 Pengwei Xu1†
Pengwei Xu1† Chenxi Zhao1†
Chenxi Zhao1† Xinxin You1,2†Fan Yang3Jieming Chen1,2Zhiqiang Ruan1,2Ruobo Gu2Junmin Xu2
Xinxin You1,2†Fan Yang3Jieming Chen1,2Zhiqiang Ruan1,2Ruobo Gu2Junmin Xu2 Chao Bian1,2*
Chao Bian1,2* Qiong Shi1,2*
Qiong Shi1,2*- 1College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 2Shenzhen Key Lab of Marine Genomics, Guangdong Provincial Key Lab of Molecular Breeding in Marine Economic Animals, BGI Academy of Marine Sciences, BGI Marine, BGI, Shenzhen, China
- 3Marine Geological Department, Marine Geological Survey Institute of Hainan Province, Haikou, China
Summary
Flying fishes are a group of Exocoetidae members with an intriguing epipelagic inhabitant. They have evolved numerous interesting characteristics. Here, we performed whole genome sequencing, de novo assembly and annotation of the representative mirrorwing flyingfish (Hirundichthys speculiger). We obtained a 1.04-Gb genome assembly using a hybrid approach from 99.21-Gb Illumina and 29.98-Gb PacBio sequencing reads. Its contig N50 and scaffold N50 values reached 992.83 and 1,152.47 kb, respectively. The assembled genome was predicted to possess 23,611 protein-coding genes, of which 23,492 (99.5%) were functionally annotated with public databases. A total of 42.02% genome sequences consisted of repeat elements, among them DNA transposons accounted for the largest proportion (24.38%). A BUSCO (Benchmarking Universal Single Copy Orthologs) evaluation demonstrated that the genome and gene completeness were 94.2% and 95.7%, respectively. Our phylogeny tree revealed that the mirrorwing flyingfish was close to Oryzias species with a divergence time of about 85.2 million years ago. Moreover, nine vison-related genes, three melatonin biosynthesis related aanat (aralkylamine N-acetyltransferase) genes, and two sunscreen biosynthesis related eevs (2-epi-5-epi-valiolone synthase) genes were identified in the assembled genome; however, the loss of SWS1 (short-wavelength sensitive opsin 1) and aanat1a in amphibious mudskippers was not presented in the mirrorwing flyingfish genome. In summary, we generate a high-quality draft genome assembly for the mirrorwing flyingfish, which provides new insights into physiology-related genes of Exocoetidae. It also serves as a powerful resource for exploring intriguing traits of Exocoetidae at a genomics level.
Introduction
Flying fishes (Exocoetidae; Beloniformes) have evolved with numerous interesting characteristics, such as gliding over water, marine- to freshwater transition, and unique craniofacial and egg buoyancy. They have been regarded as an extraordinary marine group with enlarged pelvic fins and hypocercal caudal fins, which could help to glide over water to reach a distance up to 400 m (Davenport, 1994). Although the oldest gliding fish fossil (Potanichthys xingyiensis) shares certain similar morphology with modern flying fishes, it is not the ancestor of the modern flying fishes, since they are thought to have evolved independently about 65.5 million years ago (Xu et al., 2012). Compared with tetrapod gliders, the gliding behavior of flying fishes could not be considered as an energy-saving strategy for long-distance movement (Rayner, 1986), but it may be just used for escaping from underwater predators [e.g., swordfish, tuna, dolphin, and squid (Kutschera, 2005)].
While the representative mirrorwing flyingfish (Hirundichthys speculiger; Figure 1A) traverses the air and water interface, it meets a series of challenges [such as relentless sunshine, lack of buoyancy, and high CO2 accumulation (Wright and Turko, 2016)] as amphibious fishes. The lower refractive index of air usually aggravates this situation, making fishes myopic in air (Baylor and Shaw, 1962). Duplication, loss, differential expression, and crucial tuning of opsin genes could lead to visual plasticity in vertebrates for adapting to the water-to-air environments (Hauser and Chang, 2017). Five types of opsins, including LWS (red: long wavelength-sensitive), SWS1 (UV: short wavelength-sensitive 1), SWS2 (violet/blue: short wavelength-sensitive 2), RH1 (dim vision: rhodopsin), and RH2 (green: green-sensitive), have been identified in non-mammalian vertebrates (Yokoyama, 2000). Modifications of opsin and melatonin biosynthesis-related arylalkylamine N-acetyltransferase (aanat) genes could enhance amphibious mudskippers' survival on land (You et al., 2014). When the mirrorwing flyingfish leaps out of water, whether it employs the same mechanisms as mudskippers (including crucial mutation sites of LWS, lack of SWS1, and loss of aanat1a in the giant-fin mudskipper; see more details in You et al., 2014) or not is still an open question.
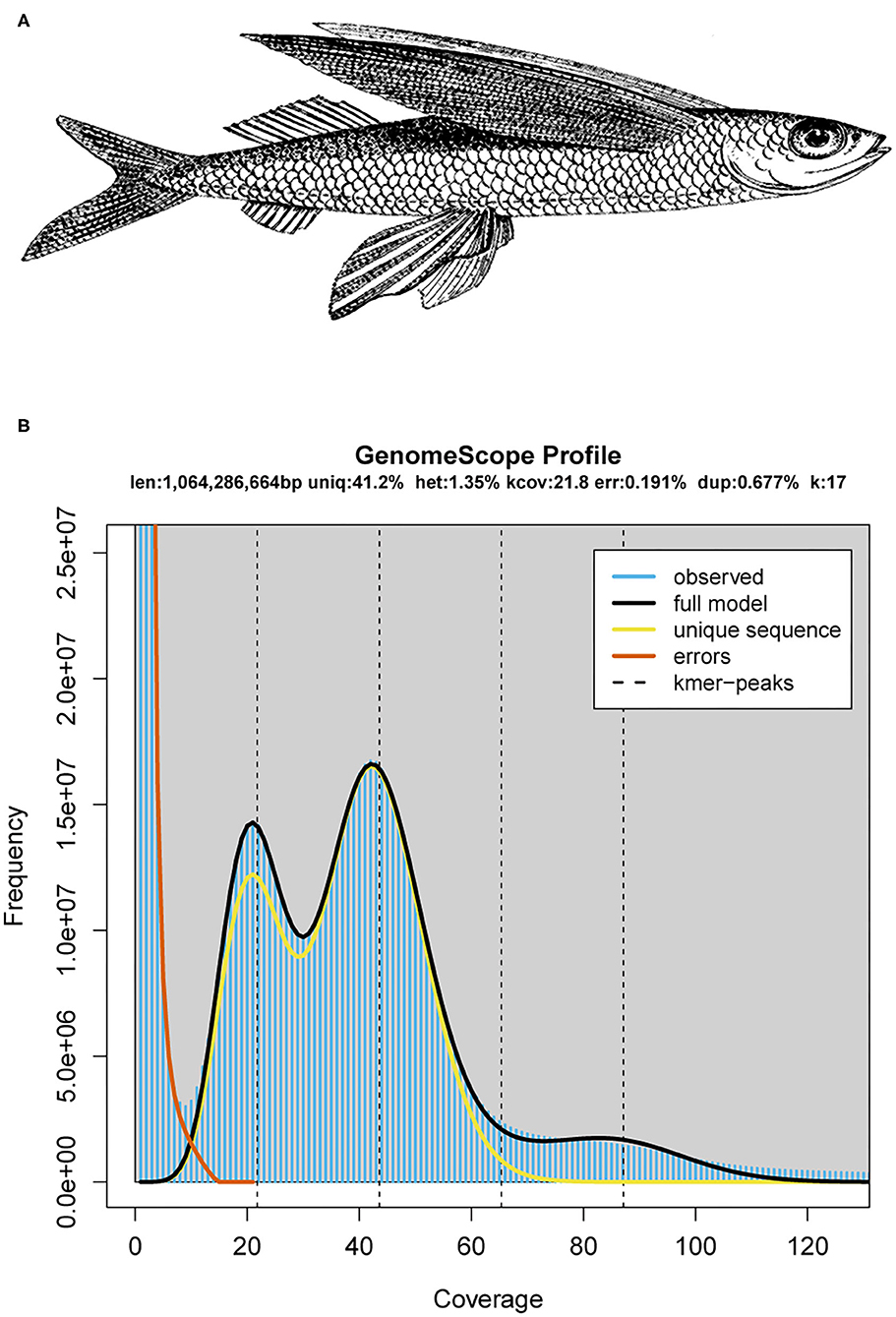
Figure 1. The schematic diagram and genomics feature of the mirrorwing flyingfish. (A) A drawing of the mirrorwing flyingfish [adopted from De Bruin et al. (1995)]. (B) A k-mer analysis of the genome sequencing reads for the mirrorwing flyingfish using GenomeScope.
Ultraviolet radiation (UVR: 280–400 nm) often causes DNA damages through oxidative stress, producing a number of disorders (such as sunburn and skin cancer risk) (Kageyama and Waditee-Sirisattha, 2019; Rosic, 2019). UV-absorbing compounds, such as mycosporine-like amino acids (MAAs) and gadusol, are commonly distributed in various marine microorganisms, invertebrates, and algae (Shick and Dunlap, 2002; Miyamoto et al., 2014). The de novo synthesis of MAA in invertebrates (such as coral and sea anemone) employed a four-step desmethyl-4-deoxygadusol synthase (DDGS) based pathway as cyanobacteria (Balskus and Walsh, 2010; Rosic and Dove, 2011; Shinzato et al., 2011), while zebrafish (Danio rerio) could convert sedoheptulose-7-phosphate (SH7P) to gadusol using 2-epi-5-epi-valiolone synthase (EEVS) and S-adenosyl-L-methionine-dependent methyltransferase [MT-Ox (Osborn et al., 2015)]. The two core genes, eevs and mt-ox, in zebrafish are flanked by four transcription factor genes [frmd4B, mitf , mdfic, and foxp1 (Osborn et al., 2015)], which is not consistent with the loss of mdfic in Japanese medaka [Oryzias latipes (Kim et al., 2018)]. Phylogenetic analysis using mitochondrial genes in Beloniformes had inferred a close relationship between the mirrorwing flyingfish and medaka (Lovejoy et al., 2004; Cui et al., 2018). Whether the mirrorwing flyingfish contains the complete gene cluster as zebrafish or incomplete cluster as medaka is valuable for checking the possible lineage-specific gene rearrangement of eevs-like cluster.
Here, we performed whole genome sequencing of the mirrorwing flyingfish and generated a draft assembly with a hybrid method (Ye et al., 2016) for the first time. Our subsequent phylogenetic and comparative genomic analyses between amphibious fishes and ordinary underwater fishes will provide insights into the evolution of vision-related genes, olfactory receptor (OR) genes, and gadusol synthesis-related genes (eevs) in the mirrorwing flyingfish. This genome assembly will serve as a valuable resource for the illumination of molecular basis for the special characteristics of flying fishes.
Value of the Data
This is the first genome report of the representative mirrorwing flyingfish. Our final assembly was 1.04 Gb, with a contig N50 of 992.83 kb and a scaffold N50 of 1,152.47 kb.
A phylogeny tree was constructed to demonstrate that the mirrorwing flyingfish was close to Oryzias species with a divergence time of about 85.2 Mya. A total of 60.71% of the mirrorwing flyingfish genome region was syntenic with O. latipes.
The genome of mirrorwing flyingfish harbored nine vision-related genes, three aanat genes, and two eevs-like genes. The existence of SWS1 and aanat1a suggests that the mirrorwing flyingfish employs different strategies for visional adaptation in air. A gene cluster of eevs-like shared the same synteny as Japanese medaka, implying a uniform gene rearrangement in Beloniformes.
Materials and Methods
Fish Sampling and Genome Sequencing
An adult mirrorwing flyingfish was captured by torch fishing in the water area of Iltis Bank, Xisha, China. Genomic DNAs were extracted from muscle tissues and purified and quality checked according to a standard protocol (Sigma-Aldrich, St. Louis, MO, USA).
Subsequently, three paired-end libraries (with insert sizes of 270, 500, and 800 bp, respectively) and three mate-pair libraries (with insert sizes of 2, 5, and 10 kb, respectively) were constructed in accordance with an Illumina standard manual before sequencing on an Illumina X-Ten platform (Illumina Inc., San Diego, CA, USA) with a PE-150 or PE-125 module. Raw reads were then processed using SOAPnuke v1.5.6 (Chen et al., 2018) with optimized parameters (“-n 0.02 -Q 2 -l 15−5 1 -d -I -q 0.4”). An additional SMART Bell library with an insert size of 20 kb was constructed based on a PacBio RS II protocol (Pacific Biosciences, Menlo Park, CA, USA). Six DNA sequencing cells were produced using the P6 polymerase/C4 chemistry (Rhoads and Au, 2015).
Genome Assembly
Distribution of k-mer frequency was constructed with jellyfish v2.0 (Marçais and Kingsford, 2011) using clean reads from short-insert libraries (270 and 500 bp). GenomeScope v1.0 (Vurture et al., 2017) was then applied to estimate the genome size and heterozygosity. A routine hybrid pipeline was employed to assemble the high heterozygous flyingfish genome (Supplementary Figure 1).
In brief, the Illumina paired-end reads were first assembled using Platanus v1.24 (Kajitani et al., 2014) with optimized parameters (assemble -k 35 -s 5 -u 0.2 -d 0.5). DBG2OLC (Ye et al., 2016) was employed to construct backbone sequences from the best overlaps between the initial contigs and raw PacBio reads. All related PacBio reads were realigned to the backbone with Sparc (Ye and Ma, 2016) to construct the most likely consensus sequences of the genome. All Illumina paired-end reads were aligned to the resulting assembly using BWA-MEM (Li, 2014). The alignments were employed for Pilon v1.24 (Walker et al., 2014) to polish the assembly. All Illumina mate-pair reads were mapped onto the corrected contigs using BWA-MEM (Li, 2014). These alignments were then processed with BESST v2.2.4 (Sahlin et al., 2014) to construct scaffolds. Completeness of the genome assembly was evaluated by BUSCO v3.0 (Simão et al., 2015) with default parameters “-l actinopterygii_odb9 -m genome -c 3 -sp zebrafish.”
Genome Annotation
Transposable elements (TEs) were identified using both homolog-based and de novo methods. For the homolog-based method, RepeatMasker v4.06 and ProteinRepeatMasker v4.06 (Chen, 2004) were employed to identify known TEs against the Repbase v21.0 (Jurka et al., 2005). For the de novo method, a de novo library was constructed using RepeatModeler v2.0 (Flynn et al., 2020) and LTR-FINDER v1.0.6 (Xu and Wang, 2007) firstly. Then, RepeatMasker v4.06 was subjected to identify the de novo TEs against the de novo library. The tandem repeat sequences were identified using Tandem Repeat Finder (Benson, 1999).
Gene models were also predicted using both homolog-based and de novo methods. For the homolog-based methods, protein sequences of zebrafish (Danio rerio), three-spined stickleback (Gasterosteus aculeatus), human (Homo sapiens), Japanese medaka (O. latipes), and green spotted pufferfish (Tetraodon nigroviridis) were derived from Ensembl-100 and aligned to our flyingfish genome using tBLASTn (Ye et al., 2006) with parameter “-e 1e-5 -m 8 -F.” Blasted hits were processed by SOLAR v0.9 (Yu et al., 2006) with parameter “-a prot2 genome2 -z” to determine the potential gene loci. We extracted the candidate gene region with 2-kb flanking sequences and employed Genewise v2.4 (Birney et al., 2004) to determine gene structures. For the de novo prediction, we trained the parameters of AUGUSTUS v3.2 (Stanke et al., 2006) using randomly selected 2,000 intact gene models that were derived from the homolog-based method. Then, we used AUGUSTUS to perform ab initio prediction on the repeat-masked genome with the trained parameters. Finally, the gene models predicted from both approaches were integrated to form non-redundant gene sets using the similar pipeline as described in a previous study (Xiong et al., 2016). Completeness of the gene sets was evaluated by BUSCO v3.0 (Simão et al., 2015) with parameters “-l actinopterygii_odb9 -m protein -c 3 -sp zebrafish.”
Gene function annotation was performed on the basis of sequence and domain similarity. The protein sequences were aligned to Kyoto Encyclopedia of Genes and Genomes (KEGG) v84.0 (Kanehisa et al., 2017), SwissProt, and TrEMBL (Uniprot release 2020-06) (Bairoch et al., 2005) using BLASTP (Ye et al., 2006) with an E-value of 1e−5. InterProScan v5.11-55.0 (Jones et al., 2014) was applied to predict domain information with public databases including Pfam (Bateman et al., 2004), SMART (Letunic et al., 2012), PANTHER (Thomas et al., 2003), PRINTS (Attwood et al., 2000), PROSITE profiles (Sigrist et al., 2010), and ProDom (Servant et al., 2002). Gene Ontology (GO) terms were predicted using the IPR entry list (Burge et al., 2012).
Four types of non-coding RNA were identified in the mirrorwing flyingfish genome. We employed tRNAscan-SE v2.0 (Lowe and Eddy, 1997) to detect transfer RNAs (tRNAs). For microRNAs (miRNAs) and small nuclear RNAs (snRNAs), the Rfam v12.0 (Nawrocki et al., 2015) database was mapped onto the assembled genome, and the matched sequences were delivered into INFERNAL v1.1.4 (Nawrocki and Eddy, 2013) to confirm structures. Ribosomal RNAs (rRNAs) in the genome were searched using animal full-length rRNAs (Quast et al., 2012) as the query.
Gene Family Prediction
To identify gene families in the mirrorwing flyingfish genome, we download protein-coding sequences of 18 representative teleost fishes from the National Center for Biotechnology Information (NCBI) databases (see more details in Supplementary Table 1), including Anabas testudineus (Ates; climbing perch), Austrofundulus limnaeus (annual killifish), Boleophthalmus pectinirostris (Bpec; great blue-spotted mudskipper), Channa argus (Carg; northern snakehead), Cyprinodon variegatus (sheepshead minnow), D. rerio (Drer; zebrafish), Fundulus heteroclitus (mummichog), Kryptolebias marmoratus (Kmar; mangrove rivulus fish), Monopterus albus (Asian swamp eel), Nothobranchius furzeri (turquoise killifish), Oreochromis aureus (Oaur; blue tilapia), O. niloticus (Onil; Nile tilapia), Oryzias latipes (Olat; Japanese medaka), O. melastigma (Omel; marine medaka), Periophthalmus magnuspinnatus (Pmag; giant-fin mudskipper), Poecilia mexicana (Atlantic molly), Xiphophorus maculatus (southern platyfish), and Maylandia zebra (Mzeb; Zebra mbuna). After removal of alternative splice variants, the protein sequences of the 18 fish species along with the mirrorwing flyingfish (H. speculiger; Hspe) were delivered to OrthoFinder v2.3.11 (Emms and Kelly, 2019) with an E-value of 1e−5 to identify orthologous groups.
Protein sequences of single-copy orthologous families were extracted and aligned using MUSCLE v3.8 (Edgar, 2004), and the alignment of protein sequences was converted to codon alignment using PAL2NAL v14 (Suyama et al., 2006). The phase 1 sites of codon aligned were extracted and concentrated to a super gene for each species. PhyML v3.0 (Guindon et al., 2010) and MrBayes v3.2 (Ronquist et al., 2012) were employed to construct a phylogenetic tree. Divergence time of these teleost fishes was estimated using MCMCTREE v4.5 in the PAML v4.5 (Yang, 2007) with five putative calibrations times, which were adapted from TIMETREE (Kumar et al., 2017). We used CAFÉ v3.0 (Han et al., 2013) with optimized parameter (-p 0.05 -t 4 -r 10000 -filter) to assess expansion and contraction of gene families. A branch specific p < 0.05 was utilized to define significance in the mirrorwing flyingfish. We employed hypergeometric tests (Falcon and Gentleman, 2008) to investigate pathway enrichments of those significantly expanded gene families, using the whole genome annotation as the background.
Synteny Analysis With Medaka and Zebrafish Genomes
After masking transposon elements of the three genomes, pairwise genome alignment among mirrorwing flyingfish, Japanese medaka, and zebrafish was carried out using LASZT v1.04.03 (Harris, 2007) with optimized parameters (T = 2 C = 2 H = 2000 Y = 3400 L = 6000 K = 2200 –format = axt). The matching length of each pairwise alignment was calculated using an in-house Perl script.
Identification of Vision-Related Genes
We applied two approaches to obtain the protein sequences of various opsins and aanat genes in 12 representative teleost fishes (with abbreviations of Ates, Bpec, Carg, Drer, Kmar, Mzeb, Oaur, Onil, Olat, Omel, Pmag, and Hspe, respectively, in Supplementary Table 1). For those with public annotations, gene sequences were directly downloaded from NCBI (Supplementary Table 2). For the mirrorwing flyingfish, however, we mapped the protein sequences of blue tilapia, zebrafish, and Japanese medaka to our assembled genome and predicted opsin and aanat genes using Exonerate v2.2.0 (Slater and Birney, 2005) with optimized parameters (-model protein2genome –showalignment false –showtargetgff true –bestn 1).
To validate the synteny of opsin genes, we downloaded those genes that have been reported to locate adjacent to an opsin gene (Lin et al., 2017) and obtained the neighboring genes from the genome annotation or using BLAST with an E-value of 1e−5 against the assembled genome. We constructed a rooted neighbor-joining (NJ) tree of opsins, using known opsin from human (ENSP00000358967.4, LWS1; ENSP00000472316.1, MWS; ENSP00000358945.4, MWS2; ENSP00000469970.1, MWS3; ENSP00000296271.3, RH1; ENSP00000249389.2, SWS1) and zebrafish (ENSDARP00000069184.5, OPN3; as the outgroup) by MEGA-X (Kumar et al., 2018) with 1,000 bootstraps.
A phylogenetic tree of aanat gene family was also constructed using the NJ method as implemented in the MEGA-X with human AANAT (NP_001079.1) and mouse AANAT (NP_033721.1) as the outgroup (Kumar et al., 2018). We applied Evolview (Subramanian et al., 2019) to edit phylogenetic trees. Five key tuning sites (including 180, 197, 277, 285, and 308) of the LWS opsins had influenced the λmax of vertebrate opsins (Bowmaker, 2008; Yokoyama, 2008). A previous report suggested that a single mutation at S180A, H197Y, Y277F, T285A, A308S, and double mutations S180A/H197Y can lead to a −7, −28, −8, −15, −27, and −11 nm shift, respectively, in the λmax of the pigments (Yokoyama and Radlwimmer, 2001). To investigate classical five key tuning sites of LWS, we obtained the global alignment of LWS in 12 teleost fishes and human being using MUSCLE v3.8 (Edgar, 2004) and highlighted the five crucial sites with Jalview v2.11.1.3 (Waterhouse et al., 2009). F86 of SWS1 opsin is crucial for UV sensing; the mutation of F86V in goldfish led to +1 nm shift in the absorption spectrum of the SWS1 opsins (Tada et al., 2009). The tuning site F86 resulting in the UV perception of SWS1 opsin in vertebrates (Hunt et al., 2007) was also checked in SWS1-containing teleost fishes.
Characterization of Gadusol Biosynthesis Genes
To identify gadusol biosynthesis related genes, we extracted the eevs-like and mt-ox genes and genes adjoined to them in zebrafish, tilapia, and medaka genomes that were collected from the NCBI database (Supplementary Table 3) as the references and employed the same method as mentioned for the vision-related genes to predict eevs-like and mt-ox in in the mirrorwing flyingfish genome. For other 11 selected teleost fishes, we retrieved eevs-like and mt-ox from the NCBI annotation. We constructed a rooted NJ tree using a dehydroquinate synthase (DHQS-like) derived from cyanobacteria (Balskus and Walsh, 2010) as the outgroup by MEGA-X with 1,000 bootstraps. Conserved domains and motifs of the candidate eevs-like genes were predicted using the NCBI Conserved Domain Database (CDD) (Lu et al., 2020) and MEME website server (Bailey et al., 2006), and then, TBtools suite was applied to illuminate the phylogenetic tree, conserved domains, and motifs (Chen et al., 2020).
Identification of Olfactory Receptor Genes
Reference sequences of olfactory receptor (OR) genes were obtained from a previous paper (Niimura, 2009). The full-length OR protein sequences were aligned to nine teleost fishes (including Ates, Bpec, Pmag, Carg, Kmar, Hspe, Drer, Oaur, and Olat) using tBLASTn (Ye et al., 2006) with an E-value of 1e−5, and the blasted hits were clustered using SOLAR v0.9 (Yu et al., 2006) to define candidate gene loci.
We extracted these candidate gene loci along with 2-kb flank region and employed GeneWise v2.4 (Birney et al., 2004) to predict gene structures. First, the potential OR genes without start/stop codons or with interrupting stop codon(s) or frameshift(s) were excluded. Second, the full-length sequences were inspected using the NCBI non-redundant database (BLASTP with an E-value of 1e−5), but those candidate OR genes with the best hit annotation of non-OR were discarded. Finally, the remaining sequences were further checked using TMHMM v2.0 (Krogh et al., 2001) to identify the putative seven transmembrane domains. We aligned the protein sequences of confirmed OR genes using MUSCLE in the MEGA-X (Kumar et al., 2018) and then constructed a rooted neighbor-joining tree using human G-protein coupled receptor 35 (NP_005292.2) and human G-protein coupled receptor 132 (NP_037477.1) as the outgroup by MEGA-X with the Poisson model and uniform rates.
Results and Discussion
Summary of the Genome Assembly and Annotation
The Illumina sequencing generated a total of ~138.13-Gb raw reads, and then, 99.21-Gb clean reads were retained after filtering low-quality sequences (Supplementary Table 4). The PacBio sequencing yielded about 29.98-Gb data, consisting of 2,785,344 reads with an N50 length of 16.5 kb (Supplementary Table 5).
A k-mer analysis predicted that the mirrorwing flyingfish had an estimated genome size of 1.06 Gb and a heterozygosity of 1.35% (Figure 1B). After contig building, consensus calling, polishing, and scaffold construction, we generated a final assembly of 1.04 Gb, which is nearly equal to the estimated genome size. The draft assembly consisted of 3,052 scaffolds (> 650 bp in length), and the contig and scaffold N50 values of our final assembly were 992.83 and 1,152.47 kb (Table 1).
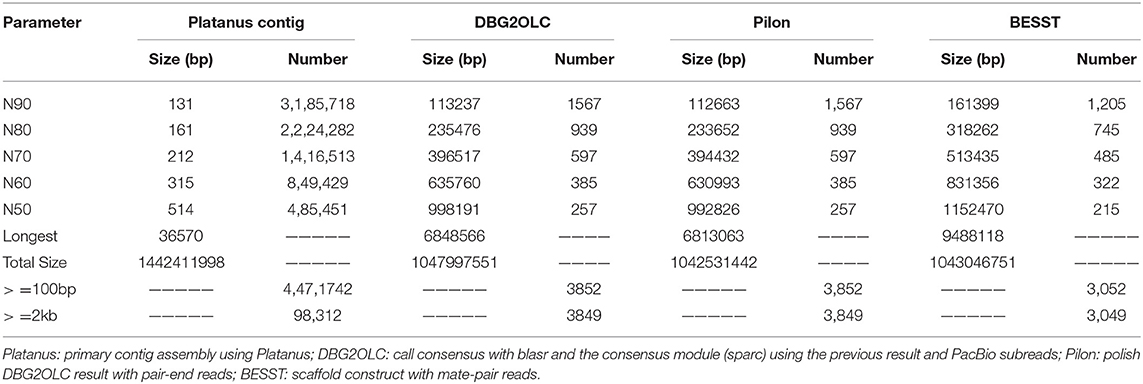
Table 1. Statistics of our genome assembly.
The BUSCO evaluation indicated that 94.2% of the Actinopterygii gene sets were identified as complete (4,317 out of 4,584, actinopterygii_odb9) in the mirrorwing flyingfish genome (Table 2). We also assessed accuracy of the draft assembly by mapping Illumina paired-end reads onto the assembled genome sequences. A total of 94.91% of the Illumina paired-end reads were properly mapped to the assembled genome, with a good coverage of 97.78% (Supplementary Table 6). The high completeness of BUSCOs and nucleotide-level accuracy, together with considerable continuity of contig sizes, suggested that our high-quality genome assembly could be qualified for further data analysis.
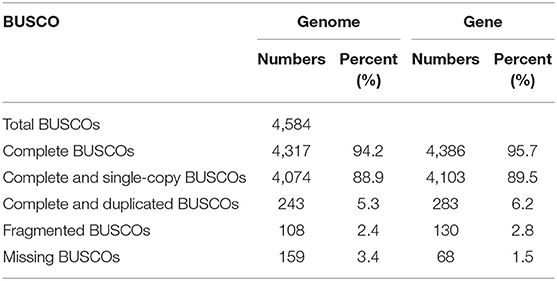
Table 2. Evaluation of the genome and gene completeness with BUSCO.
Repeat content of the mirrorwing flyingfish genome was calculated by combination of both homolog-based and de novo methods. We determined that repeat elements occupied 42.02% of the assembled genome, and DNA transposons accounted for the largest proportion (24.38%) of transposable elements (TEs; Supplementary Table 8). A total of 8.19% of the mirrorwing flyingfish genome sequences were composed of tandem repeat elements (Supplementary Table 7). Divergence rates of the TEs in the mirrorwing flyingfish genome were determined using Repbase and de novo libraries, respectively. We observed that 10.72 Mb of identified TEs had a <10% divergence rate from the Repbase consensus; 277.08 Mb of TE sequences (26.56% of the assembly genome) had a <10% divergence rate from the de novo library (Supplementary Figure 2), which were possible to be active with a recent origin.
We predicted 23,611 protein-coding genes in the mirrorwing flyingfish genome, with an average gene length of 14.35 kb. Moreover, 99.50% of these genes could be functionally annotated by at least one of the four popular databases, with 20,692 KEGG hits, 21,453 SwissProt hits, 23,477 TrEMBL hits, and 21,888 Interpro hits (Supplementary Table 9). Additionally, the BUSCO evaluation of genes demonstrated that 95.7% of the Actinopterygii gene sets were predicted as complete (4,386 out of 4,584 actinopterygii_odb9) in the mirrorwing flyingfish gene set (Table 2), suggesting high quality of our gene prediction. Furthermore, we identified four types of non-coding RNA, 247 miRNAs, 2,138 tRNAs, 538 rRNAs, and 298 snRNAs in the assembled genome (Supplementary Table 10).
Gene Families and Phylogeny
Our gene family data demonstrated that protein-coding sequences in the 19 teleost fishes were clustered into 22,669 gene families, of which 4,632 families were 1:1 single-copy orthologs. A total of 93.5% (22,083 out of 23,611) of the mirrorwing flyingfish protein-coding genes were grouped into 17,352 gene families (Supplementary Table 11), defining 7,335 single-copy orthologs and 323 unique paralogs (Supplementary Figure 3B).
Using the 4,632 1:1 single-copy orthologous genes, we established a coincident phylogenetic topology with the ML and Bayes methods (Supplementary Figures 4, 5). The divergence tree revealed that the flyingfish was close to the two medaka species with a divergence time of about 85.2 Mya (Supplementary Figure 6). A total of 60.71% (633.32 Mb) of the mirrorwing flyingfish genome was syntenic with Japanese medaka, while only 14.66% (152.94 Mb) of the mirrorwing flyingfish genome shared synteny with zebrafish (see more details in Supplementary Table 12).
We identified 1,236 expanded gene families and 1,539 contracted gene families in the mirrorwing flyingfish genome (Supplementary Figure 3A). Among them, 135 and 131 were significantly expanded and contracted (p < 0.05). The KEGG enrichment analysis demonstrated that those genes belonging to the expanded gene families were related to signaling molecules and interaction, nervous system, and immune system (Supplementary Table 13, p < 0.01).
Various Vision-Related Genes in the Mirrorwing Flyingfish
Vision plays a vital role in animal life, affording an important ability to perceive environmental stimuli. The visual ability of this animal depends on the numbers of opsin proteins (Bowmaker, 2008). Various fishes have accommodated a wide range of habitats (such as freshwater and marine, stagnant and running water, and shallow and deep sea), which provide differential vision adaptation (Hauser and Chang, 2017). We classified 12 teleost fishes into three groups in terms of living habitat, including genuine amphibious inhabitant (Ates, Bpec, Pmag, Carg, Kmar), normal underwater dweller (Drer, Oaur, Onil, Mzeb, Olat, Omel), and temporary water surface traveler (Hspe), for comparison of the variations among opsin proteins.
The mirrorwing flyingfish genome contains five types of opsins, with two LWS, two SWS2, one SWS1, one RH1, and three RH2 (Figure 2; Table 3). The maximal absorption spectra (λmax) of flyingfish LWS, based on the popular “five-sites” rule (You et al., 2014), are predicted to be 560 nm, which is similar to the parameters in climbing perch, northern snakehead, mangrove rivulus, blue tilapia, Nile tilapia, zebra mbuna, Japanese medaka, and marine medaka (Supplementary Table 14). The five crucial sites of LWS in the mirrorwing flying fish are 180S, 197H, 277Y, 285T, and 308A (Supplementary Figure 7).
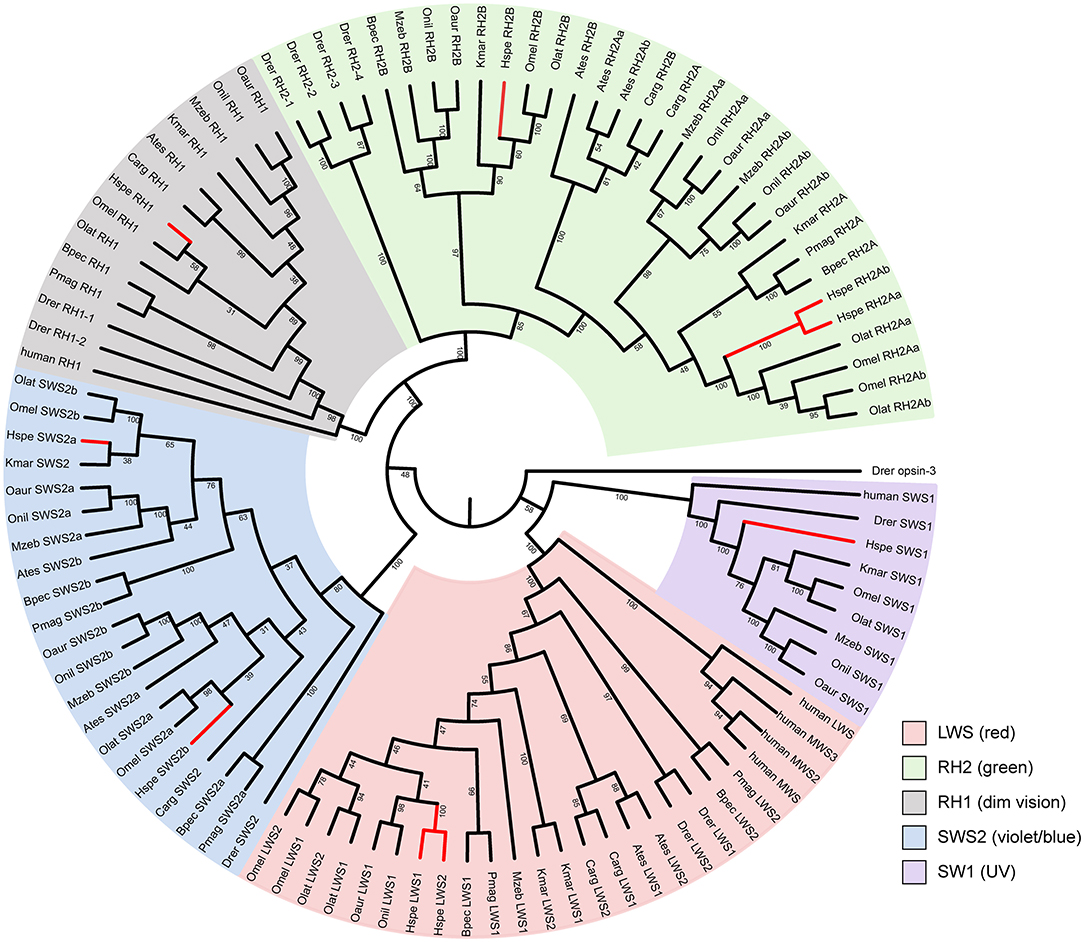
Figure 2. The phylogenetic tree of vertebrate opsin genes. A rooted neighbor-joining (NJ) tree was constructed with zebrafish opsin3 as the outgroup. Abbreviations are provided in Supplementary Table 1.

Table 3. Copy number of vison-related genes in the 12 representative teleost fishes.
The synteny of opsins in 12 teleost fishes is quite conserved except SWS1 (Supplementary Figures 8, 9). All amphibious fishes except mangrove rivulus fish have lost SWS1 (Supplementary Figure 8B), which is used for UV vision. This SWS1 missing could be related to the landing activity of these fishes. Since ultraviolet light can cause damages to the retina, the critical mutation of F86V could potentially alter absorption wave of SWS1 opsins toward violet light sensing so as to minimize the UV-induced damages (Cowing et al., 2002). These examined fishes in this study have V (valine) at 86 instead of F (phenylalanine; see Supplementary Figure 10), implying that these fishes could be UV sensing. Related amino acid numbering was based on the bovine rhodopsin sequence [GenBank accession no. M21606; (Palczewski et al., 2000)].
The five crucial sites of LWS in the mirrorwing flyingfish showed a narrow range of color sensing, demonstrating the same tendency as some amphibious fishes, such as climbing perch, northern snakehead, and mangrove rivulus fish. When these fishes move out of water, they can keep the same long-wave sensing as that in water. The SWS1 loss events in the five examined amphibious fishes in our present study may have developed for the water-to-terrestrial adaptation; however, the reservation of SWS1 in the mirrorwing flyingfish might be due to the short period of gliding in air instead of a real amphibious life (Davenport, 1994).
Low retinal dopamine levels could cause myopia (Feldkaemper and Schaeffel, 2013), and AANAT1a can reduce the dopamine content in the retina via acetylation (Zilberman-Peled et al., 2006). The loss of aanat1a in amphibious giant-fin mudskipper could be beneficial for movement in air (You et al., 2014). Interestingly, 12 teleost fishes except for giant-fin mudskipper have one copy of annat1a (see more details in Table 4; Figure 3). A previous study reported that the Atlantic flyingfish (C. heterurus) had a pyramidal shape cornea, which could assure both hypermetropic underwater vision and emmetropic vision in air (Baylor, 1967). Since the mirrorwing flyingfish owned three copies of aanat (without absence of aanat1a), its unique cornea might be responsible for a temporary air vision. Gadusol biosynthesis genes in the mirrorwing flyingfish we identify two copies of eevs-like and one copy of mt-ox in all the selected 12 fish genomes. Interestingly, the mirrorwing flyingfish has the same gene cluster as medaka, with mdfic2 missing in the gene cluster “foxp1b-mdfic2-mt-ox-eevs-a-mitfa-frmd4Ba” (see more details in Table 5). All fishes shared the gene cluster of “foxp1a-eevs-b-mitfb-frmd4Bb” except for zebrafish (Supplementary Figure 11). Perhaps, the examined zebrafish genome was modified by genetic engineering (Carpio and Estrada, 2006). The two isotypes of eevs-like gene contain five exons, conserved domain CCD, and six conserved motifs (Figure 4). It seems that this Beloniformes species had experienced the same gene loss event.
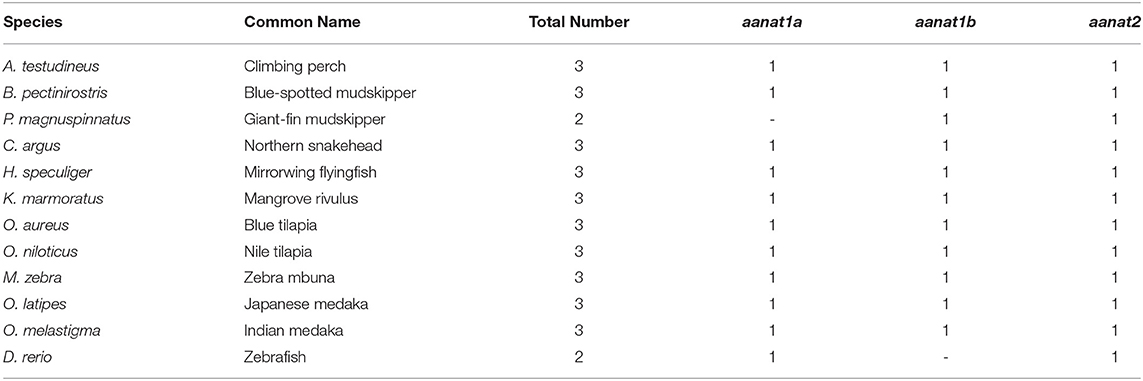
Table 4. Copy number of aanat genes in the 12 representative teleost fishes.
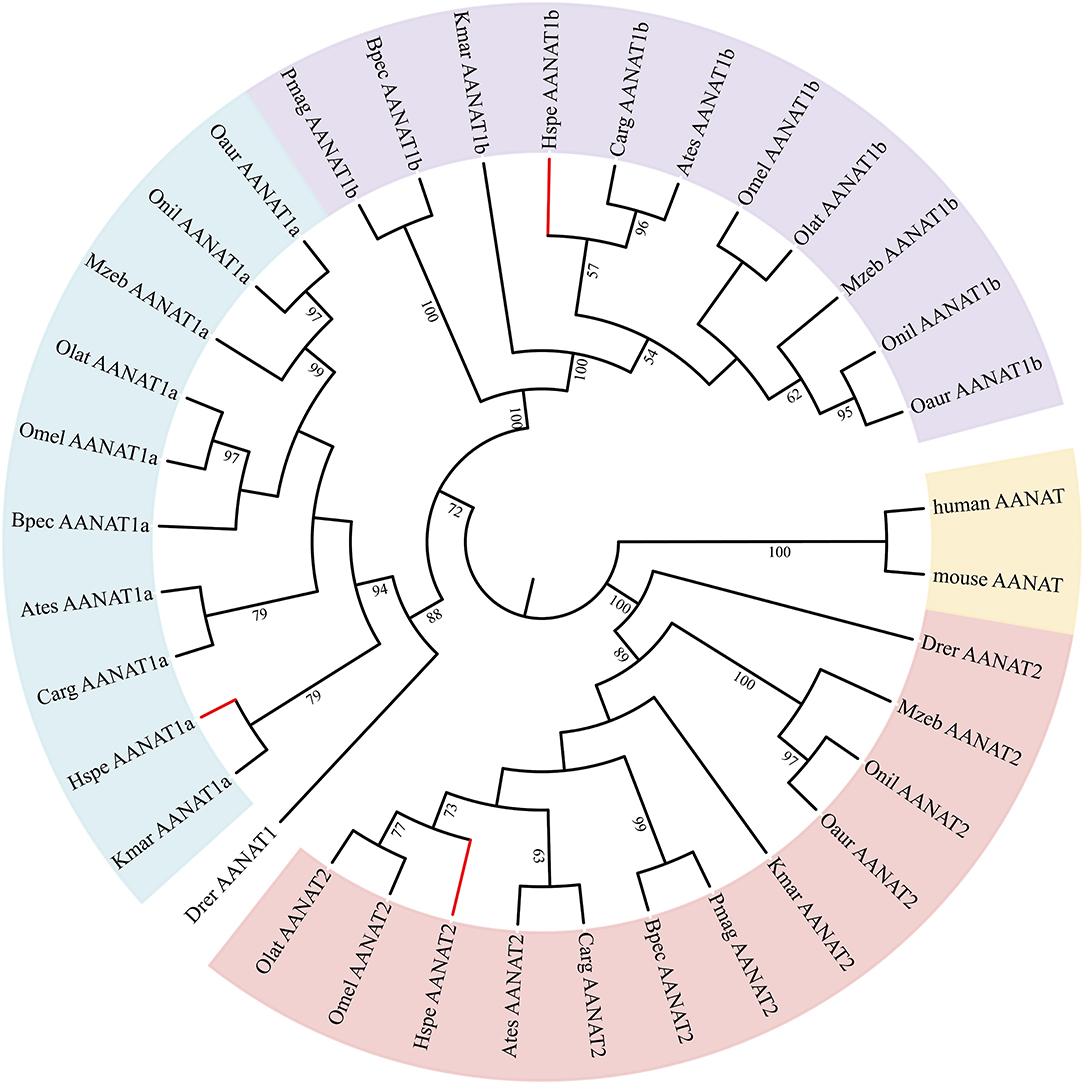
Figure 3. The rooted NJ tree of vertebrate aanat genes. It was constructed with human AANAT (NP_001079.1) and mouse AANAT (NP_033721.1) as the outgroup.
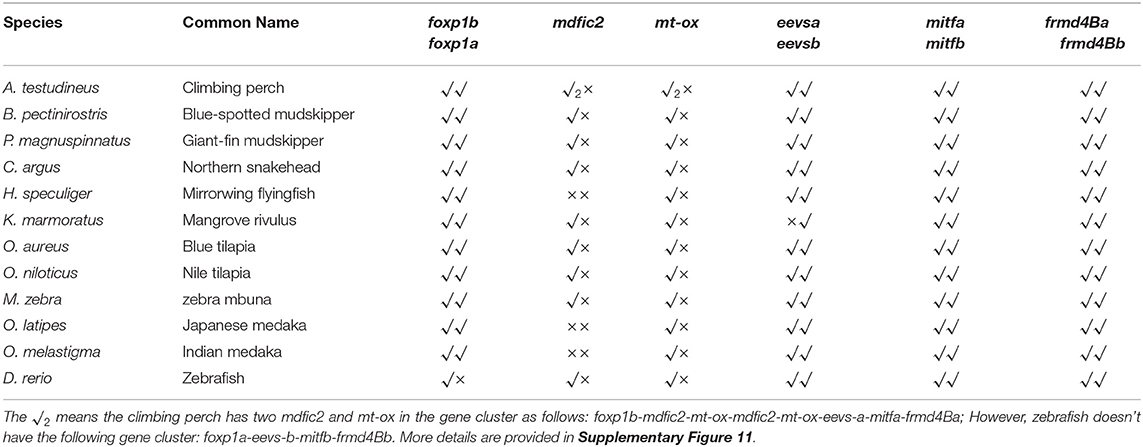
Table 5. Genetic analysis of eevs and mt-ox genes in selected fishes.
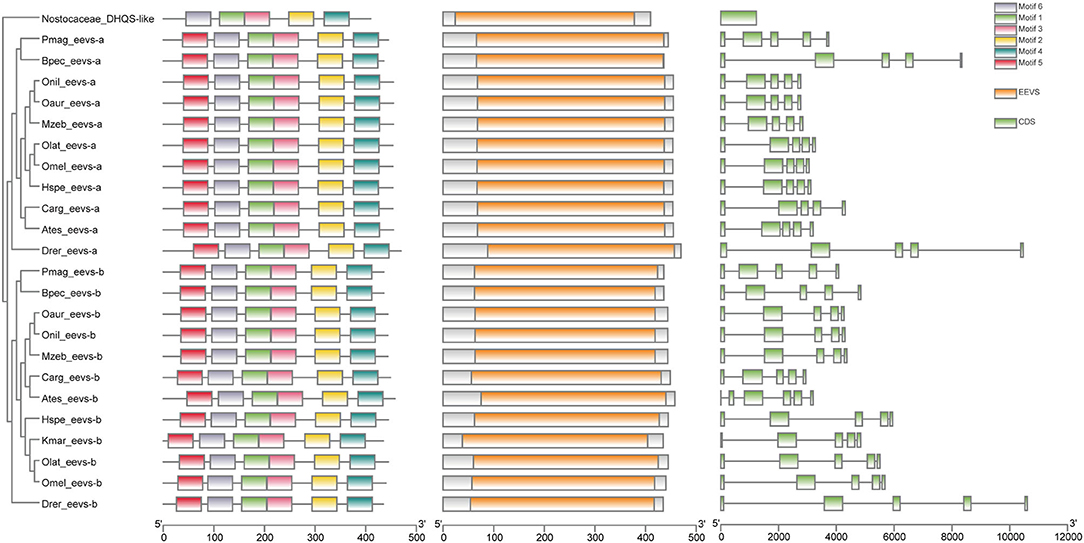
Figure 4. The rooted NJ tree of teleost eevs-like genes. It was constructed with cyanobacteria DHQS-like as the outgroup. The first column is the rooted tree, the second column is the six motifs derived from MEME web service, the third column is the conserved domain CDD derived from NCBI, and the four column is the detailed structures of eevs-like genes.
Olfactory Genes in the Mirrorwing Flyingfish
Olfaction is an essential component of the animal sensory system for perceiving water- and air-soluble chemicals that can help to localize food, predators, and spawning migration sites (Hopfield, 1991). We identified 781 intact OR genes in nine representative fishes (Supplementary Table 15). These identified ORs could be classified into five subfamilies, including delta, epsilon, zeta, eta, and beta (see more details in Supplementary Figure 12).
The mirrorwing flyingfish possessed 50 intact OR genes; among them, the number of air-/waterborne OR genes were much less than climbing perch, northern snakehead, and zebrafish. Surprisedly, we could not find any airborne OR gene in the mirrorwing flyingfish genome. Although this fish could glide a while above water, the detailed classification and copy numbers of OR genes appear to be the same as those in medaka, while they are different from amphibious fishes (such as mudskippers; see You et al., 2014).
Conclusions
We obtained a draft genome assembly for the representative mirrorwing flyingfish with a hybrid method after Illumina and PacBio sequencing. We constructed a phylogenetic tree to illuminate the relationship of the mirrorwing flyingfish and other 18 teleost fishes. We also investigated vision-related genes, olfactory receptor genes, and gadusol synthesis-related genes in representative teleost fishes. Since the mirrorwing flyingfish could leave water for a while, it may exhibit similar traits as amphibious fishes. However, our genomic comparisons of vision-related and olfactory receptor genes revealed that the mirrorwing flyingfish potentially shared the same genetic mechanisms as its phylogenetic relatives (medaka species) but different from popular amphibious fishes (such as mudskippers). This high-quality genome assembly provides a valuable genetic resource for the mirrorwing flyingfish, and it will also facilitate in-depth biomedical studies on various Exocoetoidea fishes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA714815; https://figshare.com/, https://doi.org/10.6084/m9.figshare.14600634.v1.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use Committee of BGI (approval ID: FT18134).
Author Contributions
QS conceived the project. PX, CZ, CB, and XY analyzed the data. XY, JC, ZR, FY, RG, and JX collected samples and assisted data analysis. PX and CZ wrote the manuscript. QS and CB revised the manuscript. All authors approved submission of the final manuscript for publication.
Funding
The work was financially supported by Shenzhen Science and Technology Innovation Program for International Cooperation (no. GJHZ20190819152407214) and Shenzhen Dapang Special Program for Industrial Development (no. KJYF202001-17).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.695700/full#supplementary-material
Supplementary Figure 1. Pipeline of the genome assembly.
Supplementary Figure 2. Distribution of divergence rates in each type of TEs within the mirrorwing flyingfish genome. The divergence rate was calculated between the identified TEs in the genome and the consensus sequences in the TE library used Repbase (A) or de novo libraries constructed by RepeatModeler and LTR_Finder (B).
Supplementary Figure 3. Evolution of 19 representative teleost genomes and their gene families. (A) Divergence tree with expanded and contracted gene families. (B) Statistics of single-copy orthologs, multiple-copy orthologs, unique paralogs, other orthologs, and unclustered gene numbers in the 19 teleost fishes.
Supplementary Figure 4. A rooted phylogenetic tree (constructed with Bayes).
Supplementary Figure 5. A rooted phylogenetic tree (constructed with PhyML).
Supplementary Figure 6. A fossil-calibrated phylogenetic tree. It was constructed with the following five calibrated times that were adapted from the Timetree: C. argus-A. testudineus (66~78Mya), H. speculiger-O. latipes (68~89 Mya), O. latipes-O. aureus (104~145 Mya), B. pectinirostris-P. magnuspinnatus (49~69 Mya), and D. rerio-O. aureus (149~165 Mya).
Supplementary Figure 7. The crucial tuning sites in LWS opsins of 12 teleost fishes and human. Five critical sites in the mirrorwing flyingfish include S180A, H197Y, Y277F, T285A, and A308S. Abbreviations of fish species are provided in Supplementary Table 1.
Supplementary Figure 8. The gene architecture of RH1 (A) and SWS1 (B) in 12 representative teleost fishes. Gene names were listed in the first line. Abbreviations of fish species are provided in Supplementary Table 1.
Supplementary Figure 9. The gene architecture of LWS-SWS2 (A) and RH2 (B) in 12 representative teleost fishes. Gene names were listed in the first line. Abbreviations of fish species are provided in Supplementary Table 1.
Supplementary Figure 10. The crucial tuning site of SWS1 (F86V) in 8 selected teleost fishes. The tuning site was marked in light blue. Abbreviations of fish species are provided in Supplementary Table 1.
Supplementary Figure 11. The gene architecture of eevs-likes and mt-ox in12 representative teleost fishes. Gene names were listed in the first line. Abbreviations of fish species are provided in Supplementary Table 1.
Supplementary Figure 12. A rooted neighbor-joining tree of olfactory receptor genes (ORs) in 9 selected teleost fishes. A total of 787 sequences were collected for construction of the circular cladogram tree. Various OR types were marked in different colors.
Supplementary Table 1. Information of 19 teleost fishes used in our present study.
Supplementary Table 2. Accession numbers of known aanat, opsin and neighboring genes.
Supplementary Table 3. Accession numbers of known adjacent genes of eevs and mt-ox.
Supplementary Table 4. Libraries and data yields for the whole genome shotgun sequencing.
Supplementary Table 5. Libraries and data yields for the PacBio sequencing.
Supplementary Table 6. The alignment result of paired-end reads mapping to H. speculiger genome.
Supplementary Table 7. Summary of repeat annotations.
Supplementary Table 8. Classification of repetitive elements.
Supplementary Table 9. Statistics of function annotation.
Supplementary Table 10. Statistics of Non-coding RNAs in the genome.
Supplementary Table 11. Statistics of gene families in 19 teleost fishes.
Supplementary Table 12. Statistics of pairwise alignment among mirrorwing flyingfish, medaka and zebrafish.
Supplementary Table 13. KEGG enrichment for genes of expanded gene families of the mirrorwing flyingfish.
Supplementary Table 14. Estimated maximal absorption spectrum (?max) of LWS.
Supplementary Table 15. Copy numbers of intact OR genes in each group of fishes and mammals.
References
Attwood, T. K., Croning, M. D. R., Flower, D. R., Lewis, A. P., Mabey, J. E., Scordis, P., et al. (2000). PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res. 28, 225–227. doi: 10.1093/nar/28.1.225
Bailey, T. L., Williams, N., Misleh, C., and Li, W. W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373. doi: 10.1093/nar/gkl198
Bairoch, A., Apweiler, R., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2005). The universal protein resource (UniProt). Nucleic Acids Res. 33, D154–D159. doi: 10.1093/nar/gki070
Balskus, E. P., and Walsh, C. T. (2010). The genetic and molecular basis for sunscreen biosynthesis in cyanobacteria. Science 329, 1653–1656. doi: 10.1126/science.1193637
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucleic Acids Res. 32, D138–D141. doi: 10.1093/nar/gkh121
Baylor, E. R. (1967). Air and water vision of the Atlantic flying fish, Cypselurus heterurus. Nature 214, 307–309. doi: 10.1038/214307a0
Baylor, E. R., and Shaw, E. (1962). Refractive error and vision in fishes. Science 136, 157–158. doi: 10.1126/science.136.3511.157
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580. doi: 10.1093/nar/27.2.573
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Bowmaker, J. K. (2008). Evolution of vertebrate visual pigments. Vision Res. 48, 2022–2041. doi: 10.1016/j.visres.2008.03.025
Burge, S., Kelly, E., Lonsdale, D., Mutowo-Muellenet, P., Mcanulla, C., Mitchell, A., et al. (2012). Manual GO annotation of predictive protein signatures: the InterPro approach to GO curation. Database 2012:bar068. doi: 10.1093/database/bar068
Carpio, Y., and Estrada, M. P. (2006). Zebrafish as a genetic model organism. Biotecnología Aplicada 23, 265–270.
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, N. (2004). Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4–10. doi: 10.1002/0471250953.bi0410s05
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix120
Cowing, J. A., Poopalasundaram, S., Wilkie, S. E., Robinson, P. R., Bowmaker, J. K., and Hunt, D. M. (2002). The molecular mechanism for the spectral shifts between vertebrate ultraviolet- and violet-sensitive cone visual pigments. Biochem. J. 367, 129–135. doi: 10.1042/bj20020483
Cui, L., Dong, Y., Cao, R., Gao, J., Cen, J., Zheng, Z., et al. (2018). Mitochondrial genome of the garfish Hyporhamphus quoyi (Beloniformes: Hemiramphidae) and phylogenetic relationships within Beloniformes based on whole mitogenomes. PLoS ONE 13, e0205025–e0205025. doi: 10.1371/journal.pone.0205025
Davenport, J. (1994). How and why do flying fish fly? Rev. Fish Biol. Fish. 4, 184–214. doi: 10.1007/BF00044128
De Bruin, G. H. P., Russell, B. C., and Bogusch, A. (1995). FAO Species Identification Field Guide for Fishery Purposes. The Marine Fishery Resources of Sri Lanka.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238–238. doi: 10.1186/s13059-019-1832-y
Falcon, S., and Gentleman, R. (2008). “Hypergeometric testing used for gene set enrichment analysis,” in Bioconductor Case Studies, eds F. Hahne, W. Huber, R. Gentleman, and S. Falcon (New York, NY: Springer), 207–220. doi: 10.1007/978-0-387-77240-0_14
Feldkaemper, M., and Schaeffel, F. (2013). An updated view on the role of dopamine in myopia. Exp. Eye Res. 114, 106–119. doi: 10.1016/j.exer.2013.02.007
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Han, M. V., Thomas, G. W., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. doi: 10.1093/molbev/mst100
Harris, R. S. (2007). Improved pairwise alignmnet of genomic DNA (PhD. Thesis). Pennsy lvania State University, Pennsylvania, United States.
Hauser, F. E., and Chang, B. S. W. (2017). Insights into visual pigment adaptation and diversity from model ecological and evolutionary systems. Curr. Opin. Genet. Dev.47, 110–120. doi: 10.1016/j.gde.2017.09.005
Hopfield, J. J. (1991). Olfactory computation and object perception. Proc. Natl. Acad. Sci. 88, 6462–6466. doi: 10.1073/pnas.88.15.6462
Hunt, D. M., Carvalho, L. S., Cowing, J. A., Parry, J. W. L., Wilkie, S. E., Davies, W. L., et al. (2007). Spectral tuning of shortwave-sensitive visual pigments in vertebrates. Photochem. Photobiol. 83, 303–310. doi: 10.1562/2006-06-27-IR-952
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kageyama, H., and Waditee-Sirisattha, R. (2019). Antioxidative, anti-inflammatory, and anti-aging properties of mycosporine-like amino acids: molecular and cellular mechanisms in the protection of skin-aging. Marine Drugs 17, 222. doi: 10.3390/md17040222
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–d361. doi: 10.1093/nar/gkw1092
Kim, O. T. P., Nguyen, P. T., Shoguchi, E., Hisata, K., Vo, T. T. B., Inoue, J., et al. (2018). A draft genome of the striped catfish, Pangasianodon hypophthalmus, for comparative analysis of genes relevant to development and a resource for aquaculture improvement. BMC Genomics 19, 733–733. doi: 10.1186/s12864-018-5079-x
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Kutschera, U. (2005). Predator-driven macroevolution in flyingfishes inferred from behavioural studies: historical controversies and a hypothesis. Ann. Hist. Phil. Biol. 10, 59–77.
Letunic, I., Doerks, T., and Bork, P. (2012). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305. doi: 10.1093/nar/gkr931
Li, H. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851. doi: 10.1093/bioinformatics/btu356
Lin, J. J., Wang, F. Y., Li, W. H., and Wang, T. Y. (2017). The rises and falls of opsin genes in 59 ray-finned fish genomes and their implications for environmental adaptation. Sci. Rep. 7:15568. doi: 10.1038/s41598-017-15868-7
Lovejoy, N. R., Iranpour, M., and Collette, B. B. (2004). Phylogeny and jaw ontogeny of beloniform fishes. Integr. Comp. Biol. 44, 366–377. doi: 10.1093/icb/44.5.366
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–d268. doi: 10.1093/nar/gkz991
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Miyamoto, K. T., Komatsu, M., and Ikeda, H. (2014). Discovery of gene cluster for mycosporine-like amino acid biosynthesis from Actinomycetales microorganisms and production of a novel mycosporine-like amino acid by heterologous expression. Appl. Environ. Microbiol. 80, 5028–5036. doi: 10.1128/AEM.00727-14
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Niimura, Y. (2009). On the origin and evolution of vertebrate olfactory receptor genes: comparative genome analysis among 23 chordate species. Genome Biol. Evol. 1, 34–44. doi: 10.1093/gbe/evp003
Osborn, A. R., Almabruk, K. H., Holzwarth, G., Asamizu, S., Ladu, J., Kean, K. M., et al. (2015). De novo synthesis of a sunscreen compound in vertebrates. Elife 4:e05919. doi: 10.7554/eLife.05919.028
Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., et al. (2000). Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745. doi: 10.1126/science.289.5480.739
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rayner, J. M. V. (1986). Pleuston: animals which move in water and air. Endeavour 10, 58–64. doi: 10.1016/0160-9327(86)90131-6
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rosic, N. N. (2019). Mycosporine-like amino acids: making the foundation for organic personalised sunscreens. Marine Drugs 17, 638. doi: 10.3390/md17110638
Rosic, N. N., and Dove, S. (2011). Mycosporine-like amino acids from coral dinoflagellates. Appl. Environ. Microbiol. 77, 8478–8486. doi: 10.1128/AEM.05870-11
Sahlin, K., Vezzi, F., Nystedt, B., Lundeberg, J., and Arvestad, L. (2014). BESST–efficient scaffolding of large fragmented assemblies. BMC Bioinformatics 15, 281–281. doi: 10.1186/1471-2105-15-281
Servant, F., Bru, C., Carrère, S., Courcelle, E., Gouzy, J., Peyruc, D., et al. (2002). ProDom: automated clustering of homologous domains. Brief. Bioinform. 3, 246–251. doi: 10.1093/bib/3.3.246
Shick, J. M., and Dunlap, W. C. (2002). Mycosporine-like amino acids and related Gadusols: biosynthesis, acumulation, and UV-protective functions in aquatic organisms. Annu. Rev. Physiol. 64, 223–262. doi: 10.1146/annurev.physiol.64.081501.155802
Shinzato, C., Shoguchi, E., Kawashima, T., Hamada, M., Hisata, K., Tanaka, M., et al. (2011). Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323. doi: 10.1038/nature10249
Sigrist, C. J., Cerutti, L., De Castro, E., Langendijk-Genevaux, P. S., Bulliard, V., Bairoch, A., et al. (2010). PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 38, D161–166. doi: 10.1093/nar/gkp885
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 1–11. doi: 10.1186/1471-2105-6-31
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–439. doi: 10.1093/nar/gkl200
Subramanian, B., Gao, S., Lercher, M. J., Hu, S., and Chen, W. H. (2019). Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 47, W270–w275. doi: 10.1093/nar/gkz357
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34, W609–612. doi: 10.1093/nar/gkl315
Tada, T., Altun, A., and Yokoyama, S. (2009). Evolutionary replacement of UV vision by violet vision in fish. Proc. Natl. Acad. Sci. 106, 17457–17462. doi: 10.1073/pnas.0903839106
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. doi: 10.1101/gr.772403
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963–e112963. doi: 10.1371/journal.pone.0112963
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Wright, P. A., and Turko, A. J. (2016). Amphibious fishes: evolution and phenotypic plasticity. J. Exp. Biol. 219, 2245–2259. doi: 10.1242/jeb.126649
Xiong, Z., Li, F., Li, Q., Zhou, L., Gamble, T., Zheng, J., et al. (2016). Draft genome of the leopard gecko, Eublepharis macularius. Gigascience 5, s13742–s13016. doi: 10.1186/s13742-016-0151-4
Xu, G. H., Zhao, L. J., Gao, K. Q., and Wu, F. X. (2012). A new stem-neopterygian fish from the middle triassic of China shows the earliest over-water gliding strategy of the vertebrates. Proc. Biol. Sci. 280, 20122261–20122261. doi: 10.1098/rspb.2012.2261
Xu, Z., and Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–268. doi: 10.1093/nar/gkm286
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Ye, C., Hill, C. M., Wu, S., Ruan, J., and Ma, Z. S. (2016). DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6, 31900–31900. doi: 10.1038/srep31900
Ye, C., and Ma, Z. S. (2016). Sparc: a sparsity-based consensus algorithm for long erroneous sequencing reads. Peerj 4, e2016–e2016. doi: 10.7717/peerj.2016
Ye, J., Mcginnis, S., and Madden, T. L. (2006). BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. doi: 10.1093/nar/gkl164
Yokoyama, S. (2000). Molecular evolution of vertebrate visual pigments. Prog. Retin Eye Res. 19, 385–419. doi: 10.1016/S1350-9462(00)00002-1
Yokoyama, S. (2008). Evolution of dim-light and color vision pigments. Annu. Rev. Genomics Hum. Genet. 9, 259–282. doi: 10.1146/annurev.genom.9.081307.164228
Yokoyama, S., and Radlwimmer, F. B. (2001). The molecular genetics and evolution of red and green color vision in vertebrates. Genetics 158, 1697–1710. doi: 10.1093/genetics/158.4.1697
You, X., Bian, C., Zan, Q., Xu, X., Liu, X., Chen, J., et al. (2014). Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 5, 1–8. doi: 10.1038/ncomms6594
Yu, X. J., Zheng, H. K., Wang, J., Wang, W., and Su, B. (2006). Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics 88, 745–751. doi: 10.1016/j.ygeno.2006.05.008
Zilberman-Peled, B., Ron, B., Gross, A., Finberg, J. P., and Gothilf, Y. (2006). A possible new role for fish retinal serotonin-N-acetyltransferase-1 (AANAT1): dopamine metabolism. Brain Res. 1073–1074, 220–228. doi: 10.1016/j.brainres.2005.12.028
Glossary

Keywords: flying fish, whole genome sequencing, genome assembly, eevs, vision-related gene, phylogenetic tree
Citation: Xu P, Zhao C, You X, Yang F, Chen J, Ruan Z, Gu R, Xu J, Bian C and Shi Q (2021) Draft Genome of the Mirrorwing Flyingfish (Hirundichthys speculiger). Front. Genet. 12:695700. doi: 10.3389/fgene.2021.695700
Received: 15 April 2021; Accepted: 03 June 2021;
Published: 07 July 2021.
Edited by:
Liang Guo, South China Sea Fisheries Research Institute, ChinaReviewed by:
Jitendra Kumar Sundaray, Central Institute of Freshwater Aquaculture, IndiaYou-Yi Kuang, Heilongjiang River Fisheries Research Institute, China
Copyright © 2021 Xu, Zhao, You, Yang, Chen, Ruan, Gu, Xu, Bian and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiong Shi, c2hpcWlvbmdAZ2Vub21pY3MuY24=; Chao Bian, YmlhbmNoYW9AZ2Vub21pY3MuY24=
†These authors have contributed equally to this work