 Maayan Kagan1,2,3†
Maayan Kagan1,2,3† Rotem Semo-Oz1,2,4†
Rotem Semo-Oz1,2,4† Yishay Ben Moshe1,2Danit Atias-Varon5Irit Tirosh1,2,4Michal Stern-Zimmer1,2Aviva Eliyahu2,6Annick Raas-Rothschild2,7Maayan Bivas1,2Omer Shlomovitz1,2
Yishay Ben Moshe1,2Danit Atias-Varon5Irit Tirosh1,2,4Michal Stern-Zimmer1,2Aviva Eliyahu2,6Annick Raas-Rothschild2,7Maayan Bivas1,2Omer Shlomovitz1,2 Odelia Chorin2,6,7Rachel Rock2,6,7
Odelia Chorin2,6,7Rachel Rock2,6,7 Michal Tzadok2,8
Michal Tzadok2,8 Bruria Ben-Zeev2,8Gali Heimer2,3,8Yoav Bolkier2,9Noah Gruber1,2,10Adi Dagan1,2,11Bat El Bar Aluma1,2,11Itai M. Pessach2,3,12Gideon Rechavi2,13,14Ortal Barel2,13,15
Bruria Ben-Zeev2,8Gali Heimer2,3,8Yoav Bolkier2,9Noah Gruber1,2,10Adi Dagan1,2,11Bat El Bar Aluma1,2,11Itai M. Pessach2,3,12Gideon Rechavi2,13,14Ortal Barel2,13,15 Ben Pode-Shakked1,2,3,6
Ben Pode-Shakked1,2,3,6 Yair Anikster2,13,16Asaf Vivante1,2,3,5*
Yair Anikster2,13,16Asaf Vivante1,2,3,5*- 1Department of Pediatrics B, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 2Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel
- 3Talpiot Medical Leadership Program, Sheba Medical Center, Tel-Hashomer, Israel
- 4Pediatric Rheumatology Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 5Pediatric Nephrology Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 6The Danek Gertner Institute of Human Genetics, Sheba Medical Center, Tel-Hashomer, Israel
- 7The Institute of Rare Diseases, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 8Pediatric Neurology Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 9Pediatric Heart Institute, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 10Pediatric Endocrinology and Diabetes Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 11Pediatric Pulmonology and National CF Center, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 12Department of Pediatric Intensive Care, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
- 13The Wohl Institute for Translational Medicine, Sheba Medical Center, Tel-Hashomer, Israel
- 14Sheba Cancer Research Center, Sheba Medical Center, Tel-Hashomer, Israel
- 15The Genomics Unit, Sheba Cancer Research Center, Sheba Medical Center, Tel-Hashomer, Israel
- 16Metabolic Disease Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel-Hashomer, Israel
Background: Genetic conditions contribute a significant portion of disease etiologies in children admitted to general pediatric wards worldwide. While exome sequencing (ES) has improved clinical diagnosis and management over a variety of pediatric subspecialties, it is not yet routinely used by general pediatric hospitalists. We aim to investigate the impact of exome sequencing in sequencing-naive children suspected of having monogenic disorders while receiving inpatient care.
Methods: We prospectively employed exome sequencing in children admitted to the general pediatric inpatient service at a large tertiary medical center in Israel. Genetic analysis was triggered by general and/or subspecialist pediatricians who were part of the primary inpatient team. We determined the diagnostic yield among children who were referred for exome sequencing and observed the effects of genetic diagnosis on medical care.
Results: A total of fifty probands were evaluated and exome sequenced during the study period. The most common phenotypes included were neurodevelopmental (56%), gastrointestinal (34%), and congenital cardiac anomalies (24%). A molecular diagnosis was reached in 38% of patients. Among seven patients (37%), the molecular genetic diagnosis influenced subsequent clinical management already during admission or shortly following discharge.
Conclusion: We identified a significant fraction of genetic etiologies among undiagnosed children admitted to the general pediatric ward. Our results support that early application of exome sequencing may be maximized by pediatric hospitalists’ high index of suspicion for an underlying genetic etiology, prompting an in-house genetic evaluation. This framework should include a multidisciplinary co-management approach of the primary care team working alongside with subspecialties, geneticists and bioinformaticians.
1 Introduction
Genetic etiologies are responsible for a substantial portion of pediatric diseases. Yet, in many cases genetic diagnosis is hindered or completely missed (Schroeder et al., 2021). In the past decade, owing to advancements in the field of genetics and the growing accessibility of next generation sequencing (NGS) methods, exome sequencing (ES) has become a powerful tool, shedding light on many genetic cases that were previously under-detected (Boycott et al., 2013; Yang et al., 2013).
Nowadays, the use of ES is a key for the identification of monogenic disorders which often cannot be clinically diagnosed. ES can identify single gene diseases (also called Mendelian or monogenic diseases). Thus, it encompasses a substantial portion of genetic alternations providing accurate and timely molecular diagnoses across a myriad of pediatric conditions (Singleton, 2011; Xue et al., 2015; Retterer et al., 2016).
In recent years, ES has been used more prevalently in the research as well as clinical settings, as it became more available and less costly. From a clinical perspective, it is prominently used in the setting of outpatient genetic consultations or clinics (Pode-Shakked et al., 2021). Nonetheless, recent implications of ES encompass diagnosis and management of patients in different pediatric subspecialties across a multitude of disease etiologies (Dixon-Salazar et al., 2012; Lee et al., 2014; Zhang et al., 2015; Pode-Shakked et al., 2021; Pode-Shakked et al., 2022). Early implementation of ES can shorten the lengthy odyssey of the diagnostic process, shorten hospitalizations, spare unnecessary invasive procedures, reduce medical expenses, and may alter surveillance and management at early stages, which could be crucial in certain cases (Valencia et al., 2015; Splinter et al., 2018; Wise et al., 2019; Manickam et al., 2021). Still, ES is rarely performed in the setting of general pediatric wards for hospitalized children. Consequently, data regarding its diagnostic yield, benefits and challenges during acute admission in this patient population is scarce.
Herein, we present a single center experience employing ES as part of the pediatric in-patient service for patients suspected by the pediatric hospitalist team to have an underlying and previously undiagnosed genetic condition.
The rationale behind the decision to routinely establish ES as an available medical test to the general pediatric ward team, was derived from the accumulating knowledge on the molecular basis of pediatric diseases, the growing availability of NGS techniques, and our previous experience with undiagnosed medical cases from the past (pre-ES era). In this respect, we recently sequenced multiple cases of unsolved medical mysteries from the past and from whom we had available DNA samples (Table 1). These cases highlighted the diagnostic power of ES and the need for quick and accurate molecular diagnosis. Subsequently, we initiated a designated hospitalists team working in collaboration with medical geneticists and subspecialist consultants, in order to provide a ‘one-stop-shop,’ in which patients suspected to harbor an underlying genetic etiology during their hospitalization, would undergo a timely and multifaceted genetic evaluation and analysis. Indeed, the aim of this initiative was never to transfer the routine care of these patients affected with different genetic diseases from their primary provider/geneticist, but rather to promptly initiate genetic evaluation and shorten the diagnostic odyssey for such families, based on an in-house team of experts well familiar with the genetic landscape of different pediatric disorders.
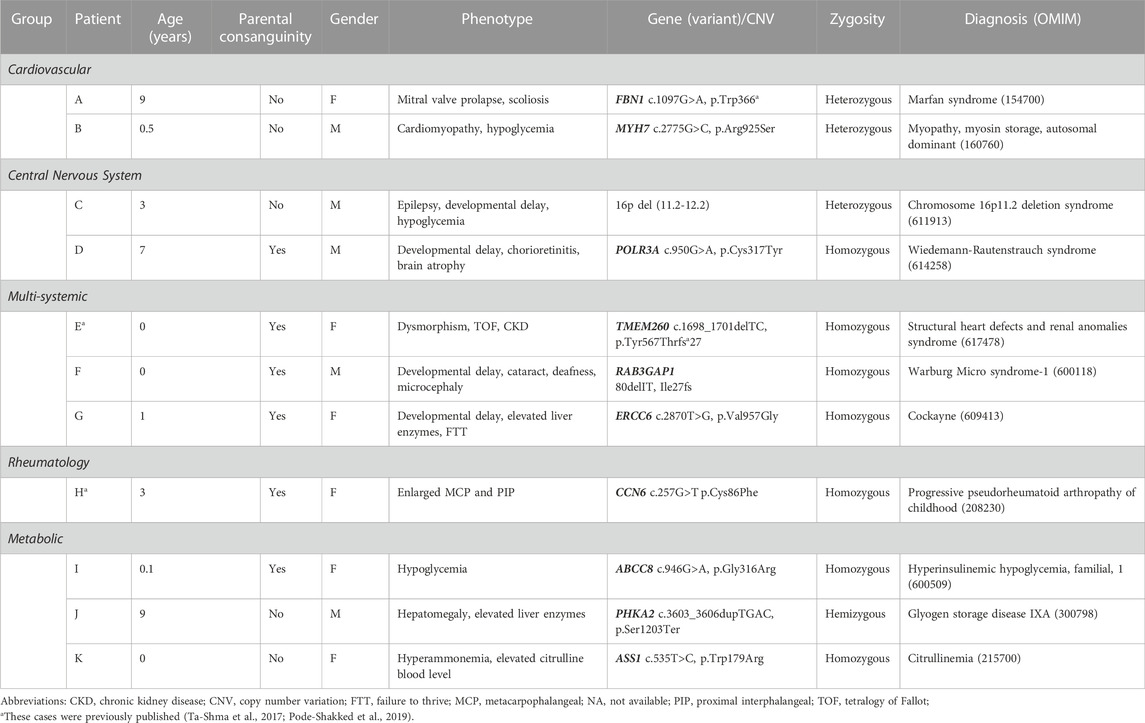
TABLE 1. Characteristics of a historical cohort of undiagnosed children which were solved following exome sequencing.
2 Materials and methods
2.1 Patients’ characteristics and the general pediatric ward
The Edmond and Lily Safra Children’s Hospital at the Sheba Medical Center (SMC) is a tertiary center accepting acute pediatric patients as well as chronic and/or complicated cases from all across the country. In addition, patients are admitted from the Palestinian National Authority (∼20% of the hospitalized children) as well as complex cases from different parts of the world. The SMC General Pediatric Ward is specialized in offering expert care to children from birth to late adolescence. The General Pediatric Ward team, with the aid of the hospital’s consulting services, is highly experienced in treating children with acute and chronic illness, complex medical conditions and syndromes, inborn errors of metabolism, diverse types of cancer, congenital anomalies and others. Children are typically admitted for ∼4 days on average during which diagnostic evaluations and treatment plans are executed.
2.2 Prospective patient selection for genetic analysis during acute hospital admission
All patients were admitted to the general pediatric ward at the Edmond and Lily Safra Children’s Hospital at SMC, Israel, between the years 2019–2022. A designated physician from the in-patient team was appointed as the genetics coordinator of the ward. Patients suspected to harbor a genetic etiology were presented daily during medical rounds to the coordinator. Subsequently, the coordinator offered each relevant family the option of genetic testing free of charge. Children for whom a specific diagnosis was highly suspected, and only a single or few genes (or mutations) were tested, were not included in this study. Each family received a detailed explanation on ES analysis, along with the advantages and pitfalls of the technique, with special attention given to secondary and incidental findings. Subsequently, a written informed consent was obtained from each interested family. The coordinator gathered probands’ and their families’ medical history and drew an annotated pedigree. Subsequently, blood samples were obtained from the proband and when feasible from additional family members, and were used for DNA extraction.
For coverage of ES costs, we used a designated fund of the children’s hospital supported by SMC. Consequently, for patients who required genetic testing, we were able to provide ES free of charge. The study was conducted in accordance with the tenets of the Declaration of Helsinki, and under approval of the Institutional Review Board at the SMC.
2.3 Clinical evaluation
Baseline characteristics were recorded for each patient upon enrollment, including current age, gender, age and phenotype at presentation, ethnic origin and parental consanguinity. The clinical symptoms were presented as HPO terms to the bioinformatics laboratory based on the clinical information. In addition, we completed a scan of the electronic medical chart for additional characteristics, such as primary clinical diagnosis and medical workup that was completed prior to the genetic testing (i.e., blood tests, imaging, etc).
2.4 Exome sequencing and bioinformatic analysis
We performed exome sequencing, variant detection, and filtering as previously described (Tirosh et al., 2019). Variant calling was performed by a team of clinician scientist bioinformaticians, who had knowledge of the clinical phenotypes and pedigree structure, as well as genetic expertise in exome evaluation as previously described (Bolkier et al., 2021). Exome data were interpreted according to the American College of Genetic and Genomic Medicine (ACMG) guidelines. During the ES initial evaluation and thereafter, the pediatrics hospitalist team was in close discussion with the in-house SMC bioinformatician team regarding the detected variants as well as filtering process. Patients who were found to harbor a variant classified as pathogenic or likely pathogenic were defined as having a positive molecular diagnosis. Patients suspected to harbor a genetic etiology and required further investigation, such as segregation analysis, were defined as having a non-conclusive ES result.
2.5 Genetic results reporting and follow up
Following ES results, families were invited for a follow-up visit with geneticists/primary provider, during which recommendations and changes to clinical management were recorded and further referral for additional consults or diagnostic tests was done. Additionally, explanations regarding risk of recurrence in future pregnancies and family planning options were conveyed.
3 Results
3.1 Patients and phenotypes
3.1.1 Historical cohort
Table 1 shows a historical cohort of eleven undiagnosed children admitted to our ward in the pre-ES era, which were solved following ES. These cases included children with ultra-rare medical conditions or children exhibiting non-specific phenotypes hampering the establishment of a single or accurate genetic diagnosis.
3.1.2 Prospective cohort
Baseline characteristics of fifty patients recruited prospectively to the study are summarized in Table 2. Patients were predominantly male and 22% were offspring of consanguineous unions. Single/duo ES was performed in 68% and trio ES was performed in 32% of cases. The most common phenotype in our cohort involved the central nervous system (CNS) (56%), gastrointestinal system (34%), and cardiovascular system (24%). In 32% dysmorphic features were noted. ES revealed a genetic diagnosis in 38% of the cohort (Table 3). Four patients (8%) had non-conclusive ES results which require further investigation (Supplementary Table S1). Notably, patients with a positive ES result had higher rates of CNS involvement compared to other phenotypes (∼63%), followed by gastrointestinal (∼42%) and facial dysmorphism (∼42%) phenotypes. Of the solved cases, three had a de novo variant detected by trio-analysis. Five out of the nineteen solved cases were from consanguineous families.
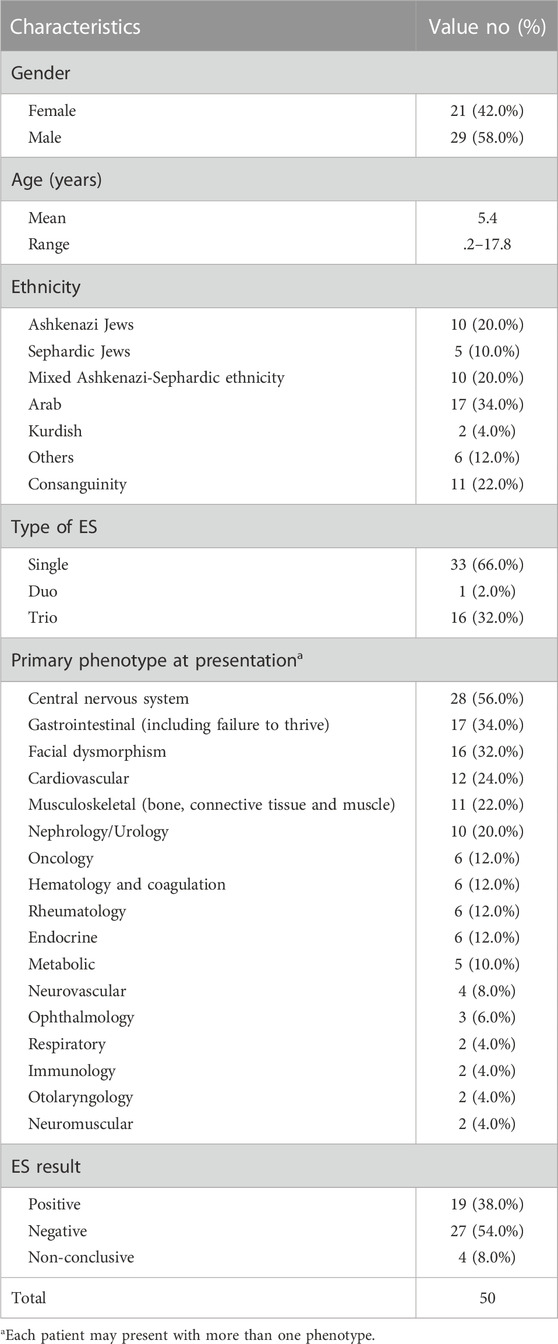
TABLE 2. Demographic and phenotypic characteristics of the fifty probands evaluated by exome sequencing (ES).
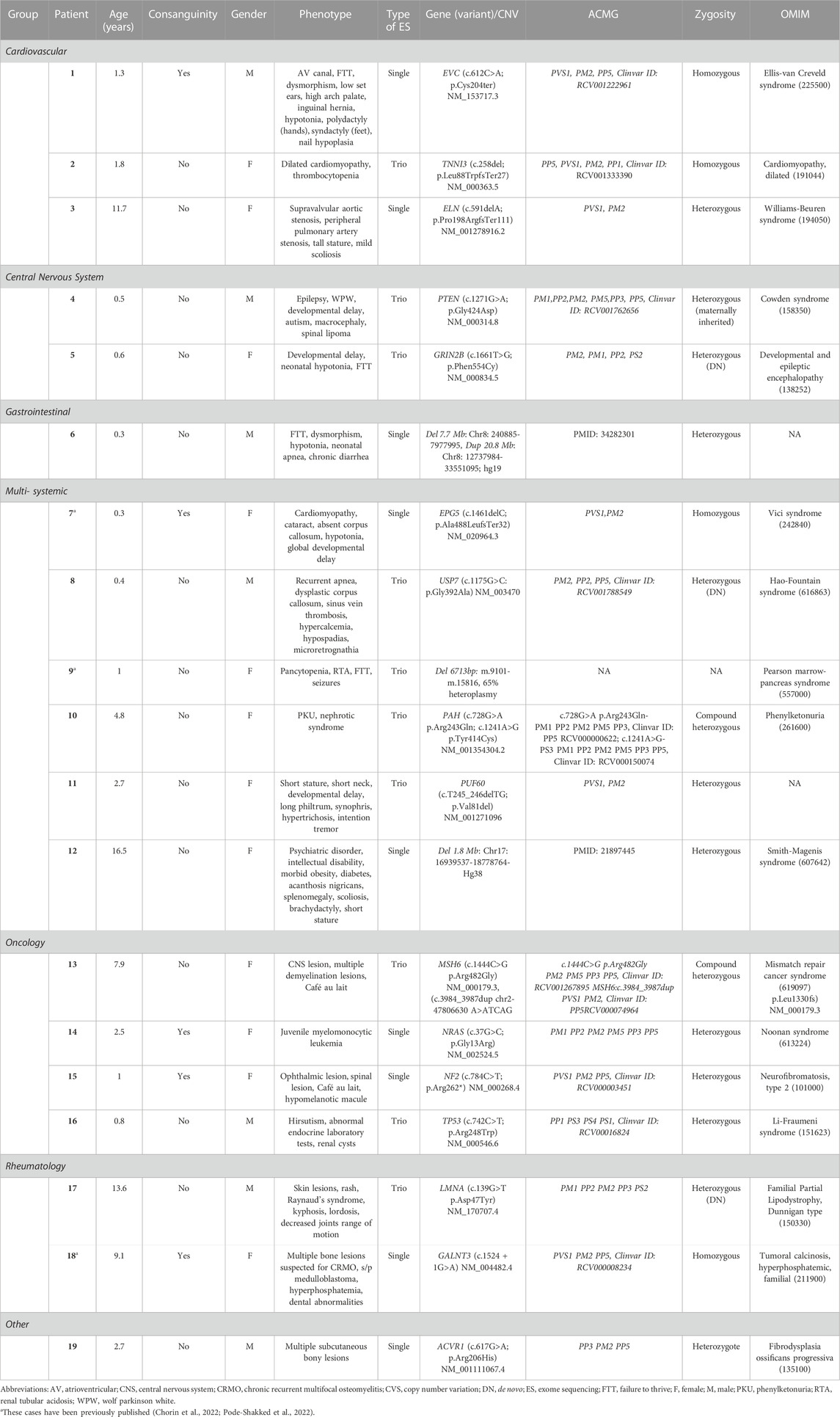
TABLE 3. Patients for whom genetic workup yielded a molecular diagnosis, shown according to clinical diagnosis and phenotype.
3.2 Effect of diagnosis on medical care
The timely genetic diagnoses affected medical care and allowed a personalized approach in a variety of ways. The establishment of accurate and correct root cause in a short period of time prevented treatment delays as well as unnecessary interventions or diagnostic procedures. Moreover, in six children (∼32%) ES opened a window for optional personalized therapy, and in several children (∼37%), ES results had a direct effect on medical management during admission or shortly following discharge as highlighted below in three representative cases:
Patient 18 (Table 3) presented with 4 months of worsening arm swelling and suspected chronic recurrent osteomyelitis (CRMO) (Figure 1). Her past medical history was notable for medulloblastoma at the age of 2 years. Prior to her hospitalization, she had an extensive diagnostic work up at the oncology clinic. MRI and PET-CT of the arm were suspected for Ewing sarcoma. Open bone biopsy ruled out malignancy and was consistent with callus formation and associated mild chronic inflammation. During her hospitalization, we noted hyperphosphatemia with normal kidney function which raised the possibility of abnormal phosphate regulation. Additionally, the fact that her parents were first degree cousins, supported the possibility of a monogenic recessive disorder. ES was completed and treatment of phosphate chelators was initiated. A homozygous GALNT3 variant which was previously reported to cause Hyperostosis Hyperphosphatemia Syndrome was detected (Topaz et al., 2004).
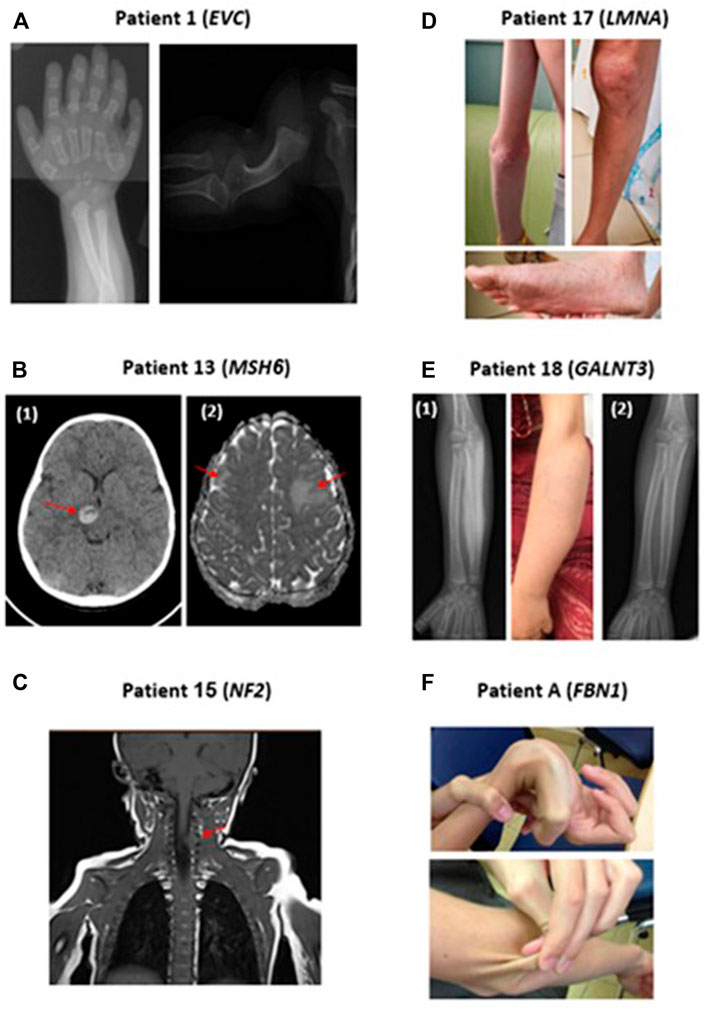
FIGURE 1. Representative clinical findings in patients with positive molecular diagnosis in our cohort. (A) Patient 1: Radiographs demonstrating polydactyly and deformation of the humerus, in a patient with Ellis-Van Creveld syndrome (EVC mutation). (B) Patient 13: Brain CT (1) and MRI (2) scans demonstrating hemorrhage of the thalamus and bilateral frontal lesions in a patient with compound heterozygous MSH6 mutations. (C) Patient 15: MRI scan preformed due to orbital lesion detected an incidental finding of a spinal lesion at C4-5 level in a patient with NF2 mutation. (D) Patient 17: Patient with LMNA mutation presenting with widespread telangiectasias and Scleroderma-like skin changes. (E) Patient 18: (1): Hyperostosis of the right ulna secondary to GALNT3 mutation demonstrated in radiographs and clearly visible in the adjacent photograph (2) Radiographs of the right Ulna following phosphorus chelator therapy, demonstrating a substantial improvement of hyperostosis. (F) Patient A: Patient with Marfan syndrome presenting with joint hypermobility and hyper elastic skin.
Elucidating the molecular diagnosis, in this case, prevented treatment with bisphosphonates for CRMO which could have worsened her condition. Subsequently, the patient’s two siblings were tested for plasma phosphate level and found to have hyperphosphatemia. In summary, this case highlights the importance of a high index of suspicion for genetic etiologies in children with parental consanguinity. Radiographs of this patient’s arm before and after treatment are presented in Figure 1.
Patient 13 (Table 3) presented with 2 weeks of headache followed by acute weakness of her left arm and leg. On examination she had hemiparesis, as well as multiple café au lait spots. Brain CT and MRI showed bleeding of her right thalamus and bilateral frontal lesions (Figure 1). Since these findings were radiologically suspected for acute disseminated encephalomyelitis (ADEM), the patient was treated with high dose intravenous methylprednisolone. The combination of brain lesions and café au lait spots, led to consideration of an underlying genetic etiology secondary to mutations in NF1 or genes coding to mismatch repair proteins such as MSH2 or MSH6. Rapid ES exam results were notable for compound heterozygous variants in MSH6, a mismatch repair gene. Pathogenic variants in this gene are known for increasing the risk of colorectal and CNS malignancies. Consequently, the patient was followed by the oncology and neurosurgery teams. Subsequent brain biopsy was performed and showed grade II astrocytoma. Segregation analysis showed that each of her parents was a carrier for one of the two variants, and therefore are also at increased risk for malignancies (Lynch syndrome), requiring additional medical follow-up. In addition, the parents decided to undergo preimplantation genetic diagnosis (PGD) for future pregnancies. This case illustrates how early suspicion and employment of ES not only changed the diagnosis and management of the patient, from a suspected autoimmune condition to malignancy, but also influenced the child’s immediate family in terms of informed anticipatory follow-up and family planning.
Patient 17 (Table 3) was admitted for evaluation of low body mass index (below first centile), Raynaud’s phenomenon, worsening sclerodermic-like skin lesions, telangiectasia and joint contractures (Figure 1). The initial differential diagnosis included autoimmune connective tissue disease versus genetic causes of lipodystrophy. A comprehensive diagnostic work-up, including gastroenterology and rheumatology testing were completed. ES revealed the rare diagnosis of heterozygous LMNA mutation causing Familial Lipodystrophy type 2 (Dunnigan type), a previously reported cause for scleroderma-like presentation (Akinci et al., 2000). In this case, establishing a molecular genetic diagnosis allowed an unequivocal disease diagnosis and opened a window for tailored medical treatment and monitoring, as well as for possible novel treatment options such as apolipoprotein C III inhibitors (Aslesh and Yokota, 2020; Lightbourne et al., 2021).
4 Discussion
In this study, we present data on the diagnostic yield of next-generation sequencing and its effect on medical care and follow-up, among sequencing-naive children suspected of having a monogenic disorder while receiving acute or chronic inpatient care at the general pediatric department. We found that ES during acute hospitalizations significantly contributed to personalized management. Moreover, we show that even though many patients had long standing conditions, only following the genetic analysis which was initiated during their hospitalization, was a molecular diagnosis reached. This was very much due to the dedication and high index of suspicion of the primary hospitalists team to pursue timely and efficient genetic analysis, and with no costs to the families, thus eliminating diagnostic disparities based on financial resources (Grant et al., 2021). The limitations of our study include a relatively small and selective cohort, a single-center based cohort with a relatively high consanguinity rate (22%), uncertainty regarding de novo mutations since in most cases we performed a proband-only (single) exome, and the lack of complimentary data obtained by chromosomal microarray for detection of copy number variations. Additionally, a designated fund enabled us to offer ES to all recruited patients at no cost to the families, eliminating disparities based on their insurance coverage or financial constraints, however we recognize that this might not be feasible in every medical center or healthcare system.
Our study highlights several important conclusions for the diagnosis of monogenic disease in the setting of the general pediatric ward. First, as mentioned above, the patient population consists of infants and children hospitalized at a general pediatric ward, rather than critically ill patients receiving intensive care. This is noteworthy as the relatively high diagnostic yield demonstrates the significant portion of genetic etiologies not only among severely affected children requiring complex care, but also those manifesting with more common general pediatric diseases. As a result, pediatric hospitalists may be the first clinicians to be approached, highlighting the importance of a high index of suspicion to the possibility of genetic etiology. Second, our results reflect the real-world clinical utility of ES, in contrast to research-based cohorts, which are more often those reported in the literature, especially for children with neurodevelopmental disorders.
While the diagnostic yield and medical implications of clinical ES in neonatal and pediatric intensive care units have been widely studied (Meng et al., 2017; Freed et al., 2020; Lunke et al., 2020; Liu et al., 2021; Ouyang et al., 2021; Scholz et al., 2021), its clinical utilization in the in-patient setting of a general pediatric ward has scarcely been reported. We identified a monogenic etiology in 38% of children suspected to have undiagnosed genetic conditions. Importantly, a significant portion of the cases we identified were related to lifelong cancer risk (e.g., TP53, NF2, NRAS, PTEN, and MSH6), affecting both informed anticipatory follow-up and anticancer treatment regimens for these children and their families (Zhang et al., 2015; Scollon et al., 2021). Of note, prior to every ES test, a formal explanation and conversation was made with the families during which the team has explained the possibility of secondary/incidental findings, as well as possible findings which may confer future cancer risk.
As the field of medical genetics continues to advance and becomes more accessible, it is growingly recognized that physicians at every stage of their career should become familiarized with the basis of genetics, which will provide them with the tools to diagnose patients with genetic diseases. Hence, we believe that proper genetic education is a steppingstone and essential at all training levels. Nonetheless, as genetics become an inherent part of in-patient care, medical genetics health providers, physicians and genetic counselors should be an integral part of the ward to provide in depth analysis within the ward, complete auxiliary testing including segregation analysis following initial results and offer novel treatments. Furthermore, following familial analysis, family planning tools and parental counselling are crucial.
A molecular genetic diagnosis can shorten the diagnostic process, prompt targeted therapy, spare unnecessary interventions and diagnostic procedures, and has the potential to lower the costs of the diagnostic process and prevent birth of additional affected family members (Lavelle et al., 2022).
In summary, we report herein the experience of using NGS tools in a pediatric ward of a tertiary hospital for rapid and accurate diagnosis of complex diseases. We identified a significant fraction of genetic etiologies among previously undiagnosed children admitted to the general pediatric ward, which impacted subsequent clinical management. The pediatric hospitalists’ high index of suspicion for an underlying genetic etiology prompted in-house genetic evaluation. Our experience portrays the important need to integrate genetics in hospital work of general pediatric hospitalists and pediatric subspecialties, requiring the current in-house pediatric hospitalist to have in-depth knowledge of genetics, along with close collaboration with bioinformaticians and geneticists allowing provision of personalized care.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Helsinki board, Sheba medical center, Tel-Hashomer, Israel. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
MK, RS-O, and AV initiated the study, recruited patients, obtained clinical data, analyzed the results, wrote the manuscript and critically reviewed it; YBM, DA-V, IT, MS-Z, AE, AR-R, MB, OS, OC, RR, MT, BB-Z, GH, YB, NG, AD, BBA, IP, GR, BP-S, and YA recruited patients, obtained clinical data, analyzed it and critically reviewed the manuscript; OB interpreted the exome sequencing data; All authors critically reviewed the manuscript.
Acknowledgments
The authors wish to thank the patients and their families for their kind assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1018062/full#supplementary-material
References
Akinci, B., Sahinoz, M., and Oral, E. (2000). Lipodystrophy syndromes: Presentation and treatment. South Dartmouth, MA: Endotext.
Aslesh, T., and Yokota, T. (2020). Development of antisense oligonucleotide gapmers for the treatment of dyslipidemia and lipodystrophy. Methods Mol. Biol. 2176, 69–85. doi:10.1007/978-1-0716-0771-8_5
Bolkier, Y., Barel, O., Marek-Yagel, D., Atias-Varon, D., Kagan, M., Vardi, A., et al. (2021). Whole-exome sequencing reveals a monogenic cause in 56% of individuals with laterality disorders and associated congenital heart defects. J. Med. Genet. 59, 691–696. doi:10.1136/jmedgenet-2021-107775
Boycott, K. M., Vanstone, M. R., Bulman, D. E., and MacKenzie, A. E. (2013). Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 14, 681–691. doi:10.1038/nrg3555
Chorin, O., Hirsch, Y., Rock, R., Salzer Sheelo, L., Goldberg, Y., Mandel, H., et al. (2022). Vici syndrome in Israel: Clinical and molecular insights. Front. Genet. 13, 991721. doi:10.3389/fgene.2022.991721
Dixon-Salazar, T. J., Silhavy, J. L., Udpa, N., Schroth, J., Bielas, S., Schaffer, A. E., et al. (2012). Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 4 (138), 138ra78. doi:10.1126/scitranslmed.3003544
Freed, A. S., Clowes Candadai, S. V., Sikes, M. C., Thies, J., Byers, H. M., Dines, J. N., et al. (2020). The impact of rapid exome sequencing on medical management of critically ill children. J. Pediatr. 226, 202–212.e1. doi:10.1016/j.jpeds.2020.06.020
Grant, P., Langlois, S., Lynd, L. D., Austin, J. C., Elliott, A. M., Dragojlovic, N., et al. (2021). Out-of-pocket and private pay in clinical genetic testing: A scoping review. Clin. Genet. 100, 504. doi:10.1111/cge.14006
Lavelle, T. A., Feng, X., Keisler, M., Cohen, J. T., Neumann, P. J., Prichard, D., et al. (2022). Cost-effectiveness of exome and genome sequencing for children with rare and undiagnosed conditions. Genet. Med. [Internet] 24 (6), 1349–1361.doi:10.1016/j.gim.2022.03.005
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA - J. Am. Med. Assoc. 312 (18), 1880–1887. doi:10.1001/jama.2014.14604
Lightbourne, M., Wolska, A., Abel, B. S., Rother, K. I., Walter, M., Kushchayeva, Y., et al. (2021). Apolipoprotein CIII and angiopoietin-like protein 8 are elevated in lipodystrophy and decrease after metreleptin. J. Endocr. Soc. 5 (2), bvaa191. doi:10.1210/jendso/bvaa191
Liu, J., Zheng, Y., Huang, J., Zhu, D., Zang, P., Luo, Z., et al. (2021). Expanding the genotypes and phenotypes for 19 rare diseases by exome sequencing performed in pediatric intensive care unit. Hum. Mutat. 42 (11), 1443–1460. doi:10.1002/humu.24266
Lunke, S., Eggers, S., Wilson, M., Patel, C., Barnett, C. P., Pinner, J., et al. (2020). Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA - J. Am. Med. Assoc. 323 (24), 2503–2511. doi:10.1001/jama.2020.7671
Manickam, K., McClain, M. R., Demmer, L. A., Biswas, S., Kearney, H. M., Malinowski, J., et al. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of medical genetics and genomics (ACMG). Genet. Med. 23 (11), 2029–2037. doi:10.1038/s41436-021-01242-6
Meng, L., Pammi, M., Saronwala, A., Magoulas, P., Ghazi, A. R., Vetrini, F., et al. (2017). Use of exome sequencing for infants in intensive care units ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171 (12), e173438. doi:10.1001/jamapediatrics.2017.3438
Ouyang, X., Zhang, Y., Zhang, L., Luo, J., Zhang, T., Hu, H., et al. (2021). Clinical utility of rapid exome sequencing combined with mitochondrial DNA sequencing in critically ill pediatric patients with suspected genetic disorders. Front. Genet. 12, 725259. doi:10.3389/fgene.2021.725259
Pode-Shakked, B., Barel, O., Singer, A., Regev, M., Poran, H., Eliyahu, A., et al. (2021). A single center experience with publicly funded clinical exome sequencing for neurodevelopmental disorders or multiple congenital anomalies. Sci. Rep. 11 (1), 19099. doi:10.1038/s41598-021-98646-w
Pode-Shakked, B., Ben-Moshe, Y., Barel, O., Regev, L. C., Kagan, M., Eliyahu, A., et al. (2022). A multidisciplinary nephrogenetic referral clinic for children and adults—Diagnostic achievements and insights. Pediatr. Nephrol. 37 (7), 1623–1646. doi:10.1007/s00467-021-05374-4
Pode-Shakked, B., Vivante, A., Barel, O., Padeh, S., Marek-Yagel, D., Veber, A., et al. (2019). Progressive Pseudorheumatoid Dysplasia resolved by whole exome sequencing: A novel mutation in WISP3 and review of the literature. BMC Medical Genetics 20 (1), 53. doi:10.1186/s12881-019-0787-x
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18 (7), 696–704. doi:10.1038/gim.2015.148
Scholz, T., Blohm, M. E., Kortüm, F., Bierhals, T., Lessel, D., Van Der Ven, A. T., et al. (2021). Whole-exome sequencing in critically ill neonates and infants: Diagnostic yield and predictability of monogenic diagnosis. Neonatology 118 (4), 454–461. doi:10.1159/000516890
Schroeder, B. E., Gonzaludo, N., Everson, K., Than, K. S., Sullivan, J., Taft, R. J., et al. (2021). The diagnostic trajectory of infants and children with clinical features of genetic disease. npj Genomic Med. 6 (1), 98. doi:10.1038/s41525-021-00260-2
Scollon, S., Eldomery, M., Reuther, J., Desrosiers, L., Lin, F., Potter, S., et al. (2021). Clinical and molecular features of pediatric cancer patients with Lynch syndrome. Mol. Genet. Metab. 69, e29859. doi:10.1002/pbc.29859
Singleton, A. B. (2011). Exome sequencing: A transformative technology. Lancet Neurology 10, 942–946. doi:10.1016/S1474-4422(11)70196-X
Splinter, K., Adams, D. R., Bacino, C. A., Bellen, H. J., Bernstein, J. A., Cheatle-Jarvela, A. M., et al. (2018). Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379 (22), 2131–2139. doi:10.1056/NEJMoa1714458
Ta-Shma, A., Khan, T. N., Vivante, A., Willer, J. R., Matak, P., Jalas, C., et al. (2017). Mutations in TMEM260 cause a pediatric neurodevelopmental, cardiac, and renal syndrome. Am. J. Hum. Genet. 100 (4), 666–675. doi:10.1016/j.ajhg.2017.02.007
Tirosh, I., Spielman, S., Barel, O., Ram, R., Stauber, T., Paret, G., et al. (2019). Whole exome sequencing in childhood-onset lupus frequently detects single gene etiologies. Pediatr. Rheumatol. 17 (1), 52. doi:10.1186/s12969-019-0349-y
Topaz, O., Shurman, D. L., Bergman, R., Indelman, M., Ratajczak, P., Mizrachi, M., et al. (2004). Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet. 36 (6), 579–581. doi:10.1038/ng1358
Valencia, C. A., Husami, A., Holle, J., Johnson, J. A., Qian, Y., Mathur, A., et al. (2015). Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: A pediatric center’s experience. Front. Pediatr. 3, 67. doi:10.3389/fped.2015.00067
Wise, A. L., Manolio, T. A., Mensah, G. A., Peterson, J. F., Roden, D. M., Tamburro, C., et al. (2019). Genomic medicine for undiagnosed diseases. Lancet 394, 533–540. doi:10.1016/S0140-6736(19)31274-7
Xue, Y., Ankala, A., Wilcox, W. R., and Hegde, M. R. (2015). Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 17, 444–451. doi:10.1038/gim.2014.122
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369 (16), 1502–1511. doi:10.1056/NEJMoa1306555
Keywords: exome sequencing (ES), general pediatrics, monogenic, hospitalized, inpatient
Citation: Kagan M, Semo-Oz R, Ben Moshe Y, Atias-Varon D, Tirosh I, Stern-Zimmer M, Eliyahu A, Raas-Rothschild A, Bivas M, Shlomovitz O, Chorin O, Rock R, Tzadok M, Ben-Zeev B, Heimer G, Bolkier Y, Gruber N, Dagan A, Bar Aluma BE, Pessach IM, Rechavi G, Barel O, Pode-Shakked B, Anikster Y and Vivante A (2023) Clinical impact of exome sequencing in the setting of a general pediatric ward for hospitalized children with suspected genetic disorders. Front. Genet. 13:1018062. doi: 10.3389/fgene.2022.1018062
Received: 12 August 2022; Accepted: 13 December 2022;
Published: 09 January 2023.
Edited by:
Keyue Ding, Mayo Clinic, United StatesReviewed by:
Xia Tang, Fudan University, ChinaMeredith Harris, Ann & Robert H. Lurie Children’s Hospital of Chicago, United States
Copyright © 2023 Kagan, Semo-Oz, Ben Moshe, Atias-Varon, Tirosh, Stern-Zimmer, Eliyahu, Raas-Rothschild, Bivas, Shlomovitz, Chorin, Rock, Tzadok, Ben-Zeev, Heimer, Bolkier, Gruber, Dagan, Bar Aluma, Pessach, Rechavi, Barel, Pode-Shakked, Anikster and Vivante. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asaf Vivante, YXNhZnZpdmFudGVAZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship