 Hoang Kim Tu Trinh
Hoang Kim Tu Trinh Le Duy Pham
Le Duy Pham Kieu Minh Le
Kieu Minh Le Hae-Sim Park
Hae-Sim Park- 1Center for Molecular Biomedicine, University of Medicine and Pharmacy at Ho Chi Minh City, Ho Chi Minh City, Vietnam
- 2Faculty of Medicine, University of Medicine and Pharmacy at Ho Chi Minh City, Ho Chi Minh City, Vietnam
- 3Department of Allergy and Clinical Immunology, Ajou University Medical Center, Suwon, South Korea
Non-steroidal anti-inflammatory drugs (NSAIDs) are extensively prescribed in daily clinical practice. NSAIDs are the main cause of drug hypersensitivity reactions all over the world. The inhibition of cyclooxygenase enzymes by NSAIDs can perpetuate arachidonic acid metabolism, shunting to the 5-lipoxygenase pathway and its downstream inflammatory process. Clinical phenotypes of NSAID hypersensitivity are diverse and can be classified into cross-reactive or selective responses. Efforts have been made to understand pathogenic mechanisms, in which, genetic and epigenetic backgrounds are implicated in various processes of NSAID-induced hypersensitivity reactions. Although there were some similarities among patients, several genetic polymorphisms are distinct in those exhibiting respiratory or cutaneous symptoms. Moreover, the expression levels, as well as the methylation status of genes related to immune responses were demonstrated to be involved in NSAID-induced hypersensitivity reactions. There is still a lack of data on delayed type reactions. Further studies with a larger sample size, which integrate different genetic pathways, can help overcome current limitations of gen etic/epigenetic studies, and provide valuable information on NSAID hypersensitivity reactions.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are extensively administered for the treatment of pain and inflammation, while they commonly induce hypersensitivity reactions as well as unexpected adverse effects (e.g., gastrointestinal hemorrhage). Indeed, 1.9-3.5% of the general adult population reported a hypersensitivity reaction to NSAIDs (Gomes et al., 2004; Zhou et al., 2016); NSAIDs are the major culprits of drug-induced hypersensitivity reactions (DHR) with the prevalence of 11.9% in patients with DHR (Sole et al., 2011; Zhao et al., 2018). Nevertheless, the prevalence seems to rise in high-risk subjects, for instance, NSAID hypersensitivity reactions occur in 7.15% of asthmatic patients exhibited, which increases to 14.89% in those with severe asthma (Thong, 2018). Thus, efforts have been made to investigate underlying mechanisms which can help predict NSAID hypersensitivity reactions.
It is well known that NSAID consumption can alter the cyclooxygenase (COX)-1 and COX-2 pathways of arachidonic acid (AA) metabolism (Doña et al., 2020), triggering symptoms in subjects with hypersensitivity reactions. Hypersensitivity to NSAIDs can be classified into cross-reactive and selective responses (Kowalski and Stevenson, 2013), which will be discussed in more detail in the following sections. Other phenotypes, including “blended reactions,” food-dependent NSAID-induced anaphylaxis, and NSAID-induced multiple selective immediate reactions were reported (Doña et al., 2020). It is crucial to understand that the risk of DHR can be affected by multiple factors such as sex, age, ethnicity, environmental factors, and genetic variants (Doña et al., 2017; Lee J.U. et al., 2019; Pérez-Sánchez et al., 2019). With advances in high-throughput sequencing technologies, many genetic studies have been conducted to elucidate genetic mechanisms of NSAID hypersensitivity phenotypes. This review aimed to summarize the current knowledge of the associations of genetic and epigenetic mechanisms in NSAID hypersensitivity.
AA and Cyclooxygenase Metabolism
Overview of AA Metabolism
At the cellular level, phospholipases are a family of enzymes responsible for phospholipid hydrolysis to liberate esterified AA (Cornejo-García et al., 2012). The initial enzyme is phospholipase A2 (PLA2) that catalyze the hydrolysis of the phospholipid sn-2 ester bond, generating a free fatty acid, a lysophospholipid, and a free AA molecule (Dennis et al., 2011). The other enzymes in the family are phospholipase C (PLC) and phospholipase D (PLD) that may also generate free AA (Dennis et al., 2011). Upon its activation, PLC catalyzes the hydrolysis of the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) on the glycerol side of the phosphodiester bond to modulate numerous PIP2-dependent cellular processes, yielding two PIP2 cleavage products, inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG) (Kano et al., 2009). In addition, PLD has proved to liberate AA by catalyzing phosphatidylcholine to generate phosphatic acid or DAG which is then hydrolyzed DAG-lipase to generate AA (Ishimoto et al., 1994).
Arachidonic acid is a membrane omega-6 fatty acid molecule released in the cytoplasm by the hydrolytic activity of the cytosolic phospholipase A2 (cPLA2) (Weller, 2016). COX and lipoxygenase (LOXs) are the 2 major enzymes in AA metabolism, which participates in the regulation of various pathophysiological processes, including inflammation and cancer (Bruno et al., 2014). COX-1 and COX-2 are the 2 main COX isoforms involved, which have a comparable molecular mass of 71 and 73 kDa, respectively (Meade et al., 1993; Hennekens et al., 1997). COX-1 enzyme is constitutively expressed in most tissues that induces inflammation in response to lipopolysaccharide stimulation as well as promotes or suppresses leukotrienes (LTs) biosynthesis (Naraba et al., 1998). Meanwhile, COX-2 is considered as an inducible enzyme expressed in inflammation (Green, 2001).
Following the COX pathway, PGG2 is formed and converted to PGH2 by the enzyme peroxygenase (Nørregaard et al., 2015). PGH2 is then transformed by proper synthases, mainly cytosolic prostaglandin E synthase (cPGES), microsomal prostaglandin E synthase-1 (mPGES-1), microsomal prostaglandin E synthase-2 (mPGES-2), prostaglandin I synthase (PGIS), and thromboxane synthase (TxS), into PGs and thromboxanes, proceeding to the synthesis of PGE2, prostacyclins (PGI2), PGD2, PGF2α, or thromboxane A2 (TXA2) (Samuelsson et al., 2007). In the 5-LOX pathway, 5-LOX catalyzes the oxidation of AA to 5-HPETE and 5-HPETE is subsequently converted into unstable intermediate cysteinyl leukotriene (LT) A4, which is hydrolyzed by LTA4 hydrolase (LTA4H) to form dihydroxy acid leukotriene LTB4 (Hedi and Norbert, 2004; Doña et al., 2020). Another path is the conversion of LTA4 to LTC4 via addition of a glutathione group by LTC4 synthase (LTC4S), which is exported by the cell and converted to downstream metabolites (Hedi and Norbert, 2004; Doña et al., 2020). Conversion of LTC4 by γ-glutamyl transferase results in LTD4 and glutamic acid release, and then dipetidase breaks the amide bond in LTD4 to synthesize LTE4 (Rouzer and Marnett, 2009; Doña et al., 2020).
The COX/5-LOX Pathway and LT Production
In susceptible subjects, non-selective NSAIDs exert effects by inhibiting COX-1 and subsequently shift the AA metabolism from PG (especially PGE2) synthesis toward pro-inflammatory cysteinyl LTs such as LTC4, LTD4, and LTE4 (Doña et al., 2020). Overproduction of LTs can trigger bronchoconstriction, recruitment of inflammatory cells (especially eosinophils) into the airways (Paruchuri et al., 2009; Pham et al., 2016; Dholia et al., 2020). Patients with NSAID hypersensitivity were demonstrated to have reduced sputum PGE2 levels but increased urinary LTE4 levels (Comhair et al., 2018; Mastalerz et al., 2019), which can explain the hypersensitivity reactions to NSAIDs. Besides increasing vascular permeability, bronchoconstriction, and mucus secretion, overproduction of LTs also leads to activation of mast cells and eosinophils. Stimulation of mast cells and eosinophils releases pro-inflammatory mediators and cytokines such as interleukin (IL)-33/thymic stromal lymphopoietin as well as facilitates the production of IL-4, IL-5, IL-9, and IL-13, which increase eosinophilic inflammation, mast cell activation, and IgE-secreting plasma cells differentiation from B cells in which close interactions with epithelial cells are involved, further enhancing type 2 airway inflammation (Buchheit et al., 2016; Choi et al., 2019).
Moreover, NSAIDs were found to elicit immune responses following either specific IgE or T cell production (Blanca et al., 1989; Kowalski and Makowska, 2015). NSAIDs can induce IgE-mediated hypersensitivity, and the symptoms range from mild urticaria and localized angioedema to anaphylaxis, within a few minutes or 1 h after NSAID consumption (Klar et al., 2019). Current studies strongly suggested that the activation of platelets as well as platelet-derived mediators play roles in the pathogenesis of NSAID hypersensitivity (Palikhe et al., 2014; Mitsui et al., 2016). Platelets are major sources of sphingolipid metabolites and increased in patients with NSAID hypersensitivity, which recruit more eosinophils and neutrophils trafficking into the airways (Trinh et al., 2016; Kim, 2019; Kim et al., 2019). Taken together, NSAIDs can interfere with AA-COX metabolism and trigger specific immune response, leading to type 2 inflammatory process in the cells and tissues involved.
Phenotypes of NSAID Hypersensitivity
Hypersensitivity reactions can be induced or triggered by multiple NSAIDs that mainly inhibit COX-1 (multiple or cross-reactive NSAIDs hypersensitivity) or by a single NSAID that elicits a humoral or cellular immune response (single or selective NSAID hypersensitivity) (Ayuso et al., 2013; Pham et al., 2016). Among patients with NSAID hypersensitivity, 76% had cross-reactions, and 24% had selective responses (Dona et al., 2011).
Non-steroidal anti-inflammatory drugs can aggravate rhinitis and/or asthmatic symptoms (NSAID-exacerbated respiratory disease, NERD), which could be considered an endotype of asthma with a unique pathophysiological mechanism (Lee et al., 2017), while they can elicit cutaneous symptoms including urticaria,/angioedema, and/or anaphylaxis in patients with chronic urticaria (CU) (called NSAID-exacerbated cutaneous disease or NECD) or those without chronic urticaria (NSAID-induced urticaria/angioedema/anaphylaxis, NIUAA). In addition, they can induce both respiratory and cutaneous symptoms (Lee Y. et al., 2019). Single NSAIDs hypersensitivity is associated with specific IgE antibody productions, which induces acute urticaria and/or angioedema as well as anaphylaxis (Single NSAID-induced urticarial/angioedema/anaphylaxis or SNIUAA) or with specific T-cell activations, which induces delayed hypersensitivity reactions (NSAID-induced delayed hypersensitivity reaction, NIDHR).
NSAID-Exacerbated Respiratory Disease
NSAID-exacerbated respiratory disease, which is characterized by a triad of asthma, chronic rhinosinusitis (CRS) with nasal polyps (NPs) and NSAID hypersensitivity, is reported in 7%-20% of asthmatic patients and 16% of patients with CRSwNP (Stevens et al., 2017; Lee, 2019). Asthma symptoms can be observed within 30-180 min after exposure to NSAIDs. Patients with NERD present more severe phenotypes with lower lung function but higher prevalence of CRS/NPs compared to aspirin/NSAID-tolerant asthmatics (ATA) (Stevens et al., 2017). Recent studies classified NERD into various subphenotypes. A Polish study demonstrated 4 subphenotypes: (1) moderate asthma with intensive upper airway symptoms and blood eosinophilia, (2) mild and well-controlled asthma with low healthcare requirement, (3) severe and poorly controlled asthma with severe exacerbations and airway obstruction, and (4) poorly controlled asthma with frequent and severe exacerbations in females (Bochenek et al., 2014). Another study in the Korean cohort revealed 4 subphenotypes with different clinical outcomes and inflammatory profiles: (1) NERD with CRS and atopy without urticaria, (2) non-atopic NERD with CRS without NECD, (3) NERD without CRS/NECD, and (4) NERD with urticaria (Lee et al., 2017). A recent study reported 3 subphenotypes: (1) mild-to-moderate asthma with equal sputum inflammatory cell distribution and the lowest concentrations of eicosanoids as well as low LTE4/logPGE2 ratio, (2) severe asthma with impaired lung function despite high-dose steroid use, high sputum eosinophilia, and LTE4 level with the highest LTE4/PGE2 ratio, and (3) mild-to-moderate asthma, sputum eosinophilia, and increased production of both LTE4 and PGE2 (Celejewska-Wojcik et al., 2020). Although further validation studies are needed, phenotypic classification of NERD will help achieve better control of asthma in clinical practice.
NSAID-Exacerbated Cutaneous Disease and NSAID-Induced Urticaria/Angioedema/Anaphylaxis
NSAID-exacerbated cutaneous disease is an exacerbation of skin symptoms in patients with a history of CU, whereas NIUA is found in patients without a history of CU. Exposure to NSAIDs could elicit urticaria/angioedema within 1 h, which could persist for several days (Dona et al., 2011). Patients with NIUA could develop NECD several years later (Asero, 2003; Dona et al., 2014; Lee Y. et al., 2019). NECD is a distinct phenotype of CU with a longer disease duration, which is more frequently associated with angioedema, atopy, and respiratory symptoms (Sanchez-Borges et al., 2015). Further studies are needed to classify NECD into subphenotypes which could help achieve better management of this disease.
Genetic Variants of NSAID Hypersensitivity
Diverse variations in genes involved in distinct steps related to NSAID hypersensitivity reactions are demonstrated as shown in Figure 1 and Supplementary Table 1. Genetic effects can directly affect either (1) the AA/COX pathway and its downstream signaling pathways; (2) intracellular activation/inactivation signaling of inflammatory cells, especially mast cells and eosinophils; (3) histamine/adenosine metabolism; and (4) activation of IgE receptors. The association of NSAID hypersensitivity with human leukocyte antigen (HLA) alleles, and drug-metabolizing enzymes were found (Fosbøl et al., 2008; Plaza-Serón et al., 2018).
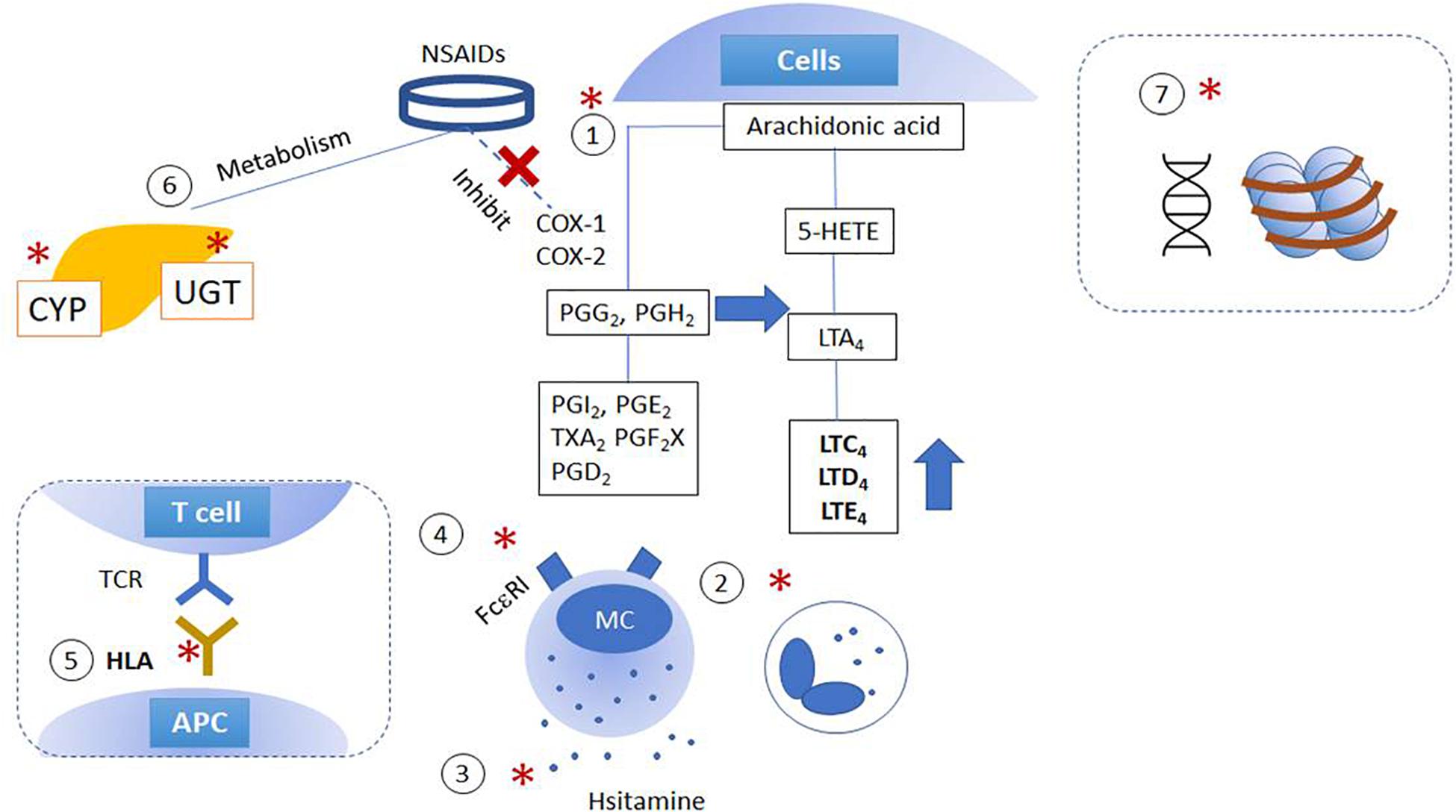
Figure 1. Schematic figures of genetic and epigenetic mechanisms of NSAID hypersensitivity. Genetic effects are suggested to affect (1) arachidonic acid/cyclooxygenase pathway; (2) cytokine and intracellular activation/inactivation signaling of inflammatory cells; (3) histamine/adenosine metabolism; (4) activation of IgE receptors, and (5) HLA and MHC class II. Moreover, further studies report the involvement of (6) mutations in enzymes involved in drug metabolism and (7) epigenetic alterations. (X): NSAIDs inhibit COX enzymes; (*) indicated the pathways which are intervened by NSAIDs. APC, antigen-presenting cells; COX, cyclooxygenase; CYP, cytochrome P450; HETE, hydroxyeicosatetraenoic acid; LT, cysteinyl leukotrienes; NSAID, non-steroidal anti-inflammatory drugs; MC, mast cells; PG, prostaglandins; TX, thromboxane; UGT, UDP-UDP-glucuronosyltransferase.
NSAID-Exacerbated Respiratory Disease
Variants in genes associated with the arachidonate 5-Lipoxygenase (5-LOX) (LT production), and COX (PG production) pathways were reported, including ALOX (Green, 2001) and LTC4S (Hedi and Norbert, 2004; Rouzer and Marnett, 2009). A variable number tandem repeats variant, in the promoter region of ALOX5 can change the transcription factor binding site and then ALOX5 down-regulation (In et al., 1997). Associations of the single SNPs of ALOX5 gene (−1780G > A, 21C > T, 270 G > A, 1782G > A) with NERD were found in Korean patients; there were no differences in allele or genotype frequencies of SNPs, but its haplotype ht1[GCGA] exhibited a higher frequency in patients with NERD than in ATA (OR = 5, 95% CI of 1.54-1.79) (Choi et al., 2004). So far, however, the data have not been replicated in other studies (Cornejo-García et al., 2012; Ayuso et al., 2015b). Up-regulation of LTC4S positive cells was found in NPs of NERD patients, which was associated with genetic polymorphisms at the LTC4 promoter region (rs730012) (Sanak et al., 1997, 2000), although it was not replicated in other populations (Ayuso et al., 2015b).
COX-1 and -2 are encoded by the prostaglandin H synthase PTGS1 and PTGS2, respectively. PTGS1 is expressed consecutively and induce the production of prostanoids, while PTGS2 is regulated by growth factors, cytokines, glucocorticoids, and bacterial endotoxin (Rouzer and Marnett, 2009). Two SNPs in PTGS1 (rs5789 and rs10306135) were significantly associated with NERD in a Spanish population (Ayuso et al., 2015b). The PTGS1 rs5789 was associated with decreased enzymatic function, while the rs10306135 variant could modulate the expression of this gene. The G-765C allele frequency of PTGS2 SNP was similar between NERD and ATA (Szczeklik et al., 2004); however, CC homozygosity was associated with the severity of NERD, which the PTGS2 −765G > C was linked with an increased production of PGD2 and PGE2 (Szczeklik et al., 2004).
Following the downstream pathway of PG, PGH2 is converted to thromboxane A2 (TBXA2) by TBXAS1, which is encoded by TBXAS1 gene. It was reported that SNP TBXAS1 (rs96229) is associated with NERD phenotype (Oh et al., 2011). TBXA2 is a platelet-derived metabolite and can cause bronchoconstriction and cell recruitment, contributing to airway hyperresponsiveness through TBXA2 receptor (TBXA2R) (Shin et al., 2003). The TBXA2R rs11085026 (795T > C) and rs4807491 (−4684C > T) were significantly associated with NERD phenotypes comparing ATA (Kim et al., 2007; Palikhe et al., 2011).
In addition, reduction in protective PGE2 is a hallmark of NSAID hypersensitivity reactions (Meade et al., 1993; Brock et al., 1999; Roca-Ferrer et al., 2011; Mastalerz et al., 2019). Previous studies demonstrated the associations of polymorphisms of PTGER2, PTGER3, and PTGER4 with NERD phenotypes. The SNPs of PTGER2 (−12813G > A, −10814T > C, −6179A > G, rs207597) were significantly associated with NERD in Japanese and Korean populations (Jinnai et al., 2004; Kim et al., 2007; Park et al., 2010). The polymorphisms of PTGER3 (rs7551789, rs7543182, and rs959) and PTGER4 −1254A > G were associated with NERD phenotypes (Park et al., 2010), in which the SNP PTGER2 −12813G > A is located in the regulatory region and assumed to involve in the down-regulation of PGE2 receptor transcription levels (Jinnai et al., 2004).
Inflammatory responses triggered by NSAID-induced reactions lead to the production of cytokines, which recruits eosinophils trafficking into the airways (Kim et al., 2010b, 2016; Bochner and Stevens, 2021). Polymorphisms of eosinophilic inflammation-related cytokines were found to play regulatory a role in PGE2 regulation. Periostin is an extracellular matrix protein and involved in cell adhesion, recruitment of inflammatory cells. Higher serum periostin levels were noted in patients with NERD (Kim et al., 2016; Wardzyńska et al., 2017). The expression of POSTN was up-regulated in CRS tissues of NERD patients (Stankovic et al., 2008). SNP −509C > T of TGFβ1was associated with NERD patients(Kim et al., 2007). NERD patients exhibited involvement of chemokine CC motif receptor 3 (CCR3)−CCR3 −520T > G and −174C > T–implicated in eosinophil recruitment, suggesting susceptibility to eosinophilic inflammation of NERD (Kim et al., 2010c).
NSAID-Exacerbated Cutaneous Disease and NSAID-Induced Urticaria/Angioedema/Anaphylaxis
Current data imply that NECD and NIUA share similar genetic backgrounds, although several distinct gene polymorphisms are discovered. Regarding the genes involved in AA metabolism, SNP LTC4S rs730012 was implicated in not only NERD patients, but also NECD and NIUA patients (Mastalerz et al., 2004; Adamjee et al., 2006).
The association between ALOX5AP rs1132340 and NIUA was confirmed (Choi et al., 2004; Cornejo-García et al., 2012). The SNP PTGER4 −1254G > A was involved in NERD and NECD phenotypes, possibly due to its location in the promoter region, leading to the down-regulation of PTGER4 in these subtypes (Park et al., 2010). Meanwhile, copy number variations in PTGER1 were detected in NIUA phenotypes with a deletion in exon 3, affected protein function, and were consecutively involve in inflammatory processes (del Carmen Plaza-Serón et al., 2016).
Mast cells are key players in patients with NSAID hypersensitivity. Cutaneous symptoms are triggered by degranulation of cutaneous or submucosal mast cells secreting histamine and other metabolites. The SNPs FCER1A −344C > T, FCER1B E237G (A > G), and FCER1G −237A > G were associated with susceptibility to atopy and higher IgE levels in NECD patients (Bae et al., 2007; Palikhe et al., 2008). Moreover, the PLA2G4A (rs12746200), PLCG1 (rs2228246), and TNFRS11A (rs1805034) were significantly associated with NIUA. The frequency of haplotype PLCG1 (rs753381-rs2228246, C-G) was lower in NIUA patients (mainly presenting angioedema), while that of TNFRS11A rs1805034-rs35211496 (C-T) was higher among patients with NIUA mainly presenting urticaria or those presenting both urticaria and angioedema, compared to control groups (Ayuso et al., 2015a). Thus, analysis of the genetic background of patients with NECD or NIUA can help identify predisposing factors.
Genes of Drug-Metabolizing Enzymes (DMEs)
Non-steroidal anti-inflammatory drugs are metabolized by phase I drug-metabolizing enzymes (DMEs) [predominantly cytochrome P450 (CYPs)] and phase II DMEs (e.g., UDP-glucuronosyltransferases) (Wyatt et al., 2012). The polymorphisms of DMEs are known to cause interindividual differences in pharmacodynamic responses, pharmacokinetics and adverse reactions, which could be related to NSAID hypersensitivity. Numerous studies have reported the associations of CYP2C polymorphisms with NSAID- induced adverse reactions and toxicity (Wyatt et al., 2012; Krasniqi et al., 2016), however, the associations of NSAID hypersensitivity and CYP2C polymorphisms are not well studied. A study in a Japanese population showed that the GA/AA genotypes of both CYP2C19 681G > A and 636G > A were associated with NERD as well as lower forced expiratory volume in the first second (FEV1%) predicted values compared to the GG genotypes (Kohyama et al., 2011). More studies are needed to understand the associations of CYP as well as other DEM polymorphisms with NSAID hypersensitivity.
New Candidate Genes
Genome-wide association studies (GWAS) provide multiple and novel candidate genes involved in NSAID hypersensitivity, as shown in Supplementary Table 1. CEP68 was found to be involved in NERD and NIUA with or without NECD phenotypes. Although the exact function of CEP68 remains unknown, its polymorphisms (rs7572857) are associated with changes in FEV1(%) after NSAID administration, and CEP68 was thus believed to be a susceptibility gene for NSAID hypersensitivity (Kim et al., 2010a). However, the association of CEP68 polymorphisms with NERD or blended reactions were indicated (Cornejo-García et al., 2014). The variant CEP68 rs1050675 (located in the 3′ UTR region) could intervene the target of recognition of some transcription factors (Kim et al., 2010a; Cornejo-García et al., 2014). In terms of NIUA phenotype, a GWAS suggested the nominal associations among live loci in the following genes: HLF, RAD51L1, COL24A1, GalNAc-T13, and FBXL17 (Park et al., 2013). Novel signatures of acute exacerbation have been identified, including EIF2AK2, MSRA, and MSRB2 in patients with asthma. EIF2AK2 is a key gene for an antiviral defense mechanism, while MRSA and MSRB2 are involved in the oxidative stress pathway (Kang et al., 2020). It has been demonstrated that patients with NERD expressed a higher level of oxidative stress, and viral infection may worsen the exacerbation of NERD (Adeli et al., 2019; Kim et al., 2019). Therefore, these genes may be potential markers predicting the respiratory exacerbation in patients with NERD. Additional studies are needed to validate the above-mentioned genes for replication, as well as to elucidate their functions.
Other Phenotypes
Some patients with NIUA develop anaphylaxis, which is called NIUAA, and those with NIUAA showed higher allelic frequency of HLA-DRB1∗11 compared to control groups (Quiralte et al., 1999). Another rare subtype is SNIUAA, in which polymorphism of NAT2 encoding for N-acetyltransferase 2 (NAT2), including NAT2∗5, ∗6, ∗7, and ∗14, was found to be associated with this phenotype. Through NAT2, blood LT can be inhibited by N-acetylation. Therefore, polymorphisms of NAT2 can increase LT levels and the risk of anaphylactoid reactions (García-Martín et al., 2015). Taken together, the genetic background plays a crucial role in NSAID hypersensitivity (Adeli et al., 2019).
Epigenetics of NSAID Hypersensitivity
Gene expressions are modulated by several mechanisms including DNA and histone modification (Figure 1). At the DNA level, CpG islands are modified by 5-methylcytosine (5mC), 5-hydroxymethycystosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). 5hmC could facilitate gene transcription, 5mc, 5fC, and 5caC decrease gene transcription by inhibiting transcription factor binding, and promote chromatin condensation (Lauschke et al., 2019). Previous studies have reported different expression levels of various genes related to immune response and/or the dysregulation of cysteinyl LTs and PG production, suggesting the involvement of epigenetic mechanisms in the pathophysiology of NSAID hypersensitivity (Roca-Ferrer et al., 2011; Lee Y. et al., 2019).
A previous study investigating genome-wide DNA methylation levels in 5 patients with NERD showed hypermethylation of 332 CpG islands in 296 genes and hypomethylation of 158 CpG islands in 141 genes. The hypomethylation was found in genes involved in lymphocyte proliferation, leukocyte activation, cytokine production, immune responses, inflammation, and immunoglobulin binding, whereas hypermethylation was found in genes involved in hemostasis, wound healing, calcium ion binding, and oxidoreductase activity. In addition, PGDS, ALOX5AP, and LTB4R were hypomethylated, while PTGES was hypermethylated in NERD compared to ATA patients. Genes related to Th2-immune response, including IL5RA and IL-10 were differently methylated between NERD and ATA groups (Cheong et al., 2011). Another study reported a lower expression of the EP2 receptor in nasal fibroblasts of NERD patients as well as EP2 mRNA expression correlated with histone acetylation (H3K27ac) levels at the EP2 promoter (Cahill et al., 2016). A recent study showed different expression levels of genes related to chemotaxis (CXCL1-CXCL3, PPBP, CXCL8, CCL18, and CCL20) and host defense (CD1A-CD1C, CLEC10A, and CLEC18B) in alveolar monocyte-derived macrophages (aMDM) from NERD patients. Moreover, aMDM of NERD patients had 3930-fold decrease and 211-fold increase at differentially methylated positions in genes involved in cell recruitment and acylcarnitine metabolism compared to controls (Haimerl et al., 2020). These findings suggest the involvement of epigenetic regulation in the pathogenesis of NERD.
Nevertheless, results of studies on epigenetic mechanisms should be interpreted with caution. First, epigenetic modification patterns vary among different types of cell and tissue; therefore, the cellular components are crucial for the investigation of the complicated pathogenesis of NSAID hypersensitivity. Secondly, epigenetic alterations could be the consequence of pharmacological effects of NSAID, rather than the cause of hypersensitivity reactions. Previous studies showed that NSAID exposure could increase the expression levels of DNMT3a and DMNT3b mRNA (Lee Y. et al., 2019), induce the promoter demethylation of Secreted Protein Acidic and Cysteine Rich (SPARC) (Pan et al., 2008), and be associated with a lower incidence of E-cadherin (CDH1) promoter methylation (Tahara et al., 2010). These findings suggest that NSAID could affect epigenetic regulations; however, the mechanisms underlying this phenomenon are not fully understood. In addition, there is a need for further studies with larger sample sizes and different populations to achieve a stronger statistical power. Functional studies of the variants should be conducted to assess which epigenetic mechanisms are significantly involved in each phenotype of NSAID hypersensitivity.
Summary and Conclusion
Non-steroidal anti-inflammatory drug hypersensitivity has different clinical phenotypes and subphenotypes, and it is the consequence of complicated pathophysiological mechanisms. Its underlying mechanisms are regulated by genetic and epigenetic variants and possible interactions between them, which could be different among populations. Genes related to the AA/COX pathway or immune cell activation are frequently candidates for studies; however, further genetic studies on other inflammatory cascades are warranted. Moreover, functional studies to determine the roles of candidate’s genetic and epigenetic polymorphisms are essential depending on various phenotypes.
Author Contributions
HT, LP, and KL searched for the literature review, summarized, and wrote the manuscript. H-SP outlined the review’s sections, revised the manuscript, and supervised the whole process. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a grant from the Korea Research-Driven Hospitals project (HR16C0001) of Korea Health Industry Development Institute and the Ministry of Health and Welfare, ROK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.647257/full#supplementary-material
Abbreviations
AA, arachidonic acid; ADORA3, adenosine A3 receptor; ALOX5 (5-LOX), arachidonate 5-lipoxygenase; aMDM, alveolar monocyte-derived macrophages; APC, antigen-presenting cells; ATA, aspirin/NSAID-tolerant asthmatics; CCR3, chemokine CC motif receptor 3; CEP68, centrosomal protein 68; COX, cyclooxygenase; cPLA2, cytosolic phospholipase; CRS, chronic rhinosinusitis; CRSwNP, chronic rhinosinusitis with nasal polyps; CTLA4, cytotoxic T-lymphocyte -associated protein 4; CU, chronic urticaria; CXCL, chemokine (C-X-C motif) ligand; CYP, cytochrome P450; DAG, 1,2-diacylglycerol; DAO, d-amino acid oxidase; DCBLD2, discoidin CUB and LCCL domain containing 2; DHR, drug-induced hypersensitivity; DME, drug-metabolizing enzymes; DPP10, dipeptidyl peptidase like 10; EMID2, emilin and multimerin domain-containing protein 2; HETE, hydroxyeicosatetraenoic acid; HLA, human leukocyte antigen; Ig, immunoglobulin; IL, interleukin; IP3, inositol 1,4,5-trisphosphate; LOX, lipoxygenase; LT, cysteinyl leukotrienes; NAT2, N-acetyltransferase 2; NECD, NSAID-exacerbated cutaneous disease; NIDHR, NSAID-induced delayed hypersensitivity reaction; NIUAA, NSAID-induced urticaria/angioedema/anaphylaxis; NP, nasal polyps; NSAID, non-steroidal anti-inflammatory drugs; MC, mast cells; MS4A2, membrane spanning 4-domains A2; MSRA(B2), methionine sulfoxide reductase A(B2); NLRP3, nucleotide-binding oligomerization domain-like receptor protein 3; PG, prostaglandins; PIP2, phospholipid phosphatidylinositol 4,5-bisphosphate; PL, phospholipase; PPBP, pro-platelet basic protein; PTGS, prostaglandin H synthase; PTGIR, prostaglandin I2 receptor; RAB1A, Ras-related protein Rab-1A; SLC6A12, solute carrier family 6 member 12; SNIUAA, single NSAID-induced urticarial/angioedema/anaphylaxis; TAPBP, transporter associated with antigen processing (TAP) binding protein; TBXAS1, thromboxane A synthase 1; TBXA2R, thromboxane A2 Receptor; TGF β 1, transforming growth factor β 1; TNFRSF1A, tumor necrosis factor receptor superfamily member 1A; TX, thromboxane; UBE3C, ubiquitin protein ligase E3C; UDP, uridine diphospho glucuronic acid; UGT, UDP-glucuronosyltransferase.
References
Adamjee, J., Suh, Y. J., Park, H. S., Choi, J. H., Penrose, J. F., Lam, B. K., et al. (2006). Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J. Pathol. 209, 392–399. doi: 10.1002/path.1979
Adeli, M., El-Shareif, T., and Hendaus, M. A. (2019). Asthma exacerbation related to viral infections: an up to date summary. J. Family. Med. Prim. Care. 8:2753. doi: 10.4103/jfmpc.jfmpc_86_19
Agúndez, J. A., Ayuso, P., Cornejo-García, J. A., Blanca, M., Torres, M. J., Doña, I., et al. (2012). The diamine oxidase gene is associated with hypersensitivity response to non-steroidal anti-inflammatory drugs. PLoS One 7:e47571.
Asero, R. (2003). Intolerance to nonsteroidal anti-inflammatory drugs might precede by years the onset of chronic urticaria. J. Allergy. Clin. Immunol. 111, 1095–1098. doi: 10.1067/mai.2003.1444
Ayuso, P., Blanca-López, N., Doña, I., Torres, M., Guéant-Rodríguez, R., Canto, G., et al. (2013). Advanced phenotyping in hypersensitivity drug reactions to NSAID s. Clin. Exp. Allergy 43, 1097–1109.
Ayuso, P., del Carmen Plaza-Serón, M., Doña, I., Blanca-López, N., Campo, P., Cornejo-García, J. A., et al. (2015a). Association study of genetic variants in PLA2G4A, PLCG1, LAT, SYK, and TNFRS11A genes in NSAIDs-induced urticaria and/or angioedema patients. Pharmacogenet. Genom. 25, 618–621. doi: 10.1097/fpc.0000000000000179
Ayuso, P., Plaza-Serón, M. D. C., Blanca-López, N., Doña, I., Campo, P., Canto, G., et al. (2015b). Genetic variants in arachidonic acid pathway genes associated with NSAID-exacerbated respiratory disease. Pharmacogenomics 16, 825–839. doi: 10.2217/pgs.15.43
Bae, J. S., Kim, S. H., Ye, Y. M., Yoon, H. J., Suh, C. H., Nahm, D. H., et al. (2007). Significant association of FcεRIα promoter polymorphisms with aspirin-intolerant chronic urticaria. J. Allergy Clin. Immnunol. 119, 449–456. doi: 10.1016/j.jaci.2006.10.006
Blanca, M., Perez, E., Garcia, J. J., Miranda, A., Terrados, S., Vega, J. M., et al. (1989). Angioedema and IgE antibodies to aspirin: a case report. Ann. Allergy 62, 295–298.
Bochenek, G., Kuschill-Dziurda, J., Szafraniec, K., Plutecka, H., Szczeklik, A., and Nizankowska-Mogilnicka, E. (2014). Certain subphenotypes of aspirin-exacerbated respiratory disease distinguished by latent class analysis. J. Allergy Clin. Immunol. 133, 98–103.e1-e6. doi: 10.1016/j.jaci.2013.07.004
Bochner, B. S., and Stevens, W. W. (2021). Biology and function of eosinophils in chronic rhinosinusitis with or without nasal polyps. Allergy Asthma Immunol. Res. 13, 8–22. doi: 10.4168/aair.2021.13.1.8
Brock, T. G., McNish, R. W., and Peters-Golden, M. (1999). Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol Chem. 274, 11660–11666. doi: 10.1074/jbc.274.17.11660
Bruno, A., Tacconelli, S., and Patrignani, P. (2014). Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic. Clin. Pharmacol. 114, 56–63.
Buchheit, K. M., Cahill, K. N., Katz, H. R., Murphy, K. C., Feng, C., Lee-Sarwar, K., et al. (2016). Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J. Allergy. Clin. Immnunol. 137, 1566–1576.e5. doi: 10.1016/j.jaci.2015.10.020.
Cahill, K. N., Raby, B. A., Zhou, X., Guo, F., Thibault, D., Baccarelli, A., et al. (2016). Impaired E prostanoid2 expression and resistance to prostaglandin e2 in nasal polyp fibroblasts from subjects with aspirin-exacerbated respiratory disease. Am. J. Respir. Cell. Mol. Biol. 54, 34–40. doi: 10.1165/rcmb.2014-0486OC
Celejewska-Wojcik, N., Wojcik, K., Ignacak-Popiel, M., Cmiel, A., Tyrak, K., Gielicz, A., et al. (2020). Subphenotypes of nonsteroidal antiinflammatory disease-exacerbated respiratory disease identified by latent class analysis. Allergy 75, 831–840. doi: 10.1111/all.14141
Cheong, H. S., Park, S. M., Kim, M. O., Park, J. S., Lee, J. Y., Byun, J. Y., et al. (2011). Genome-wide methylation profile of nasal polyps: relation to aspirin hypersensitivity in asthmatics. Allergy 66, 637–644. doi: 10.1111/j.1398-9995.2010.02514.x
Cho, S. H., Park, J. S., Park, B. L., Bae, D. J., Uh, S. T., Kim, M. K., et al. (2013). Association analysis of tapasin polymorphisms with aspirin-exacerbated respiratory disease in asthmatics. Pharmacogenet. Genom. 23, 341–348. doi: 10.1097/fpc.0b013e328361d4bb
Choi, J. H., Kim, S. H., Cho, B., Lee, S., Kim, S., Suh, C., et al. (2009). Association of TNF-α promoter polymorphisms with aspirin-induced urticaria. J. Clin. Pharm. 34, 231–238. doi: 10.1111/j.1365-2710.2008.00979.x
Choi, J. H., Park, H. S., Oh, H. B., Lee, J. H., Suh, Y. J., Park, C. S., et al. (2004). Leukotriene-related gene polymorphisms in ASA-intolerant asthma: an association with a haplotype of 5-lipoxygenase. Hum. Genet. 114, 337–344. doi: 10.1007/s00439-004-1082-1
Choi, Y., Lee, Y., and Park, H. S. (2019). Which factors associated with activated eosinophils contribute to the pathogenesis of aspirin-exacerbated respiratory disease? Allergy Asthma Immunol. Res. 11, 320–329. doi: 10.4168/aair.2019.11.3.320
Comhair, S. A. A., Bochenek, G., Baicker-McKee, S., Wang, Z., Stachura, T., Sanak, M., et al. (2018). The utility of biomarkers in diagnosis of aspirin exacerbated respiratory disease. Respir. Res. 19:210. doi: 10.1186/s12931-018-0909-6
Cornejo-García, J. A., Flores, C., Plaza-Serón, M. C., Acosta-Herrera, M., Blanca-López, N., Doña, I., et al. (2014). Variants of CEP68 gene are associated with acute urticaria/angioedema induced by multiple non-steroidal anti-inflammatory drugs. PLoS One 9:e90966. doi: 10.1371/journal.pone.0090966
Cornejo-García, J., Jagemann, L., Blanca-López, N., Doña, I., Flores, C., Guéant-Rodríguez, R., et al. (2012). Genetic variants of the arachidonic acid pathway in non-steroidal anti-inflammatory drug-induced acute urticaria. Clin. Exp. Allergy 42, 1772–1781. doi: 10.1111/j.1365-2222.2012.04078.x
Dekker, J., Nizankowska, E., Schmitz-Schumann, M., Pile, K., Bochenek, G., Dyczek, A., et al. (1997). Aspirin-induced asthma and HLA-DRB1 and HLA-DPB1 genotypes. Clin. Exp. Allergy 27, 574–577. doi: 10.1046/j.1365-2222.1997.540848.x
del Carmen Plaza-Serón, M., Ayuso, P., Pérez-Sánchez, N., Doña, I., Blanca-Lopez, N., Flores, C., et al. (2016). Copy number variation in ALOX5 and PTGER1 is associated with NSAIDs-induced urticaria and/or angioedema. Pharmacogenet. Genom. 26, 280–287. doi: 10.1097/fpc.0000000000000216
Dennis, E. A., Cao, J., Hsu, Y. H., Magrioti, V., and Kokotos, G. (2011). Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185. doi: 10.1021/cr200085w
Dholia, N., Sethi, G. S., Naura, A. S., and Yadav, U. C. S. (2020). Cysteinyl leukotriene D(4) (LTD(4)) promotes airway epithelial cell inflammation and remodelling. Inflamm. Res. 70, 109–126. doi: 10.1007/s00011-020-01416-z
Doña, I., Barrionuevo, E., Salas, M., Cornejo-García, J., Perkins, J., Bogas, G., et al. (2017). Natural evolution in patients with nonsteroidal anti-inflammatory drug-induced urticaria/angioedema. Allergy 72, 1346–1355. doi: 10.1111/all.13147
Dona, I., Blanca-Lopez, N., Cornejo-Garcia, J. A., Torres, M. J., Laguna, J. J., Fernandez, J., et al. (2011). Characteristics of subjects experiencing hypersensitivity to non-steroidal anti-inflammatory drugs: patterns of response. Clin. Exp. Allergy 41, 86–95. doi: 10.1111/j.1365-2222.2010.03651.x
Dona, I., Blanca-Lopez, N., Torres, M. J., Gomez, F., Fernandez, J., Zambonino, M. A., et al. (2014). NSAID-induced urticaria/angioedema does not evolve into chronic urticaria: a 12-year follow-up study. Allergy 69, 438–444. doi: 10.1111/all.12335
Doña, I., Pérez-Sánchez, N., Eguiluz-Gracia, I., Muñoz-Cano, R., Bartra, J., Torres, M. J., et al. (2020). Progress in understanding hypersensitivity reactions to nonsteroidal anti-inflammatory drugs. Allergy 75, 561–575. doi: 10.1111/all.14032
Esmaeilzadeh, H., Nabavi, M., Amirzargar, A. A., Aryan, Z., Arshi, S., Bemanian, M. H., et al. (2015). HLA-DRB and HLA-DQ genetic variability in patients with aspirin-exacerbated respiratory disease. J. Rhinol. Allergy. 29, e63–e69.
Fosbøl, E. L., Gislason, G. H., Jacobsen, S., Abildstrom, S. Z., Hansen, M. L., Schramm, T. K., et al. (2008). The pattern of use of non-steroidal anti-inflammatory drugs (NSAIDs) from 1997 to 2005: a nationwide study on 4.6 million people. Pharmacoepidemiol. Drug. Saf. 17, 822–833. doi: 10.1002/pds.1592
García-Martín, E., Esguevillas, G., Blanca-López, N., García-Menaya, J., Blanca, M., Amo, G., et al. (2015). Genetic determinants of metamizole metabolism modify the risk of developing anaphylaxis. Pharmacogenet. Genom. 25, 462–464. doi: 10.1097/fpc.0000000000000157
Gomes, E., Cardoso, M. F., Praca, F., Gomes, L., Marino, E., and Demoly, P. (2004). Self-reported drug allergy in a general adult Portuguese population. Clin. Exp. Allergy 34, 1597–1601. doi: 10.1111/j.1365-2222.2004.02070.x
Green, G. A. (2001). Understanding NSAIDs: from aspirin to COX-2. Clin. Cornerst. 3, 50–59. doi: 10.1016/s1098-3597(01)90069-9
Haimerl, P., Bernhardt, U., Schindela, S., Henkel, F. D. R., Lechner, A., Zissler, U. M., et al. (2020). Inflammatory macrophage memory in nonsteroidal anti-inflammatory drug-exacerbated respiratory disease. J. Allergy Clin. Immunol. 147, 587–599. doi: 10.1016/j.jaci.2020.04.064
Hedi, H., and Norbert, G. (2004). 5-Lipoxygenase pathway, dendritic cells, and adaptive immunity. Biomed. Res. Int. 2004, 99–105. doi: 10.1155/s1110724304310041
Hennekens, C. H., Dyken, M. L., and Fuster, V. (1997). Aspirin as a therapeutic agent in cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation 96, 2751–2753. doi: 10.1161/01.cir.96.8.2751
Hitomi, Y., Ebisawa, M., Tomikawa, M., Imai, T., Komata, T., Hirota, T., et al. (2009). Associations of functional NLRP3 polymorphisms with susceptibility to food-induced anaphylaxis and aspirin-induced asthma. J. Allergy Clin. Immnunol. 124, 779–785.e6.
In, K. H., Asano, K., Beier, D., Grobholz, J., Finn, P., Silverman, E., et al. (1997). Naturally occurring mutations in the human 5-lipoxygenase gene promoter that modify transcription factor binding and reporter gene transcription. J. Clin. Invest. 99, 1130–1137. doi: 10.1172/jci119241
Ishimoto, T., Akiba, S., Sato, T., and Fujii, T. (1994). Contribution of phospholipases A2 and D to arachidonic acid liberation and prostaglandin D2 formation with increase in intracellular Ca2+ concentration in rat peritoneal mast cells. Euro. J. Biochem. 219, 401–406. doi: 10.1111/j.1432-1033.1994.tb19952.x
Jinnai, N., Sakagami, T., Sekigawa, T., Kakihara, M., Nakajima, T., Yoshida, K., et al. (2004). Polymorphisms in the prostaglandin E2 receptor subtype 2 gene confer susceptibility to aspirin-intolerant asthma: a candidate gene approach. Hum. Mol. Genet. 13, 3203–3217. doi: 10.1093/hmg/ddh332
Kang, M. G., Lee, H. S., Tantisira, K. G., and Park, H. W. (2020). Genetic signatures of acute asthma exacerbation related with ineffective response to corticosteroid. Allergy. Asthma Immunol. Res. 12:626. doi: 10.4168/aair.2020.12.4.626
Kano, M., Ohno-Shosaku, T., Hashimotodani, Y., Uchigashima, M., and Watanabe, M. (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380. doi: 10.1152/physrev.00019.2008
Kim, J. H., Park, B. L., Cheong, H. S., Bae, J. S., Park, J. S., Jang, A. S., et al. (2010a). Genome-wide and follow-up studies identify CEP68 gene variants associated with risk of aspirin-intolerant asthma. PLoS One 5:e13818. doi: 10.1371/journal.pone.0013818
Kim, J. M., Park, B. L., Park, S. M., Lee, S. H., Kim, M. O., Jung, S., et al. (2010d). Association analysis of N-acetyl transferase-2 polymorphisms with aspirin intolerance among asthmatics. Pharmacogenomics 11, 951–958. doi: 10.2217/pgs.10.65
Kim, M. A., Yoon, M. K., Lee, Y. S., Izuhara, K., Ohta, S., Ono, J., et al. (2016). Clinical implication of the serum periostin level for differentiating phenotypes of NSAID hypersensitivity. Allergol. Int. 65, 492–494. doi: 10.1016/j.alit.2016.04.013
Kim, S. H. (2019). Sphingosine-1-phosphate: Biomarker, contributor, or target for asthma? Allergy Asthma Immunol. Res. 11, 299–301. doi: 10.4168/aair.2019.11.3.299
Kim, S. H., Choi, H., Yoon, M. G., Ye, Y. M., and Park, H. S. (2015). Dipeptidyl-peptidase 10 as a genetic biomarker for the aspirin-exacerbated respiratory disease phenotype. Ann. Allergy Asthma Immunol. 114, 208–213. doi: 10.1016/j.anai.2014.12.003
Kim, S. H., Choi, J. H., Lee, K. W., Kim, S. H., Shin, E. S., Oh, H. B., et al. (2005). The human leucocyte antigen-DRB1∗ 1302-DQB1∗ 0609-DPB1∗ 0201 haplotype may be a strong genetic marker for aspirin-induced urticaria. Clin. Exp. Allergy 35, 339–344. doi: 10.1111/j.1365-2222.2004.02197.x
Kim, S. H., Kang, Y. M., Kim, S. H., Cho, B. Y., Ye, Y. M., Hur, G. Y., et al. (2009a). Histamine N-methyltransferase 939A> G polymorphism affects mRNA stability in patients with acetylsalicylic acid-intolerant chronic urticaria. Allergy 64, 213–221. doi: 10.1111/j.1398-9995.2008.01795.x
Kim, S. H., Kim, Y. K., Park, H. W., Kim, S. H., Kim, S. H., Ye, Y. M., et al. (2009b). Adenosine deaminase and adenosine receptor polymorphisms in aspirin-intolerant asthma. Respir. Res. 103, 356–363. doi: 10.1016/j.rmed.2008.10.008
Kim, S. H., Nam, E. J., Kim, Y. K., Ye, Y. M., and Park, H. S. (2010b). Functional variability of the adenosine A3 receptor (ADORA3) gene polymorphism in aspirin-induced urticaria. Br. J. Dermatol. 163, 977–985. doi: 10.1111/j.1365-2133.2010.09983.x
Kim, S. H., Oh, J. M., Kim, Y. S., Palmer, L., Suh, C. H., Nahm, D. H., et al. (2006a). Cysteinyl leukotriene receptor 1 promoter polymorphism is associated with aspirin-intolerant asthma in males. Clin. Exp. Allergy 36, 433–439. doi: 10.1111/j.1365-2222.2006.02457.x
Kim, S. H., Park, H. S., Holloway, J. W., Shin, H. D., and Park, C. S. (2007). Association between a TGFβ1 promoter polymorphism and rhinosinusitis in aspirin-intolerant asthmatic patients. Respir. Res. 101, 490–495. doi: 10.1016/j.rmed.2006.07.002
Kim, S. H., Son, J., Yang, E. M., Kim, J. E., and Park, H. S. (2011). A functional promoter polymorphism of the human IL18 gene is associated with aspirin-induced urticaria. Br. J. Dermatol. 165, 976–984. doi: 10.1111/j.1365-2133.2011.10467.x
Kim, S. H., Uuganbayar, U., Trinh, H. K. T., Pham, D. L., Kim, N., Kim, M., et al. (2019). Evaluation of neutrophil activation status according to the phenotypes of adult asthma. Allergy Asthma Immunol. Res. 11, 381–393. doi: 10.4168/aair.2019.11.3.381
Kim, S. H., Yang, E. M., Lee, H. N., Choi, G.-S., Ye, Y.-M., and Park, H.-S. (2010c). Association of the CCR3 gene polymorphism with aspirin exacerbated respiratory disease. Respir. Res. 104, 626–632. doi: 10.1016/j.rmed.2009.11.024
Kim, S. H., Ye, Y. M., Lee, S. K., Choi, J. H., Holloway, J., Park, C. S., et al. (2006b). Association of TNF-α genetic polymorphism with HLA DPB1∗ 0301. Clin. Exp. Allergy 36, 1247–1253. doi: 10.1111/j.1365-2222.2006.02567.x
Klar, H., Sotošek, N., Šelb, J., and Košnik, M. (2019). Selective hypersensitivity to a single nonsteroidal anti-inflammatory drug. Acta Dermatovenerol. Alp. Pannonica. Adriat. 28, 97–101.
Kohyama, K., Abe, S., Kodaira, K., Yukawa, T., Hozawa, S., Morioka, J., et al. (2011). Polymorphisms of the CYP2C19 gene in Japanese patients with aspirin-exacerbated respiratory disease. J Allergy Clin. Immunol. 128:1117. doi: 10.1016/j.jaci.2011.07.013
Kowalski, M. L., and Makowska, J. S. (2015). Seven steps to the diagnosis of NSAIDs hypersensitivity: how to apply a new classification in real practice? Allergy Asthma Immunol. Res. 7, 312–320. doi: 10.4168/aair.2015.7.4.312
Kowalski, M. L., and Stevenson, D. D. (2013). Classification of reactions to nonsteroidal antiinflammatory drugs. Immunol. Allergy Clin. North. Am. 33, 135–145. doi: 10.1016/j.iac.2012.10.008
Krasniqi, V., Dimovski, A., Domjanović, I. K., Bilić, I., and Božina, N. (2016). How polymorphisms of the cytochrome P450 genes affect ibuprofen and diclofenac metabolism and toxicity/Kako polimorfizmi gena citokroma P450 utječu na metabolizam i toksičnost ibuprofena i diklofenaka. Arh. Hig. Rada Toksikol. 67, 1–8. doi: 10.1515/aiht-2016-67-2754
Lauschke, V. M., Zhou, Y., and Ingelman-Sundberg, M. (2019). Novel genetic and epigenetic factors of importance for inter-individual differences in drug disposition, response and toxicity. Pharmacol. Ther. 197, 122–152. doi: 10.1016/j.pharmthera.2019.01.002
Lee, H. Y., Ye, Y. M., Kim, S. H., Ban, G. Y., Kim, S. C., Kim, J. H., et al. (2017). Identification of phenotypic clusters of nonsteroidal anti-inflammatory drugs exacerbated respiratory disease. Allergy 72, 616–626. doi: 10.1111/all.13075
Lee, J. S., Kim, J. H., Bae, J. S., Kim, J. Y., Park, T. J., Pasaje, C. F., et al. (2010). Association analysis of UBE3C polymorphisms in Korean aspirin-intolerant asthmatic patients. Ann. Allergy. Asthma. Immunol. 105, 307–312.e1.
Lee, J. U., Park, J. S., Chang, H. S., and Park, C. S. (2019). Complementary participation of genetics and epigenetics in development of NSAID-exacerbated respiratory disease. Allergy Asthma Immunol. Res. 11, 779–794. doi: 10.4168/aair.2019.11.6.779
Lee, Y., Shin, Y. S., and Park, H. S. (2019). New phenotypes in hypersensitivity reactions to nonsteroidal anti-inflammatory drugs. Curr. Opin. Allergy Clin. Immunol. 19, 302–307. doi: 10.1097/ACI.0000000000000541
Mastalerz, L., Setkowicz, M., Sanak, M., and Szczeklik, A. (2004). Hypersensitivity to aspirin: common eicosanoid alterations in urticaria and asthma. J. Allergy Clin. Immnunol. 113, 771–775. doi: 10.1016/j.jaci.2003.12.323
Mastalerz, L., Tyrak, K. E., Ignacak, M., Konduracka, E., Mejza, F., Ćmiel, A., et al. (2019). Prostaglandin E(2) decrease in induced sputum of hypersensitive asthmatics during oral challenge with aspirin. Allergy 74, 922–932. doi: 10.1111/all.13671
Matsuo, H., Yokooji, T., Morita, H., Ooi, M., Urata, K., Ishii, K., et al. (2013). Aspirin augments IgE-mediated histamine release from human peripheral basophils via Syk kinase activation. Allergol. Int. 62, 503–511. doi: 10.2332/allergolint.13-oa-0536
Meade, E. A., Smith, W. L., and Dewitt, D. L. (1993). Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 268, 6610–6614. doi: 10.1016/s0021-9258(18)53294-4
Mitsui, C., Kajiwara, K., Hayashi, H., Ito, J., Mita, H., Ono, E., et al. (2016). Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immnunol. 137, 400–411. doi: 10.1016/j.jaci.2015.05.041
Mullarkey, M. F., Thomas, P. S., Hansen, J. A., Webb, D., and Nisperos, B. (1986). Association of aspirin-sensitive asthma with HLA-DQw2. Am. J. Respir. Crit. Care. Med. 133, 261–263.
Naraba, H., Murakami, M., Matsumoto, H., Shimbara, S., Ueno, A., Kudo, I., et al. (1998). Segregated coupling of phospholipases A2, cyclooxygenases, and terminal prostanoid synthases in different phases of prostanoid biosynthesis in rat peritoneal macrophages. J. Immunol. 160, 2974–2982.
Nørregaard, R., Kwon, T.-H., and Frøkićr, J. (2015). Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney. Res. Clin. Pract. 34, 194–200. doi: 10.1016/j.krcp.2015.10.004
Oh, S. H., Kim, Y. H., Park, S. M., Cho, S. H., Park, J. S., Jang, A. S., et al. (2011). Association analysis of thromboxane A synthase 1 gene polymorphisms with aspirin intolerance in asthmatic patients. Pharmacogenomics 12, 351–363. doi: 10.2217/pgs.10.181
Pacor, M. L., Di Lorenzo, G., Mansueto, P., Martinelli, N., Esposito-Pellitteri, M., Pradella, P., et al. (2006). Relationship between human leucocyte antigen class I and class II and chronic idiopathic urticaria associated with aspirin and/or NSAIDs hypersensitivity. Med. Inflamm. 2006, 62489–62489. doi: 10.1155/MI/2006/62489
Palikhe, N. S., Sin, H. J., Kim, S. H., Sin, H. J., Hwang, E. K., Ye, Y. M., et al. (2012). Genetic variability of prostaglandin E2 receptor subtype EP4 gene in aspirin-intolerant chronic urticaria. J. Hum. Genet. 57, 494–499. doi: 10.1038/jhg.2012.55
Palikhe, N., Kim, S. H., Lee, H. Y., Kim, J. H., Ye, Y. M., and Park, H. S. (2011). Association of thromboxane A2 receptor (TBXA2R) gene polymorphism in patients with aspirin-intolerant acute urticaria. Clin. Exp. Allergy 41, 179–185. doi: 10.1111/j.1365-2222.2010.03642.x
Palikhe, N., Kim, S. H., Yang, E. M., Kang, Y. M., Ye, Y. M., Hur, G. Y., et al. (2008). Analysis of high-affinity IgE receptor (FcεR1) polymorphisms in patients with aspirin-intolerant chronic urticaria. Allergy Asthma Procs. 29, 250–257. doi: 10.2500/aap.2008.29.3116
Palikhe, S., Palikhe, N. S., Kim, S. H., Yoo, H. S., Shin, Y. S., and Park, H. S. (2014). Elevated platelet activation in patients with chronic urticaria: a comparison between aspirin-intolerant and aspirin-tolerant groups. Ann. Allergy Asthma Immunol. 113, 276–281. doi: 10.1016/j.anai.2014.06.011
Pan, M. R., Chang, H. C., Chuang, L. Y., and Hung, W. C. (2008). The nonsteroidal anti-inflammatory drug NS398 reactivates SPARC expression via promoter demethylation to attenuate invasiveness of lung cancer cells. Exp. Biol. Med. (Maywood) 233, 456–462. doi: 10.3181/0709-RM-257
Park, B. L., Kim, T. H., Kim, J. H., Bae, J. S., Pasaje, C. F. A., Cheong, H. S., et al. (2013). Genome-wide association study of aspirin-exacerbated respiratory disease in a Korean population. Hum. Genet. 132, 313–321. doi: 10.1007/s00439-012-1247-2
Park, B. L., Park, S. M., Park, J. S., Uh, S. T., Kim, Y. H., Choi, I. S., et al. (2010). Association of PTGER gene family polymorphisms with aspirin intolerant asthma in Korean asthmatics. BMB Rep. 43, 445–449. doi: 10.5483/bmbrep.2010.43.6.445
Park, J. S., Chang, H. S., Park, C. S., Lee, J. H., Lee, Y. M., Choi, J. H., et al. (2005). Association analysis of cysteinyl-leukotriene receptor 2 (CYSLTR2) polymorphisms with aspirin intolerance in asthmatics. Pharmacogenet. Genom. 15, 483–492. doi: 10.1097/01.fpc.0000166456.84905.a0
Park, J. S., Heo, J. S., Chang, H. S., Choi, I. S., Kim, M. K., Lee, J. U., et al. (2014). Association analysis of member RAS oncogene family gene polymorphisms with aspirin intolerance in asthmatic patients. DNA Cell. Biol. 33, 155–161. doi: 10.1089/dna.2013.2213
Paruchuri, S., Tashimo, H., Feng, C., Maekawa, A., Xing, W., Jiang, Y., et al. (2009). Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 206, 2543–2555. doi: 10.1084/jem.20091240
Pasaje, C. F. A., Kim, J. H., Park, B. L., Cheong, H. S., Chun, J. Y., Park, T. J., et al. (2010). Association of SLC6A12 variants with aspirin-intolerant asthma in a Korean population. Ann. Hum. Genet. 74, 326–334. doi: 10.1111/j.1469-1809.2010.00584.x
Pasaje, C. F. A., Kim, J. H., Park, B. L., Cheong, H. S., Kim, M. K., Choi, I. S., et al. (2011). A possible association of EMID2 polymorphisms with aspirin hypersensitivity in asthma. Immunogenetics 63, 13–21. doi: 10.1007/s00251-010-0490-8
Pavon-Romero, G., Perez-Rubio, G., Ramirez-Jimenez, F., Ambrocio-Ortiz, E., Banuelos-Ortiz, E., Alvarado-F, N., et al. (2018). MS4A2-rs573790 is associated with aspirin-exacerbated respiratory disease: replicative study using a candidate gene strategy. Front. Genet. 9:363. doi: 10.3389/fgene.2018.00363
Pérez-Sánchez, N., Jurado-Escobar, R., Doña, I., Soriano-Gomis, V., Moreno-Aguilar, C., Bartra, J., et al. (2019). Pharmacogenomics as a tool for management of drug hypersensitivity reactions. Curr. Treat. Options. Oncol. 6, 1–17. doi: 10.1007/s40521-019-0199-3
Pham, D. L., Kim, J. H., Trinh, H. K. T., and Park, H. S. (2016). What we know about nonsteroidal anti-inflammatory drug hypersensitivity. Korean J. Internal. Med. 31, 417–432. doi: 10.3904/kjim.2016.085
Plaza-Serón, M. d. C., García-Martín, E., Agúndez, J. A., and Ayuso, P. (2018). Hypersensitivity reactions to nonsteroidal anti-inflammatory drugs: an update on pharmacogenetics studies. Pharmacogenomics 19, 1069–1086. doi: 10.2217/pgs-2018-0079
Quiralte, J., Sánchez-García, F., Torres, M.-J., Blanco, C., Castillo, R., Ortega, N., et al. (1999). Association of HLA-DR11 with the anaphylactoid reaction caused by nonsteroidal anti-inflammatory drugs. J. Allergy Clin. Immnunol. 103, 685–689. doi: 10.1016/s0091-6749(99)70243-5
Roca-Ferrer, J., Garcia-Garcia, F. J., Pereda, J., Perez-Gonzalez, M., Pujols, L., Alobid, I., et al. (2011). Reduced expression of COXs and production of prostaglandin E(2) in patients with nasal polyps with or without aspirin-intolerant asthma. J. Allergy Clin. Immunol. 128, 66–72.e1. doi: 10.1016/j.jaci.2011.01.065
Rouzer, C. A., and Marnett, L. J. (2009). Cyclooxygenases: structural and functional insights. J. Lipid. Res. 50, S29–S34.
Samuelsson, B., Morgenstern, R., and Jakobsson, P.-J. (2007). Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol. Rev. 59, 207–224. doi: 10.1124/pr.59.3.1
Sanak, M., Pierzchalska, M., Bazan-Socha, S., and Szczeklik, A. (2000). Enhanced expression of the leukotriene C4 synthase due to overactive transcription of an allelic variant associated with aspirin-intolerant asthma. Am. J. Respir. Cell Mol. Biol. 23, 290–296. doi: 10.1165/ajrcmb.23.3.4051
Sanak, M., Simon, H. U., and Szczeklik, A. (1997). Leukotriene C4 synthase promoter polymorphism and risk of aspirin-induced asthma. Lancet 350, 1599–1599. doi: 10.1016/s0140-6736(05)64015-9
Sánchez-Borges, M., Acevedo, N., Vergara, C., Jiménez, S., Zabner-Oziel, P., Monzón, A., et al. (2009). The A-444C polymorphism in the leukotriene C4 synthase gene is associated with aspirin-induced urticaria. J. Investig. Allergol. Clin. Immunol. 19, 375–382.
Sanchez-Borges, M., Caballero-Fonseca, F., Capriles-Hulett, A., and Gonzalez-Aveledo, L. (2015). Aspirin-exacerbated cutaneous disease (AECD) is a distinct subphenotype of chronic spontaneous urticaria. J. Eur. Acad. Dermatol. Venereol. 29, 698–701. doi: 10.1111/jdv.12658
Shin, H. D., Park, B. L., Jung, J. H., Wang, H. J., Park, H. S., Choi, B. W., et al. (2003). Association of thromboxane A2 receptor (TBXA2R) with atopy and asthma. J. Allergy Clin. Immnunol. 112, 454–457. doi: 10.1067/mai.2003.1641
Shin, S., Park, J. S., Kim, Y. J., Oh, T., An, S., and Park, C.-S. (2012). Differential gene expression profile in PBMCs from subjects with AERD and ATA: a gene marker for AERD. Mol. Genet. Genom. 287, 361–371. doi: 10.1007/s00438-012-0685-9
Sole, D., Ivancevich, J. C., Borges, M. S., Coelho, M. A., Rosario, N. A., Ardusso, L. R., et al. (2011). Anaphylaxis in Latin America: a report of the online Latin American survey on anaphylaxis (OLASA). Clinics (Sao Paulo) 66, 943–947. doi: 10.1590/s1807-59322011000600004
Stankovic, K. M., Goldsztein, H., Reh, D. D., Platt, M. P., and Metson, R. (2008). Gene expression profiling of nasal polyps associated with chronic sinusitis and aspirin-sensitive asthma. Laryngoscope 118, 881–889. doi: 10.1097/MLG.0b013e31816b4b6f
Stevens, W. W., Peters, A. T., Hirsch, A. G., Nordberg, C. M., Schwartz, B. S., Mercer, D. G., et al. (2017). Clinical characteristics of patients with chronic rhinosinusitis with nasal polyps, asthma, and aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. Pract. 5, 1061–1070.e3. doi: 10.1016/j.jaip.2016.12.027.
Szczeklik, W., Sanak, M., and Szczeklik, A. (2004). Functional effects and gender association of COX-2 gene polymorphism G-765C in bronchial asthma. J. Allergy Clin. Immunol. 114, 248–253. doi: 10.1016/j.jaci.2004.05.030
Tahara, T., Shibata, T., Nakamura, M., Yamashita, H., Yoshioka, D., Okubo, M., et al. (2010). Chronic aspirin use suppresses CDH1 methylation in human gastric mucosa. Dig. Dis. Sci. 55, 54–59. doi: 10.1007/s10620-008-0701-4
Thong, B. Y. H. (2018). Nonsteroidal anti-inflammatory drug hypersensitivity in the Asia-Pacific. Asia. Pac. Allergy 8, e38–e38. doi: 10.5415/apallergy.2018.8.e38
Trinh, H. K., Kim, S. C., Cho, K., Kim, S. J., Ban, G. Y., Yoo, H. J., et al. (2016). Exploration of the sphingolipid metabolite, sphingosine-1-phosphate and sphingosine, as novel biomarkers for aspirin-exacerbated respiratory disease. Sci. Rep. 6:36599. doi: 10.1038/srep36599
Vasconcelos, F., Mabel, L., Rodrigues, R. d. O, Albuquerque, A. A., Barroso, G. D., Sasahara, G. L., et al. (2018). Polymorphism of IL10, IL4, CTLA4, and DAO genes in cross-reactive nonsteroidal anti-inflammatory drug hypersensitivity. J. Clin. Pharmacol. 58, 107–113. doi: 10.1002/jcph.986
Wardzyńska, A., Makowska, J. S., Pawełczyk, M., Piechota-Polańczyk, A., Kurowski, M., and Kowalski, M. L. (2017). Periostin in exhaled breath condensate and in serum of asthmatic patients: relationship to upper and lower airway disease. Allergy Asthma Immunol. Res. 9, 126–132. doi: 10.4168/aair.2017.9.2.126
Weller, P. F. (2016). Leukocyte lipid bodies—structure and function as “eicosasomes”. Trans. Am. Clin. Climatol. Assoc. 127:328.
Wyatt, J., Pettit, W., and Harirforoosh, S. (2012). Pharmacogenetics of nonsteroidal anti-inflammatory drugs. Pharmacog. J. 12, 462–467.
Zhao, Y., Sun, S., Li, X., Ma, X., Tang, H., Sun, L., et al. (2018). Drug-induced anaphylaxis in China: a 10 year retrospective analysis of the Beijing Pharmacovigilance Database. Int. J. Clin. Pharm. 40, 1349–1358. doi: 10.1007/s11096-017-0535-2
Keywords: asthma, urticaria, epigenetic, non-steroidal anti-inflammatory drug, hypersensitivity, genetic polymorphism
Citation: Trinh HKT, Pham LD, Le KM and Park H-S (2021) Pharmacogenomics of Hypersensitivity to Non-steroidal Anti-inflammatory Drugs. Front. Genet. 12:647257. doi: 10.3389/fgene.2021.647257
Received: 29 December 2020; Accepted: 05 May 2021;
Published: 25 June 2021.
Edited by:
Tilo Grosser, University of Pennsylvania, United StatesReviewed by:
M. Isabel Lucena, University of Malaga, SpainMelih O. Babaoglu, Hacettepe University, Turkey
Copyright © 2021 Trinh, Pham, Le and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hae-Sim Park, aHNwYXJrQGFqb3UuYWMua3I=
†These authors have contributed equally to this work