 Jialing Yu
Jialing Yu Na Chen1
Na Chen1 Min Chen
Min Chen Yeqing Qian
Yeqing Qian Minyue Dong
Minyue Dong- 1Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
- 2Key Laboratory of Reproductive Genetics Zhejiang University, Ministry of Education, Hangzhou, Zhejiang, China
- 3Key Laboratory of Women’s Reproductive Health of Zhejiang Province, Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Tetrasomy 9p is a rare syndrome characterized by fetal growth restriction, Dandy-Walker malformation, cardiac anomalies, and facial abnormalities and is discovered by ultrasound during the prenatal examination. Herein, we report a fetus of tetrasomy 9p without obvious phenotypic manifestations during the first trimester that was identified by non-invasive prenatal testing (NIPT). NIPT revealed that the gain of 9p24.3–9p11 that was approximately 46.36 Mb in size. Karyotyping of amniocytes indicated an additional marker in all metaphase. Chromosome microarray and fluorescence in situ hybridization on uncultured amniocytes revealed tetrasomic of 9p24.3q13, and that the supernumerary chromosome is a dicentric isochromosome consisted of two copies of the 9p arm. Taken together, it was indicated that the fetal karyotype was 47,XY,+idic (9) (q13), and that multiple techniques are crucial to the prenatal diagnosis.
Introduction
Tetrasomy 9p (T9p), which was first defined in 1973 (Ghymers et al., 1973), is a rare abnormality typically resulting from a supernumerary isochromosome and mostly documented after birth. The phenotype of T9p varies from fetuses with multiple abnormalities to phenotypically normal adults (Bellil et al., 2020; Shu et al., 2021). Fetuses with T9p usually exhibit abnormal ultrasound findings including facial clefts, fetal growth retardation (FGR), and Dandy-Walker variant (Chen et al., 2007). So far, few cases have been diagnosed prenatally.
Non-invasive prenatal testing (NIPT) is emerging as a robust technique to screen for trisomies in 13, 18, 21, and sex chromosome (Lo et al., 1997; Lo et al., 1998; Song et al., 2013; Yin et al., 2015; Gross et al., 2016). Furthermore, its ability in screening for subchromosomal abnormalities such as Cri-du-chat deletions, 1p36 deletion syndrome, Wolf-Hirschhorn syndrome, Prader-Willi deletions, or Angelman deletions, has been proven as shown by high predictive positive value (Liu et al., 2016; Liang et al., 2019).
In the current investigation, we present a case of non-mosaic T9p that was identified through NIPT and validated by the combination of karyotyping, chromosome microarray (CMA), and fluorescence in situ hybridization (FISH).
Patients and methods
Case presentation
A healthy 37-years-old pregnant woman, who claimed no family history of genetic abnormalities and previously delivered two healthy boys, was referred to Women’s Hospital, School of medicine, Zhejiang University. She had unremarkable ultrasound screening at 12th week of gestation that revealed a normal nuchal translucency (NT) and the presence of nasal bone, while she received NIPT at the 15th week of gestation because of advanced maternal age. NIPT showed a duplication at 9p24.3p11.2. Amniocentesis was performed at the 18th week of gestation, and the fetal sample was analyzed by karyotyping, CMA, and FISH.
After genetic counseling, the woman and her husband decided to terminate the pregnancy; however, they declined to undergo a fetal MRI and fetal autopsy.
Non-invasive prenatal testing screening
NIPT procedures, including cell-free DNA extraction, library construction, and next-generation sequencing (NGS) were performed as described previously (Chen et al., 2019). Bioinformatic methods combined with a locally weighted polynomial regression were used to eliminate GC-bias, and a binary hypothesis was performed to obtain a higher accuracy for NIPT detection. Low coverage whole genome sequencing of plasma DNA was carried out on each sample, resulting in 10.19 million unique reads that corresponds to 0.1 × human genome depth. A fetal copy-number analysis (CNV) was performed to detect subchromosomal deletion and duplication, as previously described (Chen et al., 2019).
DNA extraction
DNA extraction was performed as described previously (Qian et al., 2019). Briefly, 10 ml maternal blood sample was centrifuged at 1,600 g for 10 min. The supernatant was re-centrifuged at 14,000 g for another 10 min. The plasma fraction was aliquoted and stored at −80°C for subsequent NIPT. The amniotic fluid was centrifuged at 1,600 g for 10 min. Fetal DNA from the centrifuged amniotic fluid cells were then extracted with QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany).
Amniocentesis and cytogenetic analysis
Transabdominal amniocentesis was performed under the real-time sonographic guidance. A total of 20 ml of amniotic fluid was withdrawn after discarding the first 2 ml of amniotic fluid (Jindal et al., 2022). Fetal amniocytes were cultured, and GTG-banding was performed according to standard cytogenetic procedures, yielding a 320–400 band level with a resolution of around 10 Mb. Generally, 30 metaphases were counted, and 5 metaphases were analyzed (Jin et al., 2021).
Chromosome microarray
Genomic DNA was extracted with the GentraPuregene Kit (Qiagen, Germany) from fetal amniotic cells. CytoScanTM HD array (Affymetrix, United States) was used in copy number analysis according to the manufacture’s instructions. The array is characterized by > 2,600,000 CNV markers, including 750,000 SNP probes and >1,900,000 non–polymorphism probes for comprehensive whole genome coverage. Chromosome Analysis Suite (ChAS) software (Affymetrix, United States) was used to visualize and analyze the results. The reporting threshold of the copy number result was set at 500 kb with marker count ≥50 for gains and 200 kb with a marker count of ≥50 for losses. The analysis was based on the GRCh37/hg19 assembly.
Fluorescence in situ hybridization analysis
Amniocytes were quadruple stained with the chromosome 9 subtelomeric p, q, and centromeric probe (Vysis, Downers Grove, IL) and DAPI (Vysis, Downers Grove, IL). The slides were hybridized according to the manufacture’s instruction and counterstained with DAPI, then analyzed by Zeiss Imager A2 microscope (Zeiss, France). Image acquisition was subsequently performed using a CCD camera with Isis (FISH Imaging System, MetaSystems, Germany).
Results
Non-invasive prenatal testing results
NIPT showed negative for other chromosomes expect for the chromosome 9 with a fetal fraction DNA concentration of 10.72%. Additionally, an elevated amount of DNA from the p-arm of chromosome 9 was observed with a mean t-score of 11.528, suggesting a gain of approximately 46.36 Mb that encompassed chromosome bands 9p24.3–9p11 (Figure 1).
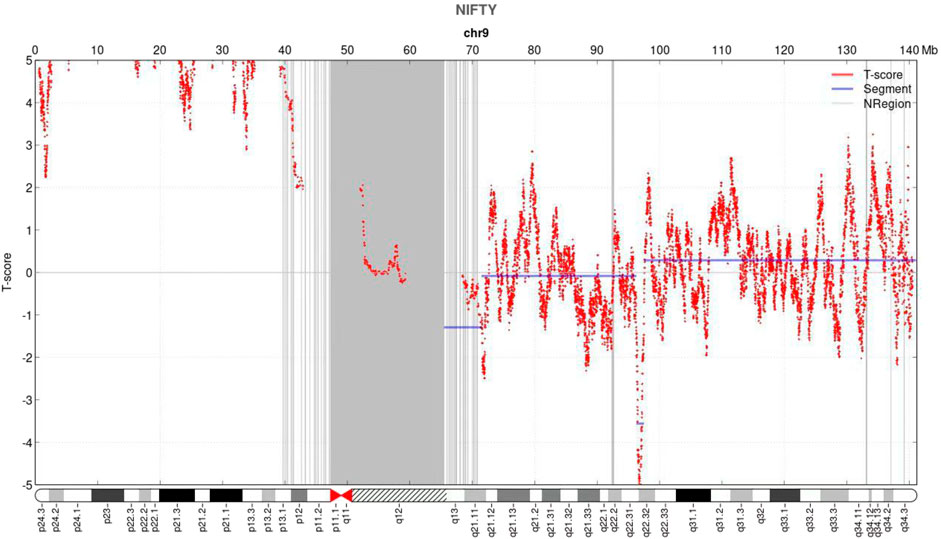
FIGURE 1. Non-invasive prenatal testing (NIPT) results of fetal chromosome 9. The horizontal axis represents genomic location (Mb), and the vertical axis represents t-score. NIPT revealed an increase in the signal of the p arm of chromosome 9.
Karyotyping, chromosome microarray, and fluorescence in situ hybridization results
Fetal karyotyping of the cultured aminocytes showed a male karyotype with the presence of supernumerary marker chromosome (SMC) (30/30) (Figure 2). CMA of the fetal DNA extracted from the uncultured aminocytes revealed a gain of approximately 68.126 Mb spanning from 9pter to 9q13 with a four-fold dose as indicated by five lines in both allele difference and BAF graphics (Figure 3). The array karyotype was: arr [GRCh37] 9p24.3q13 (203862_68330127)x4 (Figure 3).
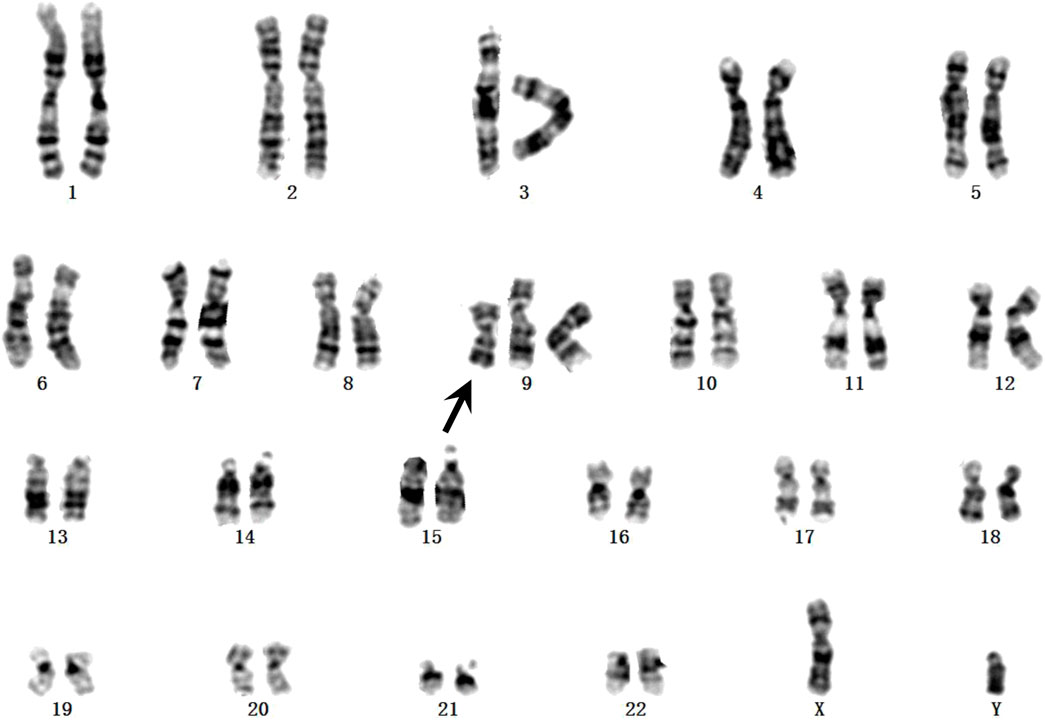
FIGURE 2. GTG-banding from the amniotic fluid sample. The supernumerary chromosome is shown by black arrow.
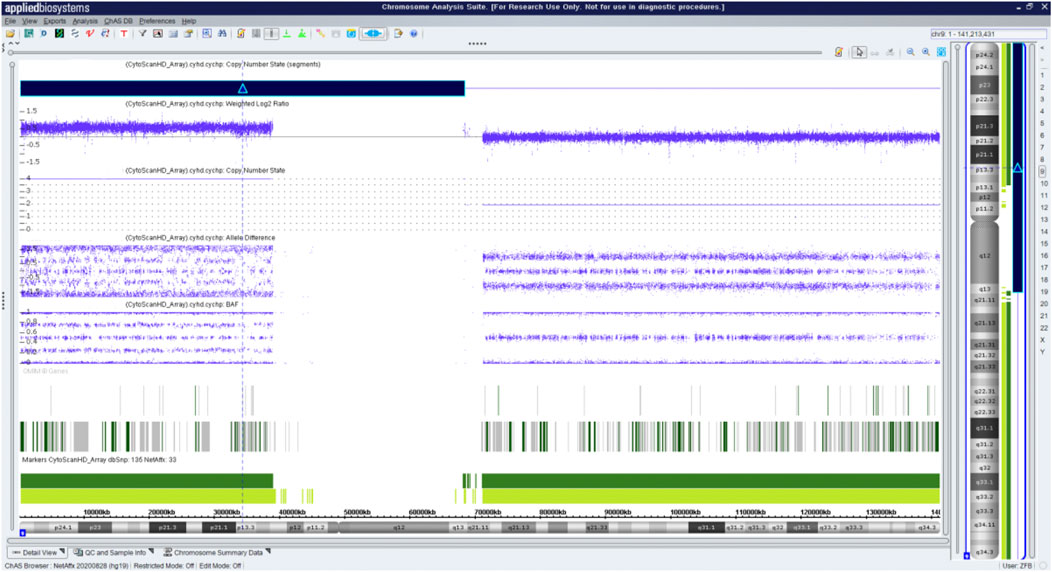
FIGURE 3. Chromosome microarray on DNA prepared from uncultured amniocytes. Results for chromosome 9 showing gain of the entire p arm and a portion of the q arm.
These findings suggest that the SMC was a tetrasomy of 9p. The gain of the short arm of chromosome 9 was confirmed by FISH, which was carried out using the chromosome 9 specific subtelomeric p, q, and centromeric probe (Figure 4). FISH showed that the SMC was an isochromosome consisting of two copies of the entire short arm and the heterochromatic region of the long arm of a chromosome 9 with two centromeres. Based on karyotyping, CMA and FISH, the fetal karyotype was 47,XY,+idic (9) (q13).ish idic (9) (q13) (305J7-T7+,D9Z1+,D9Z1+,305J7-T7+).arr [GRCh37] 9p24.3q13 (203862_68330127)x4.
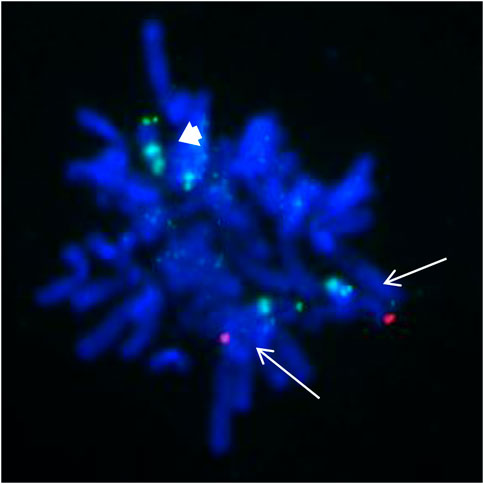
FIGURE 4. FISH from the uncultured amniocytes. Green signals indicate the subtelomeric part of chromosome 9p; red signals indicate the subtelomeric part of chromosome 9q; aqua signals indicate the centromeic of chromosome 9. Two normal chromosome 9 contain both green and red signals as showed with white arrow. The isodicentric chromosome 9p (bold white arrow) with two green signals and two aqua signals.
Discussion
Herein, we reported a prenatal case of tetrasomy 9p that presented without obvious ultrasound anomalies during the first trimester. To the best of our knowledge, our case is the first prenatal case of non-mosaic tetrasomy 9p identified through NIPT.
NIPT may detect CNVs with a size greater 10 Mb with high sensitivity and specificity (Lo et al., 1997; Lo et al., 1998; Song et al., 2013; Yin et al., 2015; Gross et al., 2016). Additionally, it has good performance in detecting known microduplication/microdeletion syndromes (MMS), which are smaller than 10 Mb, such as DiGeorge (93%) and 22q11.22 microduplication (68%) (Liang et al., 2019). Our data showed that NIPT provided quite precise duplication localization which was validated by amniocyte CMA results, suggesting the reliability of NIPT in detecting rare chromosome anomalies such as tetrasomy 9p.
Besides, NIPT can screen MMS before visible abnormalities are revealed on routine prenatal ultrasound examination. Most T9p fetuses were identified through ultrasound findings. Among the 60% of cases reviewed by Vinksel et al. (2019) fetuses with T9p were identified through ultrasound performed during the second and third trimesters. Among the cases reported in the first trimester, increased NT was the most common features, which can be present as an isolated phenotype or accompanied by other anomalies (cleft lip and/or palate, facial dysmorphism and skeletal abnormalities) (Vinksel et al., 2019; Kok Kilic et al., 2022). In our case, only the first trimester ultrasound scan was performed; therefore, other common abnormalities associated with T9p might not have displayed. In another case of mosaic T9p identified through NIPT, the pregnant women presented with a normal NT and nasal bone in the first trimester, while showed isolated persistent left superior vena cava until 25th week gestation ultrasound examination (Wang et al., 2015). Both this case and our case indicate that NIPT is superior to echography in screening for early prenatal abnormalities.
It was suggested that the degree of the mosaicism is associated with the severity of the phenotype of T9p. However, the severity of the phenotype of T9p is not linearly correlated with the mosaic level of the supernumerary chromosome in lymphocyte samples. A case with full tetrasomy 9p in the blood, 65% mosaic in the buccal mucosa, was a health woman who was accidentally identified due to her previous pregnancy with an inv (7) baby (Papoulidis et al., 2012). Among the 8 cases reported with a clinically normal phenotype, the tetrasomic clone ranged from 6% to 100% in peripheral blood lymphocytes, except in five cases that had fertility related issues (5/8) (Papoulidis et al., 2012; Bellil et al., 2020; Shu et al., 2021).
One might speculate that the tissue-limited mosaicism is related to severity of the phenotype of T9p. Supporting this, patients with i (9p) in fibroblasts tend to have more severe manifestations than those whose i (9p) is limited to lymphocytes, especially in terms of cardiac defects and viability (El Khattabi et al., 2015). Numerous data suggest that isochromosome 9p is predominantly present in peripheral blood but with a lower frequency in cell lines derived from the skin, amniotic fluid, or chorionic villus sampling. Hence, even a normal karyotype from amniocytes does not necessarily rule out the possibility of tetrasomy 9p (Cuoco et al., 1982; Papenhausen et al., 1990; Schaefer et al., 1991; Grass et al., 1993; Lloveras et al., 2004; McAuliffe et al., 2005). Additionally, the tissue-limited mosaicism mechanism at some extent unveiled that the prenatal cases of T9p showed numerous ultrasound abnormalities even with low-level mosaic status as the amniocytes are a mixture of cells derived from different germ layers (Chen et al., 2007). Based on published reports, non-mosaic fetuses have poorer prognosis, which explains the higher incidence of early death, FGR, Dandy-Walker malformation, and other congenital anomalies (Tang et al., 2004; Tan et al., 2007; El Khattabi et al., 2015). As such, the patient in the present study decided to terminate the pregnancy after genetic consultation.
Another factor that may affect the severity of the phenotype is breakpoint position; however, this remains controversial (Grass et al., 1993; Stumm et al., 1999; El Khattabi et al., 2015; Pinto et al., 2018; Vinksel et al., 2019). Some suggested that the phenotype will be more severe in patients that harbor the large portion of 9q extending to 9q21, which encompasses a large duplicated region with several OMIM morbid genes compared with those containing exclusively the entire 9p (El Khattabi et al., 2015; Pinto et al., 2018; Vinksel et al., 2019). A total of 21 prenatal cases with ultrasound anomalies have been reviewed and showed that involvement of the 9q region appears to have similar phenotypes to the p10 region in terms of facial anomalies, FGR, central nervous system dysfunction and cardiac anomalies (Table 1). Surprisingly, cardiac malformation seems to be much frequent when the region q12-q13 is involved.

TABLE 1. Malformations in relation to breakpoints among prenatal cases associated with T9p.
Due to the poor resolution of conventional cytogenetic techniques, CMA with high throughput has been widely complemented into prenatal examinations for fetuses with ultrasound anomalies as it offers several advantages, such as fast reporting (without further cell culture) and high resolution in detecting copy number changes. Furthermore, it can provide a more precise location of the breakpoint of the tetrasomy 9p, thus improving the understanding of the genotype-phenotype correlation. However, the breakpoints of 9p10, q12, and q13 are located in the heterochromatin region, wherein a few markers have been set in CMA, making it hazardous to define the precise localization. With the release of the T2T-CHM13 reference genome, long-reads based whole genome sequencing may overcome the remaining gaps and provide convincing breakpoints for the complex chromosome rearrangement, including in T9p (Aganezov et al., 2022; Nurk et al., 2022).
T9p usually occurs de novo, and recurrence has been observed in only one report, indicating gonadal mosaicism (El Khattabi et al., 2015). Most supernumerary chromosomes are of maternal origin, including T9p (Kotzot et al., 1996; Dutly et al., 1998). The supernumerary chromosome can be separated into two forms according to the number of centromeres: i (9) with a single centromeric region or two centromeres forming idic (9). Several models have been proposed to explain the formation of an isochromosome. One mechanism relies on homologous recombination (HR) when intra-chromosomal U-type recombination occurs during meiosis I followed by non-disjunction in meiosis II (Floridia et al., 1996; Kotzot et al., 1996; Dutly et al., 1998; Knijnenburg et al., 2007). This U-type exchange is more likely to result in a dicentric than in a monocentric isochromosome. Another mechanism refers to the formation of monocentric isochromosomes and relies on centromere misdivision during the premeiotic stage followed by non-disjuntion at meiosis I (Rivera et al., 1986); alternatively, a non-disjuntion at meiosis II is followed by post-zygotic centromere misdivision (de Ravel et al., 2004). However, Kotzot et al. (1996) oppose to the former hypothesis in the formation of isochromosome 18p as this would require two abnormal cell divisions.
In conclusion, tetrasomy 9p is a rare chromosome rearrangement that often occurs de novo. A prenatal case of tetrasomy 9p without any ultrasound abnormality during the first trimester was revealed by NIPT and confirmed by invasive diagnosis. NIPT can screen not only for canonical trisomy 13, 18, and 21 but also large fragment copy number changes, such as tetrasomy 9p before it manifests with a significant phenotype. Furthermore, multiple techniques, such as karyotyping, FISH and CMA, are critical for a precise prenatal diagnostic.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI repository with the Accession number PRJNA880763, with the URL: https://dataview.ncbi.nlm.nih.gov/object/PRJNA880763.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University (IRB-20220260-R). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Patient workups were contributed by MS and YQ Genetic analysis was contributed by JY and NC Pathological analysis was contributed by JY. Manuscript drafting was contributed by JY, MC, YQ, and MD. Final approval of the version to be submitted for publishing consideration was contributed by JY, NC, MC, MS, YQ, and MD.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 81900904, 82001625, and 81901382).
Acknowledgments
We thank the patients and volunteers for their supports during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aganezov, S., Yan, S. M., Soto, D. C., Kirsche, M., Zarate, S., Avdeyev, P., et al. (2022). A complete reference genome improves analysis of human genetic variation. Science 376 (6588), eabl3533. doi:10.1126/science.abl3533
Bellil, H., Herve, B., Herzog, E., Ayoubi, J. M., Vialard, F., and Poulain, M. (2020). A high level of tetrasomy 9p mosaicism but no clinical manifestations other than moderate oligozoospermia with chromosomally balanced sperm: A case report. J. Assist. Reprod. Genet. 37 (3), 573–577. doi:10.1007/s10815-020-01690-0
Cazorla Calleja, M. R., Verdú, A., and Félix, V. (2003). Dandy-Walker malformation in an infant with tetrasomy 9p. Brain Dev. 25 (3), 220–223. doi:10.1016/s0387-7604(02)00211-5
Chen, C. P., Chang, T. Y., Chern, S. R., Lee, C. C., Town, D. D., Lee, M. S., et al. (2007). Prenatal diagnosis of low-level mosaic tetrasomy 9p by amniocentesis. Prenat. Diagn. 27 (4), 383–385. doi:10.1002/pd.1678
Chen, C. P., Wang, L. K., Chern, S. R., Wu, P. S., Chen, Y. T., Kuo, Y. L., et al. (2014). Mosaic tetrasomy 9p at amniocentesis: Prenatal diagnosis, molecular cytogenetic characterization, and literature review. Taiwan. J. Obstet. Gynecol. 53 (1), 79–85. doi:10.1016/j.tjog.2013.12.002
Chen, M., Fu, X. Y., Luo, Y. Q., Qian, Y. Q., Pan, L., Wang, L. Y., et al. (2019). Detection of fetal duplication 16p11.2q12.1 by next-generation sequencing of maternal plasma and invasive diagnosis. J. Matern. Fetal. Neonatal Med. 32 (1), 38–45. doi:10.1080/14767058.2017.1369947
Cuoco, C., Gimelli, G., Pasquali, F., Poloni, L., Zuffardi, O., Alicata, P., et al. (1982). Duplication of the short arm of chromosome 9. Analysis of five cases. Hum. Genet. 61 (1), 3–7. doi:10.1007/bf00291321
de Ravel, T. J., Keymolen, K., van Assche, E., Wittevronghel, I., Moerman, P., Salden, I., et al. (2004). Post-zygotic origin of isochromosome 12p. Prenat. Diagn. 24 (12), 984–988. doi:10.1002/pd.956
Deurloo, K. L., Cobben, J. M., Heins, Y. M., de Ru, M., Wijnaendts, L. C., and van Vugt, J. M. (2004). Prenatal diagnosis of tetrasomy 9p in a 19-week-old fetus with dandy-walker malformation: A case report. Prenat. Diagn. 24 (10), 796–798. doi:10.1002/pd.850
Dhandha, S., Hogge, W. A., Surti, U., and McPherson, E. (2002). Three cases of tetrasomy 9p. Am. J. Med. Genet. 113 (4), 375–380. doi:10.1002/ajmg.b.10826
Di Vera, E., Liberati, M., Celentano, C., Calabrese, G., Guanciali-Franchi, P. E., Morizio, E., et al. (2008). Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J. Assist. Reprod. Genet. 25 (11-12), 577–580. doi:10.1007/s10815-008-9257-7
Dutly, F., Balmer, D., Baumer, A., Binkert, F., Schinzel, A., and Schinzel, A. (1998). Isochromosomes 12p and 9p: Parental origin and possible mechanisms of formation. Eur. J. Hum. Genet. 6 (2), 140–144. doi:10.1038/sj.ejhg.5200168
El Khattabi, L., Jaillard, S., Andrieux, J., Pasquier, L., Perrin, L., Capri, Y., et al. (2015). Clinical and molecular delineation of tetrasomy 9p syndrome: Report of 12 new cases and literature review. Am. J. Med. Genet. A 167 (6), 1252–1261. doi:10.1002/ajmg.a.36932
Floridia, G., Piantanida, M., Minelli, A., Dellavecchia, C., Bonaglia, C., Rossi, E., et al. (1996). The same molecular mechanism at the maternal meiosis I produces mono- and dicentric 8p duplications. Am. J. Hum. Genet. 58 (4), 785–796.
Grass, F. S., Parke, J. C., Kirkman, H. N., Christensen, V., Roddey, O. F., Wade, R. V., et al. (1993). Tetrasomy 9p: Tissue-limited idic(9p) in a child with mild manifestations and a normal CVS result. Report and review. Am. J. Med. Genet. 47 (6), 812–816. doi:10.1002/ajmg.1320470603
Gross, S. J., Stosic, M., McDonald-McGinn, D. M., Bassett, A. S., Norvez, A., Dhamankar, R., et al. (2016). Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11.2 deletion syndrome. Ultrasound Obstet. Gynecol. 47 (2), 177–183. doi:10.1002/uog.15754
Hengstschläger, M., Bettelheim, D., Drahonsky, R., Repa, C., Deutinger, J., and Bernaschek, G. (2004). Prenatal diagnosis of tetrasomy 9p with Dandy-Walker malformation. Prenat. Diagn. 24 (8), 623–626. doi:10.1002/pd.933
Jin, P., Gao, X., Wang, M., Qian, Y., Yang, J., Yang, Y., et al. (2021). Case Report: Identification of maternal low-level mosaicism in the dystrophin gene by droplet digital polymerase chain reaction. Front. Genet. 12, 686993. doi:10.3389/fgene.2021.686993
Jindal, A., Sharma, M., and Chaudhary, C. (2022). “Amniocentesis,” in StatPearls. (Treasure island (FL) (Florida,United States: StatPearls Publishing LLC.).
Knijnenburg, J., van Haeringen, A., Hansson, K. B., Lankester, A., Smit, M. J., Belfroid, R. D., et al. (2007). Ring chromosome formation as a novel escape mechanism in patients with inverted duplication and terminal deletion. Eur. J. Hum. Genet. 15 (5), 548–555. doi:10.1038/sj.ejhg.5201807
Kok Kilic, G., Pariltay, E., Karaca, E., Durmaz, B., Ekici, H., Imamoglu, M., et al. (2022). Prenatal diagnosis of a case with tetrasomy 9p confirmed by cytogenetics, FISH, microarray analysis and review. Taiwan. J. Obstet. Gynecol. 61 (1), 122–126. doi:10.1016/j.tjog.2021.10.003
Kotzot, D., Bundscherer, G., Bernasconi, F., Brecevic, L., Lurie, I. W., Basaran, S., et al. (1996). Isochromosome 18p results from maternal meiosis II nondisjunction. Eur. J. Hum. Genet. 4 (3), 168–174. doi:10.1159/000472191
Lazebnik, N., and Cohen, L. (2015). Prenatal diagnosis and findings of tetrasomy 9p. J. Obstet. Gynaecol. Res. 41 (7), 997–1002. doi:10.1111/jog.12706
Liang, D., Cram, D. S., Tan, H., Linpeng, S., Liu, Y., Sun, H., et al. (2019). Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet. Med. 21 (9), 1998–2006. doi:10.1038/s41436-019-0467-4
Liu, H., Gao, Y., Hu, Z., Lin, L., Yin, X., Wang, J., et al. (2016). Performance evaluation of NIPT in detection of chromosomal copy number variants using low-coverage whole-genome sequencing of plasma DNA. PLoS One 11 (7), e0159233. doi:10.1371/journal.pone.0159233
Lloveras, E., Pérez, C., Solé, F., Zamora, L., Lladonosa, A., Espinet, B., et al. (2004). Two cases of tetrasomy 9p syndrome with tissue limited mosaicism. Am. J. Med. Genet. A 124a (4), 402–406. doi:10.1002/ajmg.a.20447
Lo, Y. M., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W., et al. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet 350 (9076), 485–487. doi:10.1016/s0140-6736(97)02174-0
Lo, Y. M., Tein, M. S., Lau, T. K., Haines, C. J., Leung, T. N., Poon, P. M., et al. (1998). Quantitative analysis of fetal DNA in maternal plasma and serum: Implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 62 (4), 768–775. doi:10.1086/301800
McAuliffe, F., Winsor, E. J., and Chitayat, D. (2005). Tetrasomy 9p mosaicism associated with a normal phenotype. Fetal diagn. Ther. 20 (3), 219–222. doi:10.1159/000083909
McDowall, A. A., Blunt, S., Berry, A. C., and Fensom, A. H. (1989). Prenatal diagnosis of a case of tetrasomy 9p. Prenat. Diagn. 9 (11), 809–811. doi:10.1002/pd.1970091110
Nakamura-Pereira, M., Cima, L. C., Llerena, J. C., Guerra, F. A., and Peixoto-Filho, F. M. (2009). Sonographic findings in a case of tetrasomy 9p associated with increased nuchal translucency and Dandy-Walker malformation. J. Clin. Ultrasound 37 (8), 471–474. doi:10.1002/jcu.20612
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science 376 (6588), 44–53. doi:10.1126/science.abj6987
Papenhausen, P., Riscile, G., Miller, K., Kousseff, B., and Tedesco, T. (1990). Tissue limited mosaicism in a patient with tetrasomy 9p. Am. J. Med. Genet. 37 (3), 388–391. doi:10.1002/ajmg.1320370319
Papoulidis, I., Kontodiou, M., Tzimina, M., Saitis, I., Hamid, A. B., Klein, E., et al. (2012). Tetrasomy 9p mosaicism associated with a normal phenotype in two cases. Cytogenet. Genome Res. 136 (4), 237–241. doi:10.1159/000337520
Pinto, I. P., Minasi, L. B., Steckelberg, R., da Silva, C. C., and da Cruz, A. D. (2018). Mosaic tetrasomy of 9p24.3q21.11 postnatally identified in an infant born with multiple congenital malformations: A case report. BMC Pediatr. 18 (1), 298. doi:10.1186/s12887-018-1275-8
Qian, Y. Q., Wang, X. Q., Chen, M., Luo, Y. Q., Yan, K., Yang, Y. M., et al. (2019). Detection of fetal subchromosomal aberration with cell-free DNA screening led to diagnosis of parental translocation: Review of 11344 consecutive cases in a University hospital. Eur. J. Med. Genet. 62 (2), 115–123. doi:10.1016/j.ejmg.2018.06.012
Rivera, H., Rivas, F., and Cantú, J. M. (1986). On the origin of extra isochromosomes. Clin. Genet. 29 (6), 540–541. doi:10.1111/j.1399-0004.1986.tb00557.x
Schaefer, G. B., Domek, D. B., Morgan, M. A., Muneer, R. S., and Johnson, S. F. (1991). Tetrasomy of the short arm of chromosome 9: Prenatal diagnosis and further delineation of the phenotype. Am. J. Med. Genet. 38 (4), 612–615. doi:10.1002/ajmg.1320380422
Shu, W., Cheng, S. S. W., Xue, S., Chan, L. W., Soong, S. I., Kan, A. S. Y., et al. (2021). First case report of maternal mosaic tetrasomy 9p incidentally detected on non-invasive prenatal testing. Genes (Basel) 12 (3), 370. doi:10.3390/genes12030370
Song, Y., Liu, C., Qi, H., Zhang, Y., Bian, X., and Liu, J. (2013). Noninvasive prenatal testing of fetal aneuploidies by massively parallel sequencing in a prospective Chinese population. Prenat. Diagn. 33 (7), 700–706. doi:10.1002/pd.4160
Stumm, M., Tönnies, H., Mandon, U., Götze, A., Krebs, P., Wieacker, P. F., et al. (1999). Mosaic tetrasomy 9p in a girl with multiple congenital anomalies: Cytogenetic and molecular-cytogenetic studies. Eur. J. Pediatr. 158 (7), 571–575. doi:10.1007/s004310051149
Tan, Y. Q., Chen, X. M., Hu, L., Guan, X. Y., and Lu, G. X. (2007). Prenatal diagnosis of nonmosaic tetrasomy 9p by microdissection and FISH: Case report. Chin. Med. J. 120 (14), 1281–1283. doi:10.1097/00029330-200707020-00016
Tang, W., Boyd, B. K., Hummel, M., and Wenger, S. L. (2004). Prenatal diagnosis of tetrasomy 9p. Am. J. Med. Genet. A 126a (3), 328. doi:10.1002/ajmg.a.20511
Vinkšel, M., Volk, M., Peterlin, B., and Lovrecic, L. (2019). A systematic clinical review of prenatally diagnosed tetrasomy 9p. Balk. J. Med. Genet. 22 (1), 11–20. doi:10.2478/bjmg-2019-0012
Wang, H., Xie, L. S., Wang, Y., and Mei, J. (2015). Prenatal diagnosis of mosaic tetrasomy 9p in a fetus with isolated persistent left superior vena cava. Taiwan. J. Obstet. Gynecol. 54 (2), 204–205. doi:10.1016/j.tjog.2014.12.005
Keywords: tetrasomy 9p syndrome, NIPT, prenatal diagnosis, CMA, karyotyping
Citation: Yu J, Chen N, Chen M, Shen M, Qian Y and Dong M (2022) Case Report: Prenatal diagnosis of fetal tetrasomy 9p initially identified by non-invasive prenatal testing. Front. Genet. 13:1020525. doi: 10.3389/fgene.2022.1020525
Received: 16 August 2022; Accepted: 10 October 2022;
Published: 31 October 2022.
Edited by:
Can Liao, Guangzhou Women and Children’s Medical Center, ChinaReviewed by:
Horacio Rivera, University of Guadalajara, MexicoHaydee Rosas-Vargas, Unidad de Investigación Médica en Genética Humana, IMSS, Mexico
Copyright © 2022 Yu, Chen, Chen, Shen, Qian and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=