 Aleša Kristan1
Aleša Kristan1 Tadej Pajič2,3,4
Tadej Pajič2,3,4 Aleš Maver3
Aleš Maver3 Tadeja Režen5
Tadeja Režen5 Tanja Kunej6Rok Količ7Andrej Vuga7Martina Fink1,2Špela Žula2Helena Podgornik2,8Saša Anžej Doma2,9Irena Preložnik Zupan2,9
Tanja Kunej6Rok Količ7Andrej Vuga7Martina Fink1,2Špela Žula2Helena Podgornik2,8Saša Anžej Doma2,9Irena Preložnik Zupan2,9 Damjana Rozman5
Damjana Rozman5 Nataša Debeljak1*
Nataša Debeljak1*- 1Medical Centre for Molecular Biology, Institute of Biochemistry and Molecular Genetics, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 2Department of Hematology, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 3Clinical Institute of Genomic Medicine, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 4Clinical Biochemistry, Faculty of Medicine, University of Maribor, Maribor, Slovenia
- 5Centre for Functional Genomics and Bio-Chips, Institute of Biochemistry and Molecular Genetics, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 6Department of Animal Science, Biotechnical Faculty, University of Ljubljana, Ljubljana, Slovenia
- 7Kemomed Research and Development, Kemomed Ltd., Ljubljana, Slovenia
- 8Clinical Biochemistry, Faculty of Pharmacy, University of Ljubljana, Ljubljana, Slovenia
- 9Department of Internal Medicine, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
An erythrocytosis is present when the red blood cell mass is increased, demonstrated as elevated hemoglobin and hematocrit in the laboratory evaluation. Congenital predispositions for erythrocytosis are rare, with germline variants in several genes involved in oxygen sensing (VHL, EGLN1, and EPAS1), signaling for hematopoietic cell maturation (EPOR and EPO), and oxygen transfer (HBB, HBA1, HBA2, and BPGM) that were already associated with the eight congenital types (ECYT1–8). Screening for variants in known congenital erythrocytosis genes with classical sequencing approach gives a correct diagnosis for only up to one-third of the patients. The genetic background of erythrocytosis is more heterogeneous, and additional genes involved in erythropoiesis and iron metabolism could have a putative effect on the development of erythrocytosis. This study aimed to detect variants in patients with yet unexplained erythrocytosis using the next-generation sequencing (NGS) approach, targeting genes associated with erythrocytosis and increased iron uptake and implementing the diagnostics of congenital erythrocytosis in Slovenia. Selected 25 patients with high hemoglobin, high hematocrit, and no acquired causes were screened for variants in the 39 candidate genes. We identified one pathogenic variant in EPAS1 gene and three novel variants with yet unknown significance in genes EPAS1, JAK2, and SH2B3. Interestingly, a high proportion of patients were heterozygous carriers for two variants in HFE gene, otherwise pathogenic for the condition of iron overload. The association between the HFE variants and the development of erythrocytosis is not clearly understood. With a targeted NGS approach, we determined an actual genetic cause for the erythrocytosis in one patient and contributed to better management of the disease for the patient and his family. The effect of variants of unknown significance on the enhanced production of red blood cells needs to be further explored with functional analysis. This study is of great significance for the improvement of diagnosis of Slovenian patients with unexplained erythrocytosis and future research on the etiology of this rare hematological disorder.
Introduction
Erythrocytes or red blood cells are the most abundant cells in the blood with the main function of tissue oxygen delivery (Lee and Percy, 2011). An increase in red blood cell mass (RCM) for more than 125% of predicted for age and sex is defined as absolute erythrocytosis. Increased RCM leads to raised blood viscosity and manifests as elevated hemoglobin (Hb) and hematocrit (Ht). Different criteria of Hb and Ht have been published for the diagnosis of erythrocytosis, with the consensus that Hb > 185 g/L and Ht > 0.52 in men and Hb > 165 g/L and Ht > 0.48 in women in at least two separate blood counts at 2 months apart requires further investigations (Patnaik and Tefferi, 2009; Keohane et al., 2013; McMullin, 2014, 2016; Bento, 2018). Clinical signs are associated with increased blood viscosity and include non-specific signs like dizziness, itching, facial plethora, redness of the hands, pulmonary hypertension, and, in some patients, serious complications like thromboembolic events and death (Lee and Arcasoy, 2015; McMullin et al., 2019a,b).
Several factors could lead to increased RCM. The cause of erythrocytosis could be congenital or acquired and can be further divided into primary and secondary. An erythrocytosis is classified as primary, when there is an intrinsic defect in the erythroid progenitor cells, and secondary when the defect outside the erythroid compartment is driving the bone marrow to produce more red blood cells (McMullin, 2016; Bento, 2018). The most common reasons for erythrocytosis are acquired, due to somatic variants or various extrinsic factors that lead to reduced oxygen supply and thus stimulation of erythropoiesis, such as chronic pulmonary, cardiac, renal, hepatic diseases, erythropoietin (EPO)-secreting tumors, high-altitude living, smoking, sleep apnea, recombinant EPO, and androgen administration (McMullin, 2008; Lee and Percy, 2011; Bento, 2018). Genetic defects in several pathways and mechanisms that regulate erythropoiesis lead to erythrocytosis, including oxygen-sensing pathway and regulation of EPO transcription, EPO signal transduction mediated via EPOR-JAK2 signaling cascade, and regulation of hemoglobin-oxygen affinity (Gaspersic et al., 2020). Primary acquired erythrocytosis, i.e., somatic erythrocytosis (OMIM ID: 133100), is the consequence of somatic genetic variants in Janus kinase 2 (JAK2) gene and SH2B adaptor protein 3 (SH2B3) gene, which lead to constant activation of EPO signaling pathway (McMullin and Cario, 2016; Maslah et al., 2017; Bento, 2018). Somatic variant p.Val617Phe and variants in exon 12 of the JAK2 gene are the reason for the development of somatic erythrocytosis also termed polycythemia vera (PV) (OMIM ID: 263300) in 98% of erythrocytosis cases (Baxter et al., 2005; Scott et al., 2007; Bento, 2018). Cases with congenital erythrocytosis due to germline variants are much rarer. Based on the genetic origin, there are eight types of congenital (familiar) erythrocytosis, namely, ECYT1–8. Heterozygous variants in the EPO receptor gene (EPOR) are responsible for the development of ECYT1 (OMIM ID: 133100). The mechanism underlying ECYT2–5 (OMIM IDs: 263400, 609820, 611783, and 617907) is impairment of genes von Hippel–Lindau tumor suppressor (VHL), egl-9 family hypoxia-inducible factor 1 (EGLN1), endothelial PAS domain protein 1 (EPAS1), and EPO involved in the oxygen sensing. ECYT6–8 (OMIM IDs: 617980, 617981, and 222800) are developed due to variants in hemoglobin genes HBB, HBA1, and HBA2 and biphosphoglycerate mutase (BPGM), which lead to increased oxygen affinity of hemoglobin (Figure 1; McMullin, 2016; Bento, 2018). Except for VHL and BPGM-associated erythrocytosis, all other familial types have shown an autosomal-dominant type of inheritance (Amberger et al., 2019). Besides the above-mentioned genes, also other genes are included in the pathways of red blood cell production but were not yet associated with the clinical outcome of erythrocytosis (Gaspersic et al., 2020).
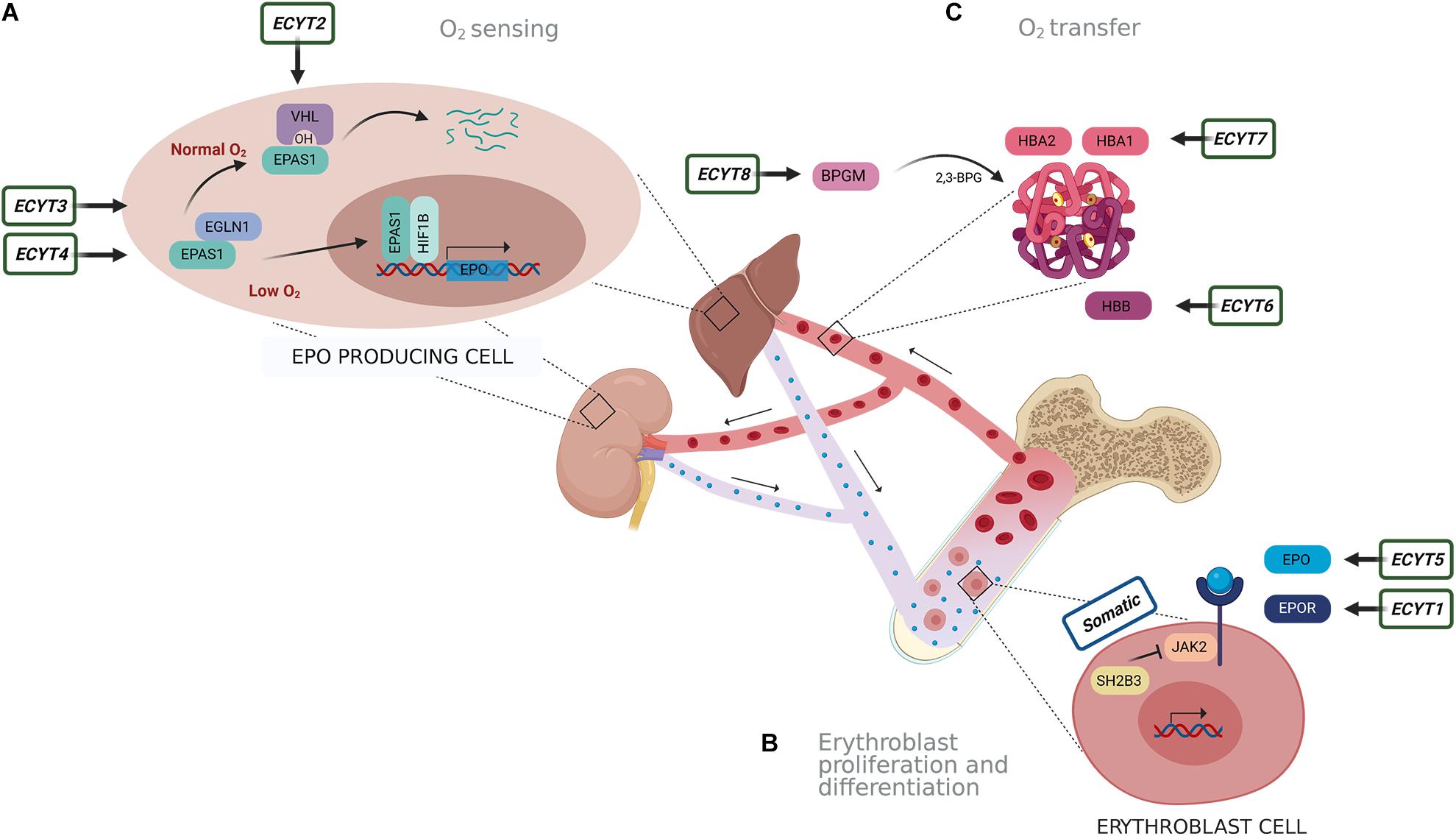
Figure 1. Schematic presentation of genes responsible for eight types of congenital erythrocytosis (ECYT1–8) and somatic erythrocytosis. Genes are involved in pathways of (A) oxygen sensing, (B) signaling for proliferation and differentiation of erythroblasts and (C) oxygen transfer. (A) Oxygen is detected in the liver and kidney. In the normal oxygen conditions, hypoxia-inducible transcription factor (HIF) (the main isoform associated with the erythropoiesis is EPAS1) is hydroxylated by prolyl hydroxylase EGLN1, following binding of VHL. This results in ubiquitination and degradation of HIF. Under hypoxic conditions (e.g., 1% O2), oxygen availability is limited; thus, hydroxylation is diminished. This results in the stabilization of HIF that is transported to the nucleus where it forms a dimer complex with its beta subunit HIF1B (officially termed ARNT) and acts as transcription factor on numerous target genes, including EPO. (B) EPO is transported to the bone marrow, where it binds with its receptor EPOR on the erythroblast cells. This subsequently activates JAK2 signaling cascade for proliferation and differentiation of erythroblasts. SH2B3 is an inhibitor of JAK2 kinase. (C) Hemoglobin alpha (HBA1 and HBA2) and beta (HBB) are important binding proteins for oxygen and are involved in oxygen transfer. The enzyme BPGM forms an allosteric effector 2,3-biphosphoglycerate (2,3-BPG) for regulation of hemoglobin oxygen affinity.
It is increasingly evident that the same genes and pathways could be involved in the pathology of different diseases. Hereditary hemochromatosis (HH) is a disorder of systemic iron overload, due to the deficiency in iron regulatory hormone hepcidin. Variants in genes encoding HH protein (HFE), hemojuvelin (HJV), hepcidin (HAMP), transferrin receptor protein 2 (TFR2), and ferroportin (SLC40A1) could lead to loss of hepcidin transcription or activity and development of five HH types. The most common causes for HH are homozygous or compound heterozygous defects (p.Cys282Tyr, p.His63Asp, and p.Ser65Cys) in gene HFE, resulting in autosomal-recessive HH type 1 (Lanktree et al., 2017; Brissot et al., 2018). Recently, several research groups found HFE variants at higher frequencies among patients with unexplained erythrocytosis, i.e., idiopathic erythrocytosis (IE), compared with the general population (Biagetti et al., 2018; Burlet et al., 2019; Gurnari et al., 2019). Similarly, patients with the HFE variants had significantly higher Hb and Ht values than normal (Barton et al., 2000; Asif et al., 2019). It was suggested that HFE mutations could induce erythropoiesis through higher iron bioavailability, since iron is dedicated to the synthesis of Hb (Hentze et al., 2010; Biagetti et al., 2018; Burlet et al., 2019). Several gene panels of hemochromatosis and iron metabolism-associated genes have been developed (Badar et al., 2016; Faria et al., 2016; Ferbo et al., 2016; Lanktree et al., 2017).
In the past, most of the laboratories and diagnostic centers included only genes for ECYT1–8 in their routine laboratory evaluation for congenital erythrocytosis, using Sanger sequencing as a predominant method (Gaspersic et al., 2020). However, the practice has been so far that only about 20–30% of patients received a proper diagnosis with screening for known variants associated with erythrocytosis. Therefore, the majority of patients remained idiopathic (Bento et al., 2013; Camps et al., 2016; Girodon et al., 2017; Bento, 2018). This implicates that several other genes and mechanisms must be involved in the disease development. In recent years, the next-generation sequencing (NGS) approach was introduced into diagnostics of patients with IE or suspected congenital erythrocytosis. The biggest advantage of NGS sequencing in rare disease diagnostics is that it allows a massive parallel sequencing of multiple genomic regions of interest at once (Fernandez-Marmiesse et al., 2018). Several researchers used whole-genome sequencing (WGS) to identify novel candidate genes and variants involved in the development of congenital erythrocytosis (Taylor et al., 2015; Lenglet et al., 2018). To overcome the drawbacks of WGS, like high cost and processing a huge amount of data, targeted NGS panels were established. Camps et al. (2016) developed the erythrocytosis gene panel of 21 candidate genes, which are involved in key disease-driven pathways or were identified in prior WGS projects (Taylor et al., 2015; Camps et al., 2016). Targeted NGS of erythrocytosis-associated genes was later also used by a French research group studying erythrocytosis (Girodon et al., 2017; Filser et al., 2021), and erythrocytosis gene panels are gradually introduced into the routine clinical practice (Gaspersic et al., 2020).
The diagnostic procedures for the evaluation of patients with abnormally high Hb and Ht had been insufficient in Slovenia so far (Mlakar, 2008). A previous genetic analysis of patients with signs of erythrocytosis included only the examination of the JAK2 gene for variants causative for PV, with allele-specific PCR, high-resolution melting (HRM) analysis, and Sanger sequencing (Baxter et al., 2005; Ugo et al., 2010; Geay et al., 2020). A huge proportion of patients with excluded PV were undiagnosed and remained idiopathic. It was urgent to modernize the diagnostic algorithm for erythrocytosis and to extend the genetic analysis for other known and novel candidate genes associated with the development of erythrocytosis. We have recently developed a national diagnostic algorithm for erythrocytosis, differentiating patients with absolute erythrocytosis, in whom acquired causes and PV are excluded (Anzej Doma et al., 2021a). The precise selection of patients to the point where genetic testing is considered is important to improve the diagnostic yield.
In the previous study, we have described and verified the targeted NGS method, which we developed for the genetic characterization of IE (Kristan et al., 2021). In the same gene panel, we included for the first time genes associated with erythrocytosis and HH. This was an important improvement to the erythrocytosis gene panels already used in international clinical practice since erythropoiesis and iron metabolism pathways are clearly intertwined. In the present study, we used targeted NGS to diagnose a group of patients with IE, which were selected over a period of 8 years (Anzej Doma et al., 2021a). The targeted NGS approach, in combination with the national diagnostic algorithm, improved the diagnostic accuracy of the Slovenian patients with IE, as we provided new explanations for the development of erythrocytosis to the patients.
Materials and Methods
Patients
Adult patients with erythrocytosis of unknown cause were selected based on the diagnostic algorithm for erythrocytosis (Anzej Doma et al., 2021a) from individuals followed up at University Medical Centre Ljubljana (UMCLj) between 2011 and 2019. All patients were of Slovenian ethnic origin. A complete hemogram was performed for the patients, with additional relevant hematological parameters like ferritin values and saturation of transferrin. The inclusion criteria were (a) confirmed absolute erythrocytosis with hemoglobin >185 g/L for men and 165 g/L for women or hematocrit >0.52 for men and >0.48 for women twice over at least 2 months; (b) absence of pathogenic JAK2 variants p.Val617Phe and exon 12 variants for PV; and (c) absence of any defined cause of secondary acquired erythrocytosis. Participants gave informed consent; the study was approved by the National Medical Ethics Committee, Ministry of Health of the Republic of Slovenia, approval no. 115/07/15 (0120-198/2015-4, 0120-287/2019-4). Patients were also reviewed for a family history of erythrocytosis. Several patients reported similar symptoms of erythrocytosis in families, and we were able to gather additional samples of other family members. Overall, 25 patients were selected for the genetic analysis with targeted NGS: 21 unrelated patients and two families, each with two participating members. Additionally, a family member without erythrocytosis from one of the families was included. DNA samples were extracted from granulocytes as described earlier (Kristan et al., 2021). We also included commercial reference DNA control NA12878, obtained from the Coriell Institute.
Targeted Next-Generation Sequencing and Data Analysis
We used targeted NGS to analyze samples from selected patients, one family member without erythrocytosis, and a reference DNA control. Library preparation and enrichment of 39 genes involved in erythropoiesis and hemochromatosis development were performed as described previously (Kristan et al., 2021). For enrichment, sample libraries were combined into multiplex pools of 12 samples pooled by mass. Enriched libraries were sequenced on MiniSeq sequencer (Illumina, San Diego, CA, United States) in 2 × 150 cycles. After duplicates were removed, the alignment of reads to UCSC hg19 reference assembly was done using BWA algorithm (v0.6.3), and variant calling was done using GATK framework (v2.8). Only variants exceeding the quality score of 30.0 and depth of 5 were used for downstream analysis. Variant annotation was performed using ANNOVAR and snpEff algorithms, with pathogenicity predictions in dbNSFPv2 database. Reference gene models and transcript sequences are based on the RefSeq database. Variants with population frequency exceeding 1% in 1000 genomes and ESP6500, synonymous variants, intronic variants, and variants outside the clinical target were filtered out during analyses (Bergant et al., 2018). Separately, we also looked for clinically significant variants in HFE gene, which has a higher population frequency than 1%. The interpretation of sequence variants was based on ACMG/AMP standards and guidelines (Richards et al., 2015), modified in accordance with the ACGS recommendations (Ellard et al., 2019). Variants are classified as follows: class 1, benign variants; class 2, possibly benign variants; class 3, variants of uncertain significance, not enough evidence; class 4, possibly pathogenic variants; and class 5, pathogenic variants. Classification of variants is supported by the evidence categories (Richards et al., 2015). A list of in silico tools with deleterious predictions of variants was obtained from the Varsome database1 (Kopanos et al., 2019). A variant was predicted to have a deleterious effect on the gene or gene product (evidence category PP3), when the majority of computational predictors or the majority of meta-predictors (REVEL, METALR, METASVM, and CADD) supported a deleterious effect. The cutoff value on deleteriousness for CADD (Combined Annotation Dependent Depletion) score was set to >20 (Rentzsch et al., 2019). The frequencies of identified variants in the general population were gathered from the GnomAD browser, version v2.1.12. We reviewed databases ClinVar (Landrum et al., 2018), LOVD (Fokkema et al., 2011), HGMD (Stenson et al., 2003), COSMIC (Tate et al., 2019), and CIViC (Griffith et al., 2017) to search for clinically relevant data of variants. Gene abbreviations are according to the official symbols in HUGO Gene Nomenclature Committee (Tweedie et al., 2021). The purpose of the reference DNA sample was to serve as a control for sequencing performance. For benchmarking the method sensitivity, reference DNA NA12878 was sequenced, and variant calls were compared against the high-confidence variant calls provided by the NIST Genome in a bottle consortium (v.3.3.2).
Validation Using Sanger Sequencing
All identified pathogenic variants were further confirmed by Sanger sequencing (GATC Biotech, Konstanz, Germany). Sanger sequencing and prior PCR amplification were performed with custom-designed primers (IDT) and are available upon request.
Results
Sequencing Performance
All 27 samples, including patients, one individual without erythrocytosis, and a reference DNA control, were sequenced by the targeted NGS. On average, 87% of mapped reads were on target regions, which indicates a successful enrichment. Targeted sequenced bases had high quality, as over 92% of aligned bases were sequenced with the minimum quality of Q30. With sequencing, we reached the median coverage of 230× (Figure 2A). The capture design encompassed 377 regions, of which we attained an average coverage of at least 10× for 362 regions (96%) across the sequenced samples, indicating that the majority of captured regions were sequenced at sufficient coverage for sensitive variant detection (Figure 2B). Only 15 target regions (4.0%) had an average coverage lower than 10× across samples, probably due to the high guanine–cytosine (GC) content of these regions (Supplementary Table 1). We cross-referenced the regions with poor coverage against the published pathogenic or likely pathogenic variants in the ClinVar database and found that these regions do not contain any known erythrocytosis-associated clinically relevant variants.
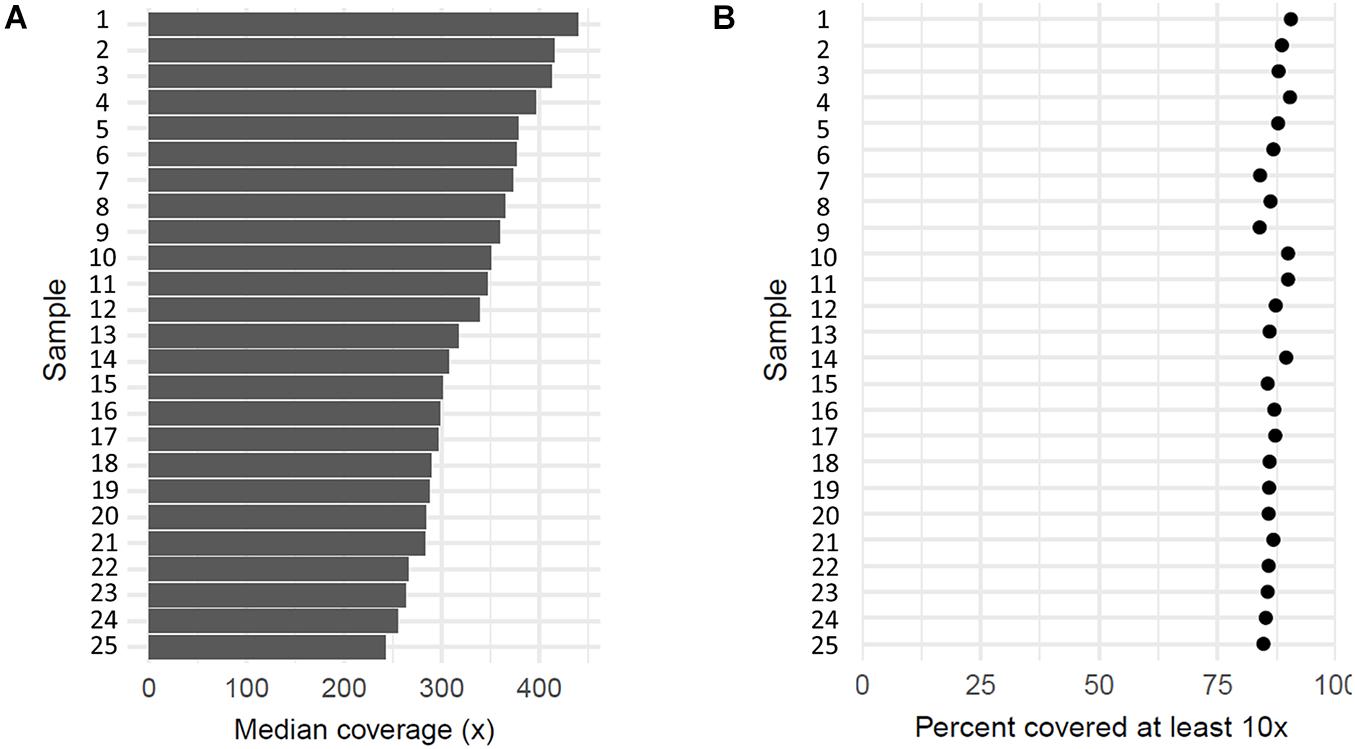
Figure 2. Coverage of 25 sequenced patient’s samples. (A) Each bar represents median coverage depth for an individual sample. (B) Each dot represents the percentage of target regions with coverage at least 10× for an individual sample.
Using our analysis approach, we detected 47 out of 51 variants within the targeted region of the NA12878 reference sample. Of these, three undetected variants were located within the known regions of poor coverage and were thus not considered in the sensitivity calculation. Apart from that, only a single variant was not detected in the sensitivity experiment due to the low coverage of the region in the control sample, corresponding to an estimated sensitivity of 97.9% for our sequencing approach.
Identified Variants
Several variants were identified using the erythrocytosis and hemochromatosis gene panel (Figure 3). Four variants were identified in the known erythrocytosis-causing genes EPAS1, JAK2, and SH2B3 (Table 1). One variant in EPAS1 gene had been already reported in the literature as causing erythrocytosis, while the remaining variants were not yet clinically associated with erythrocytosis. In addition, we also identified variants from the hemochromatosis set of genes, two HFE variants (Table 2) and one in the SEC23 homolog B (SEC23B) gene, which was found to be associated with a different condition. All the identified variants were located in the coding regions of the analyzed genes.
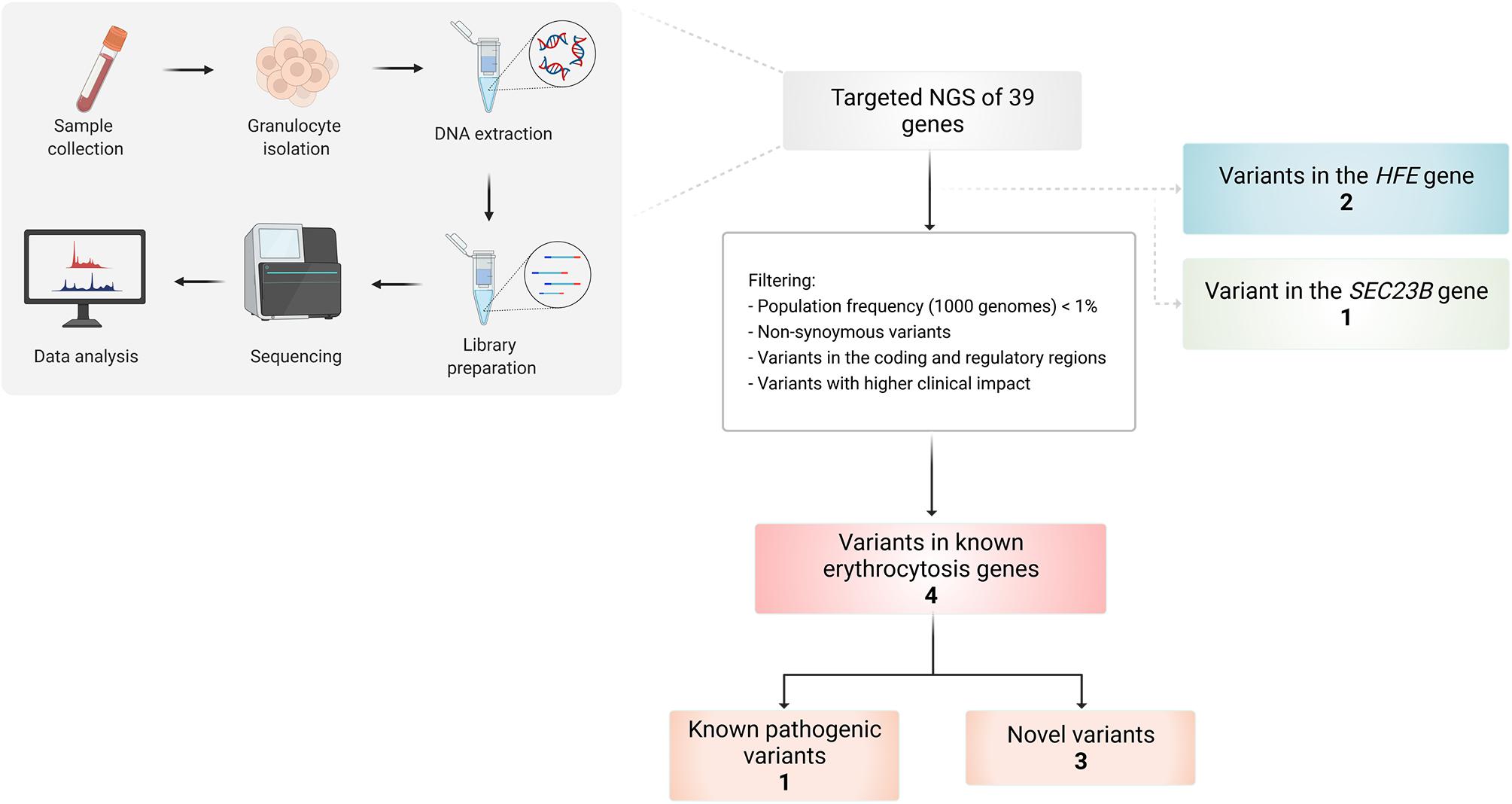
Figure 3. Workflow of the study and overview of the identified variants in selected patients using targeted NGS of 39 genes.
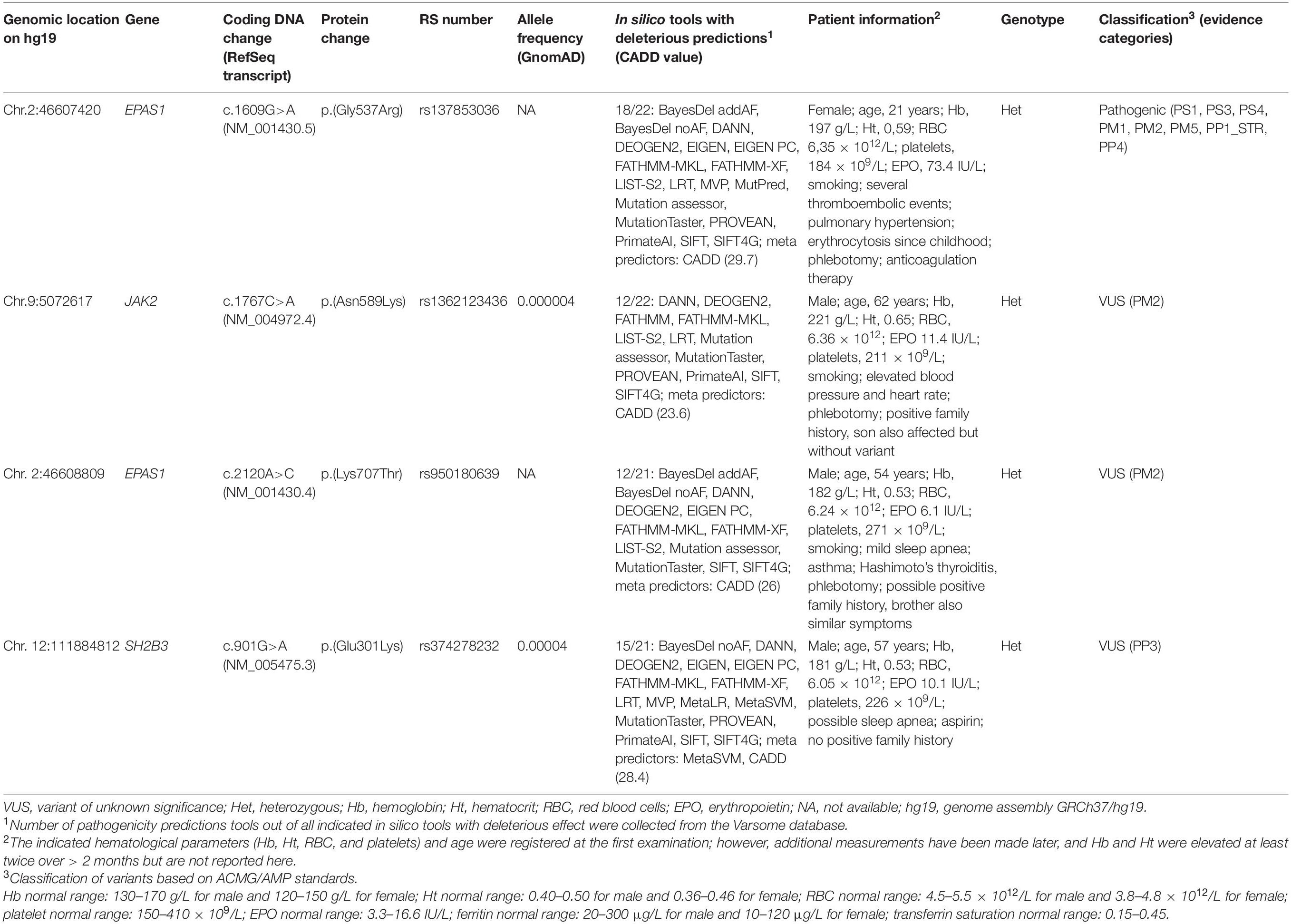
Table 1. Known and novel variants detected in the known erythrocytosis genes.
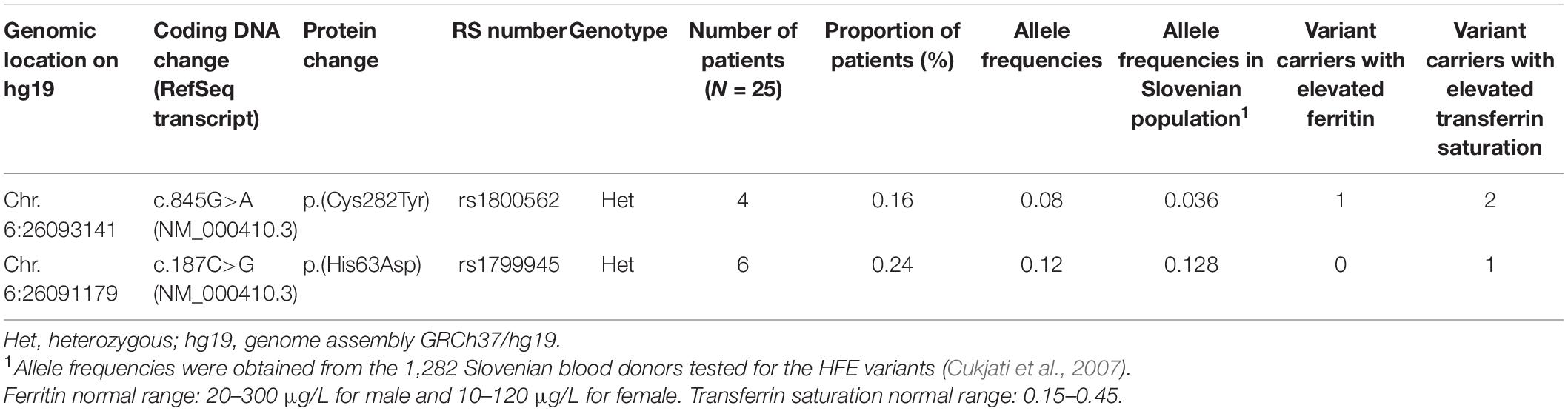
Table 2. Distribution of HFE variants among IE patients and number of variant carriers with elevated ferritin and transferrin saturation.
Known Pathogenic Variants
Heterozygous variant c.1609G>A in EPAS1 gene is a pathogenic variant that represents an established cause of familial erythrocytosis type 4 (ECYT4, OMIM ID 611783). This variant has been previously reported as pathogenic in numerous unrelated individuals with erythrocytosis (Gale et al., 2008; Percy et al., 2008a; Perrotta et al., 2013; Liu et al., 2017; Oliveira et al., 2018). In addition, it has been reported to co-segregate with erythrocytosis in multiple affected family members (Gale et al., 2008; Percy et al., 2008a; Liu et al., 2017). The variant is also classified as pathogenic in the ClinVar, LOVD, and HGMD databases (ClinVar ID: 6469, LOVD ID: EPAS1_000002, HGMD ID: CM081583); and the deleterious effect on the protein was confirmed in functional studies (Gale et al., 2008; Furlow et al., 2009; Perrotta et al., 2013; Tarade et al., 2018). The clinical presentation of the patient was highly consistent with the disease.
Novel Variants in Known Candidate Genes
Three heterozygous variants c.2120A>C, c.1767C>A, and c.901G>A with unknown clinical impact were identified in known erythrocytosis-associated genes EPAS1, JAK2, and SH2B3 genes, respectively.
Heterozygous variant c.901G>A in SH2B3 gene has been already recorded in the HGMD database (ID: CM1614293). It was previously reported in one patient with IE without a family history of erythrocytosis (Camps et al., 2016).
On the contrary, heterozygous variant c.2120A>C in EPAS1 gene has not yet been reported in association with human diseases in the literature and is not recorded among the clinically relevant variants in ClinVar, LOVD, and HGMD databases at the time of data analysis.
Similarly, heterozygous variant c.1767C>A in JAK2 gene has not been yet recorded among the clinically relevant variants in ClinVar, HGMD, COSMIC, and CIViC databases. The variant in JAK2 gene was not found in other affected or unaffected relatives, and the allele fraction [variant allele frequency (VAF)] was calculated to be 22%, which indicates its possible somatic origin.
We classified all three variants as variants with unknown significance (VUS) due to low population frequency in the GnomAD project or high pathogenicity. Variants in EPAS1 and JAK2 genes were observed at extremely low frequencies in the gnomAD project or were absent from the GnomAD controls. Variant SH2B3 c.901G>A was predicted to have a deleterious effect on the protein as the majority of meta-predictors (REVEL, METASVM, and CADD) supported its deleteriousness.
Variants in the Hemochromatosis Genes
With the targeted NGS analysis, we identified two heterozygous variants p.(Cys282Tyr) and p.(His63Asp) in HFE gene. Out of 25 patients with IE, 10 patients were carriers for variant p.(Cys282Tyr) or variant p.(His63Asp). Comparing the observed VAFs with the allele frequencies in the Slovenian population (Cukjati et al., 2007), IE patients had a higher incidence of variant p.(Cys282Tyr). Only a few patients with identified heterozygous HFE variants had elevated ferritin and saturation of transferrin > 45%, which is indicative of iron overload (Table 2).
Additionally, we identified a carriership of a pathogenic missense c.40C>T (NM_006363.4) variant in SEC23B gene, associated with the congenital dyserythropoietic anemia, type 2 (CDAII; OMIM:224100) in two unrelated patients. The significance of this finding in the development of erythrocytosis is not clear and is less likely causative concerning a high frequency of heterozygous variant carriers in the population. This variant was therefore excluded from the further report and interpretation of variants.
Discussion
With previously established targeted NGS of 39 erythrocytosis and hemochromatosis-associated genes, we identified high-confidence variants among patients with IE and discovered few potential disease-causing variants in patients who previously lacked a specific diagnosis. We were able to provide an actual genetic cause (variant p.Gly537Arg in EPAS1 gene) for erythrocytosis in one patient, which was also validated by Sanger sequencing. This is the first patient in Slovenia with a conclusive diagnosis of congenital erythrocytosis type 4 (ECYT4). Relatives of this patient will be further included in the study and genetically tested, to provide proper prognosis and genetic counseling to the family. Three novel candidate variants in the genes known to be associated with erythrocytosis, i.e., EPAS1, JAK2, and SH2B3, were identified. Interestingly, another research group studying erythrocytosis identified the majority of variants in genes EPAS1, SH2B3, JAK2, and EGLN1 (Girodon et al., 2021). Variant c.901G>A (NM_005475.2) in SH2B3 gene was the only one already identified in the erythrocytosis patient without myeloproliferative neoplasm (MPN) (Camps et al., 2016). Nevertheless, further segregation analyses and functional studies are necessary to support and ascertain its pathogenic nature.
Among all identified variants, the variants located within or in the vicinity of the important regions for protein function will have a higher likelihood of causality. The identified pathogenic EPAS1 variant c.1609G>A (p.Gly537Arg) is located six amino acids from the hydroxylation site p.Pro531, crucial for the oxygen-dependent regulation of EPAS1 stability (Kristan et al., 2019). Another amino acid change (c.1609G>T; p.Gly537Trp) (Percy et al., 2008b) (evidence category PM5) and the same amino acid change caused by a different base substitution (c.1609G>C; p.Gly537Arg) (Oliveira et al., 2018) (evidence category PS1) have been reported as pathogenic in patients with erythrocytosis. Furthermore, several amino acid substitutions c.1601C>G (p.Pro534Arg), c.1601C>T (p.Pro534Leu), c.1603A>G (p.Met535Val), c.1603A>T (p.Met535Leu), c.1604T>C (p.Met535Thr), c.1615G>A (p.Asp539Asn), c.1620C>A (p.Phe540Leu), and c.1631C>G (p.Pro544Arg) in the immediate vicinity of the identified variant have been reported in patients with erythrocytosis as pathogenic (Bento, 2018; Kristan et al., 2019), suggesting the critical importance of the identified region for the function of EPAS1 protein and severe effect on patient’s phenotype (Anzej Doma et al., 2021b). Some previous studies have shown that identified variant c.1609G>A (p.Gly537Arg), similar to other variants located C-terminal to p.Pro531, inhibits the binding of the EGLN1 hydroxylase to the EPAS1 protein. This results in impaired degradation and thus enhanced stabilization of EPAS1, which affects increased expression of target genes (Gale et al., 2008; Furlow et al., 2009; Perrotta et al., 2013; Tarade et al., 2018). Another identified variant in EPAS1 gene, c.2120A>C (p.Lys707Thr), is located far downstream of the primary hydroxylation site, unlikely to have an effect on hydroxylation and degradation. However, the variant is located within the nuclear localization signal (NLS) at the C-terminus of the EPAS1 protein. Two basic NLS motifs were shown to be present in region 705–742 amino acids and are required for localization of transcription factor EPAS1 to the nucleus under hypoxia and normoxia. Furthermore, the introduced variant p.Lys709Thr partly impaired the nuclear accumulation of EPAS1 induced by hypoxia (Luo and Shibuya, 2001). This, together with the low frequency of variant among GnomAD controls, implies the potential effect of variant p.Lys707Thr on protein function and activity. Further functional studies are needed to provide further evidence of variant causality. No variants in the NLS region were previously associated with ECYT4, indicating that analysis of this region may be of importance in further diagnoses.
Gene JAK2 codes for a non-receptor tyrosine kinase, in which its roles are to phosphorylate tyrosine residues in the EPO receptor and signal transducers and activators of transcription (STATs) and to activate the transcription of target genes for proliferation and differentiation of hematopoietic stem cells. The protein JAK2 has four major domains, with two Jak homology (JH) domains important for downstream signaling. The C-terminal JH1 domain (849–1124 aa) is a kinase domain responsible for phosphorylation, while structurally similar pseudo-kinase domain (JH2) (545–809 aa) lacks catalytic activity, and it is involved in auto-inhibition of JAK2 in the absence of ligand–receptor interaction (Gnanasambandan and Sayeski, 2011; Seif et al., 2017; UniProt Consortium., 2021). Variants in exons 12–15 were found in patients with various hematological disorders, and those exons (except for the exon 12) are located in the JH2 domain (Ma et al., 2009; Gnanasambandan and Sayeski, 2011). The most common somatic pathogenic variant in the exon 14, p.Val617Phe, which is responsible for the development of PV and other MPNs, allows the kinase to evade negative regulation and confer constitutive activation of JAK2 (Gnanasambandan and Sayeski, 2011). Identified variant p.(Asn589Lys) in the exon 13 of JAK2 gene is also located in the auto-inhibitory domain JH2; therefore, the potential pathophysiological mechanism for enhanced erythrocytes production could be the same.
SH2B3 protein (also termed LNK) is a negative regulator of erythropoiesis, as it binds to the cytokines and JAK2 and inhibits downstream signaling pathways (Maslah et al., 2017). SH2B3 has three domains: a dimerization domain, central pleckstrin homology domain (PH), and a Src homology 2 (SH2) domain. The PH domain (194–307 aa) has a role in binding the adapter protein SH2B3 to the plasma membrane, which is important for early inhibitory action during cytokine signaling. A mutational hotspot in the SH2B3 protein is present in the PH domain, encompassing exons 2, 3, and 4. Identified SH2B3 variant in our study was located in exon 4, at the C-terminus of the PH domain. The majority of the SH2B3 variants found in patients with PV and also in patients with p.Val617Phe-negative erythrocytosis were located in the PH domain, with the amino acid residue 208 (exon 2) as a preferential mutational target site (Spolverini et al., 2013; McMullin and Cario, 2016; Maslah et al., 2017).
Somatic variants in JAK2 and SH2B3 genes are the cause for somatic erythrocytosis (OMIM ID: 133100), somatic myelofibrosis (OMIM ID: 254450), and thrombocythemia (OMIM ID: 614521, 187950) (McMullin and Cario, 2016; Amberger et al., 2019). In addition, germline variants in the JAK2 gene are causative for thrombocythemia 3 (OMIM ID: 614521) (Mead et al., 2012; Amberger et al., 2019); and in several cases, germline JAK2 variants, also in combination with other variants, were responsible for the activation of JAK2/STAT signaling in hereditary erythrocytosis or PV patients (Kapralova et al., 2014, 2016; Milosevic Feenstra et al., 2016; Wu et al., 2018). Heterozygous variant identified in the JAK2 gene was present in only one affected family member and was observed in 22% of the sequence reads, as VAF was 0.22. The expected VAF for heterozygous germline variants is around 50%, with the distribution from 40 to 60%. For the somatic variants, VAF deviates from the germline and is usually lower (<40%), because the acquired variant is not present in all cells (Montgomery et al., 2018; Baer et al., 2019). This together indicates that JAK2 p.(Asn589Lys) could be of somatic origin; however, further testing of different type of sample (e.g., buccal swab) is required to confirm that. We already reported about two additional variants in the same individual with the JAK2 p.(Asn589Lys) variant, missense in the EGLN1 gene and intronic in the JAK2, that showed segregation in the family (Kristan et al., 2021). In the present study, we used different filtration parameters for the selection of variants as in the previous study by Kristan et al. (2021); therefore, those two variants were filtered out. None of the two variants were directly involved in the development of erythrocytosis; however, the correlation between the variants, together with the novel, potential somatic variant in the JAK2 gene, should be tested.
Besides known erythrocytosis genes, additional genes involved in the HIF and other erythropoiesis and iron metabolism pathways were included in the gene panel, in an attempt to discover novel candidate genes for the development of erythrocytosis. Unfortunately, we did not identify any novel disease-driver genes to be implicated in the erythropoiesis; however, we recognized a high incidence of heterozygous variants in HFE gene among the studied group of IE patients. This confirmed the observations of other researchers (Biagetti et al., 2018; Burlet et al., 2019; Gurnari et al., 2019). The difference of allele frequencies among our population of IE patients and the general population was larger for variant p.(Cys282Tyr). However, due to the small number of patients, this finding is of limited importance. Although homozygous variants p.(Cys282Tyr) have a high prevalence, the penetrance is low, as only 25–60% of patients develop clinical signs and only 55–82% of patients have increased serum ferritin level (Brissot et al., 2018). Low penetrance of causal HFE variants could indeed explain elevated ferritin values and saturation of transferrin in a small number of patients from our cohort. It would be useful to compare ferritin and saturation of transferrin values between larger cohorts of IE patients with HFE variants and IE HFE-wt patients, to see if there is any statistical significant difference. Biagetti et al. (2018) showed a higher frequency of elevated ferritin in mutated patients; however, the difference was not statistically significant due to a small group of patients (Biagetti et al., 2018). The pathophysiological mechanism between HFE variants and enhanced erythropoiesis is not clearly understood. In patients with high ferritin and transferrin values, this correlation could be explained by iron overload. However, this is not the case in patients with normal values; therefore, additional mechanisms must be involved. One possible explanation could be the linkage between the HIF pathway and iron metabolism. Schwartz et al. (2019) showed that low levels of hepatic iron regulator hepcidin lead to iron uptake and stabilization of EPAS1, through decreased prolyl-hydroxylase EGLN1 activity, as iron is an important co-factor for hydroxylation. Stabilized duodenal EPAS1 than activates target genes which regulate iron efflux (Schwartz et al., 2019). However, Schwartz et al. (2019) focused on the regulation of EPAS1 in duodenal enterocytes, and it would be interesting to study this mechanism also in other cell types and potentially find a connection with increased erythropoiesis.
With our sequencing approach, we achieved a high accuracy of sequenced bases, and high percentage of targeted regions had sufficient depth of coverage for germline variant calling. The sensitivity of our approach was high, as we accurately detected approximately 98% of variants calls in the reference NA12878 sample. Nevertheless, some limitations of the method were recognized and should be taken into consideration for future applications. The biggest limitation was insufficient coverage of some target regions. The majority of the target regions with poor coverage were seen in the first coding exons, as those regions are GC rich. Inadequate coverage in the first exon regions with high GC content is a common problem in the NGS approach, and usually other methods are applied to sufficiently sequence those regions (Chen et al., 2013). Another shortcoming of the applied method was the detection of variants in HBA1 and HBA2 genes, which was also observed by other researchers (Girodon et al., 2017; Filser et al., 2021). NGS, particularly read alignment for these two genes, is challenging, due to high sequence similarity. For future analysis of genes with high homology, a classical sequencing approach is suggested. One limitation of the study was also a low number of studied patients, but this was expected, as congenital erythrocytosis is a rare disorder. We identified only a few pathogenic variants and VUS with a likely causative mechanism in 16% (four out of 25) of patients with high Hb and Ht included in the study. This percentage is similar to the proportion of identified variants observed by other researchers, despite differences in the gene panel (Girodon et al., 2021). This indicates that the number of genes and gene regions included in the targeted panel must be extended. Also, we should consider the possibility that we missed larger structural variants, for instance, larger deletions, insertions, translocations, or copy number variations, that are harder to detect with limited targeted NGS or Sanger sequencing. To bypass this limitation, whole-exome sequencing (WES) or WGS needs to be applied in the future, especially the long-read sequencing approach for more sensitive and specific identification of larger variants.
Conclusion
Through the study, we examined the patients with suspected congenital erythrocytosis for the first time with the established targeted NGS in the Slovenian clinical setting. As far as we know, this is the first established erythrocytosis gene panel for NGS, which includes genes associated with enhanced erythropoiesis and iron uptake. We showed that targeted NGS was indeed useful to explore variants in cases with suspected congenital erythrocytosis; however, in only approximately 15% of the patients, clinically interesting variants were identified. We identified ECYT4 with a known pathogenic variant in EPAS1 gene and provided a diagnosis for one patient enabling proper decision regarding prognosis, genetic counseling, and treatment. Three VUS were identified in the known erythrocytosis genes, i.e., EPAS1, JAK2, and SH2B3. Further functional studies are necessary in order to elucidate the mechanism of action and explore the effect of variants on the development of erythrocytosis. Additionally, the germline or somatic origin should be clarified for the variants in JAK2 and SH2B3, as those two genes are commonly associated with the somatic type of erythrocytosis.
In several patients, we detected two heterozygous variants in HFE gene, which confirmed the observations of other researchers. However, for the comprehensive study of the involvement of the HFE variants in erythropoiesis, we need to perform additional research on a larger group of patients.
For the remaining patients with no identified variants, broader approaches like WGS and WES should be applied, to detect the possible genetic cause of congenital erythrocytosis.
Data Availability Statement
The datasets presented in this article are not readily available because due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Slovenian National Medical Ethics Committee, Ministry of Health of the Republic of Slovenia. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AK, ND, and IPZ contributed to conception and design of the study. IPZ, SAD, MF, TP, and HP revised clinical data. AK, TR, TK, RK, AV, and ND contributed with design of targeted NGS approach. AK, TP, AM, and ŠŽ performed the NGS analysis. AK wrote the first draft of the manuscript. TP, AM, and ND wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by the Slovenian Research Agency, Grant Nos. L3-9279 and P1-0390 and Young Researcher funding to AK and by University Medical Centre, Ljubljana, Grant Nos. 20170073 and 20200231.
Conflict of Interest
RK and AV are employees of the Kemomed Ltd., Kemomed Research and Development. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.689868/full#supplementary-material
Footnotes
References
Amberger, J. S., Bocchini, C. A., Scott, A. F., and Hamosh, A. (2019). OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038–D1043. doi: 10.1093/nar/gky1151
Anzej Doma, S., Drnovsek, E., Kristan, A., Fink, M., Sever, M., Podgornik, H., et al. (2021a). Diagnosis and management of non-clonal erythrocytosis remains challenging: a single centre clinical experience. Ann. Hematol. doi: 10.1007/s00277-021-04546-4544 [Epub ahead of print].
Anzej Doma, S., Kristan, A., Debeljak, N., and Preložnik Zupan, I. (2021b). Congenital erythrocytosis – a condition behind recurrent thromboses: a case report and literature review. Clin. Hemorheol. Microcirculat. doi: 10.3233/CH-211120 [Epub ahead of print].
Asif, S., Begemann, M., and Raza, S. (2019). Polycythemia in patients with hereditary hemochromatosis: real or myth? J. Clin. Med. Res. 11, 422–427. doi: 10.14740/jocmr3816
Badar, S., Busti, F., Ferrarini, A., Xumerle, L., Bozzini, P., Capelli, P., et al. (2016). Identification of novel mutations in hemochromatosis genes by targeted next generation sequencing in Italian patients with unexplained iron overload. Am. J. Hematol. 91, 420–425. doi: 10.1002/ajh.24304
Baer, C., Walter, W., Hutter, S., Twardziok, S., Meggendorfer, M., Kern, W., et al. (2019). “Somatic” and “pathogenic” - is the classification strategy applicable in times of large-scale sequencing? Haematologica 104, 1515–1520. doi: 10.3324/haematol.2019.218917
Barton, J. C., Bertoli, L. F., and Rothenberg, B. E. (2000). Peripheral blood erythrocyte parameters in hemochromatosis: evidence for increased erythrocyte hemoglobin content. J. Lab. Clin. Med. 135, 96–104. doi: 10.1016/s0022-2143(00)70026-70026
Baxter, E. J., Scott, L. M., Campbell, P. J., East, C., Fourouclas, N., Swanton, S., et al. (2005). Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061. doi: 10.1016/S0140-6736(05)71142-71149
Bento, C. (2018). Genetic basis of congenital erythrocytosis. Int. J. Lab. Hematol. 40, 62–67. doi: 10.1111/ijlh.12828
Bento, C., Almeida, H., Maia, T. M., Relvas, L., Oliveira, A. C., Rossi, C., et al. (2013). Molecular study of congenital erythrocytosis in 70 unrelated patients revealed a potential causal mutation in less than half of the cases (Where is/are the missing gene(s)?). Eur. J. Haematol. 91, 361–368. doi: 10.1111/ejh.12170
Bergant, G., Maver, A., Lovrecic, L., Cuturilo, G., Hodzic, A., and Peterlin, B. (2018). Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: a retrospective survey in 1,059 cases. Genet. Med. 20, 303–312. doi: 10.1038/gim.2017.142
Biagetti, G., Catherwood, M., Robson, N., Bertozzi, I., Cosi, E., McMullin, M. F., et al. (2018). HFE mutations in idiopathic erythrocytosis. Br. J. Haematol. 181, 270–272. doi: 10.1111/bjh.14555
Brissot, P., Pietrangelo, A., Adams, P. C., de Graaff, B., McLaren, C. E., and Loreal, O. (2018). Haemochromatosis. Nat. Rev. Dis. Primers 4:18016. doi: 10.1038/nrdp.2018.16
Burlet, B., Bourgeois, V., Buriller, C., Aral, B., Airaud, F., Garrec, C., et al. (2019). High HFE mutation incidence in idiopathic erythrocytosis. Br. J. Haematol. 185, 794–795. doi: 10.1111/bjh.15631
Camps, C., Petousi, N., Bento, C., Cario, H., Copley, R. R., McMullin, M. F., et al. (2016). Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 101, 1306–1318. doi: 10.3324/haematol.2016.144063
Chen, Y. C., Liu, T., Yu, C. H., Chiang, T. Y., and Hwang, C. C. (2013). Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One 8:e62856. doi: 10.1371/journal.pone.0062856
Cukjati, M., Vaupotic, T., Rupreht, R., and Curin-Serbec, V. (2007). Prevalence of H63D, S65C and C282Y hereditary hemochromatosis gene mutations in Slovenian population by an improved high-throughput genotyping assay. BMC Med. Genet. 8:69. doi: 10.1186/1471-2350-8-69
Ellard, S., Baple, E. L., Berry, I., Forrester, N., Turnbull, C., Owens, M., et al. (2019). ACGS Best Practice Guidelines for Variant Classification 2019. Available online at: https://www.acgs.uk.com/media/11285/uk-practiceguidelines-for-variant-classification-2019-v1-0-3.pdf
Faria, R., Silva, B., Silva, C., Loureiro, P., Queiroz, A., Fraga, S., et al. (2016). Next-generation sequencing of hereditary hemochromatosis-related genes: novel likely pathogenic variants found in the Portuguese population. Blood Cells Mol. Dis. 61, 10–15. doi: 10.1016/j.bcmd.2016.07.004
Ferbo, L., Manzini, P. M., Badar, S., Campostrini, N., Ferrarini, A., Delledonne, M., et al. (2016). Detection of a rare mutation in the ferroportin gene through targeted next generation sequencing. Blood Transfus 14, 531–534. doi: 10.2450/2016.0286-215
Fernandez-Marmiesse, A., Gouveia, S., and Couce, M. L. (2018). NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr. Med. Chem. 25, 404–432. doi: 10.2174/0929867324666170718101946
Filser, M., Aral, B., Airaud, F., Chauveau, A., Bruce, A., Polfrit, Y., et al. (2021). Low incidence of EPOR mutations in idiopathic erythrocytosis. Haematologica 106, 299–301. doi: 10.3324/haematol.2019.244160
Fokkema, I. F., Taschner, P. E., Schaafsma, G. C., Celli, J., Laros, J. F., and den Dunnen, J. T. (2011). LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32, 557–563. doi: 10.1002/humu.21438
Furlow, P. W., Percy, M. J., Sutherland, S., Bierl, C., McMullin, M. F., Master, S. R., et al. (2009). Erythrocytosis-associated HIF-2α mutations demonstrate a critical role for residues C-terminal to the hydroxylacceptor proline. J. Biol. Chem. 284, 9050–9058. doi: 10.1074/jbc.M808737200
Gale, D. P., Harten, S. K., Reid, C. D. L., Tuddenham, E. G. D., and Maxwell, P. H. (2008). Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 (Schwartz et al.) mutation. Blood 112, 919–921. doi: 10.1182/blood-2008-04-153718
Gaspersic, J., Kristan, A., Kunej, T., Zupan, I. P., and Debeljak, N. (2020). Erythrocytosis: genes and pathways involved in disease development. Blood Transfus doi: 10.2450/2020.0197-120 Online ahead of print.
Geay, A., Aral, B., Bourgeois, V., Martin, P., Airaud, F., Garrec, C., et al. (2020). Diagnosis of exon 12-positive polycythemia vera rescued by NGS. Clin. Case Rep. 8, 790–792. doi: 10.1002/ccr3.2720
Girodon, F., Airaud, F., Garrec, C., Bezieau, S., and Gardie, B. (2017). Gene panel sequencing in idiopathic erythrocytosis. Haematologica 102:e30. doi: 10.3324/haematol.2016.158337
Girodon, F., Aral, B., Airaud, F., Garrec, C., Bezieau, S., and Gardie, B. (2021). “NGS analysis in hereditary erythrocytosis: a french experience,” in Proceedings of the MPN&MPNr-EuroNet Fifteenth Meeting: Diagnosis and therapy of Myeloproliferative Neoplasms and Related Hereditary Diseases. Available online at: http://mpneuronet.com/events/ljubljana/
Gnanasambandan, K., and Sayeski, P. P. (2011). A structure-function perspective of Jak2 mutations and implications for alternate drug design strategies: the road not taken. Curr. Med. Chem. 18, 4659–4673. doi: 10.2174/092986711797379267
Griffith, M., Spies, N. C., Krysiak, K., McMichael, J. F., Coffman, A. C., Danos, A. M., et al. (2017). CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49, 170–174. doi: 10.1038/ng.3774
Gurnari, C., Lombardi, A. M., Cosi, E., Biagetti, G., Buccisano, F., Franceschini, L., et al. (2019). Genetic analysis of erythrocytosis reveals possible causative and modifier gene mutations. Br. J. Haematol. 186, e100–e103. doi: 10.1111/bjh.15931
Hentze, M. W., Muckenthaler, M. U., Galy, B., and Camaschella, C. (2010). Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38. doi: 10.1016/j.cell.2010.06.028
Kapralova, K., Horvathova, M., Kucerova, J., Pospisilova, D., Leroy, E., Constantinescu, S. N., et al. (2014). JAK2 E846D germline mutation associated with erythrocytosis causes increased and prolonged epo-induced activation of STAT5. Blood 124, 4008–4008. doi: 10.1182/blood.V124.21.4008.4008
Kapralova, K., Horvathova, M., Pecquet, C., Fialova Kucerova, J., Pospisilova, D., Leroy, E., et al. (2016). Cooperation of germ line JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia. Blood 128, 1418–1423. doi: 10.1182/blood-2016-02-698951
Keohane, C., McMullin, M. F., and Harrison, C. (2013). The diagnosis and management of erythrocytosis. BMJ 347:f6667. doi: 10.1136/bmj.f6667
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Kristan, A., Debeljak, N., and Kunej, T. (2019). Genetic variability of hypoxia-inducible factor alpha (HIFA) genes in familial erythrocytosis: analysis of the literature and genome databases. Eur. J. Haematol. 103, 287–299. doi: 10.1111/ejh.13304
Kristan, A., Gaspersic, J., Rezen, T., Kunej, T., Kolic, R., Vuga, A., et al. (2021). Genetic analysis of 39 erythrocytosis and hereditary hemochromatosis-associated genes in the Slovenian family with idiopathic erythrocytosis. J. Clin. Lab. Anal. 35:e23715. doi: 10.1002/jcla.23715
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi: 10.1093/nar/gkx1153
Lanktree, M. B., Sadikovic, B., Waye, J. S., Levstik, A., Lanktree, B. B., Yudin, J., et al. (2017). Clinical evaluation of a hemochromatosis next-generation sequencing gene panel. Eur. J. Haematol. 98, 228–234. doi: 10.1111/ejh.12820
Lee, F. S., and Percy, M. J. (2011). The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 6, 165–192. doi: 10.1146/annurev-pathol-011110-130321
Lee, G., and Arcasoy, M. O. (2015). The clinical and laboratory evaluation of the patient with erythrocytosis. Eur. J. Intern. Med. 26, 297–302. doi: 10.1016/j.ejim.2015.03.007
Lenglet, M., Robriquet, F., Schwarz, K., Camps, C., Couturier, A., Hoogewijs, D., et al. (2018). Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 132, 469–483. doi: 10.1182/blood-2018-03-838235
Liu, Q., Tong, D., Liu, G., Yi, Y., Zhang, D., Zhang, J., et al. (2017). HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal-cell carcinoma. Cancer Biol. Ther. 18, 944–947. doi: 10.1080/15384047.2017.1394553
Luo, J. C., and Shibuya, M. (2001). A variant of nuclear localization signal of bipartite-type is required for the nuclear translocation of hypoxia inducible factors (1alpha, 2alpha and 3alpha). Oncogene 20, 1435–1444. doi: 10.1038/sj.onc.1204228
Ma, W., Kantarjian, H., Zhang, X., Yeh, C. H., Zhang, Z. J., Verstovsek, S., et al. (2009). Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J. Mol. Diagn. 11, 49–53. doi: 10.2353/jmoldx.2009.080114
Maslah, N., Cassinat, B., Verger, E., Kiladjian, J. J., and Velazquez, L. (2017). The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 31, 1661–1670. doi: 10.1038/leu.2017.139
McMullin, M. F. (2008). The classification and diagnosis of erythrocytosis. Int. J. Lab. Hematol. 30, 447–459. doi: 10.1111/j.1751-553X.2008.01102.x
McMullin, M. F. (2014). Secondary erythrocytosis. Hematology 19, 183–184. doi: 10.1179/1024533214z.000000000263
McMullin, M. F. (2016). Investigation and management of erythrocytosis. Curr. Hematol. Malig. Rep. 11, 342–347. doi: 10.1007/s11899-016-0334-331
McMullin, M. F., and Cario, H. (2016). LNK mutations and myeloproliferative disorders. Am. J. Hematol. 91, 248–251. doi: 10.1002/ajh.24259
McMullin, M. F., Harrison, C. N., Ali, S., Cargo, C., Chen, F., Ewing, J., et al. (2019a). A guideline for the diagnosis and management of polycythaemia vera. a British society for haematology guideline. Br. J. Haematol. 184, 176–191. doi: 10.1111/bjh.15648
McMullin, M. F. F., Mead, A. J., Ali, S., Cargo, C., Chen, F., Ewing, J., et al. (2019b). A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis: a British society for haematology guideline. Br. J. Haematol. 184, 161–175. doi: 10.1111/bjh.15647
Mead, A. J., Rugless, M. J., Jacobsen, S. E., and Schuh, A. (2012). Germline JAK2 mutation in a family with hereditary thrombocytosis. N. Engl. J. Med. 366, 967–969. doi: 10.1056/NEJMc1200349
Milosevic Feenstra, J. D., Nivarthi, H., Gisslinger, H., Leroy, E., Rumi, E., Chachoua, I., et al. (2016). Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 127, 325–332. doi: 10.1182/blood-2015-07-661835
Mlakar, U. (2008). Smernice za odkrivanje in Zdravljenje prave policitemije. Zdrav Vestn. 77, 11–14.
Montgomery, N. D., Selitsky, S. R., Patel, N. M., Hayes, D. N., Parker, J. S., and Weck, K. E. (2018). Identification of germline variants in tumor genomic sequencing analysis. J. Mol. Diagn. 20, 123–125. doi: 10.1016/j.jmoldx.2017.09.008
Oliveira, J. L., Coon, L. M., Frederick, L. A., Hein, M., Swanson, K. C., Savedra, M. E., et al. (2018). Genotype–phenotype correlation of hereditary erythrocytosis mutations, a single center experience. Am. J. Hematol. doi: 10.1002/ajh.25150 Online ahead of print.
Patnaik, M. M., and Tefferi, A. (2009). The complete evaluation of erythrocytosis: congenital and acquired. Leukemia 23, 834–844. doi: 10.1038/leu.2009.54
Percy, M. J., Beer, P. A., Campbell, G., Dekker, A. W., Green, A. R., Oscier, D., et al. (2008a). Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood 111, 5400–5402. doi: 10.1182/blood-2008-02-137703
Percy, M. J., Furlow, P. W., Lucas, G. S., Li, X., Lappin, T. R. J., McMullin, M. F., et al. (2008b). A Gain-of-Function mutation in the HIF2A gene in familial erythrocytosis. New Engl. J. Med. 358, 162–168. doi: 10.1056/nejmoa073123
Perrotta, S., Stiehl, D. P., Punzo, F., Scianguetta, S., Borriello, A., Bencivenga, D., et al. (2013). Congenital erythrocytosis associated with gain-of-function HIF2A gene mutations and erythropoietin levels in the normal range. Haematologica 98, 1624–1632. doi: 10.3324/haematol.2013.088369
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi: 10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Schwartz, A. J., Das, N. K., Ramakrishnan, S. K., Jain, C., Jurkovic, M. T., Wu, J., et al. (2019). Hepatic hepcidin/intestinal HIF-2alpha axis maintains iron absorption during iron deficiency and overload. J. Clin. Invest. 129, 336–348. doi: 10.1172/JCI122359
Scott, L. M., Tong, W., Levine, R. L., Scott, M. A., Beer, P. A., Stratton, M. R., et al. (2007). JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 356, 459–468. doi: 10.1056/NEJMoa065202
Seif, F., Khoshmirsafa, M., Aazami, H., Mohsenzadegan, M., Sedighi, G., and Bahar, M. (2017). The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 15:23. doi: 10.1186/s12964-017-0177-y
Spolverini, A., Pieri, L., Guglielmelli, P., Pancrazzi, A., Fanelli, T., Paoli, C., et al. (2013). Infrequent occurrence of mutations in the PH domain of LNK in patients with JAK2 mutation-negative’idiopathic’ erythrocytosis. Haematologica 98, e101–e102. doi: 10.3324/haematol.2013.090175
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S., et al. (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581. doi: 10.1002/humu.10212
Tarade, D., Robinson, C. M., Lee, J. E., and Ohh, M. (2018). HIF-2alpha-pVHL complex reveals broad genotype-phenotype correlations in HIF-2alpha-driven disease. Nat. Commun. 9:3359. doi: 10.1038/s41467-018-05554-5551
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). COSMIC: the catalogue of somatic mutations in Cancer. Nucleic Acids Res. 47, D941–D947. doi: 10.1093/nar/gky1015
Taylor, J. C., Martin, H. C., Lise, S., Broxholme, J., Cazier, J. B., Rimmer, A., et al. (2015). Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 47, 717–726. doi: 10.1038/ng.3304
Tweedie, S., Braschi, B., Gray, K., Jones, T. E. M., Seal, R. L., Yates, B., et al. (2021). Genenames.org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. 49, D939–D946. doi: 10.1093/nar/gkaa980
Ugo, V., Tondeur, S., Menot, M. L., Bonnin, N., Le Gac, G., Tonetti, C., et al. (2010). Interlaboratory development and validation of a HRM method applied to the detection of JAK2 exon 12 mutations in polycythemia vera patients. PLoS One 5:e8893. doi: 10.1371/journal.pone.0008893
UniProt Consortium. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Keywords: NGS – next-generation sequencing, erythrocytosis, targeted panel sequencing, iron metabolism, diagnostics, rare disease (RD)
Citation: Kristan A, Pajič T, Maver A, Režen T, Kunej T, Količ R, Vuga A, Fink M, Žula Š, Podgornik H, Anžej Doma S, Preložnik Zupan I, Rozman D and Debeljak N (2021) Identification of Variants Associated With Rare Hematological Disorder Erythrocytosis Using Targeted Next-Generation Sequencing Analysis. Front. Genet. 12:689868. doi: 10.3389/fgene.2021.689868
Received: 01 April 2021; Accepted: 16 June 2021;
Published: 19 July 2021.
Edited by:
Yanling Yang, Peking University First Hospital, ChinaReviewed by:
Nancy Monroy-Jaramillo, National Institute of Neurology and Neurosurgery, MexicoPrashanth N. Suravajhala, Birla Institute of Scientific Research, India
Copyright © 2021 Kristan, Pajič, Maver, Režen, Kunej, Količ, Vuga, Fink, Žula, Podgornik, Anžej Doma, Preložnik Zupan, Rozman and Debeljak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nataša Debeljak, bmF0YXNhLmRlYmVsamFrQG1mLnVuaS1sai5zaQ==