 Qianqian Li1*
Qianqian Li1* Xiaofan Zhu1
Xiaofan Zhu1 Chenguang Yu2
Chenguang Yu2 Lin Shang3
Lin Shang3 Ranran Li4
Ranran Li4 Xia Wang5Yaping Yang5
Xia Wang5Yaping Yang5 Jingjing Meng1
Jingjing Meng1 Xiangdong Kong1*
Xiangdong Kong1*- 1Genetics and Prenatal Diagnosis Center, Department of Obstetrics and Gynecology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology and Center for Human Genome Research, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Foot and Ankle Surgery, Zhengzhou Orthopedic Hospital, Zhengzhou, China
- 4School of Life Science and Technology, Xinxiang Medical University, Xinxiang, China
- 5AiLife Diagnostics, Inc., Houston, TX, United States
External ophthalmoplegia with rib and vertebral anomalies (EORVA) is characterized by congenital nonprogressive external ophthalmoplegia, ptosis, scoliosis, torticollis, vertebral, and rib anomalies, caused by homozygous mutations in the myogenic factor 5 gene (MYF5) located on chromosome 12q21.31. Uniparental disomy (UPD) is a rare inheritance of a pair of chromosomes originating from only one parent. This study describes a case of an 8-year-old boy with ptosis, scoliosis, and dysmorphic hypoplastic ribs with fusion anomalies. Trio-based exome sequencing (trio-ES) identified a novel homozygous mutation c.191delC (p.Ala64Valfs*33) in MYF5 in the proband, with the father being heterozygous and the mother wild-type, as verified by Sanger sequencing. UPD identified from trio-ES variant call format data suggested the possibility of paternal UPD of chromosome 12 (UPD12pat) in the proband, further confirmed to be a complete isodisomy type of UPD by genome-wide single nucleotide polymorphism array. MYF5 was significantly downregulated by 69.14% (**p < 0.01) in HeLa cells transfected with mutant MYF5 containing c.191delC compared to those transfected with the wild-type MYF5, resulting in a truncated protein with a size of ∼20 kDa. In conclusion, this study identified a novel homozygous mutation in MYF5, broadening the genetic spectrum of EORVA and further deepening the understanding of this rare disease.
Introduction
External ophthalmoplegia with rib and vertebral anomalies (EORVA; MIM# 618155), an extremely rare autosomal recessive disorder, is characterized by congenital nonprogressive external ophthalmoplegia and ptosis, with torticollis and scoliosis developing during childhood. In addition, patients may present with hypoplastic or missing ribs with fusion anomalies (Di Gioia et al., 2018). EORVA is caused by homozygous mutations in the myogenic factor 5 gene (MYF5; MIM* 159990) located on chromosomal 12q21.31. MYF5, a transcriptional activator encoded by MYF5, is a member of the Myc-like basic helix-loop-helix transcription factor family that plays an important role in promoting the transcription of muscle-specific target genes and hence, muscle differentiation (Wang et al., 1996). To date, only two homozygous variations in MYF5 have been reported (Di Gioia et al., 2018).
Uniparental disomy (UPD), first introduced by Engel (1980), is a non-traditional Mendelian inheritance pattern, in which a pair of chromosomes are inherited from only one parent. It mainly consists of two subtypes: heterodisomy (hUPD), where a pair of non-identical chromosomes are contributed by one parent due to an error during meiosis I, and isodisomy (iUPD), where a chromosome from one parent is duplicated due to an error during meiosis II (Benn, 2021). Recently, Nakka et al. identified 675 instances of UPD across 4,400,363 consented research participants from the personal genetics company 23andMe, Inc., and 431,094 UK Biobank participants, and estimated that the prevalence of UPD on all chromosomes was 1:2000 (0.1%) (Nakka et al., 2019). The clinical consequences of UPD depend on the chromosomes involved, including imprinting diseases, recessive Mendelian diseases, and mosaic aneuploidy associated with diseases (Scuffins et al., 2021).
In this study, an 8-year-old boy with ptosis, scoliosis, and dysmorphic hypoplastic ribs with fusion anomalies was identified with a novel homozygous variation in MYF5 due to paternal iUPD of chromosome 12 (iUPD12pat).
Case Presentation
The proband was an 8-year-old boy with ptosis since birth (Figure 1A). At the age of 1 year, the proband was diagnosed with ptosis by magnetic resonance imaging of the ocular motor nerve. At the age of 6 years, the proband underwent reconstructive surgery, and the symptom of ptosis was improved (Figure 1B, before surgery; Figure 1C, after surgery). And at the age of 6 years, the proband was further diagnosed with scoliosis by computed tomography at a local hospital (data not shown) and further diagnosed with scoliosis and dysmorphic hypoplastic ribs with fusion anomalies, however, with no vertebral anomalies, by digital radiography (DR) at Zhengzhou Orthopedic Hospital at the age of 7 years (Figure 1D, 1 and 2). At the age of 8 years, the DR images suggested a significant aggravation of scoliosis for the proband (Figure 1D, 3 and 4).
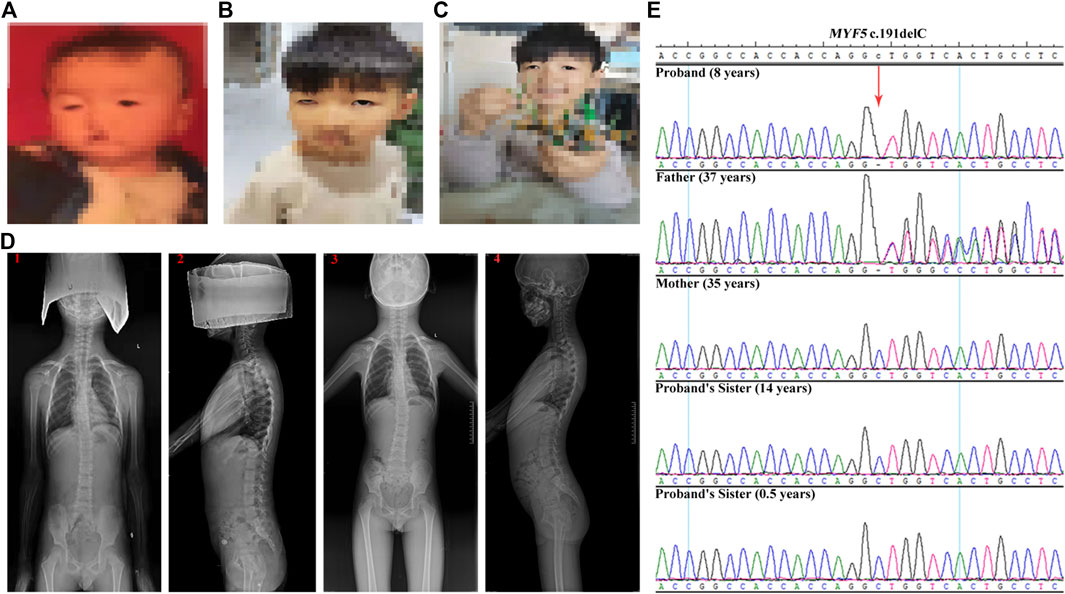
FIGURE 1. Clinical features of the proband and Sanger sequencing results of the family. (A–C) The ptosis phenotype at birth, at the age of 6 years before the reconstructive surgery, and at the age of 6 years after the reconstructive surgery, respectively, showing the improvement in ptosis symptom for the proband. (D) DR images acquired at the age of 7 years (1 and 2) and at the age of 8 years (3 and 4), showing the significant aggravation of the scoliosis condition for the proband. (E) Sanger sequencing results of c.191delC for the family. The proband is homozygous. The father is heterozygous, whereas the mother and the two sisters are wild-type.
Methods
Ethics Approval
The family provided written informed consent to carry out this study. This study was approved by the appropriate local institutional review board on human subject research at the First Affiliated Hospital of Zhengzhou University.
Quantitative Fluorescent Polymerase Chain Reaction
Genomic DNA was extracted from 500 μL of peripheral blood using the Lab-Aid Nucleic Acid (DNA) Isolation Kit (Zeesan, Xiamen, China). The genetic relationship of the proband and the parents was confirmed by QF-PCR using the Goldeneye™ DNA ID System 20A Kit (Peoplespot, Beijing, China).
Trio-Based Exome Sequencing
The methods of experiment and data analysis used for trio-ES have been described in detail in our previous study (Li et al., 2021). The candidate mutation in MYF5 was verified by Sanger sequencing using the primer pairs provided in Supplementary Table S1.
Bioinformatics Analysis
The conservation analysis was performed using the MultAlin tool (http://multalin.toulouse.inra.fr/multalin/multalin.html). The variant frequency in different populations was obtained from 1000G (https://www.internationalgenome.org/), gnomAD (http://gnomad-sg.org/), and ExAC databases (http://exac.broadinstitute.org). SIFT (http://provean.jcvi.org/protein_batch_submit.php?species=human), PolyPhen2 (http://genetics. bwh.harvard.edu/pph2/index.shtml), and MutationTaster (https://www.mutationtaster.org/) were used to predict the deleterious effects of the variant on the protein structure and function.
UPD was identified online through AilisNGS® (“https://www.ailifeus.com/#/products/ngs”. Houston, USA) from the trio-ES variant call format (vcf) data as per the method previously described (King et al., 2014).
Genome-Wide Single Nucleotide Polymorphism Array
Genome-wide SNP array, including fragmentation, labelling, and hybridization, was carried out using DNA samples from the proband and the father using Illumina HumanCytoSNP-12 v2.1 BeadChip (California, USA) on Illumina iScan. The methods of experiment and data analysis have been previously described (Bruno et al., 2011).
Cell Line, Plasmids, and Transfection
HeLa cells (human epithelial cervix carcinoma cells, American Type Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium (Gibco Life Technologies, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco Life Technologies) and 5% CO2 at 37°C.
The full-length coding region of MYF5 was sub-cloned into p3×FLAG-CMV-10 (p3×FLAG-CMV-10-MYF5-WT), whereas the mutant plasmid was constructed using the PCR-based site-directed mutagenesis (p3×FLAG-CMV-10-MYF5-MU) using the primers given in Supplementary Table S1.
HeLa cells were cultured for 24 h and then transfected with 2 μg of p3×FLAG-CMV-10-MYF5-WT, p3×FLAG-CMV-10-MYF5-MU, or p3×FLAG-CMV-10.
Western Blotting
Following 48 h incubation with 5% CO2 at 37°C, the cells were collected and lysed in lysis buffer. Then, the cell lysate was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After blocking with 5% skimmed milk powder in 1 × TBST buffer (10 mM Tris, 150 mM NaCl, and 0.05% Tween-20, pH7.5), the membrane was incubated with the primary antibody (anti-DDDDK tag, M185-3L, Medical & Biological Laboratories Co., Ltd, Nagoya, Japan) at 4°C overnight. Following washing with 1 × TBST buffer, the membrane was incubated with the secondary antibody (BL001A, Biosharp, Anhui, China). β-Actin (GB11001, Servicebio, Wuhan, China) was used as a loading control.
Statistical Analysis
Data are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 6. Student’s two-tailed t-test was used for between-group comparisons. Statistically significant differences were considered at p < 0.05 (**p < 0.01).
Results
The genetic relationship between the proband and the parents was confirmed by QF-PCR (Supplementary Table S2 and Supplementary Figure S1). The quality control of the trio-ES data is summarized in Supplementary Table S3. After filtering, only the homozygous candidate variant c.191delC (p.Ala64Valfs*33) in MYF5 (NM_005593.3) was identified in the proband (reference allele/alternative allele, ref/alt: 0/78), with the father heterozygous (ref/alt: 40/32) and the mother wild-type (Supplementary Figures S2–4), as confirmed by Sanger sequencing (Figure 1E).
Online prediction using MultAlin indicated that p.Ala64 was highly conservative across different species (Figure 2A, black box), and c.191delC was absent in 1000G, ExAC, and gnomAD databases (Supplementary Table S4). UPD identified based on the trio-ES vcf data suggested the possibility of UPD12pat in the proband (Supplementary Figure S5).
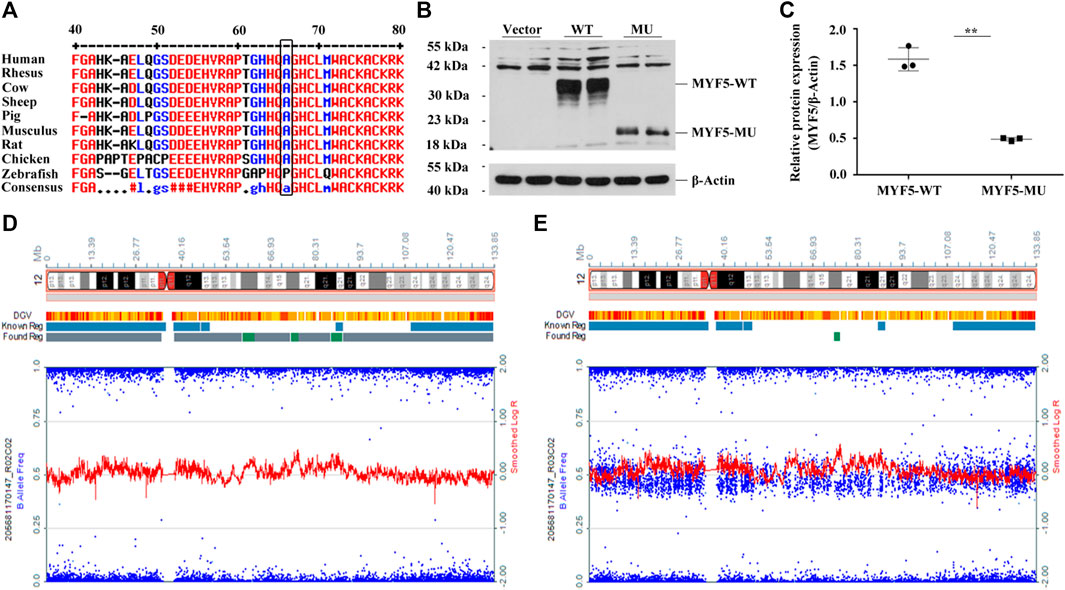
FIGURE 2. Functional study of c.191delC and genome-wide SNP array results of the proband and the father. (A) p.Ala64 is highly conservative across different species. (B), (C) Western blotting indicated the significant downregulation of MYF5 by 69.14% (**p < 0.01) in cells transfected with p3×FLAG-CMV-10-MYF5-MU compared to those transfected with p3×FLAG-CMV-10-MYF5-WT, resulting in a truncated protein with a size of ∼20 kDa. Vector: p3×FLAG-CMV-10. (D), (E) Genome-wide SNP array results of the proband and the father, respectively (only chromosome 12 shown).
For the proband, log R ratio and B allelic frequency confirmed that UPD was a complete paternal UPD12 spanning 12p13.33-q24.33, named arr [GRCh37/hg19]12p13.33q24.33 (413,635-133,272,968)×2 hmz according to the International System for Human Cytogenomic Nomenclature (ISCN 2016), including the paternal mutant allele of MYF5 (Figure 2D, the proband; 2E, the father). All SNPs are listed in Supplementary Excel S1 (205681170147_R02C02, the proband; 205681170147_R03C02, the father).
Western blotting confirmed the significant downregulation of MYF5 by 69.14% (**p < 0.01) in cells transfected with p3×FLAG-CMV-10-MYF5-MU compared to those transfected with p3×FLAG-CMV-10-MYF5-WT, resulting in a truncated protein with a size of ∼20 kDa (Figures 2B,C). These findings suggested that the mutation of c.191delC drastically affects the biological function of MYF5.
Discussion
External ophthalmoplegia with rib and vertebral anomalies is a rare autosomal recessive disorder characterized by congenital ophthalmoplegia with scoliosis and vertebral and rib anomalies, with only five individuals (three families) reported to date (Di Gioia et al., 2018). In this study, we report a novel homozygous variant in MYF5 (c.191delC) due to iUPD.
The proband in this study was an 8-year-old boy diagnosed with ptosis, scoliosis, and dysmorphic hypoplastic ribs with fusion anomalies. To the best of our knowledge, individuals with EORVA reported previously (Traboulsi et al., 2000; Di Gioia et al., 2018) and the patient in this study had similar phenotypes regarding external ophthalmoplegia, rib, and vertebral anomalies. In most cases, unilateral or bilateral ptosis was present, including our current case. Additionally, delayed motor development can occur, which was previously reported in one EORVA case but not observed in our case. Cognition was normal in all cases.
For the proband, trio-ES suggested a novel homozygous variant c.191delC in exon 1 of MYF5. This variant was heterozygous in the father and absent in the mother (Supplementary Figures S2–4), as further confirmed by Sanger sequencing (Figure 1E). To identify this non-Mendelian inheritance, UPD was analyzed based on the trio-ES vcf data. The result indicated that the homozygous state of c.191delC in the proband may be presumably attributed to UPD12pat (Supplementary Figure S5A), which was finally confirmed to be a complete iUPD by genome-wide SNP array (Figures 2D,E and Supplementary Excel S1).
The novel variant c.191delC results in p.Ala64Valfs*33, causing a frameshift mutation and premature termination of translation at codon 96. To our knowledge, this is the third pathogenic homozygous mutation reported in EORVA patients and the first mutation caused by UPD in MYF5. The biological function of MYF5 was severely affected because of the mutant-type (Figures 2B,C). Therefore, regular clinical observations need to be conducted on the proband to evaluate if the clinical symptoms of EOVAR will worsen in the future, though undergoing reconstructive surgery for ptosis.
UPD is the inheritance in which both homologous chromosomes are from one parent with no representative copy from the other, occurring as iUPD, hUPD, a combination of both, or only chromosome segment(s) (Schroeder et al., 2014). In 2020, a UPD study of trios in a clinical cohort of suspected genetic diseases revealed that the incidence of UPD was about 2:1,000 (0.2%) (Yauy et al., 2020). Later in 2021, in a population of 32,067 clinical exome trios, 16 cases were related to a positive test result through homozygous sequence variations (Scuffins et al., 2021).
Complete iUPD, an underestimated cause of recessive Mendelian disorders, is the inheritance of two identical chromosomes that are the same at all polymorphic sites along their length (Middleton et al., 2006), and is the predominant UPD type observed in the largest chromosomes (Del Gaudio et al., 2020). Roberts et al. identified a homozygous nonsense mutation c.1618A > T (p.Lys540*) in CD45 (NM_002838) in a severe combined immunodeficiency patient caused by maternal iUPD1 (iUPD1mat) (Roberts et al., 2012). Chen et al. represented a case of epileptic disorder associated with a novel mutation c.2873_c.2874delCT (p.Thr958Thrfs*17) in homozygosity in CNTN2 (NM_005076) due to UPD1mat (Chen et al., 2021). Several autosomal recessive disorders have been reported to be owing to the homozygous mutations on chromosome 2 disclosed by iUPD2pat (Kantarci et al., 2008; Dasi et al., 2016; Hara-Isono et al., 2021). Sasaki et al. reported a 3M syndrome patient with a homozygous mutation c.2975G > C (p.Arg992Pro) in CUL7 (NM_014780) attributed to iUPD6mat (Sasaki et al., 2011). Cho et al. unmasked a homozygous mutation c.1120C > G (p.Tyr400*) in SUOX (NC_000012.11) in a patient with isolated sulfite oxidase deficiency resulting from UPD12pat (Cho et al., 2013). Wiszniewski et al. revealed a homozygous mutation c.1148delC (p.Thr383Ilefs*13) in CNGB3 (NM_019098) in a case with achromatopsia associated with UPD14mat (Wiszniewski et al., 2007). SoehnSoehn et al. described four novel homozygous mutations in FA2H (NM_024306) in four unrelated families with spastic paraplegia type 35 because of UPD16 (SoehnSoehn et al., 2016).
In conclusion, to the best of our knowledge, for the first time, we identified a novel homozygous mutation in MYF5 due to iUPD12pat in a case of EORVA, a rare event and enriching MYF5 gene variation spectrum, further suggesting that UPD of any chromosome is associated with an increased risk of recessive disease because it may affect children when only one parent is a carrier of a pathogenic variant. Additionally, the results re-emphasized the clinical importance of UPD in case the results are inconsistent with recessive inheritance.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the guidelines set forth by the Declaration of Helsinki. This study was also conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS). The family provided written informed consent to participate in this research. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
QL, XZ, CY, and XK designed the research. QL, XZ, CY, and JM performed the experiments. LS supported the DR images of the proband. QL, XZ, CY, XW, and YY analyzed the data. QL wrote the manuscript. XZ, RL, XW, YY, and XK revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (no. 82000321) and the Henan Educational Committee Program for Science and Technology Development of Universities (22A310022).
Conflict of Interest
XW and YY were employed by AiLife Diagnostics, Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors were very grateful to the family for participating in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.780363/full#supplementary-material
References
Benn, P. (2021). Uniparental Disomy: Origin, Frequency, and Clinical Significance. Prenatal Diagn. 41 (5), 564–572. doi:10.1002/pd.5837
Bruno, D. L., White, S. M., Ganesamoorthy, D., Burgess, T., Butler, K., Corrie, S., et al. (2011). Pathogenic Aberrations Revealed Exclusively by Single Nucleotide Polymorphism (SNP) Genotyping Data in 5000 Samples Tested by Molecular Karyotyping. J. Med. Genet. 48 (12), 831–839. doi:10.1136/jmedgenet-2011-100372
Chen, W., Chen, F., Shen, Y., Yang, Z., and Qin, J. (2021). Case Report: A Case of Epileptic Disorder Associated with a Novel CNTN2 Frameshift Variant in Homozygosity Due to Maternal Uniparental Disomy. Front. Genet. 12, 743833. doi:10.3389/fgene.2021.743833
Cho, S. Y., Goh, D. L.-M., Lau, K.-C., Ong, H. T., and Lam, C.-w. (2013). Microarray Analysis Unmasked Paternal Uniparental Disomy of Chromosome 12 in a Patient with Isolated Sulfite Oxidase Deficiency. Clinica Chim. Acta 426, 13–17. doi:10.1016/j.cca.2013.08.013
Dasi, M. A., Gonzalez-Conejero, R., Izquierdo, S., Padilla, J., Garcia, J. L., Garcia-Barberá, N., et al. (2016). Uniparental Disomy Causes Deficiencies of Vitamin K-dependent Proteins. J. Thromb. Haemost. 14 (12), 2410–2418. doi:10.1111/jth.13517
Del Gaudio, D., Shinawi, M., Astbury, C., Tayeh, M. K., Deak, K. L., and Raca, G. (2020). Diagnostic Testing for Uniparental Disomy: a Points to Consider Statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22 (7), 1133–1141. doi:10.1038/s41436-020-0782-9
Di Gioia, S. A., Shaaban, S., Elcioglu, B. N. H., Chan, W.-M., Robson, C. D., Ecklund, K., et al. (2018). Recessive MYF5 Mutations Cause External Ophthalmoplegia, Rib, and Vertebral Anomalies. Am. J. Hum. Genet. 103 (1), 115–124. doi:10.1016/j.ajhg.2018.05.003
Engel, E. (1980). A New Genetic Concept: Uniparental Disomy and its Potential Effect, Isodisomy. Am. J. Med. Genet. 6 (2), 137–143. doi:10.1002/ajmg.1320060207
Hara-Isono, K., Matsubara, K., Hamada, R., Shimada, S., Yamaguchi, T., Wakui, K., et al. (2021). A Patient with Silver-Russell Syndrome with Multilocus Imprinting Disturbance, and Schimke Immuno-Osseous Dysplasia Unmasked by Uniparental Isodisomy of Chromosome 2. J. Hum. Genet. 66 (11), 1121–1126. doi:10.1038/s10038-021-00937-7
Kantarci, S., RaggeRagge, N. K., Thomas, N. S., Robinson, D. O., Noonan, K. M., Russell, M. K., et al. (2008). Donnai-Barrow Syndrome (DBS/FOAR) in a Child with a homozygousLRP2mutation Due to Complete Chromosome 2 Paternal Isodisomy. Am. J. Med. Genet. 146A (14), 1842–1847. doi:10.1002/ajmg.a.32381
King, D. A., Fitzgerald, T. W., Miller, R., Canham, N., Clayton-Smith, J., Johnson, D., et al. (2014). A Novel Method for Detecting Uniparental Disomy from Trio Genotypes Identifies a Significant Excess in Children with Developmental Disorders. Genome Res. 24 (4), 673–687. doi:10.1101/gr.160465.113
Li, Q., Zhu, X., Wang, C., Meng, J., Chen, D., and Kong, X. (2021). Identification of a Rare Case with Nagashima-type Palmoplantar Keratoderma and 18q Deletion Syndrome via Exome Sequencing and Low-Coverage Whole-Genome Sequencing. Front. Genet. 12, 707411. doi:10.3389/fgene.2021.707411
Middleton, F. A., Trauzzi, M. G., Shrimpton, A. E., Gentile, K. L., Morley, C. P., Medeiros, H., et al. (2006). Complete Maternal Uniparental Isodisomy of Chromosome 4 in a Subject with Major Depressive Disorder Detected by High Density SNP Genotyping Arrays. Am. J. Med. Genet. 141B (1), 28–32. doi:10.1002/ajmg.b.30250
Nakka, P., Pattillo Smith, S., O’Donnell-LuriaDonnell-Luria, A. H., McManus, K. F., Mountain, J. L., Ramachandran, S., et al. (2019). Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 105 (5), 921–932. doi:10.1016/j.ajhg.2019.09.016
Roberts, J. L., Buckley, R. H., Luo, B., Pei, J., Lapidus, A., Peri, S., et al. (2012). CD45-deficient Severe Combined Immunodeficiency Caused by Uniparental Disomy. Proc. Natl. Acad. Sci. 109 (26), 10456–10461. doi:10.1073/pnas.1202249109
Sasaki, K., Okamoto, N., Kosaki, K., Yorifuji, T., Shimokawa, O., Mishima, H., et al. (2011). Maternal Uniparental Isodisomy and Heterodisomy on Chromosome 6 Encompassing a CUL7 Gene Mutation Causing 3M Syndrome. Clin. Genet. 80 (5), 478–483. doi:10.1111/j.1399-0004.2010.01599.x
Schroeder, C., Ekici, A. B., Ekici, A. B., Moog, U., Grasshoff, U., Mau-Holzmann, U., et al. (2014). Genome-wide UPD Screening in Patients with Intellectual Disability. Eur. J. Hum. Genet. 22 (10), 1233–1235. doi:10.1038/ejhg.2014.63
Scuffins, J., Keller-Ramey, J., Dyer, L., Douglas, G., Torene, R., Gainullin, V., et al. (2021). Uniparental Disomy in a Population of 32,067 Clinical Exome Trios. Genet. Med. 23 (6), 1101–1107. doi:10.1038/s41436-020-01092-8
SoehnSoehn, A. S., RattayRattay, T. W., Beck-Wödl, S., Schäferhoff, K., Monk, D., Döbler-Neumann, M., et al. (2016). Uniparental Disomy of Chromosome 16 Unmasks Recessive Mutations of FA2H/SPG35 in 4 Families. Neurology 87 (2), 186–191. doi:10.1212/wnl.0000000000002843
Traboulsi, E. I., Lee, B. A., Mousawi, A., Khamis, A. R., and Engle, E. C. (2000). Evidence of Genetic Heterogeneity in Autosomal Recessive Congenital Fibrosis of the Extraocular Muscles. Am. J. Ophthalmol. 129 (5), 658–662. doi:10.1016/s0002-9394(99)00467-5
Wang, Y., Schnegelsberg, P. N. J., Dausman, J., and Jaenisch, R. (1996). Functional Redundancy of the Muscle-specific Transcription Factors Myf5 and Myogenin. Nature 379 (6568), 823–825. doi:10.1038/379823a0
Wiszniewski, W., Lewis, R. A., and Lupski, J. R. (2007). Achromatopsia: the CNGB3 p.T383fsX Mutation Results from a Founder Effect and Is Responsible for the Visual Phenotype in the Original Report of Uniparental Disomy 14. Hum. Genet. 121 (3-4), 433–439. doi:10.1007/s00439-006-0314-y
Keywords: external ophthalmoplegia with rib and vertebral anomalies, trio-based exome sequencing, paternal uniparental isodisomy, chromosome 12, the myogenic factor 5 gene, homozygous mutation
Citation: Li Q, Zhu X, Yu C, Shang L, Li R, Wang X, Yang Y, Meng J and Kong X (2022) Case Report: A Novel Homozygous Mutation in MYF5 Due to Paternal Uniparental Isodisomy of Chromosome 12 in a Case of External Ophthalmoplegia With Rib and Vertebral Anomalies. Front. Genet. 12:780363. doi: 10.3389/fgene.2021.780363
Received: 21 September 2021; Accepted: 30 December 2021;
Published: 03 February 2022.
Edited by:
Kathleen M. Gorman, Temple Street Children’s University Hospital, IrelandCopyright © 2022 Li, Zhu, Yu, Shang, Li, Wang, Yang, Meng and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianqian Li, MzcwMzA4MTBAcXEuY29t; Xiangdong Kong, a29uZ3hkQDI2My5uZXQ=