 Rosangela Ferese1
Rosangela Ferese1 Rosa Campopiano1
Rosa Campopiano1 Simona Scala1Carmelo D’Alessio1Marianna Storto1
Simona Scala1Carmelo D’Alessio1Marianna Storto1 Fabio Buttari1
Fabio Buttari1 Diego Centonze1,2
Diego Centonze1,2 Giancarlo Logroscino3,4
Giancarlo Logroscino3,4 Chiara Zecca3Stefania Zampatti1,5
Chiara Zecca3Stefania Zampatti1,5 Francesco Fornai1,6Vittoria Cianci7Elisabetta Manfroi8
Francesco Fornai1,6Vittoria Cianci7Elisabetta Manfroi8 Emiliano Giardina5,9
Emiliano Giardina5,9 Mauro Magnani10Antonio Suppa1,11Giuseppe Novelli1,9
Mauro Magnani10Antonio Suppa1,11Giuseppe Novelli1,9 Stefano Gambardella1,10*
Stefano Gambardella1,10*- 1IRCCS Neuromed, Pozzilli, Italy
- 2Laboratory of Synaptic Immunopathology, Department of Systems Medicine, Tor Vergata University, Rome, Italy
- 3Center for Neurodegenerative Diseases and the Aging Brain, Department of Clinical Research in Neurology, The University of Bari “Aldo Moro,” “Pia Fondazione Card G. Panico” Hospital Tricase, Lecce, Italy
- 4Department of Basic Medicine Neuroscience and Sense Organs, University “Aldo Moro” Bari, Bari, Italy
- 5Genomic Medicine Laboratory, IRCCS Fondazione Santa Lucia, Rome, Italy
- 6Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
- 7Regional Epilepsy Centre, Great Metropolitan Hospital Bianchi-Melacrino-Morelli, Reggio Calabria, Italy
- 8Department of Neuroscience- Neurogenetics, Santa Maria Hospital, Terni, Italy
- 9Department of Biomedicine and Prevention, University of Rome “Tor Vergata,” Rome, Italy
- 10Department of Biomolecular Sciences, University of Urbino “Carlo Bo,” Urbino, Italy
- 11Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy
Charcot-Marie-Tooth (CMT) disease is the most prevalent inherited motor sensory neuropathy, which clusters a clinically and genetically heterogeneous group of disorders with more than 90 genes associated with different phenotypes. The goal of this study is to identify the genetic features in the recruited cohort of patients, highlighting the role of rare variants in the genotype-phenotype correlation. We enrolled 67 patients and applied a diagnostic protocol including multiple ligation-dependent probe amplification for copy number variation (CNV) detection of PMP22 locus, and next-generation sequencing (NGS) for sequencing of 47 genes known to be associated with CMT and routinely screened in medical genetics. This approach allowed the identification of 26 patients carrying a whole gene CNV of PMP22. In the remaining 41 patients, NGS identified the causative variants in eight patients in the genes HSPB1, MFN2, KIF1A, GDAP1, MTMR2, SH3TC2, KIF5A, and MPZ (five new vs. three previously reported variants; three sporadic vs. five familial variants). Familial segregation analysis allowed to correctly interpret two variants, initially reported as “variants of uncertain significance” but re-classified as pathological. In this cohort is reported a patient carrying a novel familial mutation in the tail domain of KIF5A [a protein domain previously associated with familial amyotrophic lateral sclerosis (ALS)], and a CMT patient carrying a HSPB1 mutation, previously reported in ALS. These data indicate that combined tools for gene association in medical genetics allow dissecting unexpected phenotypes associated with previously known or unknown genotypes, thus broadening the phenotype expression produced by either pathogenic or undefined variants.
Clinical trial registration: ClinicalTrials.gov (NCT03084224).
Introduction
Charcot-Marie-Tooth (CMT) disease, also known as hereditary motor and sensory neuropathy, is a common, clinically heterogeneous group of inherited peripheral neuropathies with an estimated prevalence of one in 2,500 individuals (Wiszniewski et al., 2013; Di Vincenzo et al., 2014).
CMT is clinically, neurophysiologically, and genetically heterogeneous. It is most commonly characterized by sensory loss that starts in the lower limbs and progresses slowly in a length-dependent manner. This produces progressive distal muscle atrophy, weakness, distal sensory loss, foot deformities, and depressed tendon reflexes (Shy et al., 2005; Rossor et al., 2013).
The clinical classification is based on age at onset, distribution of muscle weakness, sensory loss, walking difficulties, and foot deformities (Rossor et al., 2013). Neurophysiology allows subdividing the disease into a demyelinating (CMT1) and axonal (CMT2) forms depending on whether the median motor nerve conduction velocity (NCV) is below or above 38 m/s, respectively. A third form, intermediate CMT, has both demyelinating and axonal features and NCV between 25 and 45 m/s (Reilly and Shy, 2009; Berciano et al., 2012).
The duplication of PMP22 is the most common cause of CMT, with a prevalence up to 40% in some populations (Reilly and Shy, 2009; Abe et al., 2011; Saporta et al., 2011; Murphy et al., 2012; Østern et al., 2013; Sivera et al., 2013). Then, approximately 100 different genes have been linked to CMT-like phenotypes which are associated with related conditions involved in axonal transport, myelin structure, and membrane metabolism that have been found in multiple unrelated families or confirmed by functional studies (Saifi et al., 2003; Saporta et al., 2011; Azzedine et al., 2012; Vallat et al., 2013; IPNMDB, 2014; OMIM, 2014; Washington University, 2014).
Therefore, the large spectrum of genetically identifiable disease alleles complicates the molecular diagnosis. Genetic heterogeneity is associated with a wide spectrum of phenotypes, complicated by the fact that mutations in the same gene cause different phenotypes (Reilly and Shy, 2009; Azzedine et al., 2012; OMIM, 2014). Furthermore, sporadic cases of CMT are not uncommon due to autosomal recessive inheritance, reduced penetrance, late-onset, small family size, and de novo mutations (Braathen et al., 2011; Saporta et al., 2011; Vallat et al., 2013).
In this study, we enrolled 67 patients and applied a diagnostic protocol including multiple ligation-dependent probe amplification (MLPA) for copy number variation (CNV) detection of PMP22 locus, and next-generation sequencing (NGS) for sequencing of 47 genes known to be associated with CMT and routinely screened in medical genetics.
The goal of this study is to identify the genetic features in the recruited cohort of patients, highlighting the role of rare variants in the genotype-phenotype correlation.
Materials and Methods
Study Population
We collected blood or DNA samples from 67 unrelated patients with a clinical diagnosis of CMT from January 2013 to December 2019 at IRCCS Neuromed Institute (Italy). Genomic DNA was isolated from peripheral blood leukocytes according to standard procedures (QIAamp DNA Blood Mini Kit – QIAGEN).
Clinical Data
Diagnosis of CMT includes the presence of slowly progressive neuropathy with or without family history and after exclusion of other common causes of acquired neuropathy. CMT subtype was classified as CMT if both motor and sensory nerves were similarly affected, and dHMN or HSN if the neuropathy showed exclusive or predominant involvement of motor or sensory nerves, respectively. CMT patients were further subdivided into demyelinating CMT if conduction velocity of the nondominant median or ulnar nerve was ≤38 m/s and axonal or intermediate CMT if >38 m/s.
Literature Review
A systematic review of the literature was conducted to identify the detection rate of genetic variants and the clinical phenotype of CMT patients. Pubmed, Medline, and Embase database identified 23 cohort analysis studies consisting of Italian and European CMT patients in the period between 1997 and 2020 (Supplementary Table S1).
Multiple Ligation-Dependent Probe Amplification
The commercially available kit P405 (MRC-Holland, Amsterdam, Netherlands) was used for the multiplex dosage. This SALSA MLPA Probemix contains 42 MLPA probes with amplification products between 130 and 445 nt: 15 probes located in the 17p12 region (PMP22 gene), two flanking probes, seven probes in the MPZ gene, five probes in the GJB1 gene, 10 reference probes detecting autosomal chromosomes, and three probes on the X-chromosome. The MLPA was performed on DNA from patients, and four normal subjects were used as internal controls.
Next-Generation Sequencing Panel
The NGS analysis was performed using the Seq Cap EZ Choice Enrichment Kits (Hoffmann-La Roche, Basel) on an Illumina MiSeq (San Diego, CA). A full list of genes sequenced is provided in Table 1. All coding exons of the RefSeq transcripts of the genes and 15 base pairs of the flanking introns were targeted, except for GJB1, for which the target region is extended 860 bases upstream of the ATG start codon to include the nerve-specific promoter region. 99% of the coding exons were sequenced with a minimal read depth of 30X.
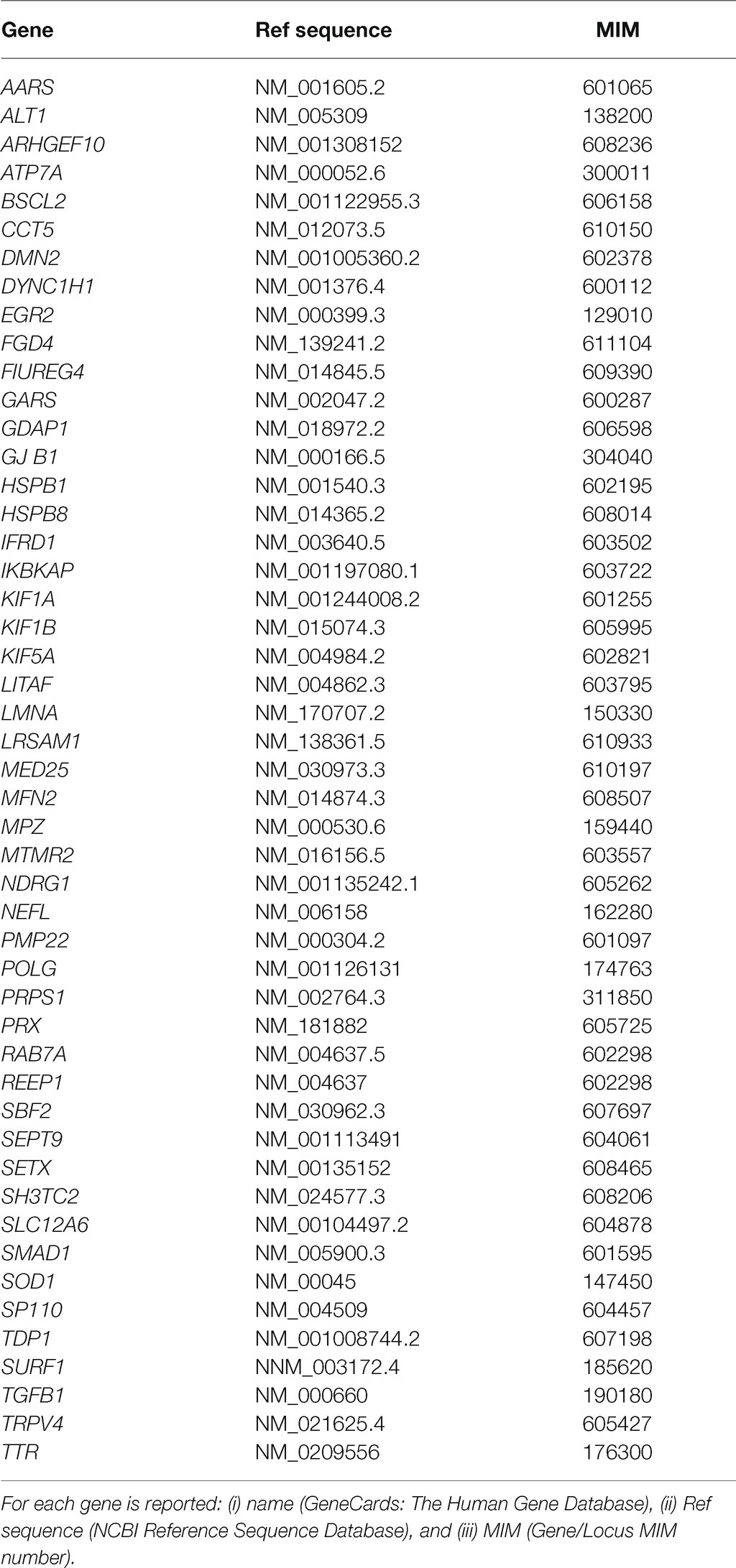
Table 1. Target genes included in NGS Panel.
GenomeUp software1 was used for data analysis. It provides automated annotation (Best Practices workflows of GATK v4.1 for germline variant calling), alignment of sequence reads to the reference genome GRCh37/hg19, and selection of potentially pathogenic variants. Direct evaluation of data sequence was performed by the Integrative Genomics Viewer v.2.3. Mutation re-sequencing and segregation analysis were performed by the Sanger sequencing ABI 3130xl Genetic Analyzer (Applied Biosystems).
Data Analysis and Variant Interpretation
Variants were classified as pathogenic (class 5), likely pathogenic (class 4), and variants of uncertain significance (VoUS; class 3) according to American College of Medical Genetics Guideline for germline variant classification (Li et al., 2017). To this aim, public databases were used (VarSome https://varsome.com; GnomAD https://gnomad.broadinstitute.org). In silico analyses of variants were performed using SIFT,2 PolyPhen2,3 PROVEAN,4 and Mutation Assessor.5 The new mutations identified have been submitted in ClinVar database.6
Results
Multiple Ligation-Dependent Probe Amplification Analysis
The variation of the whole PMP22 gene CNV was assessed through MLPA, confirming the clinical diagnosis in 26 out of 67 patients (38.8%). In detail, 20/67 (30%) were carriers of the heterozygote whole gene duplication, thus confirming the clinical diagnosis of CMT1A; 5/67 (7.4%) were carriers of the heterozygote whole gene deletion, confirming HNPP clinical diagnosis; and one was a carrier of a rare whole gene mosaic duplication of 1.5-Mb in heterozygous in PMP22 gene (MIM 601097), considered as pathogenic (Rautenstrauss et al., 1998) and thus responsible for CMT disease 1A, autosomal dominant (MIM 118220). (Family ID 564, II:1; Figure 1A), classified as sporadic on anamnesis.
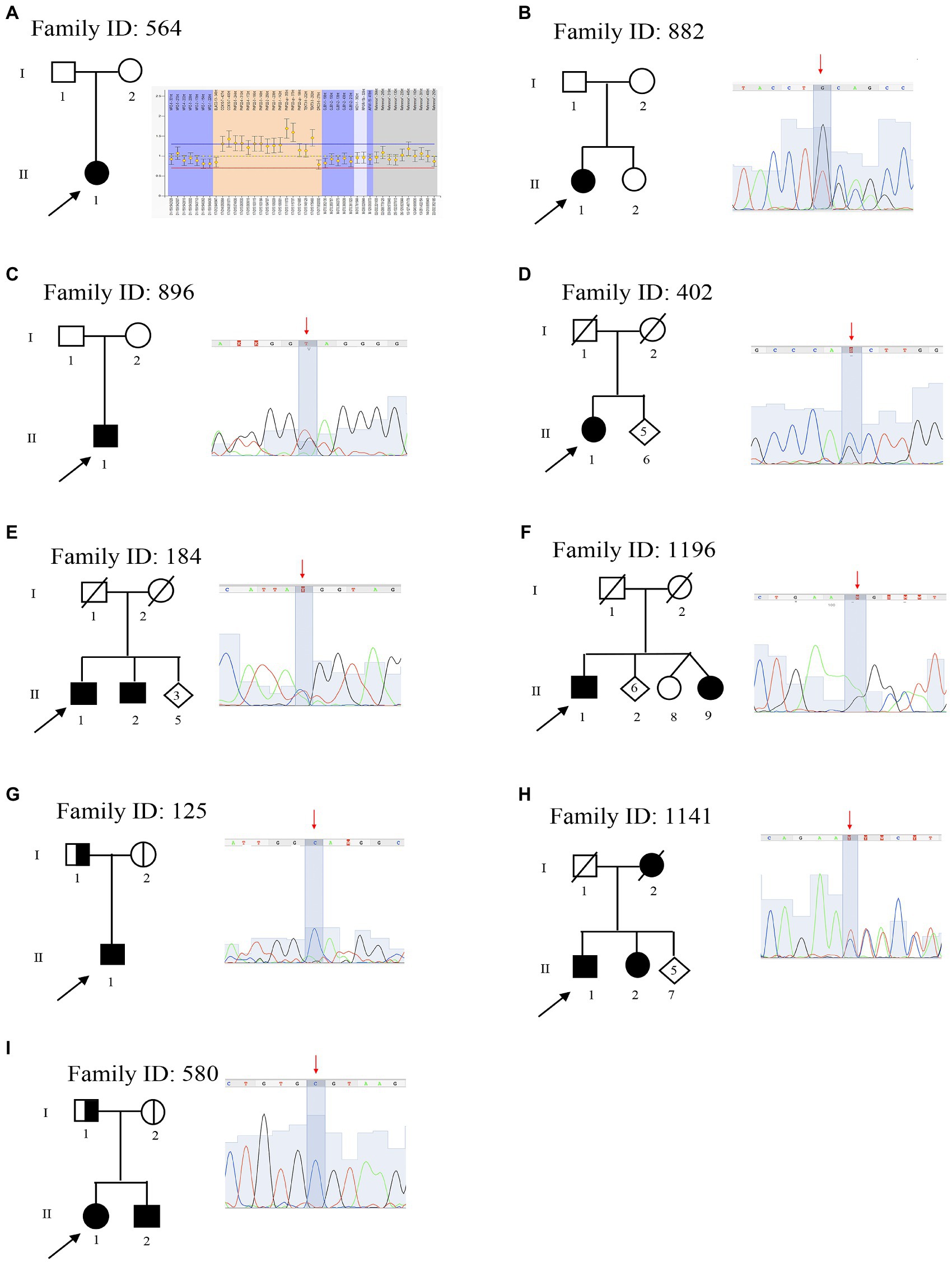
Figure 1. Pedigree and electropherograms of class 4 and 5 mutations identified by next-generation sequencing (NGS). (A) Family ID: 564, MPLA analysis and identified a mosaic duplication of 1.5-Mb (17p11.2–12); (B) Family ID: 882, heterozygous stop mutation in mitofusin-2 (MFN2) gene: NM_001127660.1:c.[2258dupT], NP_001121132.1:p.(Gln754AlafsTer9; rs773371488); (C) Family ID: 896, heterozygous stop mutation in MPZ gene: NM_000530.8: c.[306delA], NP_000521: p.(Asp104ThrfsTer14), (rs281865125); (D) Family ID: 402, heterozygous missense mutation in HSPB1 gene: NM_001540.3:[c.570G > C], NP_001531.1:p.(Gln190His); (E) Family ID: 184, heterozygous missense mutation in KIF1A gene: NM_001244008.2:c.[5332C > T], NP_ p.(Arg1778Trp), (rs765668490); (F) Family ID: 1196, heterozygous stop mutation in GADP1 gene: NM_018972.2: c.[140delA], NP_061845.2: p.(Lys47ArgfsTer3); (G) Family ID: 1251, homozygous splicing mutations in SH3TC2 gene: NM_024577.3: c.[805 + 2T > C], (rs139052887); (H) Family ID: 1141, heterozygous deletion mutation in KIF5A gene: NM_004984.2:c.[2868_2870delTCT], NP_004975.2:p.(Leu957del), (rs575223790); and (I) Family ID: 580, homozygous missense mutation in MTMR2 gene: NM_016156.5:c.[463T > C], NP_057240.3:p.(Cys155Arg).
Next-Generation Sequencing Analysis
The remaining 41 patients were tested by NGS using a target panel that considers 49 genes associated with CMT (Table 1). This approach identified the causative variants (pathogenic or likely pathogenic), in 8/41 patients (19.5%; Figure 2A). Thus, NGS improved the detection rate to 50.8% (38.8% MLPA + 12% NGS). The onset of all patients with a genetic diagnosis ranges from 7 to 57 years old, and the clinical features, consistent with phenotypes reported in OMIM database, are summarized in Table 2.
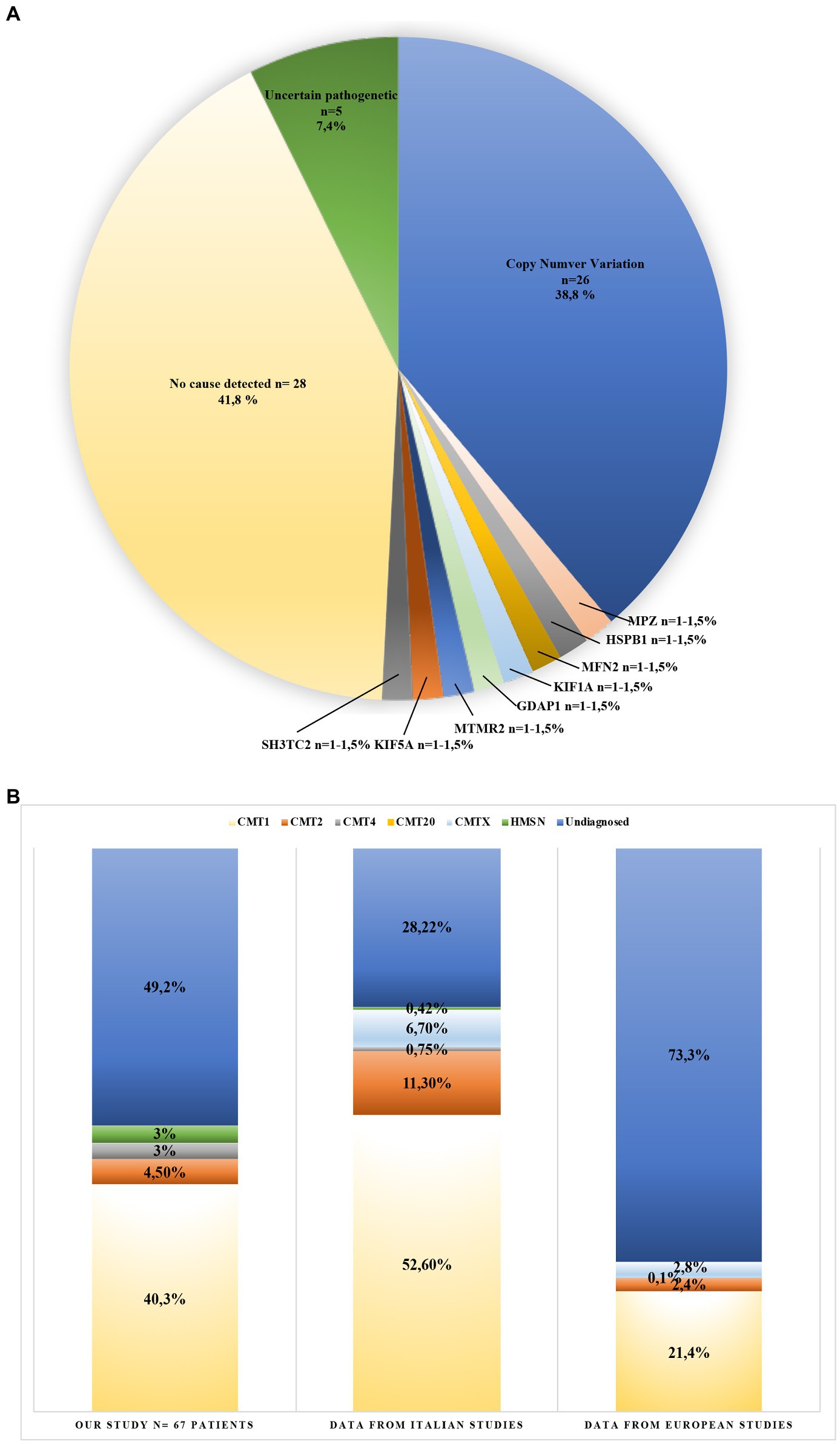
Figure 2. Genotype and phenotype of Charcot-Marie-Tooth (CMT) patients from this study (A), and CMT patients reviewed from the literature (B). (A) Variants identified in 67 CMT patients (blue: copy number variation 38.8%; orange: MPZ mutation 1.5%; gray: HSPB1 mutation 1.5%; light yellow: MFN2 mutation 1.5%; light blue: KIF1A mutation 1.5%; green: GAPD1 mutation 1.5%; dark blue: MTMR2 mutation 1.5%; brown: KIF5A mutation 1.5%; dark gray: SH3TC2 mutation 1.5%; yellow: no cause detected 41.8%; and dark green: uncertain pathogenetic 7.4%). (B) This graphic compares detection yield in CMT phenotypes obtained from the literature. (light yellow: CMT1; orange: CMT2; gray: CMT4; light blue: CMTX; green: HMSN; and blue: undiagnosed). Bar 1: data obtained in this study; Bar 2: data obtained from three Italian studies; and Bar 3: data obtained from 20 European studies (see Supplementary Material).
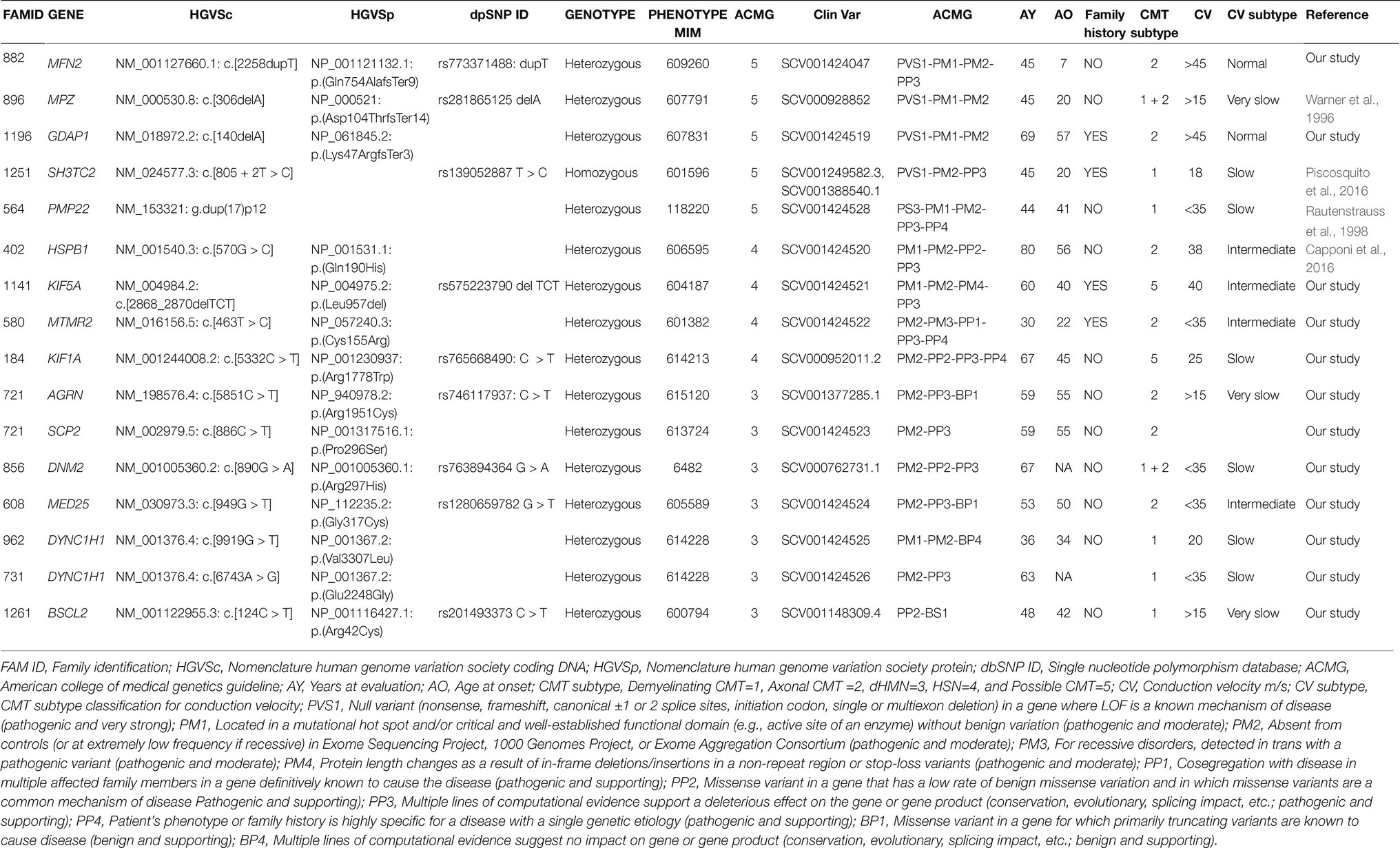
Table 2. Genetic and clinical data in the cohort of patients analyzed. Clinical (A) and genetic (B) features of the 15 CMT patients (16 variants) identified by NGS.
These variants fall in eight different genes: MFN2, MPZ, GDAP1, SH3TC2, HSPB1, KIF5A, MTMR2, KIF1A, responsible for demyelinating CMT (5/64, 8%), and axonal or intermediate CMT (3/64, 4.7%; Table 2).
Of these variants, three have been previously reported as causative of CMT (Families ID 896, 125, 402; Warner et al., 1996; Capponi et al., 2016; Piscosquito et al., 2016), five are new variants (Figure 1).
In six out of eight probands, familiar members were available for segregation analysis. Of these, four variants were already classified as pathological (Families ID 882, 1141, 721, 961), while two patients were carriers of variants classified as VoUS, but reclassified as pathological through data obtained from familiar segregation (Families ID 580, 184; Figure 1). The variants identified have been classified as sporadic (3/8, 37.5%) or familiar (5/8, 62.5%).
Causative Variants Identified as Sporadic
Family ID 882
The proband, a 45 years old female (II:1; Figure 1B), onset at 7 years old, presented with a moderate hyposthenia, axonal neuropathy, and normal conduction velocity (>45 m/s). She is a carrier of the new heterozygous stop mutation: NM_001127660.1:c.[2258dupT], NP_001121132.1:p.(Gln754AlafsTer9; rs773371488) in mitofusin-2 (MFN2; MIM 608507) considered as pathogenic and thus responsible for CMT disease 2A2A and autosomal dominant (MIM 609260). The variant was considered as sporadic since it is not present in the mother (I:2), and her father (I:1) is reported as neurological healthy.
Family ID 896
The proband, a 45 years old male (II:1; Figure 1C), onset at 20 years old, presented with axonal and demyelinating neuropathy with a very slow conduction velocity (>15 m/s). Molecular analysis identified the known heterozygous stop mutation: NM_000530.8: c.[306delA], NP_000521: p.(Asp104ThrfsTer14), (rs281865125) in MPZ (MIM 159440) considered as pathogenic (Warner et al., 1996) and thus responsible for CMT disease-dominant intermediate D (MIM 607791). The variant was considered sporadic. Family members were not available for testing, but both his mother I:2 and father I:1 were reported as neurological healthy before the age of 20 years old.
Family ID 402
The proband, a 76 years old female (II:1; Figure 1D), onset at 56, presented with progressive ankle instability and gait difficulties with and foot drop. She had a diagnosis of axonal neuropathy with an intermediate conduction velocity (38 m/s). Molecular analysis identified the known heterozygous missense mutation: NM_001540.3:c.[570G > C], NP_001531.1:p.(Gln190His) in HSPB1 (MIM 602195) considered as likely pathogenic (Capponi et al., 2016) and thus responsible for CMT disease, axonal, and type 2F autosomal dominant (MIM 606595). Family members were not available for testing.
Causative Variants Identified as Familiar
Family ID 184
The proband, a 67 years old male (II:1; Figure 1E), onset at 45 years old, presented with neuropathy, mild hyposthenia of the distal musculature, and hearing loss. He showed a slow conduction velocity (25 m/s) and signs of pyramidal tract dysfunction. Molecular analysis identified a new heterozygous missense mutation: NM_001244008.2:c.[5332C > T], NP_ 001230937:p.(Arg1778Trp), (rs765668490: C > T) in KIF1A (MIM 601255), initially considered as VoUS, but classified as likely pathogenic because of its presence in his affected brother (II:2). This variant is responsible for neuropathy, hereditary sensory, type IIC, and autosomal recessive (MIM 614213).
Family ID 1196
The proband, 69 years old male (II:1; Figure 1F), onset at 57 years old, presented with axonal neuropathy. He showed a normal conduction velocity. Molecular analysis identified a new heterozygous stop mutation: NM_018972.2: c.[140delA], NP_061845.2: p.(Lys47ArgfsTer3) in GADP1 (MIM 606598) considered as pathogenic and thus responsible for CMT disease axonal, autosomal dominant, and type 2K (MIM 607831). His sister (II:9) has a similar condition but was not available for testing.
Family ID 125
The proband, a 45 years old male (II:1; Figure 1G), onset at 20 years old, presented with early onset demyelinating neuropathy with slow conduction velocity (18 m/s). Molecular analysis identified the known homozygous splicing mutations: NM_024577.3: c.[805 + 2T > C], (rs139052887T > C) in SH3TC2 (MIM 608206) considered as pathogenic (Piscosquito et al., 2016) and thus responsible for CMT disease, autosomal recessive, and type 4C (MIM 601596). Both parents (I:1 and I:2) are heterozygote healthy carriers of this variant.
Family ID 1141
The proband a 60 years old male (II:1; Figure 1H), onset at 40 years old, presented with neuropathy and spastic paraplegia. He showed an intermediate conduction velocity (40 m/s). Molecular analysis identified a new heterozygous deletion mutation: NM_004984.2:c.[2868_2870delTCT], NP_004975.2:p.(Leu957del), (rs575223790) in KIF5A (MIM 602821) considered as likely pathogenic and thus responsible for spastic paraplegia 10 with neuropathy and autosomal dominant (MIM 604187). This variant is familiar since it is present in his affected sister (II:2) and absent in his healthy sister (II:4).
Family ID 580
The proband is a 30 years old female (II:1; Figure 1I), onset at 22, presented with axonal neuropathy and an intermediate conduction velocity (<35 m/s). Molecular analysis identified the new homozygous missense mutation: NM_016156.5:c.[463T > C], NP_057240.3:p.(Cys155Arg) in MTMR2 (MIM 603557) initially considered as VoUS, but classified as likely pathogenic because of its presence in homozygous in her affected brother (II:2), and in heterozygous in her parents (I:1 and I:2). Thus, this variant is responsible for CMT disease, type 4B1, and autosomal recessive (MIM 601382).
Variants Identified as VoUs
Seven VoUS were found in 6/67 patients (7%; Table 2; Figure 2A). Heterozygous variants identified in AGRN, SCP2, DNM2, MED25, DYNC1H1, and BSCL2 genes associated with autosomal dominant and recessive CMT.
Family ID 721
The proband a 59 years old male (II:1; Figure 3A), onset at 55 years old, presented with axonal neuropathy. He showed a very slow conduction velocity (>15 m/s). He had clear psychic slowness, cognitive impairment, and floating paresthesia in the upper limbs. Molecular analysis identified two new heterozygous missense mutations: (a) in AGRN (MIM 103320) NM_198576.4:c.[5851C > T], NP_940978.2: p.(Arg1951Cys), (rs746117937) responsible for myasthenic syndrome, congenital, 8, with pre- and postsynaptic defects, autosomal recessive (MIM 615120), and SCP2 (MIM 184755) NM_002979.5 c.[886C > T] and NP_001317516.1:p.(Pro296Ser) responsible for leukoencephalopathy with dystonia and motor neuropathy, autosomal recessive (MIM 613724). Both variants are not present in the healthy sister (II:2), but these data are not sufficient to consider this variant as likely pathogenic.
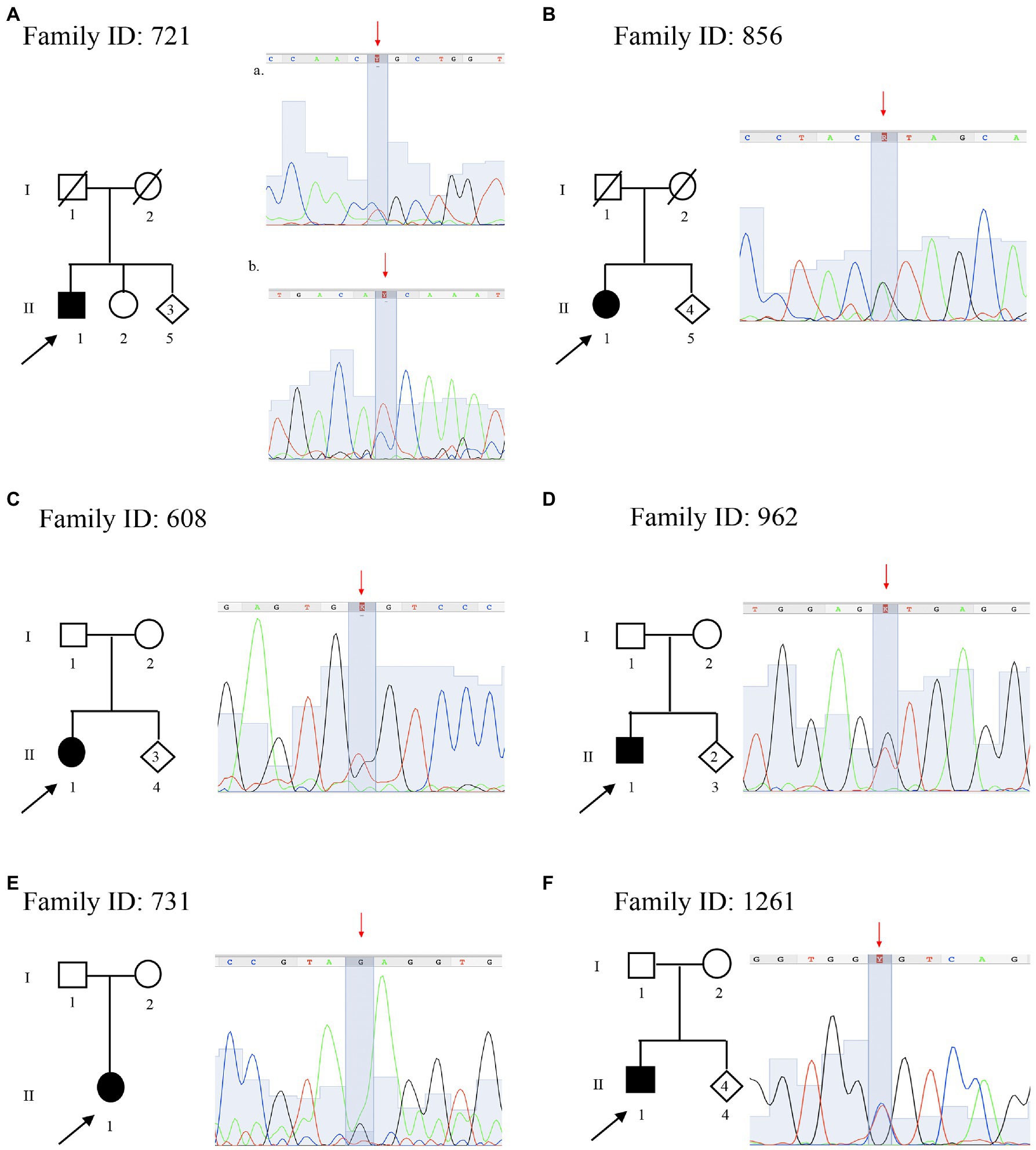
Figure 3. Pedigree and electropherograms of variants of uncertain significance identified by NGS. (A) Family ID: 721, two missense variants in (a) AGRN gene NM_198576.4:c.[5851C > T], NP_940978.2:p.(Arg1951Cys), (rs746117937) and (b) SCP2 gene NM_002979.5 c.[886C > T], p.(Pro296Ser); (B) Family ID: 856, missense variant in DNM2 gene NM_001005360.2:c.[890G > A], NP_001005360.1:p.(Arg297His); (rs763894364); (C) Family ID: 608, missense variant in MED25 gene NM_030973.3:c.[949G > T], NP_112235.2: p.(Gly317Cys); (rs1280659782); (D) Family ID: 962, N missense variant in DYNC1H1 gene NM_001376.4:c.[9919G > T], NP_001367.2:p.(Val3307Leu); (E) Family ID: 731, missense variant in DYNC1H1 gene NM_001376.4:c.[6743A > G], NP_001367.2:p.(Glu2248Gly); and (F) Family ID: 1261, missense variant in BSCL2 gene NM_001122955.3:c.[124C > T], NP_001116427.1:p.(Arg42Cys); (rs201493373).
Family ID 856
The proband, a 67 years old female (II:1; Figure 3B), presented with axonal neuropathy. She showed a slow conduction velocity (<35 m/s). Molecular analysis identified a new heterozygous missense mutation: NM_001005360.2:c.[890G > A], NP_001005360.1:p.(Arg297His; rs763894364) in DNM2 (MIM 602378) responsible for CMT disease, axonal type 2M, and autosomal dominant (MIM 606482). Family members were not available for testing.
Family ID 608
The proband, a 53 years old female (II:1; Figure 3C), onset at 50, presented with axonal neuropathy. She showed an intermediate conduction velocity (<35 m/s). Molecular analysis identified a new heterozygous missense mutation: NM_030973.3;c.[949G > T], NP_112235.2: p.(Gly317Cys; rs1280659782 G > T) in MED25 (MIM 610197) responsible for CMT disease, type 2B2, and autosomal recessive (MIM 605589). Family members were not available for testing.
Family ID 962
The proband, 36 years old male (II:1; Figure 3D), onset at 34, presented with demyelinating neuropathy. He showed a slow conduction velocity (20 m/s). Molecular analysis identified a new heterozygous missense mutation: NM_001376.4:c.[9919G > T], NP_001367.2:p.(Val3307Leu) in DYNC1H1 (MIM 600112) responsible for CMT disease, type 20, and autosomal dominant (MIM 614228). Family members were not available for testing.
Family ID 731
The proband, 63 years old female (II:1; Figure 3E), presented with axonal neuropathy. She showed a slow conduction velocity (<35 m/s). Molecular analysis identified a new heterozygous missense mutation: NM_001376.4:c.[6743A > G], NP_001367.2:p.(Glu2248Gly) in DYNC1H1 (MIM 600112) responsible for CMT disease, type 20, and autosomal dominant (MIM 614228). Family members were not available for testing.
Family ID 1261
The proband, 48 years old male (II:1; Figure 3F), onset a 42 years old, presented with demyelinating neuropathy. He showed a very slow conduction velocity (>15 m/s) and hollow foot. Molecular analysis identified a new heterozygous missense mutation: NM_001122955.3:c.[124C > T], NP_001116427.1:p.(Arg42Cys); (rs201493373) in BSCL2 (MIM 606158), responsible for neuropathy, distal hereditary motor, and type VA (MIM 600794) autosomal dominant. Family members were not available for testing.
Discussion
In this study, 67 patients with a clinical diagnosis of CMT were selected for molecular analysis of genes known to be related to the disease. Although the genetic of CMT is featured by a strong genetic heterogeneity, most of the patients carry 17p CNV of PMP22. Among this CNV, almost all cases carry the whole gene duplication of PMP22. The prevalence of the duplication varies among populations, ranging from 15% in Norwegian studies, up to more than 50% in Italy and Spain (Abe et al., 2011; Saporta et al., 2011; Murphy et al., 2012; Østern et al., 2013; Sivera et al., 2013; Høyer et al., 2014). In this study, only 30% of Italian patients (20/67) carry the whole gene duplication of PMP22, thus under-representing other Italian studies though still being in line with prevalence measured in European studies (Figure 2B).
When ruling out carriers of 17p CNV in PMP22, NGS identified the causative variants in 8/41 patients, which correspond only to 12% of the patients analyzed in this study (8 out of 67), being over 50% of the total of causative variants confirming the clinical diagnosis detected in the present analysis. Thus, combining various genetic tools using a target panel of 49 genes allowed to cover to a greater extent the complex clinical setting.
Several cohort studies tried to identify the diagnostic yield in CMT patients. To this aim, we conducted a systematic review of 23 cohort analysis studies consisting of Italian and European CMT patients in the period between 1997 and 2020. This data, summarized in Figure 2B and Supplementary data, identified a diagnostic yield of 71.7% in Italy (Mostacciuolo et al., 2001; Manganelli et al., 2014; Gentile et al., 2020) and 26.7% in Europe (Bort et al., 1997; Saporta et al., 2011; Murphy et al., 2012; Gess et al., 2013; Østern et al., 2013; Sivera et al., 2013; Høyer et al., 2014; Antoniadi et al., 2015; Laššuthová et al., 2016; Lupo et al., 2016; Dohrn et al., 2017; Marttila et al., 2017; Bacquet et al., 2018; Hoebeke et al., 2018; Milley et al., 2018; Nicolas et al., 2018; Lerat et al., 2019; Vaeth et al., 2019; Cortese et al., 2020; Figure 2B). Therefore, the prevalence of genetic cases of CMT provided by the present data represents an average compared with the general prevalence measured in Italy and Europe. The differences in the diagnostic rate may be explained by the differences in the specific features of the cohorts being analyzed, the number of demyelinating CMT cases enrolled, and the heterogeneous inclusion criteria considering the common causative, which were genes adopted by the previous MLPA and Sanger sequencing.
In our cohort, NGS approach identified variants in eight different genes: MFN2, MPZ, GDAP1, SH3TC2, HSPB1, KIF5A, MTMR2, and KIF1A (Table 2), five responsible for demyelinating CMT, and three for axonal or intermediate CMT. These NGS-identified variants appeared as sporadic (3/8) or familial (5/8).
A frequency of sporadic or de novo mutations is reported in 28–34% (Marques et al., 2005; Ando et al., 2017), although this percentage may vary and some studies report up to a half of the patients as sporadic (Ando et al., 2017). In this study, variants considered as sporadic are reported in three out of eight patients with a NGS genetic diagnosis (37.5%). This percentage, in line with other studies, may represent an overestimation since this trend cannot be verified in a few families where a full familial anamnesis is lacking. This point is crucial since it demonstrates how a careful, multifaceted genetic approach with a complete familial anamnesis and genetic segregation analysis could provide a reduction of sporadic CMT.
Among sporadic variants, the proband Family ID 564 (Figure 1A) is a carrier of a whole gene mosaic in the PMP22 gene. This is consistent with the high instability of the PMP22 gene which undergoes de novo PMP22 duplications in up to 90% of sporadic CMT1 (Boerkoel et al., 2002; Marques et al., 2005).
The other probands carry sporadic variants in the gene MFN2, MPZ, and HSPB1. Mutations in MFN2 cause CMT type 2A by altering mitochondrial fusion and trafficking along with the axonal microtubule system (Guerriero et al., 2020). Pathogenic variants in MFN2 are typically inherited as autosomal dominant and are usually missense variants (Di Vincenzo et al., 2014).
The sporadic variant identified, Family ID 883 (Figure 1B), consists of a single base insertion in the coiled-coil domains producing a stop mutation p.(Gln754AlafsTer9). This could be a de novo variant. De novo MFN2 mutations are regularly found in patients with a classical CMT2 phenotype (Østern et al., 2013), and some MFN2 mutations have also been reported as de novo in several patients (Züchner et al., 2004; Chung et al., 2006; Verhoeven et al., 2006; Cho et al., 2007). The early onset of the proband, at 7 years old, is in line with data reporting that mutations in MPZ, MFN2, or NEFL are the most frequent disease causes of patients with infantile-onset CMT (Hsu et al., 2019).
Thanks to NGS studies, several data are broadening the phenotype spectrum produced by mutations in some CMT genes. For example, mutations in the N-terminal motor domain of KIF5A are responsible for hereditary spastic paraplegia (SPG10 MIM 604187) and CMT type 2 (CMT2), clustered in the switch regions SWI (199–204) and SWII (232–237) necessary for microtubules interaction (Filosto et al., 2018). On the other hand, mutations in the C-terminal cargo-binding tail domain (Tail domain) are related to a specific ALS phenotype (ASL MIM 617921), with an early disease onset but a longer survival compared with typical amyotrophic lateral sclerosis (ALS; Brenner et al., 2018; Nicolas et al., 2018). Beyond these main phenotypes, other complex and overlapping phenotypes are emerging, which demonstrates that KIF5A mutations may be responsible for a wide range and heterogeneous disease spectrum (Schüle et al., 2008; Tessa et al., 2008). For example, in a patient, a mixed slowly progressive disease was described resembling ALS as well as HSP and axonal neuropathy. This is caused by a mutation within the terminal region of the stalk domain (Cho et al., 2007). Additionally, the patient Family ID 1141 (Figure 1H) reported in this paper represents the first case of a familial mutation, the new heterozygous deletion p.Leu957del, falling in the tail domain of KIF5A producing spastic paraplegia 10 with autosomal dominant neuropathy (MIM 604187).
Taken together, these results broaden the phenotypic spectrum of KIF5A mutations and confirm the importance of cytoskeletal defects in the pathogenesis of both ALS and CMT.
In line with this, the phenotype related to heat shock protein 27 (HSP27) is broadening. For instance, mutations in the HSPB1 gene have been reported to cause autosomal dominant CMT with minimal sensory involvement (CMT 2F MIM 606595). Then, two independent studies reported different HSPB1 mutations in two sporadic cases and one consanguineous family with ALS, suggesting that the disease spectrum of HSP27 may not be limited to CMT2/dHMN (Scarlato et al., 2015; Capponi et al., 2016; Adriaenssens et al., 2017). In this study, we identified the known heterozygous missense mutation p.Gln190H in Family ID 402 (Figure 1D), which falls in the C-terminal domain involved in the control of its chaperone-like activity, responsible for CMT, axonal, and type 2F autosomal dominant (MIM 606595; Lelj-Garolla and Mauk, 2012). This variant falls in the C-terminal domain involved in the control of chaperone-like activity (Lelj-Garolla and Mauk, 2012; Capponi et al., 2016). It has been previously reported in a sporadic ALS patient with onset at 58 yo, and axonal neuropathy with an intermediate conduction velocity (38 m/s; Capponi et al., 2016). Therefore, considering this study in which the same variant has been identified in a CMT patient, the genotype-phenotype correlation is very complex. Functional characterization of different mutations located in the C-terminal domain did not show a clear correlation between the location of the mutation and associated cellular phenotype. This suggested that the genomic variant, more than its location, determines the cell pathology and phenotype, However, the present study contradicts such a conclusion which is not consistent with the present data which indicate how the same substitution has been identified in patients owing to a different disease (ALS in one case CMT in another; Capponi et al., 2016). This suggests the importance of how specific genetic backgrounds may alter the expression of the genetic variants while explaining how rare variants, like VoUS may be responsible for the disease.
In this study, seven VoUS were identified in six (7%) patients. These variants, all in heterozygous status, all fall in one of those genes associated with autosomal dominant and recessive CMT: AGRN, SCP2, DNM2, MED25, DYNC1H1, and BSCL2. This high number of VoUS is in line with the previous studies reporting a higher number of VoUS in neuropathy-associated genes, including single mutations in autosomal recessive CMT genes compared with the control population, with some patients presenting more than one VoUS in different CMT genes (Gonzaga-Jauregui et al., 2015). For instance, here, we report multiple VoUS in the proband of Family ID 721 (Figure 3A) characterized by two VoUS in AGRN and SCP2. A recent hypothesis suggests that a combinatorial effect of rare variants contributes to the disease burden in CMT and partly explains its various phenotypes. This confirms what was demonstrated by using in vivo experimental models in zebrafish (Cortese et al., 2020). Therefore, the interpretation of VoUS remains a key diagnostic challenge in the current NGS era.
Conclusion
This cohort analysis demonstrates the importance of combining different molecular approaches to identify the causative variant in CMT patients. The use of NGS target panel consisting of 49 genes identified the causative variants in eight patients, improving the detection rate to 50.8%. Although this seems to represent only a small fraction of patients, the identification of rare mutations allows dissecting of unexpected phenotypes associated with previously known or unknown genotypes, thus broadening the phenotype expression produced by variants. This is the example of a patient carrying a novel familial mutation in the tail domain of KIF5A (a protein domain previously associated with familial ALS), and a CMT patient carrying an HSPB1 mutation, previously reported in ALS.
In this cohort, the higher frequency of VoUS identified is in line with the previous studies, confirming that the interpretation of VoUS remains a key diagnostic challenge in the current NGS era. In line with this, in this cohort, the segregation analysis allowed to correctly interpret two variants, initially reported as VoUS, but re-classified as pathological.
To improve the detection rate in CMT patients, whole-genome sequencing (WGS) and whole-exome sequencing (WES) are strongly required. While the use of WES is now accepted for diagnostic purposes, WGS is able to both identify novel and rare variants in coding as well as noncoding regions. Therefore, these approaches allow the identification of new genes and rare variants, thus improving the genetic detection rate.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by IRCCS Neuromed Ethical Committees. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CD, FB, DC, GL, CZ, SZ, AS, FF, VC, EM, EG, MM, and LS performed the recruitment and clinical evaluations of patients. RF, RC, and SS performed the genetic analyses. DC, FF, GN, MS, and SG supervised the work. RF and SG wrote the first draft of the manuscript. All authors read the manuscript, contributed to the manuscript revision, and approved the final version.
Funding
This work was supported by the Italian Ministry of Health (current research 2019–2023: Identification of New Variants and/or New Genes Responsible for Ataxia and Spastic Paraplegia), (5XMille – 2018).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the patients and their relatives participating in this study.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fgene.2021.682050/full#supplementary-material
Footnotes
1. ^https://platform.genomeup.com
3. ^http://genetics.bwh.harvard.edu/pph2
4. ^http://provean.jcvi.org/index.php
References
Abe, A., Numakura, C., Kijima, K., Hayashi, M., Hashimoto, T., and Hayasaka, K. (2011). Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J. Hum. Genet. 56, 364–368. doi: 10.1038/jhg.2011.20
Adriaenssens, E., Geuens, T., Baets, J., Echaniz-Laguna, A., and Timmerman, V. (2017). Novel insights in the disease biology of mutant small heat shock proteins in neuromuscular diseases. Brain 140, 2541–2549. doi: 10.1093/brain/awx187
Ando, M., Hashiguchi, A., Okamoto, Y., Yoshimura, A., Hiramatsu, Y., Yuan, J., et al. (2017). Clinical and genetic diversities of Charcot-Marie-Tooth disease with MFN2 mutations in a large case study. J. Peripher. Nerv. Syst. 22, 191–199. doi: 10.1111/jns.12228
Antoniadi, T., Buxton, C., Dennis, G., Forrester, N., Smith, D., Lunt, P., et al. (2015). Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med. Genet. 16:84. doi: 10.1186/s12881-015-0224-8
Azzedine, H., Senderek, J., Rivolta, C., and Chrast, R. (2012). Molecular genetics of Charcot-Marie-Tooth disease: from genes to genomes. Mol. Syndromol. 3, 204–214. doi: 10.1159/000343487
Bacquet, J., Stojkovic, T., Boyer, A., Martini, N., Audic, F., Chabrol, B., et al. (2018). Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation. BMJ Open 8:e021632. doi: 10.1136/bmjopen-2018-021632
Berciano, J., Sevilla, T., Casasnovas, C., Sivera, R., Vílchez, J. J., Infante, J., et al. (2012). Illa I; Programa 3 (Enfermedades Neuromusculares) del Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) del Instituto de Salud Carlos III. Guía diagnóstica en el paciente con enfermedad de Charcot-Marie-Tooth [Guidelines for molecular diagnosis of Charcot-Marie-Tooth disease]. Neurologia 27, 169–178. doi: 10.1016/j.nrl.2011.04.015
Boerkoel, C. F., Takashima, H., Garcia, C. A., Olney, R. K., Johnson, J., Berry, K., et al. (2002). Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann. Neurol. 51, 190–201. doi: 10.1002/ana.10089
Bort, S., Nelis, E., Timmerman, V., Sevilla, T., Cruz-Martínez, A., Martínez, F., et al. (1997). Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum. Genet. 99, 746–754. doi: 10.1007/s004390050442
Braathen, G. J., Sand, J. C., Lobato, A., Høyer, H., and Russell, M. B. (2011). Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur. J. Neurol. 18, 39–48. doi: 10.1111/j.1468-1331.2010.03037.x
Brenner, D., Yilmaz, R., Müller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Capponi, S., Geuens, T., Geroldi, A., Origone, P., Verdiani, S., Cichero, E., et al. (2016). Molecular chaperones in the pathogenesis of amyotrophic lateral sclerosis: the role of HSPB1. Hum. Mutat. 37, 1202–1208. doi: 10.1002/humu.23062
Cho, H. J., Sung, D. H., Kim, B. J., and Ki, C. S. (2007). Mitochondrial GTPase mitofusin 2 mutations in Korean patients with Charcot-Marie-Tooth neuropathy type 2. Clin. Genet. 71, 267–272. doi: 10.1111/j.1399-0004.2007.00763.x
Chung, K. W., Kim, S. B., Park, K. D., Choi, K. G., and Lee, J. H. (2006). Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain 129, 2103–2118. doi: 10.1093/brain/awl174
Cortese, A., Wilcox, J. E., Polke, J. M., Poh, R., and Skorupinska, M. (2020). Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 94, e51–e61. doi: 10.1212/WNL.0000000000008672
Di Vincenzo, C., Elzinga, C. D., Medeiros, A. C., Karbassi, I., and Jones, J. R. (2014). The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol. Genet. Genomic. Med. 2, 522–529. doi: 10.1002/mgg3.106
Dohrn, M. F., Glöckle, N., Mulahasanovic, L., Heller, C., Mohr, J., Bauer, C., et al. (2017). Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J. Neurochem. 143, 507–522. doi: 10.1111/jnc.14217
Filosto, M., Piccinelli, S. C., Palmieri, I., Necchini, N., and Valente, M. (2018). A novel mutation in the stalk domain of KIF5A causes a slowly progressive atypical motor syndrome. J. Clin. Med. 8:17. doi: 10.3390/jcm8010017
Gentile, L., Russo, M., Fabrizi, G. M., Taioli, F., Ferrarini, M., Testi, S., et al. (2020). Charcot-Marie-Tooth disease: experience from a large Italian tertiary neuromuscular center. Neurol. Sci. 41, 1239–1243. doi: 10.1007/s10072-019-04219-1
Gess, B., Schirmacher, A., Boentert, M., and Young, P. (2013). Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul. Disord. 23, 647–651. doi: 10.1016/j.nmd.2013.05.005
Gonzaga-Jauregui, C., Harel, T., Gambin, T., Kousi, M., Griffin, L. B., Francescatto, L., et al. (2015). Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 12, 1169–1183. doi: 10.1016/j.celrep.2015.07.023
Guerriero, S., D'Oria, F., Rossetti, G., Favale, R. A., Zoccolella, S., Alessio, G., et al. (2020). CMT2A harboring mitofusin 2 mutation with optic nerve atrophy and normal visual acuity. Int. Med. Case Rep. J. 13, 41–45. doi: 10.2147/IMCRJ.S237620
Hoebeke, C., Bonello-Palot, N., Audic, F., Boulay, C., Tufod, D., Attarian, S., et al. (2018). Retrospective study of 75 children with peripheral inherited neuropathy: genotype-phenotype correlations. Arch. Pediatr. 25, 452–458. doi: 10.1016/j.arcped.2018.09.006
Høyer, H., Braathen, G. J., Busk, Ø. L., Holla, Ø. L., Svendsen, M., Hilmarsen, H. T., et al. (2014). Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. Biomed. Res. Int. 2014:210401. doi: 10.1155/2014/210401
Hsu, Y. H., Lin, K. P., Guo, Y. C., Tsai, Y. S., Liao, Y. C., and Lee, Y. C. (2019). Mutation spectrum of Charcot-Marie-Tooth disease among the Han Chinese in Taiwan. Ann. Clin. Transl. Neurol. 6, 1090–1101. doi: 10.1002/acn3.50797
IPNMDB (2014). The mutation database of inherited peripheral neuropathies. Available at: http://www.molgen.ua.ac.be/CMTMutations/Home/Default.cfm
Laššuthová, P., Šafka Brožková, D., Krůtová, M., Neupauerová, J., Haberlová, J., Mazanec, R., et al. (2016). Improving diagnosis of inherited peripheral neuropathies through gene panel analysis. Orphanet J. Rare Dis. 11:118. doi: 10.1186/s13023-016-0500-5
Lelj-Garolla, B., and Mauk, A. G. (2012). Roles of the N- and C-terminal sequences in Hsp27 self-association and chaperone activity. Protein Sci. 21, 122–133. doi: 10.1002/pro.761
Lerat, J., Magdelaine, C., Roux, A. F., Darnaud, L., Beauvais-Dzugan, H., Naud, S., et al. (2019). Hearing loss in inherited peripheral neuropathies: molecular diagnosis by NGS in a French series. Mol. Genet. Genomic. Med. 7:e839. doi: 10.1002/mgg3.839
Li, M. M., Datto, M., and Duncavage, E. J. (2017). Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 19, 4–23. doi: 10.1016/j.jmoldx.2016.10.002
Lupo, V., García-García, F., Sancho, P., Tello, C., García-Romero, M., Villarreal, L., et al. (2016). Assessment of targeted next-generation sequencing as a tool for the diagnosis of Charcot-Marie-Tooth disease and hereditary motor neuropathy. J. Mol. Diagn. 18, 225–234. doi: 10.1016/j.jmoldx.2015.10.005
Manganelli, F., Tozza, S., Pisciotta, C., Bellone, E., Iodice, R., Nolano, M., et al. (2014). Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 19, 292–298. doi: 10.1111/jns.12092
Marques, W. J., Freitas, M. R., Oliveira, A. S. B., Calia, L., Melo, A., Lucena, R., et al. (2005). 17p duplicated Charcot–Marie–Tooth 1A: characteristics of a new population. J. Neurol. 252, 972–979. doi: 10.1007/s00415-005-0797-9
Marttila, M., Kytövuori, L., Helisalmi, S., Kallio, M., Laitinen, M., Hiltunen, M., et al. (2017). Molecular epidemiology of Charcot-Marie-Tooth disease in Northern Ostrobothnia, Finland: a population-based study. Neuroepidemiology 49, 34–39. doi: 10.1159/000478860
Milley, G. M., Varga, E. T., Grosz, Z., Nemes, C., Arányi, Z., Boczán, J., et al. (2018). Genotypic and phenotypic spectrum of the most common causative genes of Charcot-Marie-Tooth disease in Hungarian patients. Neuromuscul. Disord. 28, 38–43. doi: 10.1016/j.nmd.2017.08.007
Mostacciuolo, M. L., Righetti, E., Zortea, M., Bosello, V., and Schiavon, F. (2001). Charcot-Marie-Tooth disease type I and related demyelinating neuropathies: mutation analysis in a large cohort of Italian families. Hum. Mutat. 18, 32–41. doi: 10.1002/humu.1147
Murphy, S. M., Laura, M., Fawcett, K., Pandraud, A., Liu, Y. T., Davidson, G. L., et al. (2012). Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 83, 706–710. doi: 10.1136/jnnp-2012-302451
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283. doi: 10.1016/j.neuron.2018.02.027
OMIM (2014). Online Mendelian Inheritance in Man, OMIM (TM). Available at: http://www.ncbi.nlm.nih.gov/omim/
Østern, R., Fagerheim, T., Hjellnes, H., Nygård, B., Mellgren, S. I., and Nilssen, Ø. (2013). Diagnostic laboratory testing for Charcot Marie Tooth disease (CMT): the spectrum of gene defects in Norwegian patients with CMT and its implications for future genetic test strategies. BMC Med. Genet. 14:94. doi: 10.1186/1471-2350-14-94
Piscosquito, G., Saveri, P., and Magri, S. (2016). Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot-Marie-Tooth disease (CMT4). J. Peripher. Nerv. Syst. 21, 142–149. doi: 10.1111/jns.12175
Rautenstrauss, B., Liehr, T., Fuchs, C., Bevot, A., Bornemann, A., Postler, E., et al. (1998). Mosaicism for Charcot-Marie-Tooth disease type 1A: onset in childhood suggests somatic reversion in early developmental stages. Int. J. Mol. Med. 1, 333–337. doi: 10.3892/ijmm.1.2.333
Reilly, M. M., and Shy, M. E. (2009). Diagnosis and new treatments in genetic neuropathies. J. Neurol. Neurosurg. Psychiatry 80, 1304–1314. doi: 10.1136/jnnp.2008.158295
Rossor, A. M., Polke, J. M., Houlden, H., and Reilly, M. M. (2013). Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 9, 562–571. doi: 10.1038/nrneurol.2013.179
Saifi, G. M., Szigeti, K., Snipes, G. J., Garcia, C. A., and Lupski, J. R. (2003). Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Investig. Med. 51, 261–283. doi: 10.1136/jim-51-05-14
Saporta, A. S., Sottile, S. L., Miller, L. J., Feely, S. M., Siskind, C. E., and Shy, M. E. (2011). Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 69, 22–33. doi: 10.1002/ana.22166
Scarlato, M., Viganò, F., Carrera, P., Previtali, S. C., and Bolino, A. (2015). A novel heat shock protein 27 homozygous mutation: widening of the continuum between MND/dHMN/CMT2. J. Peripher. Nerv. Syst. 20, 419–421. doi: 10.1111/jns.12139
Schüle, R., Kremer, B. P., Kassubek, J., Auer-Grumbach, M., and Kostic, V. (2008). SPG10 is a rare cause of spastic paraplegia in European families. J. Neurol. Neurosurg. Psychiatry 79, 584–587. doi: 10.1136/jnnp.2007.137596
Shy, M. E., Lupski, J. R., and Chance, P. F. (2005). “Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic and pathologic features,” in Peripheral Neuropathy. 4th Edn. eds. P. J. Dyck and P. K. Thomas (Philadelphia: Elsevier Saunders), 1623–1658.
Sivera, R., Sevilla, T., Vílchez, J. J., Martínez-Rubio, D., and Chumillas, M. J. (2013). Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81, 1617–1625. doi: 10.1212/WNL.0b013e3182a9f56a
Tessa, A., Silvestri, G., de Leva, M. F., Modoni, A., Denora, P. S., and Masciullo, M. (2008). A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. J. Neurol. 255, 1090–1092. doi: 10.1007/s00415-008-0840-8
Vaeth, S., Christensen, R., Dunø, M., Lildballe, D. L., and Thorsen, K. (2019). Genetic analysis of Charcot-Marie-Tooth disease in Denmark and the implementation of a next generation sequencing platform. Eur. J. Med. Genet. 62, 1–8. doi: 10.1016/j.ejmg.2018.04.003
Vallat, J. M., Mathis, S., and Funalot, B. (2013). The various Charcot-Marie-Tooth diseases. Curr. Opin. Neurol. 26, 473–480. doi: 10.1097/WCO.0b013e328364c04b
Verhoeven, K., Claeys, K. G., Züchner, S., Schröder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 129, 2093–2102. doi: 10.1093/brain/awl126
Warner, L. E., Hilz, M. J., Appel, S. H., Killian, J. M., Kolodry, E. H., Karpati, G., et al. (1996). Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron 17, 451–460. doi: 10.1016/S0896-6273(00)80177-4
Washington University (2014). Neuromuscular disease center: hereditary motor sensory neuropathies. Available at: http://neuromuscular.wustl.edu/time/hmsn.html
Wiszniewski, W., Szigeti, K., and Lupski, J. R. (2013). “Chapter 126 – hereditary motor and sensory neuropathies,” in Emery and Rimoin’s Principles and Practice of Medical Genetics. 6th Edn. eds. L. R. David, E. P. Reed, and K. Bruce (Oxford, UK: Academic Press), 1–24.
Züchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., et al. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451. doi: 10.1038/ng1341
Glossary

Keywords: neurogenetics, Charcot-Marie-Tooth disease, multiple ligation dependent probe amplification, next-generation sequencing, diagnosis
Citation: Ferese R, Campopiano R, Scala S, D’Alessio C, Storto M, Buttari F, Centonze D, Logroscino G, Zecca C, Zampatti S, Fornai F, Cianci V, Manfroi E, Giardina E, Magnani M, Suppa A, Novelli G and Gambardella S (2021) Cohort Analysis of 67 Charcot-Marie-Tooth Italian Patients: Identification of New Mutations and Broadening of Phenotype Expression Produced by Rare Variants. Front. Genet. 12:682050. doi: 10.3389/fgene.2021.682050
Edited by:
Xiu-An Yang, Chengde Medical College, ChinaReviewed by:
Rincic Martina, University of Zagreb, CroatiaRaji Grewal, Saint Francis Medical Center, United States
Copyright © 2021 Ferese, Campopiano, Scala, D’Alessio, Storto, Buttari, Centonze, Logroscino, Zecca, Zampatti, Fornai, Cianci, Manfroi, Giardina, Magnani, Suppa, Novelli and Gambardella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Gambardella, c3RlZmFuby5nYW1iYXJkZWxsYUB1bml1cmIuaXQ=