 Qi Tian1†
Qi Tian1† Yang Cao2†
Yang Cao2† Li Shu1,3,4
Li Shu1,3,4 Yongjun Chen5Ying Peng1Yaqin Wang6Yuanyuan Chen7Hua Wang1,3*
Yongjun Chen5Ying Peng1Yaqin Wang6Yuanyuan Chen7Hua Wang1,3* Xiao Mao1,3*
Xiao Mao1,3*- 1Department of Medical Genetics, Maternal and Child Health Hospital of Hunan Province, Changsha, China
- 2Department of Radiology, Chenzhou First People's Hospital, Chenzhou, China
- 3National Health Commission Key Laboratory of Birth Defects Research, Prevention and Treatment, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 4Department of School of Life Sciences, Central South University, Changsha, China
- 5Department of Neurology, Nanhua Affiliated Hospital, University of South China, Hengyang, China
- 6Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 7Reproductive Center of Maternal and Child Health Hospital of Hunan Province, Changsha, China
Background: The molybdenum cofactor (Moco) deficiency in humans results in the inactivity of molybdenum-dependent enzymes and is caused by pathogenic variants in MOCS1 (Molybdenum cofactor synthesis 1), MOCS2 (Molybdenum cofactor synthesis 2), and GPHN (Gephyrin). These genes along with MOCS3 (Molybdenum cofactor synthesis 3) are involved in Moco biosynthesis and providing cofactors to Moco-dependent enzymes. Until now, there was no study to confirm that MOCS3 is a causative gene of Moco deficiency.
Methods: Detailed clinical information was collected in the pedigree. The Whole-exome sequencing (WES) accompanied with Sanger sequencing validation were performed.
Results: We described the clinical presentations of an infant, born to a non-consanguineous healthy family, diagnosed as having MOCS3 variants caused Moco deficiency and showing typical features of Moco deficiency including severe neurologic symptoms and cystic encephalomalacia in the brain MRI, resulting in neonatal death. Compound heterozygous variants in the MOCS3 gene were identified by WES. Positive sulfite and decreased levels of uric acid in plasma and urine were detected.
Conclusion: To our knowledge, this is the first case of MOCS3 variants causing Moco deficiency. Our study may contribute to genetic diagnosis of Moco deficiency and future genetic counseling.
Introduction
The molybdenum cofactor (Moco) deficiency in humans results in the inactivity of molybdenum-dependent enzymes including sulfite oxidase, xanthine oxidoreductase, and aldehyde oxidase (Mayr et al., 2021). The inactivation of these enzymes in neurons was related to severe neurological symptoms and residual diseases (Zaki et al., 2016). The etiology of Moco deficiency was caused by pathogenic variants in MOCS1 (Molybdenum cofactor synthesis 1), MOCS2 (Molybdenum cofactor synthesis 2), and GPHN (Gephyrin). The typical clinical features of Moco deficiency patients with these variants are characterized by neonatal-onset intractable seizures, followed by feeding difficulty, developmental delay, and neonatal deaths (Mechler et al., 2015).
The signs and symptoms of patients with Moco deficiency are attributed to the pathogenesis of inactivating the molybdenum-dependent enzymes (Atwal and Scaglia, 2016). The enzyme inactivation subsequently leads to the accumulation of metabolites including sulfite, xanthine, hypoxanthine, etc. It is believed that the pathogenesis is caused by accumulated metabolites, and it may exert toxic effects on neurons and lead to seizures, encephalopathy, and other neurological symptoms (Mechler et al., 2015).
Until now, there have been no cases reported with the MOCS3 (Molybdenum cofactor synthesis 3) variation that presented with the typical features of Moco deficiency. The only reported case related to MOCS3 showed a slight change in sulfite metabolites, and as a result the patient only presented mild neurologic disorder symptoms such as high muscular tone and limited speech ability. Except for the thin splenium of corpus callosum, the neurologic imaging was also unremarkable (Huijmans et al., 2017). Therefore, there was not enough evidence to support the pathogenesis of MOCS3 variation and studies illustrating the phenotypic spectrum of MOCS3-induced Moco deficiency are needed.
Here we described a case of Moco deficiency identified with compound heterozygous variations in the MOCS3 gene. The infant presented aggressive phenotypes of Moco deficiency including early onset of neonatal seizures, hypertonia, feeding difficulty, and severe developmental delay. The MRI indicated global cystic encephalomalacia and cortical necrosis. The patient showed poor reaction to treatment and resulted in infant death. To the best of our knowledge, our report is probably the first study to prove that MOCS3 is a pathogenic gene of Moco deficiency. Our study may contribute to genetic diagnosis of Moco deficiency and future genetic counseling.
Case Description
Our patient is the second child born to the non-consanguineous Chinese parents. The first male child presented neonatal seizures, hypotonia, and developmental delay. He died at the age of 8 months and the suspected diagnosis was hypoxic-ischemic encephalopathy, which was made by Magnetic resonance imaging (MRI) presentation.
The second female child was born at full term (40 weeks) with no abnormality in pregnancy. Without any inducement, generialized tonic-clonic seizures were observed within 24 h after birth and seizures were not controlled after multiple treatments. Her after birth physical examination indicated microcephaly [occipitofrontal head circumference 30.0 cm, < −3 Standard deviation (SD)], hypotonia, and hyper-reflexia. At the age of 5 months the patient was admitted to hospital for the reoccurrence of the seizures. She did not develop the ability to lift her head and trace. MRI showed frontotemporal, parietal, and occipital lobes cystic encephalomalacia and bilateral basal ganglia atrophy (Figure 1). The patient showed severe neurodevelopmental delay. She was still unable to trace, lift her head, roll over, and sit at 10 months. The patient had feeding difficulties resulting in low breast milk intake and died at age of 12 months because of lung infection.
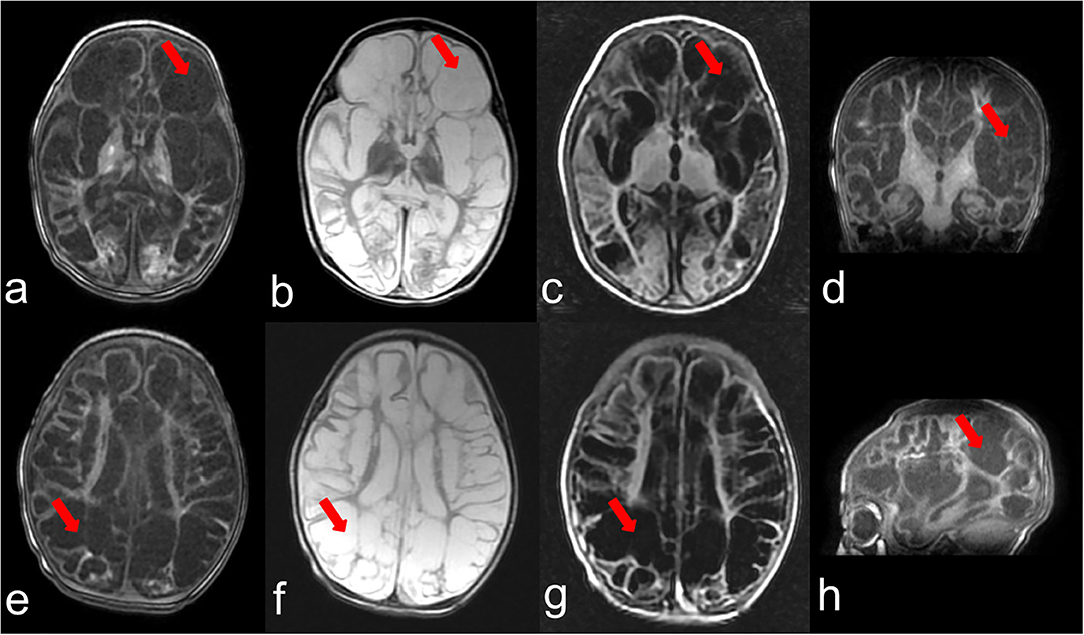
Figure 1. Brain MRI at the age of 3 months. (a–c) represent T1,T2,T2-FLAIR weighted images of one section and (e–g) indicate T1,T2,T2-FLAIR weighted images of another section, respectively. (d,h) show T1 weighted images of the coronal plane and sagittal plane. The arrows point out the representing lesions in each picture.
Diagnostic Assessment
Whole-exome sequencing (WES) accompanied with Sanger sequencing validation was performed in the proband and her parents (Figure 2). The variants were firstly filtered according to HGMD and ACMG guidelines Disease-causing mutations (DM) and probable/possible pathological mutation (DM) in the HGMD (Professional version 2019.1) database, and pathogenic (P) and likely pathogenic (LP) variants interpreted by ACMG guidelines for interpretation of genetic variants were included. Secondly, variants were filtered according to allele frequency, variant type, and inheritance mode. Variants with minor allele frequencies (MAF) <0.1%, variant depth of coverage ≥20 and alteration base depth of coverage ≥4 were taken on for further analyses. Filtering was conducted on the remaining variants according to variant type and inheritance model of the associated disease. The proband was identified with compound heterozygous variants in NM_014484.49 (MOCS3): c.1375C>T; p.Gln459Ter (chr20:49576754-49576754) and NM_014484.49 (MOCS3): c.325C>G; p.Leu109Val (chr20:49575704-49575704). Her father was a heterozygous carrier of the MOCS3 c.1375C>T variant and the mother carried another variant in the MOCS3 c.325C>G. All the segregated variants of this case were listed in the Supplementary Table 1 and the annotation of each variant is given.
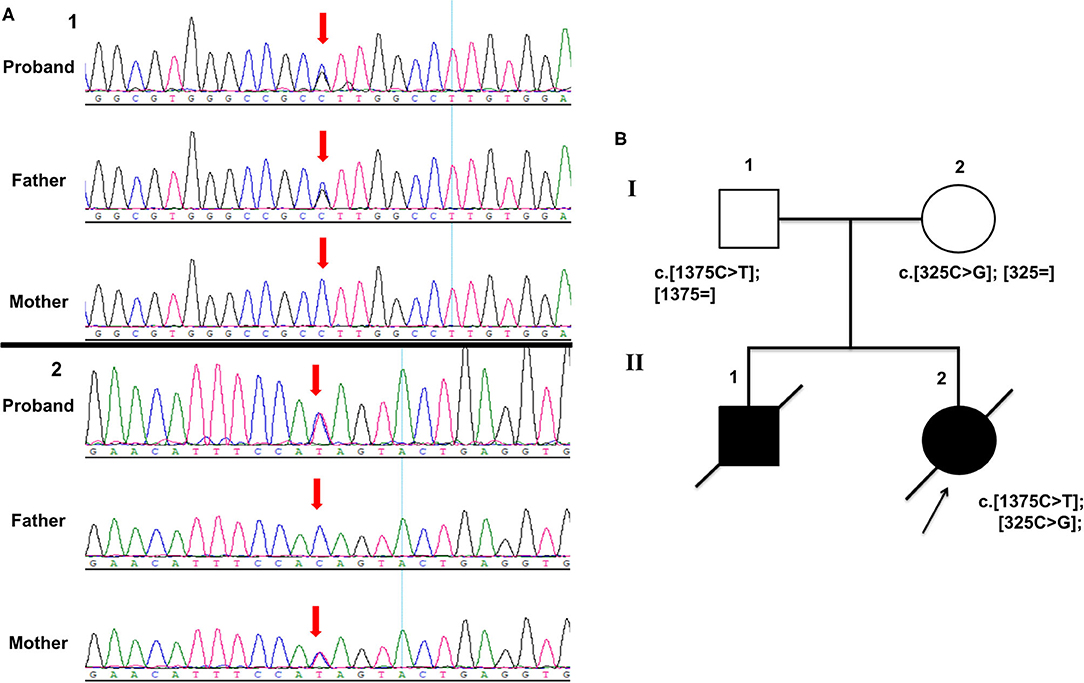
Figure 2. (A) Sanger sequencing electropherograms showing compound heterozygous variants in MOCS3 (A showing c.1375C>T and B showing c.325C>G). (B) Pedigree chart of the family.
The variants are evaluated by MutationTaster (v2.0), CADD (v1.6), FATHMM (v2.3). The scores of each variant of MOCS3 from the bioinformatic prediction tools are as follows: for c.325C>G the scores are 1.000, 22.8, and 1.19 and for c.1375C>T (premature termination mutation) the scores are 0.958, 36, NA, respectively. The allele frequency of c.325C>G was 6.181e−05 in gnomAD database, 5.182e−05 in the ExAC and 0 in the 1,000 genomes database. The allele frequency of c.1375C>T was 0 in gnomAD the ExAC and the 1,000 genomes databases.
Moco deficiency-related laboratory tests were conducted. The urine and plasma uric acid levels were detected by Beckman Coulter AU680 and enzymatic colorimetric assay on the Cobas 8000 system. The urine sulfite was tested by a urine sulfite test strip. The urine uric acid level was 32 mmol/mol creatinine (normal range 350–2,500 mmol/mol creatinine) and plasma uric acid level was 0.01 mmol/l (normal range 0.08–0.37 mmol/l). The urine sulfite test strip was positive.
Discussion
Moco deficiency is characterized by severe neonatal neuropathologic symptoms, typical neuroimaging findings and variants in causative genes MOCS1, MOCS2, and GPHN (Atwal and Scaglia, 2016). These genes along with MOCS3 are involved in Moco biosynthesis and providing cofactors to Moco dependent enzymes. In reported Moco deficiency cases, two thirds are caused by MOCS1 variants, followed by MOCS2 and GPHN. Most of these cases presented typical symptoms of Moco deficiency (Reiss and Johnson, 2003; Zaki et al., 2016; Mayr et al., 2021). Until now there was no evidence that MOCS3 was one of the causative genes of Moco deficiency. In 2017 there was one case with the MOCS3 variant who showed a slight disorder of sulfite metabolites and near to normal neurologic symptoms (Huijmans et al., 2017). In our study, we identified a Moco deficiency patient with variants in MOCS3 presented typical features of Moco deficiency including early onset severe neurological symptoms accompanied with typical MRI manifestations and biochemical changes. The published variant in 2017 was a missense variant and the variants in our study included a premature termination variation, and it could be a possible reason for the severity of our case. To our knowledge, our study is the first study to show that MOCS3 is a pathogenic gene of Moco deficiency.
The clinical diagnosis of Moco deficiency is often supported by typical MRI readings (brain edema, cystic encephalomalacia, atrophy of cortex and white matter, focal or bilateral changes of globus pallidus, thalamus, etc.), and it has to be confirmed by genetic and biochemical testing (Durmaz and Ozbakir, 2018; Arican et al., 2019). The primary features of the disease were reported to be seizures (72%), feeding difficulties (26%), hypotonia (11%), and developmental delay (9%). The median onset age was the first day of life and the median survival was 36 months (Mechler et al., 2015). In our study, the proband presented similar symptoms as the first infant, and the inherited diseases were considered and further tests were performed. Although the first infant was suspected as hypoxic-ischemic encephalopathy, in some cases the neurological manifestations of Moco deficiency may overlap with severe hypoxic-ischemic encephalopathy, and shows seizures, developmental delay, and cystic encephalomalacia in MRI etc. (Zaki et al., 2016; Yoganathan et al., 2018). However, Moco deficiency usually presents more aggressive neurological symptoms. The positive urine test for sulfite with decreased plasma and urine uric acid would assist diagnosis (Yoganathan et al., 2018; Bender et al., 2019).
MOCS3 might involve in the pathogenesis of neurodevelopmental diseases by impacting the Moco biosynthesis pathway, affecting neuronal receptors and causing neuronal death. MOCS3 knocked out cells were reported to cause combined deficiency of sulfite oxidase, xanthine oxidoreductase, and aldehyde oxidase (Chowdhury et al., 2012). This resulted in accumulation of many metabolites, especially sulfite, a neurotoxin proved to react with cystin to form the S-sulfocysteine and thiosulfate (Neukranz et al., 2019). The structure of S-sulfocysteine highly resembled the excitatory neurotransmitter glutamate that could bind and stimulate glutamatergic receptors. It has been proved in a zebrafish model that S-sulfocysteine could stimulate glutamatergic receptors, induce seizure-like movements, and increase cell death in the central nervous system (Zaki et al., 2016; Plate et al., 2019). Accumulation of S-sulphocysteine and thiosulfate would activate the N-methyl-D-aspartate receptor and increase intracellular magnesium and calcium. This could enhance the permeability of the mitochondria, compromise the mitochondrial energy supply, and reduce the cell viability in cerebral cortex of rat brain (Grings et al., 2014; Marelja et al., 2018; Bender et al., 2019; Plate et al., 2019). All these processes may be the possible mechanisms underlying the neurological dysfunction observed in our patient.
In summary, we reported a novel causative gene MOCS3 in a Moco deficiency patient. Our study may contribute to genetic diagnosis of Moco deficiency and future genetic counseling.
Materials and Methods
Leukocyte DNA was extracted from peripheral blood using the phenol-chloroform method. DNA was then sheared to ~200 bp by the Biorupter UCD-200 (Diagenode). The DNA fragments were then repaired at the end, and one A base was added to the 3′ end. The DNA fragments were connected with sequencing adaptors, and fragments of ~320 bp were collected by XP beads. After PCR amplification, the DNA fragments were hybridized and captured by IDT's xGen Exome Research Panel (Integrated DNA Technologies, San Diego, USA) according to the manufacturer's protocol. The hybrid products were eluted and collected. Then, DNA was PCR amplified and purified. The libraries were tested for enrichment by qPCR, and size distribution and concentration were determined using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). WES was performed on the HiSeq 2500 system (Illumina) with an average coverage depth of 100 × of the variants for sequencing the genomic DNA of the family. Raw sequence reads were mapped to the hg19/GRCh37 using Elandv2e (CASAVA1.8.2; Illumina) to remove duplicated reads and CASAVA1.8.2 was further used to call single-nucleotide variants and short insertions/deletions (Markus et al., 2020; Tessarech et al., 2020). The variants were annotated using ANNOVAR (Wang et al., 2010).
A public database (1,000 Genomes Project, ExAC, gnomAD) was used to detect variants frequencies. The pathogenicity of variants was predicted using following software programs:
Combined Annotation Dependent Depletion [CADD] [https://cadd.gs.washington.edu/] (Kircher et al., 2014; Rentzsch et al., 2019).
MutationTaster [http://www.mutationtaster.org/] (Schwarz et al., 2014).
FATHMM [http://fathmm.biocompute.org.uk/fathmmMKL.htm] (Shihab et al., 2013a,b).
Sanger sequencing was performed on the DNA of the proband's parents to validate the variants found in WES using standard methods on ABI 3730 automated sequencer with BigDye™ Terminator v3.1 Cycle Sequencing Kit, as described previously (Stockley et al., 2013). The primers are as follows: forward primer (5′-CTCTGTCCCGAGATGAGATTCT-3′) and reverse primer (5′-CGTTGTCCGAGCAGTCAGC-3'), forward primer (5′-AAGAAGCAATCTGGGAAGAGAAG) and reverse primer (5′-ATGACCCTCTTTCTGAATAATTAAA).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The study was approved by the Ethics Committee of the Maternal and Child Health Hospital of Hunan Province (2020-S003). Informed consent was obtained from the legal guardian of the participant for the publication of this case report (including all data and images).
Author Contributions
XM and HW designed the research. QT and YaC interpreted the data and wrote the manuscript. HW, LS, YoC, YW, and YuC did the follow-up study and collected, evaluated the clinical, and genetic evidence. QT and HW revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81801136), Major Scientific and Technological Projects for Collaborative Prevention and Control of Birth Defects in Hunan Province (2019SK1010, 2019SK1012, 2019SK1014), the National Key R&D Program of China (No. 2019YFC1005100), the China Postdoctoral Science Foundation (2019M662804), and the Changsha Municipal Natural Science Foundation (kq2007048).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the families and clinical staff for participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.651878/full#supplementary-material
References
Arican, P., Gencpinar, P., Kirbiyik, O., Bozkaya Yilmaz, S., Ersen, A., Oztekin, O., et al. (2019). The clinical and molecular characteristics of molybdenum cofactor deficiency due to MOCS2 mutations. Pediatr. Neurol. 99, 55–59. doi: 10.1016/j.pediatrneurol.2019.04.021
Atwal, P. S., and Scaglia, F. (2016). Molybdenum cofactor deficiency. Mol. Genet. Metab. 117, 1–4. doi: 10.1016/j.ymgme.2015.11.010
Bender, D., Kaczmarek, A. T., Santamaria-Araujo, J. A., Stueve, B., Waltz, S., Bartsch, D., et al. (2019). Impaired mitochondrial maturation of sulfite oxidase in a patient with severe sulfite oxidase deficiency. Hum. Mol. Genet. 28, 2885–2899. doi: 10.1093/hmg/ddz109
Chowdhury, M. M., Dosche, C., Lohmannsroben, H. G., and Leimkuhler, S. (2012). Dual role of the molybdenum cofactor biosynthesis protein MOCS3 in tRNA thiolation and molybdenum cofactor biosynthesis in humans. J. Biol. Chem. 287, 17297–17307. doi: 10.1074/jbc.M112.351429
Durmaz, M. S., and Ozbakir, B. (2018). Molybdenum cofactor deficiency: neuroimaging findings. Radiol. Case Rep. 13, 592–595. doi: 10.1016/j.radcr.2018.02.025
Grings, M., Moura, A.P., Amaral, A. U., Parmeggiani, B., Gasparotto, J., Moreira, J. C, et al. (2014). Sulfite disrupts brain mitochondrial energy homeostasis and induces mitochondrial permeability transition pore opening via thiol group modification. Biochim. Biophys. Acta 1842, 1413–1422. doi: 10.1016/j.bbadis.2014.04.022
Huijmans, J. G. M., Schot, R., de Klerk, J. B. C., Williams, M., de Coo, R. F. M., Duran, M., et al. (2017). Molybdenum cofactor deficiency: identification of a patient with homozygote mutation in the MOCS3 gene. Am. J. Med. Genet. A 173, 1601–1606. doi: 10.1002/ajmg.a.38240
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Marelja, Z., Leimkuhler, S., and Missirlis, F. (2018). Iron sulfur and molybdenum cofactor enzymes regulate the drosophila life cycle by controlling cell metabolism. Front. Physiol. 9:50. doi: 10.3389/fphys.2018.00050
Markus, F., Angelini, C., Trimouille, A., Rudolf, G., Lesca, G., Goizet, C., et al. (2020). Rare variants in the GABAA receptor subunit epsilon identified in patients with a wide spectrum of epileptic phenotypes. Mol. Genet. Genomic Med. 8:e1388. doi: 10.1002/mgg3.1388
Mayr, S. J., Mendel, R. R., and Schwarz, G. (2021). Molybdenum cofactor biology, evolution and deficiency. Biochim. Biophys. Acta Mol. Cell. Res. 1868:118883. doi: 10.1016/j.bbamcr.2020.118883
Mechler, K., Mountford, W. K., Hoffmann, G. F., and Ries, M. (2015). Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet. Med. 17, 965–970. doi: 10.1038/gim.2015.12
Neukranz, Y., Kotter, A., Beilschmidt, L., Marelja, Z., Helm, M., Graf, R., et al. (2019). Analysis of the cellular roles of MOCS3 identifies a MOCS3-independent localization of NFS1 at the tips of the centrosome. Biochemistry 58, 1786–1798. doi: 10.1021/acs.biochem.8b01160
Plate, J., Sassen, W. A., Hassan, A. H., Lehne, F., Koster, R. W., and Kruse, T. (2019). S-sulfocysteine induces seizure-like behaviors in zebrafish. Front. Pharmacol. 10:122. doi: 10.3389/fphar.2019.00122
Reiss, J., and Johnson, L. J. (2003). Mutations in the molybdenum cofactor biosynthetic genes MOCS1, MOCS2, and GEPH. Hum. Mutat. 21, 569–576. doi: 10.1002/humu.10223
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi: 10.1093/nar/gky1016
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Shihab, H. A., Gough, J., Cooper, D. N., Day, I. N., and Gaunt, R. T. (2013b). Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29, 1504–1510. doi: 10.1093/bioinformatics/btt182
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013a). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Stockley, J., Morgan, N. V., Bem, D., Lowe, G. C., Lordkipanidze, M., Dawood, B., et al. (2013). Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood 122, 4090–4093. doi: 10.1182/blood-2013-06-506873
Tessarech, M., Gorce, M., Boussion, F., Bault, J. P., Triau, S., Charif, M., et al. (2020). Second report of RING finger protein 113A (RNF113A) involvement in a Mendelian disorder. Am. J. Med. Genet. A 182, 565–569. doi: 10.1002/ajmg.a.61384
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Yoganathan, S., Sudhakar, S., Thomas, M., Kumar Dutta, A., Danda, S., and Chandran, M. (2018). Novel Imaging finding and novel mutation in an infant with molybdenum cofactor deficiency, a mimicker of hypoxic-ischaemic encephalopathy. Iran. J. Child Neurol. 12, 107–112. doi: 10.22037/ijcn.v12i2.12671
Keywords: MOCS3, neurodevelopmental outcome, sulfite oxidase, whole exome sequencing, molybdenum cofactor deficiency
Citation: Tian Q, Cao Y, Shu L, Chen Y, Peng Y, Wang Y, Chen Y, Wang H and Mao X (2021) Case Report: Compound Heterozygous Variants in MOCS3 Identified in a Chinese Infant With Molybdenum Cofactor Deficiency. Front. Genet. 12:651878. doi: 10.3389/fgene.2021.651878
Received: 11 January 2021; Accepted: 12 March 2021;
Published: 08 April 2021.
Edited by:
Can Liao, Prenatal Diagnostic Center, Guangzhou Women and Children's Medical Center, ChinaReviewed by:
James A. Poulter, University of Leeds, United KingdomShabeesh Balan, RIKEN Center for Brain Science (CBS), Japan
Copyright © 2021 Tian, Cao, Shu, Chen, Peng, Wang, Chen, Wang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Mao, Z2J0ZWNoaWVzQG91dGxvb2suY29t; Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship