 Pan Wang
Pan Wang Guiyang Zhang
Guiyang Zhang Zu-Guo Yu2
Zu-Guo Yu2 Guohua Huang
Guohua Huang- 1School of Electrical Engineering, Shaoyang University, Shaoyang, China
- 2Key Laboratory of Intelligent Computing and Information Processing of Ministry of Education and Hunan Key Laboratory for Computation and Simulation in Science and Engineering, Xiangtan University, Xiangtan, China
Knowledge about protein-protein interactions is beneficial in understanding cellular mechanisms. Protein-protein interactions are usually determined according to their protein-protein interaction sites. Due to the limitations of current techniques, it is still a challenging task to detect protein-protein interaction sites. In this article, we presented a method based on deep learning and XGBoost (called DeepPPISP-XGB) for predicting protein-protein interaction sites. The deep learning model served as a feature extractor to remove redundant information from protein sequences. The Extreme Gradient Boosting algorithm was used to construct a classifier for predicting protein-protein interaction sites. The DeepPPISP-XGB achieved the following results: area under the receiver operating characteristic curve of 0.681, a recall of 0.624, and area under the precision-recall curve of 0.339, being competitive with the state-of-the-art methods. We also validated the positive role of global features in predicting protein-protein interaction sites.
Introduction
Proteins are one of the most important components of the cell, and also are the principal undertaker of the activities of life. The functions of proteins are manifested mainly by interacting with various molecules such as DNA/RNA, proteins, or other ligands (Dias and Kolaczkowski, 2017). The protein-protein interaction (PPI) plays a key role in the cellular process such as signal transduction, transport, and metabolism (Li et al., 2019) and also is involved in the pathogenesis of diseases such as Alzheimer’s cervical cancer, bacterial infection, and prion diseases (Cohen and Prusiner, 1998; Selkoe, 1998; Loregian et al., 2002). Therefore, knowledge of PPI is critical for understanding the molecular mechanisms hidden in the phenomenon of life (Das and Chakrabarti, 2021). Many experimentally verified or computationally predicted PPIs have been hosted for scientific research in public databases such as the Human Protein Reference Database (Keshava Prasad et al., 2009), STRING (Von Mering et al., 2005), the database of interacting proteins (Salwinski et al., 2004), and the protein interaction database (Kerrien et al., 2007). The protein-protein interaction site (PPIS) is defined as surface residues where proteins interact with each other (Aumentado-Armstrong et al., 2015). The identification of PPIS is the premise for determining PPI (Wang et al., 2019). The knowledge about PPIS holds vast potential to infer cell regulatory mechanisms, locate drug targets, identify structures and functions of protein complexes (Deng et al., 2009; Orii and Ganapathiraju, 2012), and uncover disease pathogenesis (Kuzmanov and Emili, 2013). Drug discovery and development are also closely associated with PPIS (Sperandio, 2012; Petta et al., 2016). Therefore, identifying PPIS is of great importance in the field of molecule biology.
It is not only costly but also time-consuming and labor-intensive to identify PPIS by experimental methods such as alanine scanning mutagenesis and crystallographic complex determination (Aumentado-Armstrong et al., 2015; Krï¿ ½ger and Gohlke, 2010; Bradshaw et al., 2011). Since Jones and Thornton pioneered a computational method for predicting and analyzing PPIS in 1997 (Jones and Thornton, 1997; Jones and Thornton, 1997), more than thirty other computational methods have been developed (Zhou and Shan, 2001; Fernandez-Recio et al., 2004; Neuvirth et al., 2004; Bradford and Westhead, 2005; Chen and Zhou, 2005; Chung et al., 2006; Liang et al., 2006; Patel et al., 2006; Li et al., 2007; Ofran and Rost, 2007; Porollo and Meller, 2007; Qin and Zhou, 2007; Tjong et al., 2007; Chen and Jeong, 2009; Dosztányi et al., 2009; Du et al., 2009; Engelen et al., 2009; Šikić et al., 2009; Fiorucci and Zacharias, 2010; Murakami and Mizuguchi, 2010; Shoemaker et al., 2010; Segura et al., 2011; Xue et al., 2011; Zhang et al., 2011; Chen et al., 2012; Jordan et al., 2012; La and Kihara, 2012; Li et al., 2012; Qiu and Wang, 2012; Zellner et al., 2012; Bendell et al., 2014; de Moraes et al., 2014; Singh et al., 2014; Wang et al., 2014; Aumentado-Armstrong et al., 2015; Bagchi, 2015; Dayal et al., 2015; Maheshwari and Brylinski, 2015; Dick and Green, 2016; Jia et al., 2016; Kuo and Li, 2016; Wei et al., 2016; Hou et al., 2017; Zhao et al., 2017; Guo et al., 2018; Northey et al., 2018; Wang et al., 2019; Wang et al., 2019; Zhang and Kurgan, 2019; Zhang and Kurgan, 2019; Deng et al., 2020; Li, 2020; Zeng et al., 2020; Zhu et al., 2020; Wang et al., 2021; Wang et al., 2021). Due to their efficiency, computational methods are becoming essentially complementary to experimental methods. Most computational methods for identifying PPIS are based on machine learning algorithms where the prediction performance depends heavily on learning algorithms and feature extractions. The learning algorithms used for PPIS prediction generally include conditional random fields (Li et al., 2007), support vector machines (Bradford and Westhead, 2005), random forest (Chen and Jeong, 2009), XGBoost (Deng et al., 2020), logistic regression (Zhang and Kurgan, 2019), Bayes method (Murakami and Mizuguchi, 2010), and artificial neural networks (Singh et al., 2014). These learning algorithms are not suitable for enough large number of training samples. Recently, deep learning algorithms have been developed that have achieved significant superiority over traditional learning algorithms, especially in many difficult cases such as image classification (Krizhevsky et al., 2012; He et al., 2016) and protein structure prediction (Callaway, 2020). Features used for PPIS prediction generally include evolutionary information (Caffrey et al., 2004; Carl et al., 2008; Choi et al., 2009), secondary structure (Guharoy and Chakrabarti, 2007; Ofran and Rost, 2007; Li et al., 2012) and physicochemical, biophysical and statistical features such as accessible surface area (de Vries and Bonvin, 2008; Hou et al., 2017) and backbone flexibility (Bendell et al., 2014). According to its source, features are divided into sequence-based, structure-based, and hybrid features, which are a combination of sequence and structure features (Zeng et al., 2020). The sequence-based feature is cheaper to calculate but does not contain any information from structures that might be responsible for protein functions. The structures of most proteins are not available, while structural information generally obtained by computational prediction contain noise, which sometimes heavily effected subsequent discrimination. Information from neighboring residues of interaction sites is important to determine protein-protein interaction sites. In addition, there exists binding signals far from interaction sites. Zeng et al. (2020) demonstrated that inclusion of global features increased the performance of predicting protein-protein interaction sites. Both the local and the global features were obtained by non-linear degeneration. That is to say, during the transformation from proteins to features, information is lost. In addition, the local and the global features also contained noise. The deep learning-based encoder answers these issues above. Inspired by this, we used the DeepPPISP proposed by Zeng et al. (2020) to refine features of protein-protein interaction sites, Extreme Gradient Boosting (XGBoost) to learn a classifier for unknown PPIS prediction.
Datasets
For a fair comparison with other state-of-the-art methods, we used the same three datasets as in the literature (Zeng et al., 2020). These datasets are named respectively Dset_186, Dset_72 (Murakami and Mizuguchi, 2010), and Dset_164 (Singh et al., 2014). The procedure of collecting them is briefly described as follows. All the data originated from the PDB database (Berman et al., 2000). Dset_186, Dset_72 and Dset_164 consisted of 186, 72, and 164 non-repetitive protein sequences with the resolution less than 3.0 Å, respectively. In each dataset, sequence homology between any two sequences was less than 25%. Three datasets were integrated, containing in total 422 protein sequences. Two proteins had no definition of secondary structure of proteins (DSSP) file without which their features cannot be computed. Thus these two protein sequences were removed by Zeng et al. (2020). Finally, the remaining 420 protein sequences were used.
Protein-protein interaction binding sites are determined by the absolute solvent accessibility of amino acids. If the absolute solvent accessibility was less than 1 Å2, the amino acid was considered to be a binding site, and otherwise it was a non-interaction site. There were 5,517, 6,096, and 1,923 binding sites, as well as 30,702, 27,585, and 16,217 non-interaction sites in the Dset_186, Dset_164, and Dset_72 datasets respectively. 83.3% of the protein sequences were randomly selected as the training set and 16.7% of the protein sequences as the testing set. The training set was further divided into two parts: 90% of the training set was used for training and 10% was used for verification. Finally, 300 protein sequences were used for training (containing 65,869 amino acid residues), 50 protein sequences for verification (containing 7,319 amino acid residues), and 70 protein sequences for independent testing (containing 11,791 amino acid residues) (Zeng et al., 2020).
Methods
The proposed method called DeepPPISP-XGB consisted of three main steps: extracting features, training a classifier, and predicting PPIS (Figure 1A). The DeepPPISP was a deep learning model proposed by Zeng et al. (Zeng et al., 2020) for PPIS (Figure 1B). Here, we used it as an encoder of amino acid sequences, because the deep learning algorithms have a powerful ability to represent objects. We trained the DeepPPISP model with the training set. The input of the first fully connected layer in the trained DeepPPISP was used as a representation of the input. The XGBoost classifier was trained by the preprocessing features of the encoder. For unknown protein sequences which have secondary structure, raw protein sequence, and position-specific scoring matrix feature, the trained DeepPPISP extracted preprocessing features firstly and then the trained XGBoost classifier predicted PPIS.
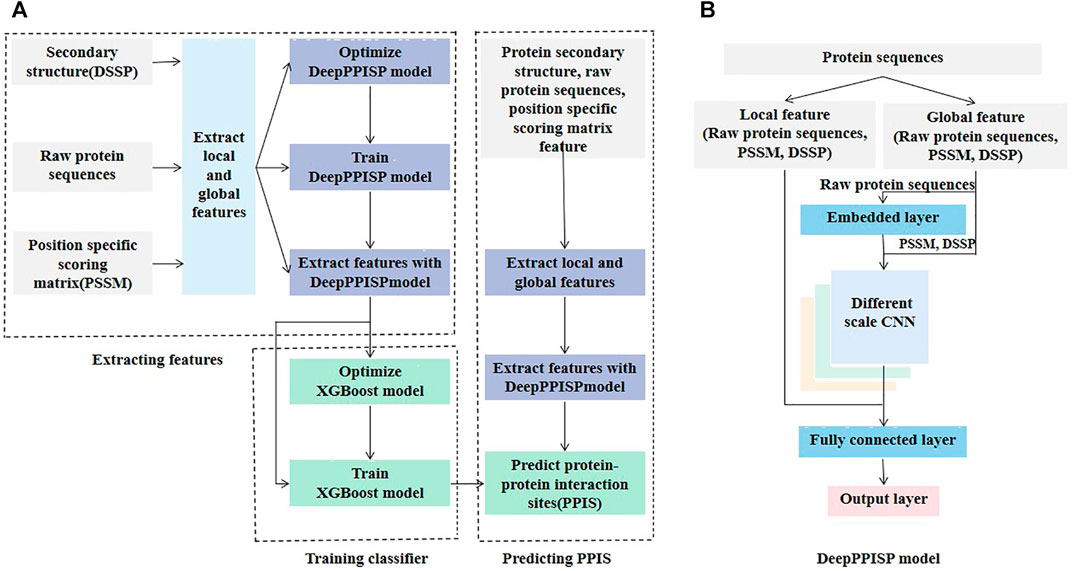
FIGURE 1. The architecture of DeepPPISP-XGB model. (A) Illustration of the DeepPPISP-XGB workflow, which consists of three modules: extracting feature, training classifier, predicting PPIS. (B) The architecture of DeepPPISP model, which contain embedding layer, different scale convolutions, fully connected layers and output layer.
DeepPPISP
As shown in Figure 1B, the DeepPPISP proposed by Zeng et al. (Zeng et al., 2020) for PPIS prediction had three types of input: position-specific scoring matrix (PSSM), secondary structure, and raw protein sequences. The PSSM is an excellent feature extractor for protein sequences and thus have widely been applied to problems in the field of computational biology, such as predicting protein post-translational modification (Huang et al., 2013; Huang et al., 2014; Dehzangi et al., 2017), membrane type (Wang et al., 2019), protein-RNA binding site (Liu et al., 2021), and structure (Guo et al., 2021). The quality of PSSM features is closely associated with the underlying multiple sequence alignments. Although there are many multiple sequence alignment algorithms including HIMMER (Eddy, 2011; Wheeler and Eddy, 2013) (Johnson et al., 2010) and Hhbilits (Remmert et al., 2012), PSI-BLAST (Altschul et al., 1997) is still a popular multiple sequence alignment and homology search algorithm. Here, PSI-BLAST was used to search NCBI’s non-redundant (NR) sequence database with three iterations and an E-value threshold of 0.001.
Many protein-protein interfaces are related to secondary structures (Taechalertpaisarn et al., 2019). Information about protein secondary structure is helpful to predict PPIS. The DSSP program (Touw et al., 2015) was used to generate nine state secondary structures: α-helix, 310- helix, π-helix, β-bridge, β-strand, β-turn, bend, loop or irregular, and no secondary structure. Therefore, each amino acid residue corresponded to a 9-dimensional vector. The primary protein sequence is valuable information and thus is essential to predict protein properties. One-hot encoding was used to encode the protein sequences. There are 20 kinds of common amino acids in the protein sequences, so each amino acid residue corresponds to a 20-dimensional 0/1 vector. The protein-protein interaction is closely associated with neighboring residues of interaction sites. The local feature of interaction sites contributes to the identification of PPIS. The sliding window method was used to collect the neighboring residues of the interaction sites. The size of the sliding window was seven. For example, if the interaction site was at position i, residues at position i-3, i-2, i-1, i, i+1, i+2, and i+3 were separated. Because each residue corresponds to a 20-dimensional PSSM feature, a 9-dimensional secondary structure feature, and a 20-dimensional one-hot feature vector, a window of seven amino acid residues was encoded into a 343-dimensional vector which was called the local feature.
Protein-protein interaction is not only linked to the local information of interacting sites, but also to global information. Zeng et al. (2020) demonstrated that the inclusion of global information improved the performance of predicting PPIS. A 500-residue peptide was used to represent the global feature of PPIS. If the number of amino acid residues in the protein sequence was less than 500, it was padded with a 0. Each peptide corresponds to a 500*49-dimensional vector called a global feature.
The local and the global features were fed into the DeepPPISP (Zeng et al., 2020). The DeepPPISP was made up of one embedding layer, three different scale convolutions, two fully connected layers, and an output layer (Figure 1B). For more detail, readers can refer to the reference (Zeng et al., 2020).
Both the local features or global features would contain a certain degree of noise. The dimension is large, especially for global features. The DeepPPISP was used to extract a more informative representation. The DeepPPISP was trained on the training data in a supervised manner. The local and global features were fed into the trained DeepPPISP, and the input to the first fully connected layer was the abstract representation of the raw features. Compared with the raw features, the abstract representation was of low dimension and had low noise.
XGBoost Algorithm
The XGBoost proposed by Chen and Guestrin, 2016 belongs to Gradient Boosting Decision Tree (GBDT) (Ke et al., 2017), and both are tree boosting algorithms. Compared with traditional tree boosting, the XGBoost used a theoretically justified weighted quantile sketch for approximate learning, a novel sparsity aware algorithm for handling sparse data, and an effective cache-aware block structure for out-of-core tree learning (Chen and Guestrin, 2016). In addition, the XGBoost performed faster as it exploited parallel and distributed computing. The XGBoost has such a significant superiority that it has widely been used in many areas including machine learning and data mining challenges.
The XGBoost is an addition model. At each iteration, the XGBoost learns a new tree that fits the residual between the predicted result of the previous trees and the true values of the training samples.
Assume that
where
where
where
where T was the number of leaf nodes and
The set of instances of the leaf node j was defined by
The objective function was further represented as
Given a fixed tree
and the optimal value of the whole tree was calculated by
It was expensive and impossible to exhaust all the possible trees for the training data. In practice, the greedy algorithm was used, which started from one node and iteratively split the node. Assume that before the node was split, the objective function of the tree was
After the node k was split into the left tree
The gain of node splitting was calculated by
The gain was used to assess the split candidates.
Evaluation Metrics
In the area of machine learning, the frequently used evaluation metrics include accuracy (ACC), Recall, Precision, F1-score (F1), and Matthews correlation coefficient (MCC) which are respectively calculated by the following formulas:
where TP and TN denote respectively the numbers of the true positive and the true negative samples, and FP and FN denote the numbers of the false positive and false negative samples. The F1-score ranges from 0 to 1. F1-score values close to 1 indicated the best prediction. The MCC represents the correlation coefficient between the actual classification and the predicted classification. The range of MCC values is −1 to 1, where 1 meant perfect prediction, and −1 indicated the worst prediction. The area under the receiver operating characteristic curve (AUROC) and area under the precision-recall curve (AUPRC) were also used to evaluate the performances.
Experiments
Visualization of Preprocessing Features
To investigate the ability of the features to discriminate protein-protein interaction sites from non-interaction sites, we used the Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2020) to depict the first two principal components. The UMAP is a powerful tool for dimension reduction and visualization. As shown in Figure 2, the features processed by the DeepPPISP demonstrated a tighter cluster than the raw features, indicating that features generated by the DeepPPISP were more discriminative. To further evaluate the performance of the preprocessed features, we performed 5-fold cross-validation and independent tests. Figure 3A showed the ROC curves of the 5-fold cross-validation over both the preprocessing features and raw features, while Figure 3B depicted the ROC curves of the independent tests. The performance of preprocessed features is equivalent to or better than those of raw features. It must be pointed out that the user-defined parameters were identical in the XGBoost classifiers. Comparison with other methods
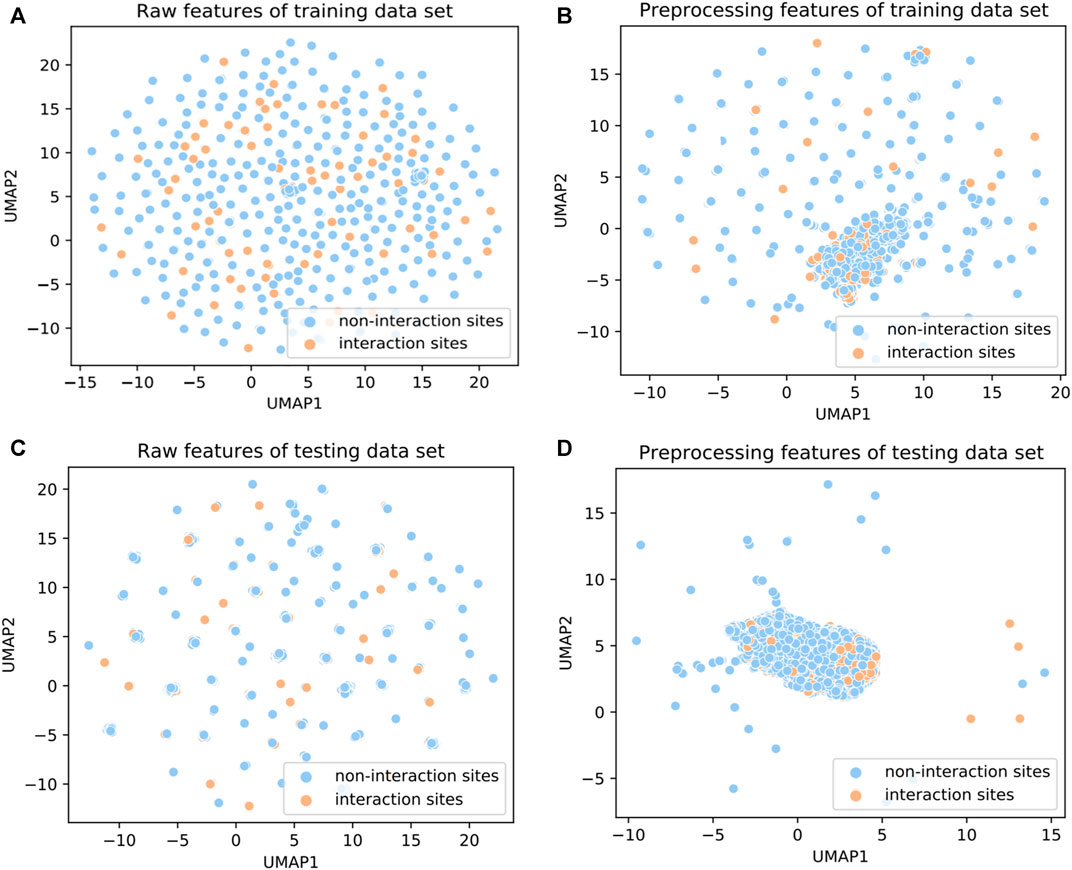
FIGURE 2. UMAP diagrams of (A) raw features of the training set, (B) preprocessing features of the training set, (C) raw features of the testing set, and (D) preprocessing feature of the testing set.
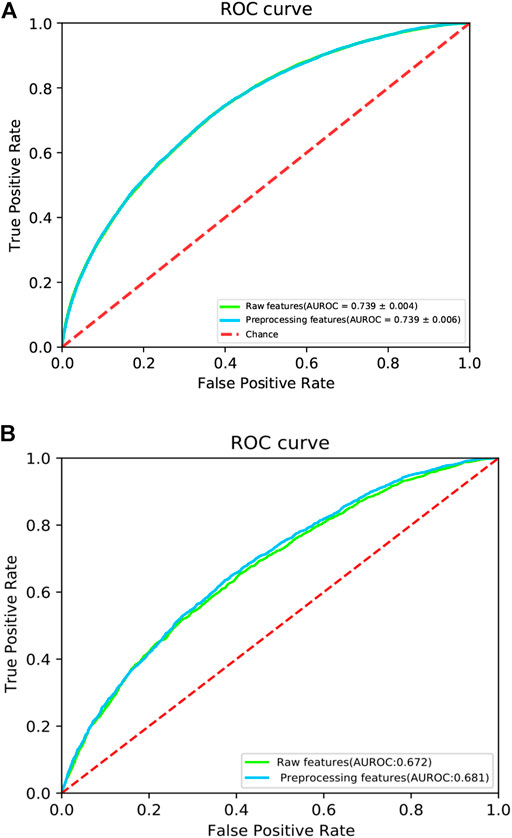
FIGURE 3. The ROC curves of (A) 5-fold cross validation and (B) independent test. The red dotted line is a control line on which AUROC = 0.5.
Due to its versatile roles in the cellular process, the identification of protein-protein interaction sites is increasingly becoming a hot topic and is also a challenging task. Over the past decades, more than 10 methods have been proposed to predict protein-protein interaction sites (Patel et al., 2006; Du et al., 2009; Murakami and Mizuguchi, 2010; Wang et al., 2014; Zhang and Kurgan, 2019; Northey et al., 2018; Zeng et al., 2020; Chen et al., 2012; Šikić et al., 2009; Fiorucci and Zacharias, 2010; Dosztányi et al., 2009; La and Kihara, 2012; Bradford and Westhead, 2005; Chen and Jeong, 2009; Chung et al., 2006; Fernandez-Recio et al., 2004; Shoemaker et al., 2010; Ofran and Rost, 2007; Qin and Zhou, 2007; Liang et al., 2006; Li et al., 2007; Zhou and Shan, 2001; Neuvirth et al., 2004; Porollo and Meller, 2007; Segura et al., 2011; Qiu and Wang, 2012; Wei et al., 2016; Zhu et al., 2020; Guo et al., 2018; Kuo and Li, 2016; Wang et al., 2021; Maheshwari and Brylinski, 2015; Li, 2020; Dick and Green, 2016; Wang et al., 2019; Zhao et al., 2017; Jia et al., 2016; Deng et al., 2020; Singh et al., 2014; Hou et al., 2017; Li et al., 2012; Wang et al., 2019; Bagchi, 2015 #412; Zhang and Kurgan, 2019). We compared the proposed method with six other state-of-the-art methods. These six competing methods were DeepPPISP (Zeng et al., 2020), SCRIBER (Zhang et al., 2019), IntPred (Northey et al., 2018), RF_PPI (Hou et al., 2017), SPRINGS (Singh et al., 2014), PSIVER (Murakami and Mizuguchi, 2010), ISIS (Ofran and Rost, 2007), and SPPIDER (Porollo and Meller, 2007). PSIVER was a Naïve Bayes-based classifier that used features from PSSM and accessibility, while SPPIDER combined fingerprints with information from the sequences and structures for PPIS predictio. Both SPRINGS and ISIS were neural network-based methods. The former used evolutionary information, averaged cumulative hydropathy, and predicted relative solvent accessibility, while the latter used structural features and evolutionary information. RF_PPI was a random forest-based classifier for PPIS prediction, while the DeepPPISP was a deep learning-based classifier. The performances of these seven methods over the independent test were listed in Table 1.
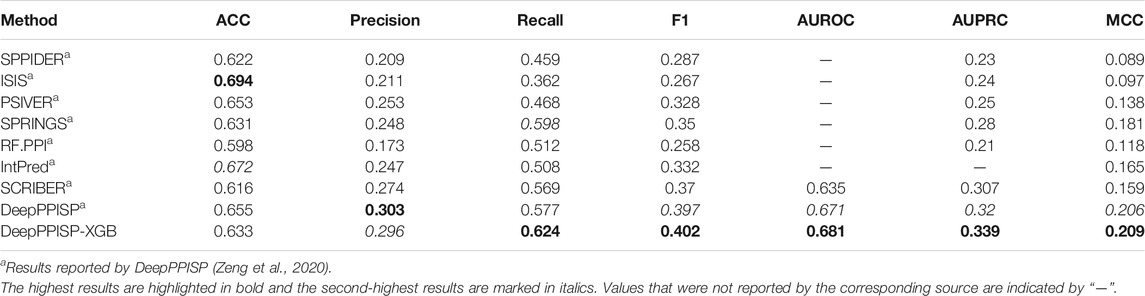
TABLE 1. Comparison with other state-of-the-art methods.
The DeepPPISP-XGB method achieved the highest value in terms of Recall, F1-score, AUROC, AUPRC, and MCC, and it reached the second-highest performance in terms of Precision. Although ISIS got the best ACC, its performance in other respects was lower than those of DeepPPISP-XGB. The DeepPPISP-XGB method improved the Recall by 4.7%, 5.5%, 11.6%, 11.2%, 2.6%, 15.6%, 26.2%, and 16.5%, in comparison with DeepPPISP, SCRIBER, IntPred, RF.PPI, SPRINGS, PSIVER, ISIS, and SPPIDER, respectively. The DeepPPISP-XGB method increased F1-score and MCC by 0.5% and 0.3%, and the AUROC by 1%, in comparison with DeepPPISP.
K-fold cross-validation is a common method in regression or classification questions. In the k-fold cross-validation, the training set was split into k parts. One part was tested and other k−1 parts were trained. The procedure was performed k times. We carried out 10-fold cross-validations, and the principle was shown (Supplementary Figure S1). Figure 4 showed ROC curves for the 10-fold cross-validations. The mean and the standard deviation of the AUROCs were 0.741 and 0.006, respectively. Supplementary Table S1 lists the ACC, Precision, Recall, F1-score, AUROC, AUPRC, and MCC for each cross-validation.
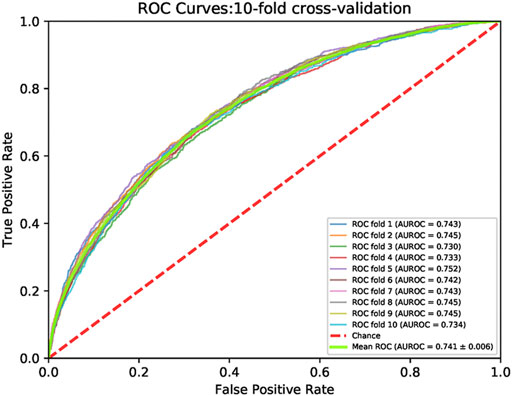
FIGURE 4. The ROC curves of 10-fold cross validation on the train set. The minimum AUROC value cross validation is 0.730 at the first fold. The maximum value of the cross validation is 0.752 at the ten-th fold. The green line represents the ROC curve of the cross validation mean. The mean value of AUROC is 0.741. The red dotted line is a control line on which AUROC = 0.5.
To further evaluate the predictive performance of the DeepPPISP-XGB method, four machine learning algorithms were used for PPIS prediction. Decision tree (Safavian and Landgrebe, 1991) is a widely utilized classification algorithm, which is made up of the root node, internal nodes, and leaf node. Random forest (RF) (Breiman, 2001) is an ensemble learning algorithm. It consists of many weak classifiers which determine the sample category. Extremely randomized tree (ERT) (Geurts et al., 2006) is similar to RF but the decision tree of ERT is randomly divided. Support vector machine (SVM) is a statistical algorithm proposed by Boser et al. (Boser et al., 1992). These classifiers were implemented in the Scikit-Learn package (v0.24.2) which has been widely utilized in computational biology. The ROC curves and the precision-recall curves are shown in Figure 5. The XGBoost classifier obtained an AUROC value of 0.681 and an AUPRC value of 0.339 on the independent test, significantly better than four classifiers.
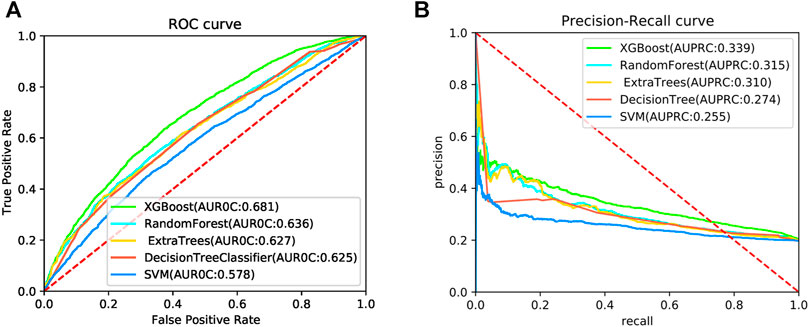
FIGURE 5. The ROC curves (A) and precision-recall curves (B) for 5 algorithms on the independent test.
The Effects of the Global Features
After removing global features, we trained DeepPPISP-XGB. The user-defined parameters of the DeepPPISP-XGB were the same as the previous. Table 2 shows the performance of predicting PPIS by using local features alone. The ROC and the precision-recall curves were displayed in Figure 6. The experimental results showed that the inclusion of the global features was beneficial to improve PPIS prediction, which was in agreement with the findings of Zeng et al. (2020).
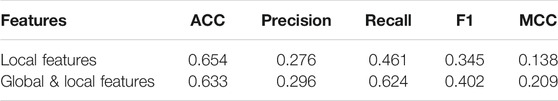
TABLE 2. Predictive performance when using local features and using combined local and global features with the DeepPPISP-XGB model.
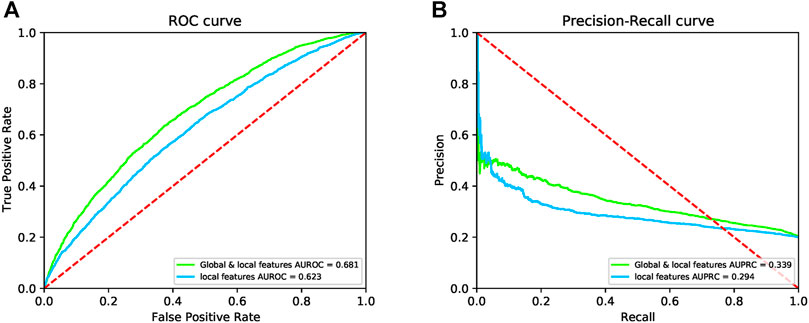
FIGURE 6. The ROC curves (A) and the precision-recall curves (B) for both local and global & local features on the independent test.
Conclusion
We presented a PPIS prediction algorithm based on the DeepPPISP and the XGBoost. The DeepPPISP served as a feature extractor to remove redundant information of the protein sequences. The XGBoost was used to construct a classifier for predicting PPIS. The DeepPPISP-XGB achieved competitive performances with other state-of-the-art methods.
Source Code
Source code is available at: https://github.com/fatancy2580/DeepPPISPXGB-master.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
GH and Z-GY conceived a concept and methodology. PW collected data, conducted the experiments, analyzed the results, and wrote the manuscript. GZ analyzed results. GH revised the manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (11871061), by the Natural Science Foundation of Hunan Province (2020JJ4034), by the Scientific Research Fund of Hunan Provincial Education Department (19A215), by the open project of Hunan Key Laboratory for Computation and Simulation in Science and Engineering (2019LCESE03), and by Shaoyang University Innovation Foundation for Postgraduate (CX2021SY041, CX2021SY001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.752732/full#supplementary-material
References
Altschul, S., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi:10.1093/nar/25.17.3389
Aumentado-Armstrong, T. T., Istrate, B., and Murgita, R. A. (2015). Algorithmic approaches to protein-protein interaction site prediction. Algorithms Mol. Biol. 10, 1–21. doi:10.1186/s13015-015-0033-9
Bagchi, A. (2015). Use of Machine Learning Features to Detect Protein-Protein Interaction Sites at the Molecular Level. Inf. Syst. Des. Intell. Appl., 49–54. Springer. doi:10.1007/978-81-322-2247-7_6
Bendell, C. J., Liu, S., Aumentado-Armstrong, T., Istrate, B., Cernek, P. T., Khan, S., et al. (2014). Transient protein-protein interface prediction: datasets, features, algorithms, and the RAD-T predictor. BMC bioinformatics 15, 1–12. doi:10.1186/1471-2105-15-82
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Boser, B. E., Guyon, I. M., and Vapnik, V. N. (1992). A training algorithm for optimal margin classifiers. Proc. fifth Annu. Workshop Comput. Learn. Theor., 144–152. doi:10.1145/130385.130401
Bradford, J. R., and Westhead, D. R. (2005). Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 21, 1487–1494. doi:10.1093/bioinformatics/bti242
Bradshaw, R. T., Patel, B. H., Tate, E. W., Leatherbarrow, R. J., and Gould, I. R. (2011). Comparing experimental and computational alanine scanning techniques for probing a prototypical protein-protein interaction. Protein Eng. Des. Selection 24, 197–207. doi:10.1093/protein/gzq047
Caffrey, D. R., Somaroo, S., Hughes, J. D., Mintseris, J., and Huang, E. S. (2004). Are protein-protein interfaces more conserved in sequence than the rest of the protein surface. Protein Sci. 13, 190–202. doi:10.1110/ps.03323604
Callaway, E. (2020). 'It will change everything': DeepMind's AI makes gigantic leap in solving protein structures. Nature 588, 203–204. doi:10.1038/d41586-020-03348-4
Carl, N., Konc, J., and Janežič, D. (2008). Protein surface conservation in binding sites. J. Chem. Inf. Model. 48, 1279–1286. doi:10.1021/ci8000315
Chen, C.-T., Peng, H.-P., Jian, J.-W., Tsai, K.-C., Chang, J.-Y., Yang, E.-W., et al. (2012). Protein-protein interaction site predictions with three-dimensional probability distributions of interacting atoms on protein surfaces. PloS one 7, e37706. doi:10.1371/journal.pone.0037706
Chen, H., and Zhou, H.-X. (2005). Prediction of interface residues in protein-protein complexes by a consensus neural network method: Test against NMR data. Proteins 61, 21–35. doi:10.1002/prot.20514
Chen, T., and Guestrin, C. (2016). “Xgboost: A scalable tree boosting system,” in Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. New York, NY: ACM, 785–794.
Chen, X.-w., and Jeong, J. C. (2009). Sequence-based prediction of protein interaction sites with an integrative method. Bioinformatics 25, 585–591. doi:10.1093/bioinformatics/btp039
Choi, Y. S., Yang, J.-S., Choi, Y., Ryu, S. H., and Kim, S. (2009). Evolutionary conservation in multiple faces of protein interaction. Proteins 77, 14–25. doi:10.1002/prot.22410
Chung, J.-L., Wang, W., and Bourne, P. E. (2006). Exploiting sequence and structure homologs to identify protein-protein binding sites. Proteins 62, 630–640. doi:10.1002/prot.20741
Cohen, F. E., and Prusiner, S. B. (1998). Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67, 793–819. doi:10.1146/annurev.biochem.67.1.793
Das, S., and Chakrabarti, S. (2021). Classification and prediction of protein-protein interaction interface using machine learning algorithm. Sci. Rep. 11, 1–12. doi:10.1038/s41598-020-80900-2
Dayal, P. V., Singh, H., Busenlehner, L. S., and Ellis, H. R. (2015). Exposing the Alkanesulfonate Monooxygenase Protein-Protein Interaction Sites. Biochemistry 54, 7531–7538. doi:10.1021/acs.biochem.5b00935
de Moraes, F. R., Neshich, I. A. P., Mazoni, I., Yano, I. H., Pereira, J. G. C., Salim, J. A., et al. (2014). Improving predictions of protein-protein interfaces by combining amino acid-specific classifiers based on structural and physicochemical descriptors with their weighted neighbor averages. Plos one 9, e87107. doi:10.1371/journal.pone.0087107
de Vries, S., and Bonvin, A. (2008). How proteins get in touch: interface prediction in the study of biomolecular complexes. Cpps 9, 394–406. doi:10.2174/138920308785132712
Dehzangi, A., López, Y., Lal, S. P., Taherzadeh, G., Michaelson, J., Sattar, A., et al. (2017). PSSM-suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 425, 97–102. doi:10.1016/j.jtbi.2017.05.005
Deng, A., Zhang, H., Wang, W., Zhang, J., Fan, D., Chen, P., et al. (2020). Developing computational model to predict protein-protein interaction sites based on the XGBoost algorithm. Ijms 21, 2274. doi:10.3390/ijms21072274
Deng, L., Guan, J., Dong, Q., and Zhou, S. (2009). Prediction of protein-protein interaction sites using an ensemble method. BMC bioinformatics 10, 1–15. doi:10.1186/1471-2105-10-426
Dias, R., and Kolaczkowski, B. (2017). Improving the accuracy of high-throughput protein-protein affinity prediction may require better training data. BMC bioinformatics 18, 7–18. doi:10.1186/s12859-017-1533-z
Dick, K., and Green, J. (2016). Comparison of sequence-and structure-based protein-protein interaction sites. IEEE EMBS Int. Student Conf. (Isc), 1–4. IEEE. doi:10.1109/embsisc.2016.7508605
Dosztányi, Z., Mészáros, B., and Simon, I. (2009). ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 25, 2745–2746. doi:10.1093/bioinformatics/btp518
Du, X., Cheng, J., and Song, J. (2009). Improved prediction of protein binding sites from sequences using genetic algorithm. Protein J. 28, 273–280. doi:10.1007/s10930-009-9192-1
Eddy, S. R. (2011). Accelerated profile HMM searches. Plos Comput. Biol. 7, e1002195. doi:10.1371/journal.pcbi.1002195
Engelen, S., Trojan, L. A., Sacquin-Mora, S., Lavery, R., and Carbone, A. (2009). Joint evolutionary trees: a large-scale method to predict protein interfaces based on sequence sampling. Plos Comput. Biol. 5, e1000267. doi:10.1371/journal.pcbi.1000267
Fernández-Recio, J., Totrov, M., and Abagyan, R. (2004). Identification of Protein-Protein Interaction Sites from Docking Energy Landscapes. J. Mol. Biol. 335, 843–865. doi:10.1016/j.jmb.2003.10.069
Fiorucci, S., and Zacharias, M. (2010). Prediction of protein-protein interaction sites using electrostatic desolvation profiles. Biophysical J. 98, 1921–1930. doi:10.1016/j.bpj.2009.12.4332
Geurts, P., Ernst, D., and Wehenkel, L. (2006). Extremely randomized trees. Mach. Learn. 63, 3–42. doi:10.1007/s10994-006-6226-1
Guharoy, M., and Chakrabarti, P. (2007). Secondary structure based analysis and classification of biological interfaces: identification of binding motifs in protein-protein interactions. Bioinformatics 23, 1909–1918. doi:10.1093/bioinformatics/btm274
Guo, H., Liu, B., Cai, D., and Lu, T. (2018). Predicting protein-protein interaction sites using modified support vector machine. Int. J. Mach. Learn. Cyber. 9, 393–398. doi:10.1007/s13042-015-0450-6
Guo, Y., Wu, J., Ma, H., Wang, S., and Huang, J. (2021). EPTool: A New Enhancing PSSM Tool for Protein Secondary Structure Prediction. J. Comput. Biol. 28, 362–364. doi:10.1089/cmb.2020.0417
He, K., Zhang, X., Ren, S., and Sun, J. (2016). “Deep residual learning for image recognition,” in Proceedings of the IEEE conference on computer vision and pattern recognition, 770–778. doi:10.1109/cvpr.2016.90
Hou, Q., De Geest, P., Vranken, W. F., Heringa, J., and Feenstra, K. A. (2017). Seeing the Trees through the Forest: Sequence-based Homo- and Heteromeric Protein-protein Interaction sites prediction using Random Forest. Bioinformatics 33, btx005–1487. doi:10.1093/bioinformatics/btx005
Huang, G., Lu, L., Feng, K., Zhao, J., Zhang, Y., Xu, Y., et al. (2014). Prediction of S-nitrosylation modification sites based on kernel sparse representation classification and mRMR algorithm. Biomed. Research International 2014, 1–10. doi:10.1155/2014/438341
Huang, G., Zhou, Y., Zhang, Y., Li, B.-Q., Zhang, N., and Cai, Y.-D. (2013). Prediction of carbamylated lysine sites based on the one-class k-nearest neighbor method. Mol. Biosyst. 9, 2729–2740. doi:10.1039/c3mb70195f
Jia, J., Liu, Z., Xiao, X., Liu, B., and Chou, K.-C. (2016). iPPBS-Opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 21, 95. doi:10.3390/molecules21010095
Johnson, L. S., Eddy, S. R., and Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC bioinformatics 11, 1–8. doi:10.1186/1471-2105-11-431
Jones, S., and Thornton, J. M. (1997). Analysis of protein-protein interaction sites using surface patches 1 1Edited by G.Von Heijne. J. Mol. Biol. 272, 121–132. doi:10.1006/jmbi.1997.1234
Jones, S., and Thornton, J. M. (1997). Prediction of protein-protein interaction sites using patch analysis 1 1Edited by G. von Heijne. J. Mol. Biol. 272, 133–143. doi:10.1006/jmbi.1997.1233
Jordan, R. A., el-Manzalawy, Y., Dobbs, D., and Honavar, V. (2012). Predicting protein-protein interface residues using local surface structural similarity. BMC bioinformatics 13, 1–14. doi:10.1186/1471-2105-13-41
Ke, G., Meng, Q., Finley, T., Wang, T., Chen, W., Ma, W., et al. (2017). Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 30, 3146–3154.
Kerrien, S., Alam-Faruque, Y., Aranda, B., Bancarz, I., Bridge, A., Derow, C., et al. (2007). IntAct--open source resource for molecular interaction data. Nucleic Acids Res. 35, D561–D565. doi:10.1093/nar/gkl958
Keshava Prasad, T. S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., Mathivanan, S., et al. (2009). Human Protein Reference Database--2009 update. Nucleic Acids Res. 37, D767–D772. doi:10.1093/nar/gkn892
Krizhevsky, A., Sutskever, I., and Hinton, G. E. (2017). Imagenet classification with deep convolutional neural networks. Commun. ACM 60, 84–90. doi:10.1145/3065386
Kr�ger, D. M., and Gohlke, H. (2010). DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 38, W480–W486. doi:10.1093/nar/gkq471
Kuo, T.-H., and Li, K.-B. (2016). Predicting Protein-Protein Interaction Sites Using Sequence Descriptors and Site Propensity of Neighboring Amino Acids. Ijms 17, 1788. doi:10.3390/ijms17111788
Kuzmanov, U., and Emili, A. (2013). Protein-protein interaction networks: probing disease mechanisms using model systems. Genome Med. 5, 37–12. doi:10.1186/gm441
La, D., and Kihara, D. (2012). A novel method for protein-protein interaction site prediction using phylogenetic substitution models. Proteins 80, 126–141. doi:10.1002/prot.23169
Li, B.-Q., Feng, K.-Y., Chen, L., Huang, T., and Cai, Y.-D. (2012). Prediction of Protein-Protein Interaction Sites by Random Forest Algorithm with mRMR and IFS. PLoS ONE 7, e43927. doi:10.1371/journal.pone.0043927
Li, M.-H., Lin, L., Wang, X.-L., and Liu, T. (2007). Protein protein interaction site prediction based on conditional random fields. Bioinformatics 23, 597–604. doi:10.1093/bioinformatics/btl660
Li, M., Gao, H., Wang, J., and Wu, F.-X. (2019). Control principles for complex biological networks. Brief. Bioinformatics 20, 2253–2266. doi:10.1093/bib/bby088
Li, Y. (2020). Computational Methods for Predicting Protein-protein Interactions and Binding Sites. London: Western University.
Liang, S., Zhang, C., Liu, S., and Zhou, Y. (2006). Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 34, 3698–3707. doi:10.1093/nar/gkl454
Liu, Y., Gong, W., Yang, Z., and Li, C. (2021). SNB‐PSSM : A spatial neighbor‐based PSSM used for protein-RNA binding site prediction. J. Mol. Recognit 34, e2887. doi:10.1002/jmr.2887
Loregian, A., Marsden, H. S., and Palù, G. (2002). Protein-protein interactions as targets for antiviral chemotherapy. Rev. Med. Virol. 12, 239–262. doi:10.1002/rmv.356
Maheshwari, S., and Brylinski, M. (2015). Prediction of protein-protein interaction sites from weakly homologous template structures using meta-threading and machine learning. J. Mol. Recognit. 28, 35–48. doi:10.1002/jmr.2410
McInnes, L., Healy, J., and Melville, J. (2020). UMAP: uniform manifold approximation and projection for dimension reduction. ArXiv [Preprint]. arXiv:1802.03426.
Murakami, Y., and Mizuguchi, K. (2010). Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics 26, 1841–1848. doi:10.1093/bioinformatics/btq302
Neuvirth, H., Raz, R., and Schreiber, G. (2004). ProMate: A Structure Based Prediction Program to Identify the Location of Protein-Protein Binding Sites. J. Mol. Biol. 338, 181–199. doi:10.1016/j.jmb.2004.02.040
Northey, T. C., Barešić, A., and Martin, A. C. R. (2018). IntPred: a structure-based predictor of protein-protein interaction sites. Bioinformatics 34, 223–229. doi:10.1093/bioinformatics/btx585
Ofran, Y., and Rost, B. (2007). ISIS: interaction sites identified from sequence. Bioinformatics 23, e13–e16. doi:10.1093/bioinformatics/btl303
Orii, N., and Ganapathiraju, M. K. (2012). Wiki-pi: a web-server of annotated human protein-protein interactions to aid in discovery of protein function. PloS one 7, e49029. doi:10.1371/journal.pone.0049029
Patel, T., Pillay, M., Jawa, R., and Liao, L. (2006) Information of binding sites improves prediction of protein-protein interaction. In 2006 5th International Conference on Machine Learning and Applications (ICMLA06) pp. 205–212. IEEE. doi:10.1109/icmla.2006.29
Petta, I., Lievens, S., Libert, C., Tavernier, J., and De Bosscher, K. (2016). Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 24, 707–718. doi:10.1038/mt.2015.214
Porollo, A., and Meller, J. (2007). Prediction-based fingerprints of protein-protein interactions. Proteins 66, 630–645. doi:10.1002/prot.21248
Qin, S., and Zhou, H.-X. (2007). meta-PPISP: a meta web server for protein-protein interaction site prediction. Bioinformatics 23, 3386–3387. doi:10.1093/bioinformatics/btm434
Qiu, Z., and Wang, X. (2012). Prediction of protein-protein interaction sites using patch-based residue characterization. J. Theor. Biol. 293, 143–150. doi:10.1016/j.jtbi.2011.10.021W
Remmert, M., Biegert, A., Hauser, A., and Söding, J. (2012). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175. doi:10.1038/nmeth.1818
Safavian, S. R., and Landgrebe, D. (1991). A survey of decision tree classifier methodology. IEEE Trans. Syst. Man. Cybern. 21, 660–674. doi:10.1109/21.97458
Salwinski, L., Miller, C. S., Smith, A. J., Pettit, F. K., Bowie, J. U., and Eisenberg, D. (2004). The database of interacting proteins: 2004 update. Nucleic Acids Res. 32, 449D–451D. doi:10.1093/nar/gkh086
Segura, J., Jones, P. F., and Fernandez-Fuentes, N. (2011). Improving the prediction of protein binding sites by combining heterogeneous data and Voronoi diagrams. BMC bioinformatics 12, 1–9. doi:10.1186/1471-2105-12-352
Selkoe, D. (1998). The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biology 8, 447–453. doi:10.1016/s0962-8924(98)01363-4
Shoemaker, B. A., Zhang, D., Thangudu, R. R., Tyagi, M., Fong, J. H., Marchler-Bauer, A., et al. (2010). Inferred Biomolecular Interaction Server-a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 38, D518–D524. doi:10.1093/nar/gkp842
Šikić, M., Tomić, S., and Vlahoviček, K. (2009). Prediction of Protein-Protein Interaction Sites in Sequences and 3D Structures by Random Forests. Plos Comput. Biol. 5, e1000278. doi:10.1371/journal.pcbi.1000278
Singh, G., Dhole, K., Pai, P. P., and Mondal, S. (2014). SPRINGS: prediction of protein-protein interaction sites using artificial neural networks. PeerJ PrePrints. doi:10.13188/2572-8679.1000001
Sperandio, O. (2012). Editorial: [Hot Topics: Toward the Design of Drugs on Protein-Protein Interactions]. Cpd 18, 4585. doi:10.2174/138161212802651661
Taechalertpaisarn, J., Lyu, R.-L., Arancillo, M., Lin, C.-M., Perez, L. M., Ioerger, T. R., et al. (2019). Correlations between secondary structure- and protein-protein interface-mimicry: the interface mimicry hypothesis. Org. Biomol. Chem. 17, 3267–3274. doi:10.1039/c9ob00204a
Tjong, H., Qin, S., and Zhou, H.-X. (2007). PI2PE: protein interface/interior prediction engine. Nucleic Acids Res. 35, W357–W362. doi:10.1093/nar/gkm231
Touw, W. G., Baakman, C., Black, J., te Beek, T. A. H., Krieger, E., Joosten, R. P., et al. (2015). A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 43, D364–D368. doi:10.1093/nar/gku1028
Von Mering, C., Jensen, L. J., Snel, B., Hooper, S. D., Krupp, M., Foglierini, M., et al. (2004). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437. doi:10.1093/nar/gki005
Wang, B., Mei, C., Wang, Y., Zhou, Y., Cheng, M.-T., Zheng, C.-H., et al. (2021). Imbalance data processing strategy for protein interaction sites prediction. Ieee/acm Trans. Comput. Biol. Bioinf. 18, 985–994. doi:10.1109/TCBB.2019.2953908
Wang, D. D., Wang, R., and Yan, H. (2014). Fast prediction of protein-protein interaction sites based on Extreme Learning Machines. Neurocomputing 128, 258–266. doi:10.1016/j.neucom.2012.12.062
Wang, S., Li, M., Guo, L., Cao, Z., and Fei, Y. (2019). Efficient utilization on PSSM combining with recurrent neural network for membrane protein types prediction. Comput. Biol. Chem. 81, 9–15. doi:10.1016/j.compbiolchem.2019.107094
Wang, X., Yu, B., Ma, A., Chen, C., Liu, B., and Ma, Q. (2019). Protein-protein interaction sites prediction by ensemble random forests with synthetic minority oversampling technique. Bioinformatics 35, 2395–2402. doi:10.1093/bioinformatics/bty995
Wang, X., Zhang, Y., Yu, B., Salhi, A., Chen, R., Wang, L., et al. (2021). Prediction of protein-protein interaction sites through eXtreme gradient boosting with kernel principal component analysis. Comput. Biol. Med. 134, 104516. doi:10.1016/j.compbiomed.2021.104516
Wang, Y., Mei, C., Zhou, Y., Wang, Y., Zheng, C., Zhen, X., et al. (2019). Semi-supervised prediction of protein interaction sites from unlabeled sample information. BMC bioinformatics 20, 1–10. doi:10.1186/s12859-019-3274-7
Wang, Y., Xu, Y., Yang, Z., Liu, X., and Dai, Q. (2021). Using Recursive Feature Selection with Random Forest to Improve Protein Structural Class Prediction for Low-Similarity Sequences. Comput. Math. Methods Med., 2021. doi:10.1155/2021/5529389
Wei, Z.-S., Han, K., Yang, J.-Y., Shen, H.-B., and Yu, D.-J. (2016). Protein-protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 193, 201–212. doi:10.1016/j.neucom.2016.02.022
Wheeler, T. J., and Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi:10.1093/bioinformatics/btt403
Xue, L. C., Dobbs, D., and Honavar, V. (2011). HomPPI: a class of sequence homology based protein-protein interface prediction methods. BMC bioinformatics 12, 1–24. doi:10.1186/1471-2105-12-244
Zellner, H., Staudigel, M., Trenner, T., Bittkowski, M., Wolowski, V., Icking, C., et al. (2012). Prescont: Predicting protein-protein interfaces utilizing four residue properties. Proteins 80, 154–168. doi:10.1002/prot.23172
Zeng, M., Zhang, F., Wu, F.-X., Li, Y., Wang, J., and Li, M. (2020). Protein-protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 36, 1114–1120. doi:10.1093/bioinformatics/btz699
Zhang, B., Li, J., Quan, L., Chen, Y., and Lü, Q. (2019). Sequence-based prediction of protein-protein interaction sites by simplified long short-term memory network. Neurocomputing 357, 86–100. doi:10.1016/j.neucom.2019.05.013
Zhang, J., and Kurgan, L. (2019). SCRIBER: accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics 35, i343–i353. doi:10.1093/bioinformatics/btz324
Zhang, Q. C., Deng, L., Fisher, M., Guan, J., Honig, B., and Petrey, D. (2011). PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 39, W283–W287. doi:10.1093/nar/gkr311
Zhao, X., Bao, L., Zhao, X., and Yin, M. (2017). PPIs Meta: A Meta-predictor of Protein-Protein Interaction Sites with Weighted Voting Strategy. Cp 14, 186–193. doi:10.2174/1570164614666170306164127
Zhou, H.-X., and Shan, Y. (2001). Prediction of protein interaction sites from sequence profile and residue neighbor list. Proteins 44, 336–343. doi:10.1002/prot.1099
Keywords: protein-protein interaction, deep learning, machine learning, extreme gradient boosting, protein functions
Citation: Wang P, Zhang G, Yu Z-G and Huang G (2021) A Deep Learning and XGBoost-Based Method for Predicting Protein-Protein Interaction Sites. Front. Genet. 12:752732. doi: 10.3389/fgene.2021.752732
Received: 03 August 2021; Accepted: 20 September 2021;
Published: 26 October 2021.
Edited by:
Lei Wang, Changsha University, ChinaCopyright © 2021 Wang, Zhang, Yu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guohua Huang, Z3VvaHVhaGhuQDE2My5jb20=