western poland. BMC Pediatr. 10:88. doi: 10.1186/1471-2431-10-88
Yu, A., Turbiville, D., Xu, F., Ray, J. W., Britt, A. D., Lupo, P. J., et al. (2019). Genotypic and phenotypic variability of 22q11.2 microduplications: an institutional experience. Am. J. Med. Genet. A 179, 2178–2189. doi: 10.1002/ajmg.a.61345
Keywords: 22q11.2 deletion syndrome, TBX1 gene, multiplex ligation-dependent probe amplification, copy number variations, droplet digital PCR, syndromic and non-syndromic congenital heart defects, chromosomal microarray analysis
Citation: Zodanu GKE, Oszlánczi M, Havasi K, Kalapos A, Rácz G, Katona M, Ujfalusi A, Nagy O, Széll M and Nagy D (2021) Systemic Screening for 22q11.2 Copy Number Variations in Hungarian Pediatric and Adult Patients With Congenital Heart Diseases Identified Rare Pathogenic Patterns in the Region. Front. Genet. 12:635480. doi: 10.3389/fgene.2021.635480
Received: 30 November 2020; Accepted: 07 April 2021;
Published: 29 April 2021.
Edited by:
Katalin Komlosi, Medical Center University of Freiburg, GermanyReviewed by:
Rincic Martina, University of Zagreb, CroatiaTársis Paiva Vieira, State University of Campinas, Brazil
Rafael Rosa, Federal University of Health Sciences of Porto Alegre, Brazil
Copyright © 2021 Zodanu, Oszlánczi, Havasi, Kalapos, Rácz, Katona, Ujfalusi, Nagy, Széll and Nagy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dóra Nagy, bmFneWRvckBnbWFpbC5jb20=