 Allan Bayat1,2*
Allan Bayat1,2* Manuela Pendziwiat3,4
Manuela Pendziwiat3,4 Ewa Obersztyn5
Ewa Obersztyn5 Paula Goldenberg6
Paula Goldenberg6 Pia Zacher7Jan Henje Döring8Steffen Syrbe8Amber Begtrup9
Pia Zacher7Jan Henje Döring8Steffen Syrbe8Amber Begtrup9 Artem Borovikov10Artem Sharkov11Aneta Karasińska12
Artem Borovikov10Artem Sharkov11Aneta Karasińska12 Maria Giżewska13Wendy Mitchell14
Maria Giżewska13Wendy Mitchell14 Eva Morava15
Eva Morava15 Rikke S. Møller1,2
Rikke S. Møller1,2 Guido Rubboli2,16
Guido Rubboli2,16- 1Institute for Regional Health Services, University of Southern Denmark, Odense, Denmark
- 2Department of Epilepsy Genetics and Personalized Medicine, Danish Epilepsy Centre, Dianalund, Denmark
- 3Department of Neuropediatrics, Children’s Hospital, University Medical Center Schleswig-Holstein, University of Kiel, Kiel, Germany
- 4Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, Kiel, Germany
- 5Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland
- 6Division of Medical Genetics, Massachusetts General Hospital, Boston, MA, United States
- 7The Saxon Epilepsy Center Kleinwachau, Radeberg, Germany
- 8Department of General Pediatrics, Division of Child Neurology and Inherited Metabolic Diseases, Center for Pediatrics and Adolescent Medicine, University Hospital Heidelberg, Heidelberg, Germany
- 9GeneDx, Gaithersburg, MD, United States
- 10Research Centre for Medical Genetics, Moscow, Russia
- 11Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, Moscow, Russia
- 12Department of Dermatology, The Nicolas Copernicus State Hospital, Koszalin, Poland
- 13Department of Pediatrics, Endocrinology, Diabetology, Metabolic Diseases and Cardiology of the Developmental Age, Pomeranian Medical University, Szczecin, Poland
- 14Department of Neurology, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
- 15Department of Clinical Genomics, Laboratory of Medicine and Pathology, Center for Individualized Medicine, Mayo Clinic, Rochester, MN, United States
- 16Department of Clinical Medicine, Faculty of Medical and Health Sciences, University of Copenhagen, Copenhagen, Denmark
The two aims of this study were (i) to describe and expand the phenotypic spectrum of PIGT deficiency in affected individuals harboring the c.1582G>A; p.Val528Met or the c.1580A > G; p.Asn527Ser variant in either homozygous or compound heterozygous state, and (ii) to identify potential genotype-phenotype correlations and any differences in disease severity among individuals with and without the PIGT variants. The existing literature was searched to identify individuals with and without the two variants. A detailed phenotypic assessment was performed of 25 individuals (both novel and previously published) with the two PIGT variants. We compared severity of disease between individuals with and without these PIGT variants. Twenty-four individuals carried the PIGT variant Val528Met in either homozygous or compound heterozygous state, and one individual displayed the Asn527Ser variant in a compound heterozygous state. Disease severity in the individual with the Asn527Ser variant was compatible with that in the individuals harboring the Val528Met variant. While individuals without the Asn527Ser or Val528Met variant had focal epilepsy, profound developmental delay (DD), and risk of premature death, those with either of the two variants had moderate to severe DD and later onset of epilepsy with both focal and generalized seizures. Individuals homozygous for the Val528Met variant generally became seizure-free on monotherapy with antiepileptic drugs, compared to other PIGT individuals who were pharmaco-resistant. Two patients were diagnosed with myoclonic-atonic seizures, and a single patient was diagnosed with eyelid myoclonia. Our comprehensive analysis of this large cohort of previously published and novel individuals with PIGT variants broadens the phenotypical spectrum and shows that both Asn527Ser and Val528Met are associated with a milder phenotype and less severe outcome. Our data show that PIGT is a new candidate gene for myoclonic atonic epilepsy. Our genotype-phenotype correlation will be useful for future genetic counseling. Natural history studies of this mild spectrum of PIGT-related disorder may shed light on hitherto unknown aspects of this rare disorder.
Introduction
Glycosylphosphatidylinositol (GPI) is a glycolipid that is synthetized and transferred to proteins in the membrane of the endoplasmic reticulum (Fujita and Kinoshita, 2012). The biogenesis of GPI-anchored proteins (GPI-APs) is a conserved post-translational mechanism in eukaryotes and is important for the attachment of these proteins to the cell membrane and for protein sorting, trafficking, and dynamics (Fujita and Kinoshita, 2012; Kinoshita and Fujita, 2016; Bayat et al., 2020b). GPI synthesis and GPI-AP modification are mediated by at least 31 genes, and pathogenic variants in 22 of these genes have so far been associated with human disease (Bellai-Dussault et al., 2019; Bayat et al., 2020b).
The PIGT gene encodes the phosphatidylinositol glycan class T protein that is part of the subunit of the heteropentameric GPI transamidase complex with PIGK, PIGS, PIGT, PIGU, and PGAA1 that facilitates the attachment of GPI anchors to proteins (Ohishi et al., 2001, 2003).
Pathogenic germline missense and truncating variants in PIGT have been associated with a condition named “multiple congenital anomalies – hypotonia - seizures syndrome 3 (MCAHS3)” (OMIM 615398). Affected individuals share features such as epileptic seizures, severe to profound developmental delay (DD), intellectual disability (ID), and multiple congenital malformations of internal organs. Most individuals are non-verbal, non-ambulatory, and suffer from intractable epilepsy (Bayat et al., 2019). Generalized myoclonic and focal motor seizures with impaired awareness associated with various type of EEG epileptic abnormalities (focal sharp-slow wave, focal sharp spike/polyspikes wave, and generalized polyspikes-wave complexes) have been observed (Jezela-Stanek et al., 2020). While focal seizures with or without bilateral spreading have been described on several occasions (Bayat et al., 2019; Jezela-Stanek et al., 2020), reports of generalized seizures remain limited. Primary generalized seizures have so far only been described in a small number of individuals with a compound heterozygous variant at position Val528Met (Jezela-Stanek et al., 2020).
Individuals that are homozygous or compound heterozygous for the missense variant c.1582G > A; p.Val528Met have been reported to have a less severe phenotype (Pagnamenta et al., 2017; Bayat et al., 2019; Jezela-Stanek et al., 2020). This is currently the only variant reported to result in a milder phenotype. Amongst the 37 individuals described with PIGT deficiency (Kvarnung et al., 2013; Nakashima et al., 2014; Lam et al., 2015; Skauli et al., 2016; Knaus et al., 2018; Kohashi et al., 2018; Yang et al., 2018; Bayat et al., 2019; Mason et al., 2019; Jezela-Stanek et al., 2020; Jiao et al., 2020), only 13 individuals have been reported to harbor this variant in a homozygous or compound heterozygous state (Pagnamenta et al., 2017; Bayat et al., 2019; Jezela-Stanek et al., 2020). Our knowledge remains limited about the extent of DD and ID associated with this variant and about the natural history in adolescence and adulthood. Furthermore, a detailed comparison of symptoms in affected individuals with and without the p.Val528Met variant has yet to be done. More individuals are needed to better delineate the genotype-phenotype correlation and to investigate if other pathogenic variants can also be associated with a milder phenotype.
Here, we describe 13 novel individuals with a PIGT deficiency and 12 previously reported individuals (Bayat et al., 2019; Jezela-Stanek et al., 2020). We also present follow-up data on two of the 12 previously published individuals [P4 and P5 from Bayat et al. (2019)]. Of the 25 individuals, 24 harbored the Val528Met variant in either homozygous or compound heterozygous state, while one individual had a previously unreported c.1580A > G; p.Asn527Ser variant. We present a detailed phenotyping of the affected individuals and expand the known epilepsy semiology to include myoclonic-atonic seizures. We also compare the findings with those seen in previously published affected individuals without the two variants.
Materials and Methods
Individual Analysis
Subjects were identified and recruited either through their treating clinicians, self-referral, or GeneMatcher (Sobreira et al., 2015). Basic medical information on birth parameters, epilepsy, developmental history, and physical examinations was collected from healthcare providers and/or families of affected individuals. Molecular testing results from exome sequencing were submitted by healthcare providers. In eight cases, individual videos provided by caregivers were also used to evaluate motor skills. All the videos submitted and evaluated were used with the written consent of guardians. Videos were evaluated by authors AB and GR.
The clinical findings of this cohort were compared to previously published individuals with PIGT deficiency who did not harbor either of the two variants. As the access to clinical data was limited in a substantial number of published individuals, we chose only to compare data on seizures and neurological development.
Standard Protocol Approvals, Registrations, and Individual Consent
The study was conducted in agreement with the Declaration of Helsinki and approved by the local ethics committee of the referring center. As all individials were either minors or had cognitive impairment, their parents or legal guardians gave informed consent. The clinical information was collected through interviews with the families and/or from hospital records of the individuals and their family members. Videos were collected from either caregivers or healthcare providers.
Literature Search
We searched MEDLINE (PubMed) with the keywords epilepsy, GPI, GPI-AP, or GPI-anchored protein in combination with PIGT or phosphatidylinositol glycan class T protein (last PubMed search: January 2021). Any relevant references in the assessed articles, which were not found in the MEDLINE search, were further investigated. Only articles written in English and published after 1980 were included to ensure optimal data collection. Only cases with a confirmed molecular diagnosis were included.
Data Availability
Anonymized data, including data not published in this article, will be made available on request from any qualified investigator.
Genetic Identification and Analysis
All affected individuals were investigated by gene panels or exome sequencing ordered by healthcare providers. Variants were classified based on the 2015 joint guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015). The protein-coding transcript used was NM_015937.6. We predicted the functional alteration of novel variants using polymorphism phenotyping-2 (PolyPhen-2) (Adzhubei et al., 2010) and SIFT (Sorting intolerant from tolerant) (Farheen et al., 2017), CADD1, and mutationTaster2.
Results
Individuals and Families
We collected 13 living, novel PIGT-deficient individuals (8 males and 5 females) harboring either the Val528Met or the Asn527Ser variant. In addition, we present follow-up data for two previously published individuals [P4 and P5 published by Bayat et al. (2019)] 2 years after the initial publication. Fourteen individuals carried the Val528Met variant in either homozygous (n = 6) or compound heterozygous (n = 8) state, while a single individual had the Asn527Ser variant in a compound heterozygous state.
From the available literature, we identified ten previously reported individuals with the Val528Met variant [Decipher ID 258094 by Pagnamenta et al. (2017), P3 and P10 by Bayat et al. (2019), and P1-P7 by Jezela-Stanek et al. (2020)]. Table 1 provides an overview of the clinical and genetic findings.
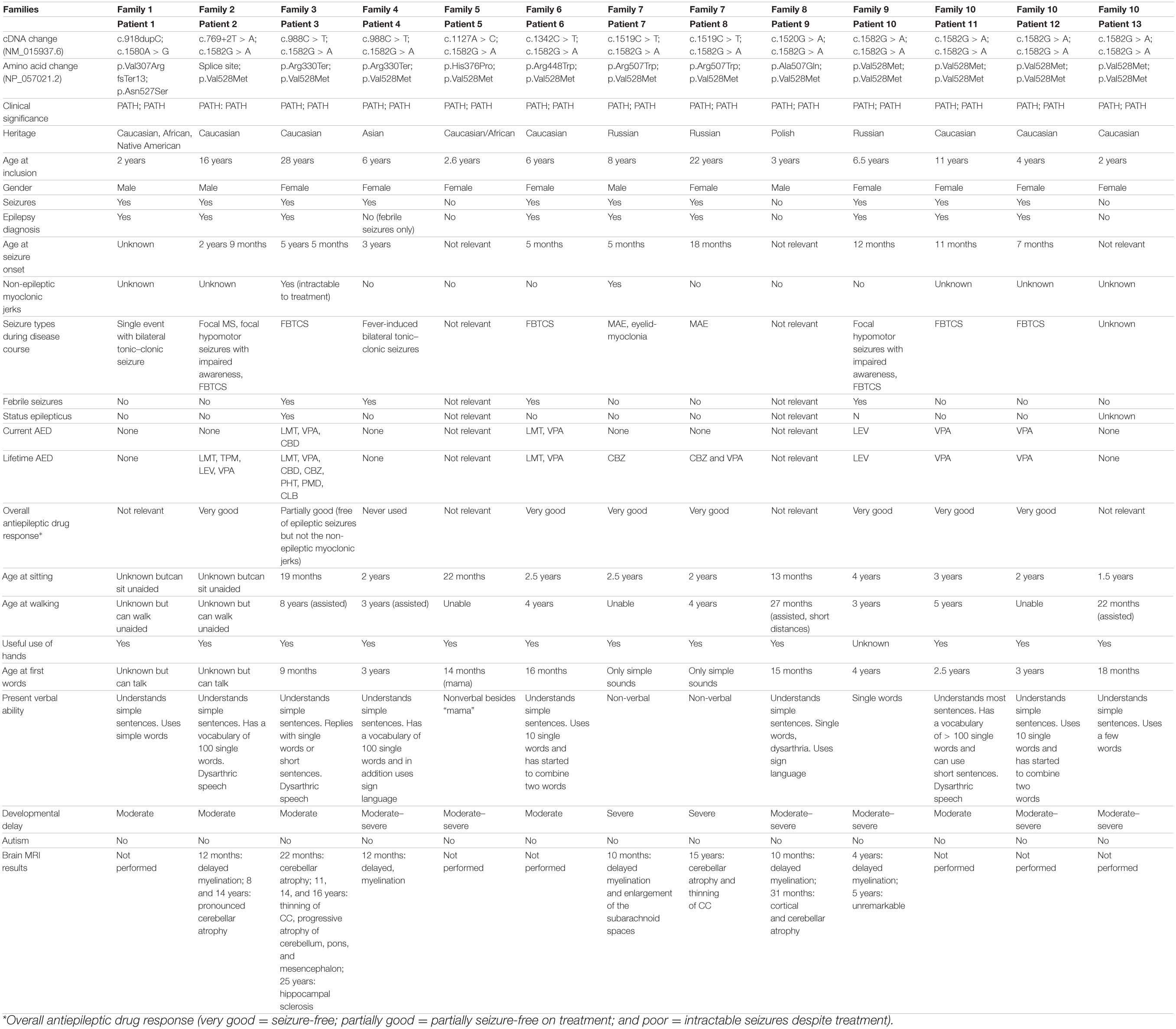
Table 1A. Clinical and genetic findings in 25 novel and previously published individuals with PIGT deficiency harboring the p.Asn527Ser or p.Val528Met variant in either homozygous or compound heterozygous state.
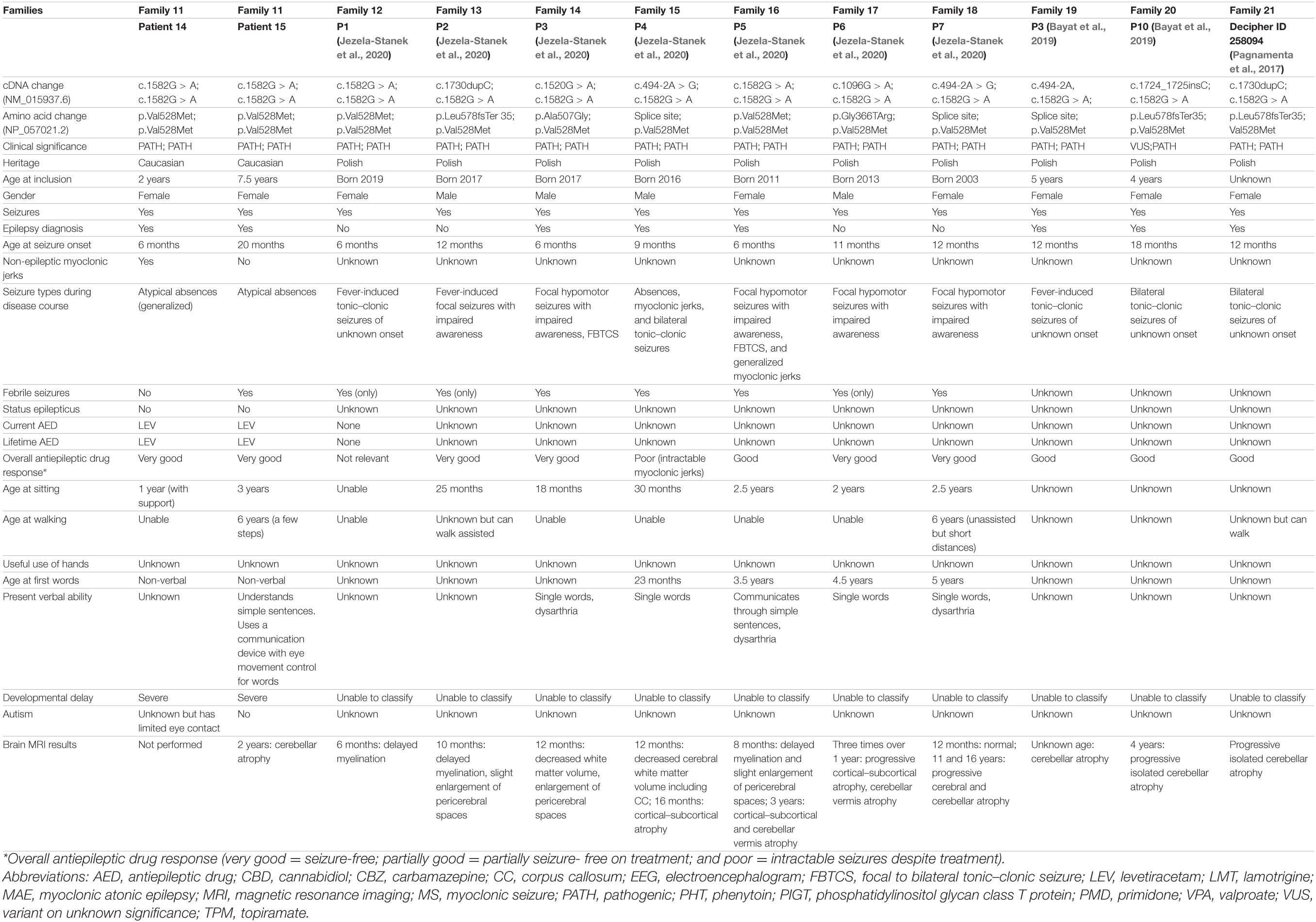
Table 1B. Clinical and genetic findings in novel and previously published individuals with PIGT deficiency harboring the p.Asn527Ser or p.Val528Met variant in either homozygous or compound heterozygous state.
Part 1 of Table 2 compares the clinical findings in the 25 individuals with the Val528Met or the Asn527Ser variant with those of 24 published individuals without these two PIGT variants (Kvarnung et al., 2013; Nakashima et al., 2014; Lam et al., 2015; Skauli et al., 2016; Knaus et al., 2018; Kohashi et al., 2018; Yang et al., 2018; Bayat et al., 2019; Mason et al., 2019; Jiao et al., 2020).
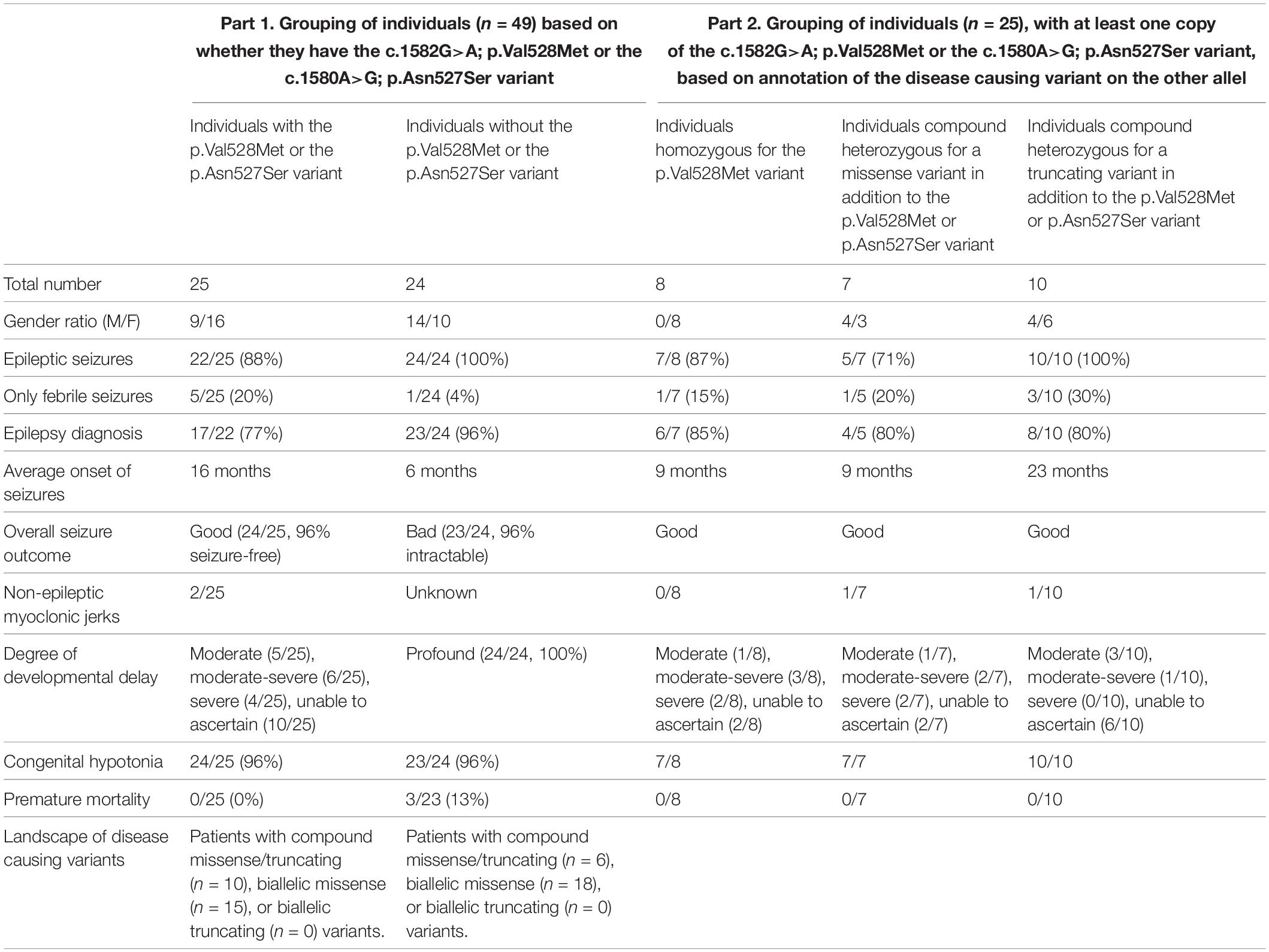
Table 2. Brief clinical overview of all published individuals with PIGT deficiency.
Phenotypic Analysis
In total, we evaluated the phenotypical features of 25 living individuals, including 15 novel and 10 previously published individuals (Bayat et al., 2020b; Jezela-Stanek et al., 2020).
Perinatal History
Few complications occurred during pregnancy. The 20-week ultrasound of one individual (P13) showed cardiac septal defects and decreased fetal movements while the other 24 patients did not exhibit any malformations on prenatal ultrasonography. The gestation length was at least 36 weeks (36–41 weeks) in all cases and was at term for the vast majority of deliveries. All children had either normal or increased birth parameters. The mean weight and length at birth were 3.9 kg (3.2–4.5 kg) and 54 cm (49–60 cm).
There were no reported stillbirth cases and no terminated pregnancies. Data on prenatal history are not shown.
Ophthalmological and Otological Features
Data on ophthalmological features were available for 22/25 (88%) individuals. Strabismus was diagnosed in 15/25 (60%), refractive error in 10/25 (40%), and cortical visual impairment (CVI) was evident in 7/25 (28%).
Individual P1 with the Asn527Ser variant had neither CVI, strabismus, nor a refractive error. Instead, he was diagnosed with features such as microphthalmia, posterior synechiae of the anterior chamber, partial angle closure, tight falciform retinal folds, peripheral avascular retina, retinal hemorrhage, and extensive vitreous membranes. Although one individual with the Asn527Ser variant exhibited ophthalmological features not previously described in PIGT deficiency, her overall phenotype was compatible with those found in affected individuals with the Val528Met variant. Exome sequencing found no additional explanation for the ophthalmological features. Functional testing by flow cytometry to determine the surface levels of GPI-APs (Knaus et al., 2018) was unfortunately unavailable for this subject. Such testing is needed in future patients with Asn527Ser variants to prove pathogenicity and to perform deep-phenotyping of the clinical features including any anomalies of the eyes.
Otological features have only rarely been addressed previously. We identified 2/25 (8%) individuals who had suffered from hearing disorders; one presented with a sensorineural deficit (P1), and the other with an unspecified impairment (P4) (no further data were available).
Cardiovascular and Gastrointestinal Features
While congenital heart defects have been associated with other pathogenic variants of the PIGT gene (Bayat et al., 2019), we cannot report any heart anomalies in individuals with Asn527Ser and Val528Met. Echocardiography was available in 10/25 individuals and was normal in all probands.
Data on gastrointestinal disturbances were available in 16/25 individuals. None of the individuals required a gastrostomy tube. Three individuals experienced feeding difficulties during the first 2 months of life, and four experienced gastroesophageal reflux and recurrent constipation.
Neurological and Behavioral Phenotype
Epileptic seizures were reported in 22/25 individuals (88%) [12 individuals from our cohort, Decipher ID 258094 (Pagnamenta et al., 2017) P1-P7 (Jezela-Stanek et al., 2020), P3 and P10 (Bayat et al., 2019)], but only 18 individuals fulfilled the diagnostic criteria for having epilepsy [11 individuals from our cohort, Decipher ID 258094 (Pagnamenta et al., 2017), P3-5 and P7 (Jezela-Stanek et al., 2020), P3 and P10 (Bayat et al., 2019)]. In 1/18 individuals (P1) only a single afebrile seizure was reported, but an epilepsy diagnosis was given due to the additional finding of interictal epileptic discharges on electroencephalogram (EEG). Four individuals [P1, P2, and P6 (Jezela-Stanek et al., 2020), and our P4] had febrile seizures as the only seizure type and did not reach an epilepsy diagnosis. Finally, febrile seizures were present in 13/25 (52%) individuals.
Age at seizure onset was available in all but one individual. The average time of onset was around 16 months of age (from 5 months to 5.5 years). Twelve individuals experienced their first epileptic seizure during the 5th to 12th month of life while the remaining children had an onset around the 2nd to 5th year of life (Table 1).
Seizure types were either focal (12/25) or primary generalized (9/25). The predominant seizure type was focal to bilateral tonic-clonic seizures, followed by primary generalized bilateral tonic-clonic seizures. Other generalized seizure types included atypical absences (3/25), myoclonic seizures (2/25), myoclonic-atonic seizures (2/25), and eyelid myoclonia (1/25). Only one individual experienced status epilepticus (P3).
Antiepileptic drugs (AEDs) were prescribed in 18/25 individuals. Pagnamenta et al. (2017) provided no data on AED treatment. Several of the individuals described by Jezela-Stanek et al. (2020) were taking AEDs despite only having febrile seizures and thereby not fulfilling the criteria for epilepsy; these authors provided no data on the type of AEDs used. Thus, a detailed description of types of AEDs prescribed in the 18 individuals was only possible for 10 individuals, all of whom fulfilled an epilepsy diagnosis. Six of these ten individuals became seizure-free on monotherapy with either valproate (VPA) or levetiracetam. Three of the ten individuals (P2, P3, and P6) became free of epileptic seizures on a combination of VPA and lamotrigine, but individual P3 had persistent non-epileptic myoclonic jerks. One individual (P7) became seizure-free on monotherapy with carbamazepine. Three of the ten individuals had been tapered off from their AED(s) and have been seizure-free for several years.
Comparison of symptoms seen in individuals homozygous for the Val528Met variant to those found in individuals compound heterozygous for Asn527Ser or Val528Met, revealed no convincing difference regarding onset of seizures, seizure types, and cognitive and developmental outcome. Although focal and primary generalized seizures were present in both subgroups, we noticed that atypical absence seizures were only present among the homozygous individuals and that homozygous individuals became seizure-free on monotherapy with AED. Compound heterozygous individuals typically needed a combination of AEDs to achieve seizure freedom. We also explored if it was possible to draw further genotype-phenotype conclusions within the group of 25 individuals (carrying the Asn527Ser or Val528Met variant) based on the annotation of the disease causing variant found on the other allel. In part 2 of Table 2 the 25 individuals were divided in three groups: (i) individuals homozygous for the p.Val528Met variant, (ii) individuals compound heterozygous for a missense variant in addition to the p.Val528Met or p.Asn527Ser variant, and (iii) individuals compound heterozygous for a truncating variant in addition to the p.Val528Met or p.Asn527Ser variant. While the majority of individuals in the two first groups experienced seizures this was the case for all individuals in the third group. A small subset of individuals in all groups only experienced fever induced seizures and thus did not reach a formal diagnosis of epilepsy (Table 2, part 2). In all three groups 80–85% of individuals with seizures reached a formal diagnosis of epilepsy. In the first two groups the age of onset of seizures was around 9 months of life while seizures started around 2 years of age in the third group. Individuals carrying a disease causing missense variant on both allels were compatible in regard of the degree of DD. The overall seizure outcome across all groups was good as all but one individual became seizure free after treatment initiation of AEDs (Table 2).
Massive action myoclonia involving all four limbs and the trunk were observed in individual P3 without any clinical and polygraphic evidence of myoclonus at rest (Supplementary Video 1). This phenomenon started around the age of 8 years and became progressively worse and disabling during the course of the disease. No epileptic abnormalities were associated with the myoclonic phenomena on EEG (Figure 1).
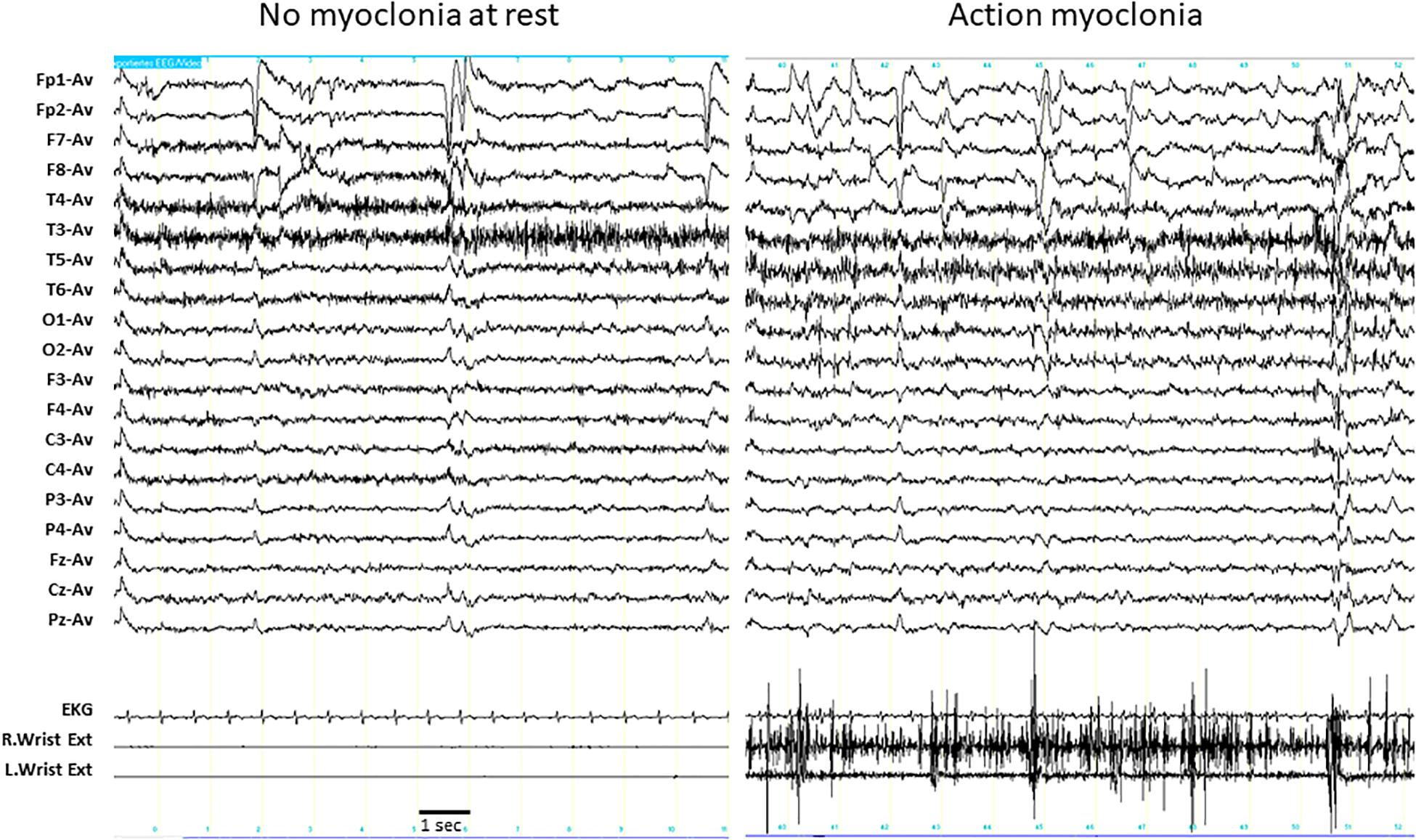
Figure 1. EEG polygraphic recording in individual P3. Left panel: the individual is at rest. EEG does not show any epileptic abnormalities, and the myoclonic EMG activity is detectable in both wrist extensors. Right panel: the individual is maintaining a posture (outstretching her arms). The muscular contraction in both wrist extensors are fragmented by irregular, high amplitude, myoclonic potentials that are intermixed with brief silent periods, reproducing the EMG pattern of action myoclonus. No epileptic discharges associated with the myoclonic potentials were detectable in the EEG. Wrist Ext, wrist extensor; R, right; L, left.
The age range of the 25 individuals spanned from 27 months to 29 years. Degree of DD was as follows: moderate (5/25), moderate-severe (6/25), and severe (4/25). In 10 individuals [Decipher ID 258094 (Pagnamenta et al., 2017), P1–7 (Jezela-Stanek et al., 2020), and P3 and P10 (Bayat et al., 2019)], we were unable to obtain enough clinical data to assess the degree of DD, notably none of the individuals had profound DD (Table 2, part 1). Of the 25 individuals, 13 eventually learned to walk: 11 could walk short distances with walking aids or support from others, and two individuals learned to walk independently. The average age of independent walking was between the 3rd and 6th year of life. We were able to collect data on manual dexterity in 13 individuals. All individuals had purposeful use of hands and could eat autonomously using cutlery, play with bricks or puzzles, or use sign language. Information on verbal capabilities was obtained from 19 individuals. All 19 individuals comprehended simple sentences and 15/19 replied using single words. Two individuals, both homozygous for the Val528Met variant, could communicate with very simple sentences: one individual (P11) was even able to educate her younger affected sibling (P12) (Supplementary Video 2). Two individuals (currently 2 and 7.5 years of age) remained non-verbal. None of the patients were diagnosed with an autism spectrum disorder.
Electroencephalogram
EEG findings were available in five individuals with epilepsy (P2, P3, P7, P8, and P10). A poorly organized background activity was reported in two individuals, with burst of theta activity intermixed. Generalized epileptic discharges with frontal predominance were reported in two individuals (P8 and P10) at the age of 14 and 3 years, respectively. Occasional focal epileptic discharges were observed in individual P2 at the age of 3 years but were not detected after the age of 14 years, whereas isolated focal spikes were observed at the age of 10 months in individual P7. Individual P3, who exhibited action myoclonus, did not exhibit interictal epileptic abnormalities on routine EEG.
Brain Imaging
At least one brain MRI scan was performed in 18/25 individuals (Table 1), and 12 of those underwent repeated MRI scans. In six individuals, the first cerebral MRI was performed during the first year of life (in all cases between the 6th and 12th month of life). Delayed myelination was noted in all six individuals, while enlargement of the subarachnoid spaces was detected in three of them. There were no follow-up data on the delayed myelination described in our individuals P4 and P7, but the myelination was age-appropriate in the remaining four individuals.
MRI was performed in twelve individuals after the age of 1 year. Scans showed age-appropriate myelination associated with several developmental anomalies such as cerebellar atrophy (11/12) occasionally involving the brainstem (1/12), and abnormal corpus callosum (3/12). Additional MRI findings included prominent cortical and subcortical volume loss (7/12), enlargement of lateral and third ventricles (3/12), white matter immaturity (2/12), and a hippocampal sclerosis (1/12) (P3). In individual P10, the MRI detected delayed myelination at the age of 4 years, but the MRI scan a year later was unremarkable (Table 1).
We have previously shown that cerebral MRI findings of individuals with PIGT deficiency include delayed myelination, prominent cortical and subcortical volume loss with brainstem atrophy, atrophy of the cerebellum, and an abnormal corpus callosum (Bayat et al., 2019). This is compatible with the current results from our individuals with Asn527Ser and Val528Met. The natural history of the delayed myelination in other PIGT variants has to be delineated further, but the present study indicates that the myelination eventually becomes age-appropriate in individuals carrying Asn527Ser and Val528Met variants. Only one individual exhibited an unremarkable MRI at 5 years of age (P9). Only few PIGT-deficient individuals without those variants have been reported to have normal cerebral MRIs (Bayat et al., 2019). The examinations were done at a very early age, however, which might explain why no structural anomalies were found.
Molecular Results
From the complete dataset of the cohort of 25 individuals, 13 different variant sites emerged: seven missense variants, two splice site variants, and four truncating variants. The Val528Met variant has been reported in 29 out of 282.654 samples (0.0001026 allele frequency)3. In comparison, the Asn527Ser variant has only been reported once in 251.292 samples (0.000003979 allele frequency) and is predicted to be damaging by all four prediction tools (Supplementary Table 1) and likely to be damaging according to the ACMG guidelines (Richards et al., 2015). The clinical findings in the individual with the Asn527Ser was compatible with those harboring the Val528Met variant. While the previously published variant c.1724_1725insC; p.Leu578fsTer35 (Bayat et al., 2019) was classified as a variant of unknown significance, all remaining variants were predicted as either likely pathogenic or pathogenic (Supplementary Table 1).
Discussion
In 2017, Pagnamenta et al. (2017) reported two individuals with PIGT deficiency: one was compound heterozygous for the variants Val528Met and Leu578∗35, while the second was homozygous for the Glu237Gln. Western blot analysis showed that the expression of the mutant protein appeared to be reduced only for the truncating variant (8). PIGT-knockout HEK293 cells were then transfected with wild-type or mutant PIGT cDNA. The largest fraction of CD59-rescued cells was observed with Val528Met, indicating highest residual functional activity (Pagnamenta et al., 2017). While the individual with the Glu237Gln was profoundly affected, the individual with the Val528Met and Leu578∗35 variants was significantly less affected. This individual walked unaided but with an ataxic gate, understood spoken language but exhibited dysarthric speech, and attended a mainstream school with one-to-one support. This was the first evidence suggesting that the Val528Met is associated with a less severe phenotype, and our data give further support to this claim. Bayat et al. (2019) described four individuals harboring the Val528Met variant in either homozygous or compound heterozygous state, and in 2020 an additional seven individuals were described (Jezela-Stanek et al., 2020). Our knowledge of the phenotypical spectrum associated with homozygous and compound heterozygous Val528Met variants remains limited. We also have little insight into the natural history of this rare disorder. This is a challenge when counseling families with newly diagnosed children with Val528Met-related PIGT deficiency.
We compared the group of PIGT individuals with the Asn527Ser or the Val528Met (in either homozygous or compound heterozygous state) to a group of PIGT individuals without such variants (Table 2). In both groups (Table 2, part 1), the majority of individuals carried biallelic missense variants while there were no cases with biallelic truncations as this would likely not be compatible with life. We also explored potential genotype-phenotype correlations within the group of individuals with the Asn527Ser or Val528Met variant and grouped individuals based on the annotation of the disease causing variant found on the other allel (Table 2, part 2). In the group of individuals that were compound heterozygous for a truncating variant in addition to the p.Val528Met or p.Asn527Ser variant all experienced epileptic seizures with an average onset around 2 years of age. We were unable to detect other genotype-phenotype correlations based on the present dataset (Table 2, part 2). Studies of rare diseases are often conducted through a multicenter approach, and researchers are dependent on data provided by caregivers and other health care providers. This gives an unavoidable data collection bias as patients are evaluated by diverse health care providers with different medical backgrounds and levels of experience. In the current study, we aimed to reduce this bias by collecting videos of individuals with the two variants. These videos of eight individuals were used to give a more uniform evaluation of motor, verbal social, and cognitive skills. We found a more favorable outcome in our individuals, with less severe global DD, a considerably later onset of epilepsy, and no premature mortality (Table 2). While no individuals with the Asn527Ser and Val528Met variants were diseased, 3/24 (13%) individuals without the variants in question died prematurely (Bayat et al., 2019). Although the cumulative mortality rate is lower compared to what has been described in other genes involved in the GPI anchor synthesis (Bayat et al., 2020a), this finding gives more strength to the claim that the two PIGT variants are associated with a milder phenotype and a better prognosis.
Epilepsy was not a constant feature in our cohort although when present, it was treatment-responsive. While all previously identified individuals with different pathogenic variants had epilepsy and a more profound global DD, the individuals with Asn527Ser or Val528Met ranged from moderate to severe global DD (Table 2, part 1). We could also show that a minority of individuals with the Val528Met variant experienced only febrile seizures or had never had epileptic seizures (Table 1). Individuals with the two variants generally became seizure-free on monotherapy with AEDs, compared with other PIGT individuals that were drug-refractory (Table 2).
The comparison of individuals with homozygous Val528Met variants and those carrying compound heterozygous variants Asn527Ser and Val528Met lead to the conclusion that homozygosity is associated with a better outcome. Our study gives evidence to the claim that Asn527Ser- and Val528Met-related PIGT deficiency is associated with a mild to moderate “developmental and epileptic encephalopathy (DEE)”, as compared to PIGT-related DEE caused by other pathogenic variants. A similar spectrum of symptoms ranging from mild to severe DEE is seen in the X-linked phosphatidylinositol glycan class A protein gene that is involved in one of the first steps of GPI anchor biosynthesis (Bayat et al., 2020b). As in other DEEs, co-existence of focal and generalized seizures in the same individual during a lifetime is a feature of PIGT deficiency. We presented five novel and two previously reported individuals [P4 and P5 (Jezela-Stanek et al., 2020)] with generalized seizures, such as myoclonic, atypical absence and tonic seizures. A detailed analysis of seizure semiology showed atypical absence seizures and bilateral tonic seizures, but it also revealed previously undetected seizure types such as myoclonic atonic seizures and eyelid-myoclonia. Myoclonic-atonic seizures are the distinguishing feature of myoclonic-atonic epilepsy (MAE), a rare epilepsy syndrome that occurs in 0.3–2.2% of children with epilepsy (Tang et al., 2020). Children with MAE usually have normal development prior to seizure onset at between 7 months and 6 years of age (Tang et al., 2020). Seizure types include myoclonic atonic, atonic, myoclonic, generalized tonic-clonic, atypical absence, and tonic seizures (Tang et al., 2020). Tang et al. (2020) showed that pathogenic genetic variants in SYNGAP1, NEXMIF, SLC6A1, KCNA2, SCN2A, STX1B, KCNB1, MECP2, ASH1L, CHD4, and SMARCA2 are the cause of seizures in 14% of MAE individuals. Our data suggest that PIGT is a new candidate gene for MAE.
The eldest proband, a 29-year-old female individual with compound heterozygous PIGT variants (P3), exhibited action myoclonia. She first showed this symptom when she was 8 years old, and it has progressively worsened and has now become very disabling. Action myoclonia may have varying etiologies, including diseases such as Lance-Adams syndrome and the large group of progressive myoclonus epilepsies. It can be associated with epileptic EEG abnormalities (Tassinari et al., 1998), and its pathophysiological mechanisms suggest a cortical-subcortical dysfunction (Avanzini et al., 2016). It can also be associated with a cerebellar syndrome and be extremely disabling and difficult to treat, as described in individual P3. As this individual was the oldest in our cohort, we cannot dismiss the possibility that myoclonus is a later manifestation in progression of PIGT deficiency syndrome.
While 22 individuals experienced seizures, only 18 of them fulfilled the clinical diagnosis of epilepsy (Fisher et al., 2014). Apart from our individual P5, who had the His376Pro and Val528Met variants and never had seizures, we found no differences in the PIGT variants among individuals with and without seizures. Our study provides a detailed and in-depth description of developmental features in patients with the less severe spectrum of PIGT deficiency, and we also offer a glimpse into the potential natural history in adulthood. Our data are important for caregivers and health care providers when dealing with uncertainties about what to expect in the future, but also when counseling families with infants newly diagnosed with PIGT deficiency related to the Val528Met variant. Further investigation is needed to better understand the natural history and to elucidate the relationship between the presence of seizures and seizure semiology and the pathogenic variants in the milder spectrum of PIGT deficiency.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by ethical committee of region Sjaelland. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ABa and RM had the idea to write the article. ABa was responsible for collecting data from the medical records and writing the first draft of the manuscript. ABa was responsible for gathering relevant literature. GR evaluated the neurophysiological data. All authors were involved in the ongoing revision of the manuscript. All authors contributed to the revision of the final manuscript.
Conflict of Interest
ABe was employed by the company GeneDx.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the author MG.
Acknowledgments
The authors would like to thank the individuals and their families for their participation in our research. The authors also thank Claire Gudex for her contribution to editing the final draft of the article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.663643/full#supplementary-material
Supplementary Table 1 | Genetic overview, annotation, and classification of novel and previously published pathogenic variants in the phosphatidylinositol glycan class T protein gene.
Supplementary Video 1 | Video of individual P3 displaying action myoclonia of limbs and the whole body during motor tasks and the absence of myoclonia during rest.
Supplementary Video 2 | Video of individuals P11 and 12 showing motor, cognitive, and verbal capabilities.
Abbreviations
AED, antiepileptic drugs; DD, developmental delay; EEG, electroencephalogram; ER, endoplasmatic reticulum; GPI, glycosylphosphatidylinositol; GPI-AP, glycosylphosphatidylinositol anchored protein; GPIBD, biosynthesis defects of the GPI anchor; ID, intellectual disability; MRI, magnetic resonance imaging; PIGT, phosphatidylinositol glycan class T protein; VPA, valproate.
Footnotes
- ^ https://cadd.gs.washington.edu
- ^ http://www.mutationtaster.org
- ^ https://gnomad.broadinstitute.org/
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Avanzini, G., Shibasaki, H., Rubboli, G., Canafoglia, L., Panzica, F., Franceschetti, S., et al. (2016). Neurophysiology of myoclonus and progressive myoclonus epilepsies. Epileptic Disord 18, 11–27. doi: 10.1684/epd.2016.0835
Bayat, A., Klovgaard, M., Johannesen, K. M., Stefan Barakat, T., Kievit, A., Montomoli, M., et al. (2020a). Deciphering the premature mortality in PIGA-CDG - An untold story. Epilepsy Res. 170:106530. doi: 10.1016/j.eplepsyres.2020.106530
Bayat, A., Knaus, A., Juul, A. W., Dukic, D., Gardella, E., Charzewska, A., et al. (2019). PIGT-CDG, a disorder of the glycosylphosphatidylinositol anchor: description of 13 novel patients and expansion of the clinical characteristics. Genet. Med. 21, 2216–2223. doi: 10.1038/s41436-019-0512-3
Bayat, A., Knaus, A., Pendziwiat, M., Afenjar, A., Barakat, T. S., Bosch, F., et al. (2020b). Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 61, 1142–1155. doi: 10.1111/epi.16545
Bellai-Dussault, K., Nguyen, T. T. M., Baratang, N. V., Jimenez-Cruz, D. A., and Campeau, P. M. (2019). Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 95, 112–121. doi: 10.1111/cge.13425
Farheen, N., Sen, N., Nair, S., Tan, K. P., and Madhusudhan, M. S. (2017). Depth dependent amino acid substitution matrices and their use in predicting deleterious mutations. Prog. Biophys. Mol. Biol. 128, 14–23. doi: 10.1016/j.pbiomolbio.2017.02.004
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Fujita, M., and Kinoshita, T. (2012). GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta 1821, 1050–1058. doi: 10.1016/j.bbalip.2012.01.004
Jezela-Stanek, A., Szczepanik, E., Mierzewska, H., Rydzanicz, M., Rutkowska, K., Knaus, A., et al. (2020). Evidence of the milder phenotypic spectrum of c.1582G >>A PIGT variant - delineation based on seven novel Polish patients. Clin. Genet. 98, 468–476. doi: 10.1111/cge.13822
Jiao, X., Xue, J., Gong, P., Bao, X., Wu, Y., Zhang, Y., et al. (2020). Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS). Orphanet. J. Rare Dis. 15:78. doi: 10.1186/s13023-020-01365-0
Kinoshita, T., and Fujita, M. (2016). Biosynthesis of GPI-anchored proteins: special emphasis on GPI lipid remodeling. J. Lipid Res. 57, 6–24. doi: 10.1194/jlr.R063313
Knaus, A., Pantel, J. T., Pendziwiat, M., Hajjir, N., Zhao, M., Hsieh, T. C., et al. (2018). Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 10:3. doi: 10.1186/s13073-017-0510-5
Kohashi, K., Ishiyama, A., Yuasa, S., Tanaka, T., Miya, K., Adachi, Y., et al. (2018). Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. 40, 53–57. doi: 10.1016/j.braindev.2017.06.005
Kvarnung, M., Nilsson, D., Lindstrand, A., Korenke, G. C., Chiang, S. C., Blennow, E., et al. (2013). A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 50, 521–528. doi: 10.1136/jmedgenet-2013-101654
Lam, C., Golas, G. A., Davids, M., Huizing, M., Kane, M. S., Krasnewich, D. M., et al. (2015). Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol. Genet. Metab. 115, 128–140. doi: 10.1016/j.ymgme.2015.04.007
Mason, S., Castilla-Vallmanya, L., James, C., Andrews, P. I., Balcells, S., Grinberg, D., et al. (2019). Case report of a child bearing a novel deleterious splicing variant in PIGT. Med. (Baltimore) 98:e14524. doi: 10.1097/MD.0000000000014524
Nakashima, M., Kashii, H., Murakami, Y., Kato, M., Tsurusaki, Y., Miyake, N., et al. (2014). Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics 15, 193–200. doi: 10.1007/s10048-014-0408-y
Ohishi, K., Inoue, N., and Kinoshita, T. (2001). PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 20, 4088–4098. doi: 10.1093/emboj/20.15.4088
Ohishi, K., Nagamune, K., Maeda, Y., and Kinoshita, T. (2003). Two subunits of glycosylphosphatidylinositol transamidase, GPI8 and PIG-T, form a functionally important intermolecular disulfide bridge. J. Biol. Chem. 278, 13959–13967. doi: 10.1074/jbc.M300586200
Pagnamenta, A. T., Murakami, Y., Taylor, J. M., Anzilotti, C., Howard, M. F., Miller, V., et al. (2017). Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur. J. Hum. Genet. 25, 669–679. doi: 10.1038/ejhg.2017.32
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Skauli, N., Wallace, S., Chiang, S. C., Baroy, T., Holmgren, A., Stray-Pedersen, A., et al. (2016). Novel pigt variant in two brothers: expansion of the multiple congenital anomalies-hypotonia seizures syndrome 3 phenotype. Genes (Basel) 7:108. doi: 10.3390/genes7120108
Sobreira, N., Schiettecatte, F., Valle, D., and Hamosh, A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930. doi: 10.1002/humu.22844
Tang, S., Addis, L., Smith, A., Topp, S. D., Pendziwiat, M., Mei, D., et al. (2020). Phenotypic and genetic spectrum of epilepsy with myoclonic atonic seizures. Epilepsia 61, 995–1007. doi: 10.1111/epi.16508
Tassinari, C. A., Rubboli, G., and Shibasaki, H. (1998). Neurophysiology of positive and negative myoclonus. Electroencephalogr. Clin. Neurophysiol. 107, 181–195. doi: 10.1016/s0013-4694(98)00058-3
Keywords: myoclonic-atonic seizures, generalized seizures, disease severity, developmental delay, glycosylphosphatidylinositol biosynthesis defects, inherited glycosylphosphatidylinositol-anchored protein (GPI-AP) deficiency
Citation: Bayat A, Pendziwiat M, Obersztyn E, Goldenberg P, Zacher P, Döring JH, Syrbe S, Begtrup A, Borovikov A, Sharkov A, Karasińska A, Giżewska M, Mitchell W, Morava E, Møller RS and Rubboli G (2021) Deep-Phenotyping the Less Severe Spectrum of PIGT Deficiency and Linking the Gene to Myoclonic Atonic Seizures. Front. Genet. 12:663643. doi: 10.3389/fgene.2021.663643
Received: 03 February 2021; Accepted: 29 March 2021;
Published: 11 May 2021.
Edited by:
Karolina Stepien, Salford Royal NHS Foundation Trust, United KingdomReviewed by:
Magdalena Sandu, Spitalul Clinic de Copii Doctor Victor Gomoiu, RomaniaAna Westenberger, University of Lübeck, Germany
Copyright © 2021 Bayat, Pendziwiat, Obersztyn, Goldenberg, Zacher, Döring, Syrbe, Begtrup, Borovikov, Sharkov, Karasińska, Giżewska, Mitchell, Morava, Møller and Rubboli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Allan Bayat, YWJheWFAZmlsYWRlbGZpYS5kaw==