 Qingming Wang1,2†
Qingming Wang1,2† Haiming Yuan
Haiming Yuan- 1Dongguan Maternal and Child Healthcare Hospital, Dongguan, China
- 2Dongguan Institute of Reproductive and Genetic Research, Dongguan, China
Pathogenic variants in the RASopathy-causing SHOC2 gene have been suggested to cause Noonan syndrome-like with loose anagen hair (NS/LAH). This condition is characterized by facial features resembling Noonan syndrome (NS), short stature, growth hormone deficiency (GHD), cognitive deficits, cardiac defects, and ectodermal abnormalities, including easily pluckable, sparse, thin, slow-growing hair, hyperpigmented skin and hypernasal voice. The mutation spectrum of SHOC2 is narrow, and only 8 pathogenic variants have been identified. Here, we report a 5-year-3-month-old Chinese female who displays characteristics typical of NS and has normal neurodevelopment. Trio-based whole-exome sequencing (WES) revealed a de novo variant (c.1231A>G, p.Thr411Ala) in SHOC2. This variant has been recently reported in one subject in the literature who displayed facial features typical of NS and also presented with significant speech delays, moderate intellectual disabilities, epilepsy, bilateral sensorineural deafness and renal dysplasia. The differential phenotypes between these subjects deserve to be further investigated. Next, we reviewed the clinical pictures of NS/LAH and noticed that a recurrent SHOC2 Ser2Gly variant was more likely to result in delayed neurodevelopment and short stature, compared to other SHOC2 variants. And growth hormone (GH) therapy could improve height prognosis. It was noticed that the slight sleep problems and friendly and relatively mature personality observed in our patient may be a novel phenotype of NS/LAH. Our study reconfirms the pathogenic nature of the SHOC2 Thr411Ala variant. It also provides insights into the genotype-phenotype relationship in NS/LAH and a foundation for its genetic counseling, diagnosis and treatment.
Introduction
Noonan syndrome-like with loose anagen hair (NS/LAH) (MIM:607721) is a very rare autosomal dominant RASopathy. This condition was characterized by facial anomalies similar to those observed in NS, short stature frequently with growth hormone deficiency (GHD), intellectual disability, ectodermal abnormalities, including easily pluckable, sparse, thin slow-growing hair, recurrent eczema and ichthyosis, dark pigmented skin, hypernasal voice and cardiac defects (especially dysplasia of the mitral valve and septal defects) (Cordeddu et al., 2009; Hoban et al., 2012; Capalbo et al., 2012b; Gripp et al., 2013; Baldassarre et al., 2014; Hannig et al., 2014; Motta et al., 2019; Motta et al., 2022). Currently, only 8 pathogenic SHOC2 variants have been reported to cause NS/LAH through gain-of-function mechanisms (Cordeddu et al., 2009; Hannig et al., 2014; Young et al., 2018; Motta et al., 2019; Motta et al., 2022). SHOC2 is a scaffold protein comprising an N-terminal lysine-rich region and subsequent 19 leucine-rich repeats (LRRs) that plays a crucial role in the activation of the ERK1 (MAPK3; 601795)/ERK2 (MAPK1; 176948) signaling pathway (Cordeddu et al., 2009; Rodriguez-Viciana et al., 2006; Matsunaga-Udagawa et al., 2010). Among all variants, a recurrent activating mutation in SHOC2 (p.Ser2Gly) has been frequently reported in NS/LAH patients with homogeneous clinical manifestations (Cordeddu et al., 2009; Gripp et al., 2013). The remaining pathogenic variants in SHOC2 might cause a milder phenotype (Hannig et al., 2014; Motta et al., 2019; Motta et al., 2022). Remarkably, a de novo SHOC2 variant (Thr411Ala) was previously reported in Subject 3 in the literature. This subject presented with facial features typical of NS and also displayed severe phenotypes including neurodevelopmental delay, epilepsy, bilateral sensorineural deafness and renal dysplasia (Motta et al., 2022). Here, we again identified the de novo SHOC2 variant (c.1231A>G, p.Thr411Ala) in a 5-year-3-month-old girl who only displayed characteristics typical of NS/LAH. The report reconfirms the pathogenic nature of the variant. Whereas, the differential phenotypes between the two subjects deserve to be further investigated. Next, we systematically reviewed the clinical characteristics of individuals with NS/LAH.
Materials and methods
Ethical compliance
This study was approved by the Ethics Committee of Dongguan Maternal and Child Healthcare Hospital (DMCH 2020-6). Written informed consent was obtained from the legal guardians for the publication of any potentially identifiable images or data included in this article.
Trio-based whole-exome sequencing (WES)
Trio-based whole-exome sequencing (Illumina, San Diego, CA, United States) was performed for the family. Sequencing was performed with an Illumina NovaSeq 6,000 (Illumina, San Diego, CA, United States). The bcl2fastq2 Conversion Software (v2.20) was employed for extracting Fastq files, and all reads were mapped to the human genome (GRCh37/hg19) by using BWA (v0.2.10) with default parameters. The Genome Analysis Toolkit (GATK; v.3.7) HaplotypeCaller was applied for identifying variants. The aligned reads were visualized by using the Integrated Genome Viewer (IGV). Common variants were filtered based on their frequencies in the databases of the Exome Sequencing Project (https://esp.gs.washington.edu), the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org), or 1,000G (http://www.1000genomes.org), and our internal database. The suspected variant was verified by Sanger sequencing. The pathogenicity of the sequence variants was interpreted according to ACMG/AMP guidelines (Richards et al., 2015).
Results
Clinical report
The proband was the first female child of unrelated Chinese parents. Her 2-year-old younger sister was apparently healthy. Her birth measurements were normal: length 49 cm, weight 2.85 kg and head circumference 34.5 cm.
Failure to thrive prompted hospitalization at the age of 4 years and 2 months. At this time, her height was 91.5 cm (<-3.3 SD), her weight was 12 kg (-3 SD) and her head circumference was 50.8 cm (1 SD). She had normal motor development. She raised her head at 3 months, sat alone at 7 months and independently walked at 1 year 2 months. She had craniofacial anomalies characterized by relatively macrocephaly, hypertelorism, long eyelashes, unilateral left ptosis, downslanting palpebral fissures, low-set posteriorly angulated ears, overfolded pinnae, teeth dysplasia, and hair anomalies, including sparse, thin, slow-growing hair that was not loose. She had mild frontal bossing, and her forehead appeared broad and square (Figure 1). Mild aortic and tricuspid regurgitation was revealed by echocardiography. Deep palmar lines and hypotonia were noted. No feeding difficulties were recorded, but she was a picky eater. She displayed slight sleep problems. She was friendly and seemed to have a relatively mature personality beyond her age. She had recurrent eczema and darkly pigmented skin. Her neck, chest, and back were non-dysmorphic. Her bone age was delayed by 2 years at chronological age 4 years 2 months. An endocrinological evaluation revealed partial GHD for IGF1 (23.0 ng/ml, n: 41.3-229.2) and IGFBP-3 (1.78 ng/ml, n: 1.0-4.7). GH therapy (50 μg/kg/day) was initiated at the age of 4 years and 5 months with height 92.0 cm (−3.5 SD) and weight 12.5 Kg (−2.8 SD). This resulted in increased growth velocity, with a height of 101 cm (−2.5 SD) and a weight of 14.5 kg (− 2.0 SD) at 5 years and 3 months of age. IGF1 level was remarkably improved (IGF1: 101 ng/ml). No adverse events occurred. She displayed normal cognitive and language abilities through professional neurological assessments. Brain magnetic resonance imaging was normal at this age. The patient was clinically diagnosed with NS assessed by experienced clinical specialists.
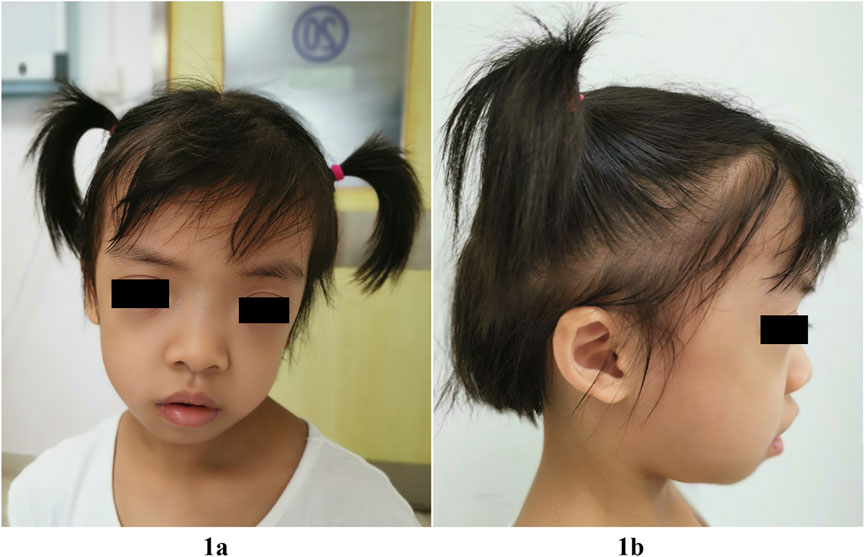
FIGURE 1. Facial features of our patient with the SHOC2 variant (p.Thr411Ala) at 5-year-3-month of age. Note relatively macrocephaly, broad and square forehead, frontal bossing, hypertelorism, long eyelashes, unilateral left ptosis, downslanting palpebral fissures (1A), low-set posteriorly angulated ears, and overfolded pinnae (1B), sparse, thin, slow-growing hair, but not loose (1A,1B).
Genetic analysis
Trio-based WES identified a heterozygous variant, c.1231A>G, p.Thr411Ala, in the SHOC2 gene in our patient. The variant was validated by bidirectional Sanger sequencing, which also demonstrated its de novo event (Figure 2A) (PS2). The Thr411 residue is located in LRR14 and is highly conserved among different species (PM1) (Figure 2B). This variant had not been reported in the public databases (ExAC, gnomAD, 1000 Genomes Project) or our internal database (PM2). This variant was predicted to have a deleterious effect on the gene product by multiple in silico prediction tools (SIFT, MutationTaster, PolyPhen-2). In addition, the patient’s phenotypes were highly consistent with those of NS/LAH (PP4), and WES also excluded other possible known genetic causes. Furthermore, this variant has been previously reported to be a de novo event in Subject 3 with NS-like facial features (Motta et al., 2022). Thus, this variant was categorized as clinically pathogenic according to ACMG/AMP guidelines (PS2+PM1+PM2+PP3+PP4) (PS: pathogenic strong; PM: pathogenic moderate; PP: pathogenic supporting).
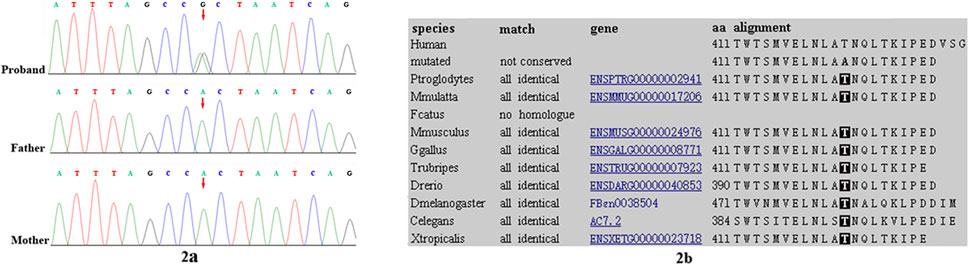
FIGURE 2. Sanger sequencing results for (A) our patient, and the patient’s (B) mother and (C) father. The analysis demonstrated the presence of a missense SHOC2 variant (c.1231A>G, p.Thr411Ala; red arrow) in our patient and the absence of the variant in her parents (2A). Location of patient’s SHOC2 missense variant (p. Thr411Ala) on a highly conserved multi-species alignment of SHOC2 protein sequence, shaded area stands for the residue Thr411 (2B).
Discussion
The SHOC2 gene was infrequently reported in NS patients. Currently, only 8 pathogenic variants in SHOC2 were identified to cause NS/LAH through gain-of-function mechanisms (Cordeddu et al., 2009; Hannig et al., 2014; Young et al., 2018; Motta et al., 2019; Motta et al., 2022). Here, we reported a 5-year-3-month-old Chinese female who has distinctive facial features (relatively macrocephaly, hypertelorism, long eyelashes, unilateral left ptosis, downslanting palpebral fissures, low-set posteriorly angulated ears, overfolded pinnae, teeth dysplasia, and sparse, thin, slow-growing hair), short stature, partial GHD, recurrent eczema and darkly pigmented skin, deep palmar lines and hypotonia, but she demonstrated normal cognitive and language development. WES identified a de novo missense variant (p.Thr411Ala) in SHOC2, and WES excluded other possible causal variants. This variant was classified as clinically pathogenic and was likely to be responsible for our patient’s clinical phenotypes.
Next, we reviewed the clinical features of individuals with pathogenic SHOC2 variants to comprehensively profile the condition (Table 1). All patients had the typical facial gestalt and sparse eyebrows, and easily pluckable, sparse, thin, slow-growing hair of NS/LAH. Most patients had short/broad neck (7/9), motor delay (6/9) and cardiac defects (6/9). The other phenotypes include short stature (4/9), darkly pigmented skin (4/9), recurrent eczema or ichthyosis (4/9) and feeding difficulties (3/9). Furthermore, the features that hypotonia and deep palmar lines observed in our patient and Subject 1 may be uncommon characteristics of NS/LAH. A recurrent activating mutation in SHOC2 (p.Ser2Gly) has been frequently reported in NS/LAH patients with a higher prevalence of speech delays, cognitive deficits and abnormal intracranial structures (Cordeddu et al., 2009; Capalbo et al., 2012a; Gripp et al., 2013; Baldassarre et al., 2014; Li et al., 2019; Lores, 2020). Pathogenic variants other than p.Ser2Gly cause milder phenotypes with normal or mildly impaired speech or cognitive development (Hannig et al., 2014; Motta et al., 2022). Severe short stature is frequently reported in SHOC2 Ser2Gly variant patients who exhibit mild to moderate GHD and growth hormone insensitivity (GHI) and thus require a higher dose of GH therapy (e.g., 35–40 μg/kg/day) (Cordeddu et al., 2009; Capalbo et al., 2012a; Capalbo et al., 2012b; Mazzanti et al., 2013). Whereas, a previously reported patient with Ser2Gly variant who had severe short stature was remarkably improved by low-dose GH therapy (25 μg/kg/day) (Takasawa et al., 2015). Currently, short stature was only described in two subjects with pathogenic variants (Met173Val and Gln269Arg respectively) other than Ser2Gly, in the recently published literature. The two subjects have GHD and have good response to GH therapy (Motta et al., 2022). Our patient carrying the SHOC2 Thr411Ala variant had obvious short stature with partial GHD. The GH therapy (50 μg/kg/day) obviously improved the linear growth of our patient, with her height reaching -2.5 SD after 11 months of treatment. These findings suggest that short stature occurred in four of eight variants, and GH therapy would be beneficial to the improvement of height.
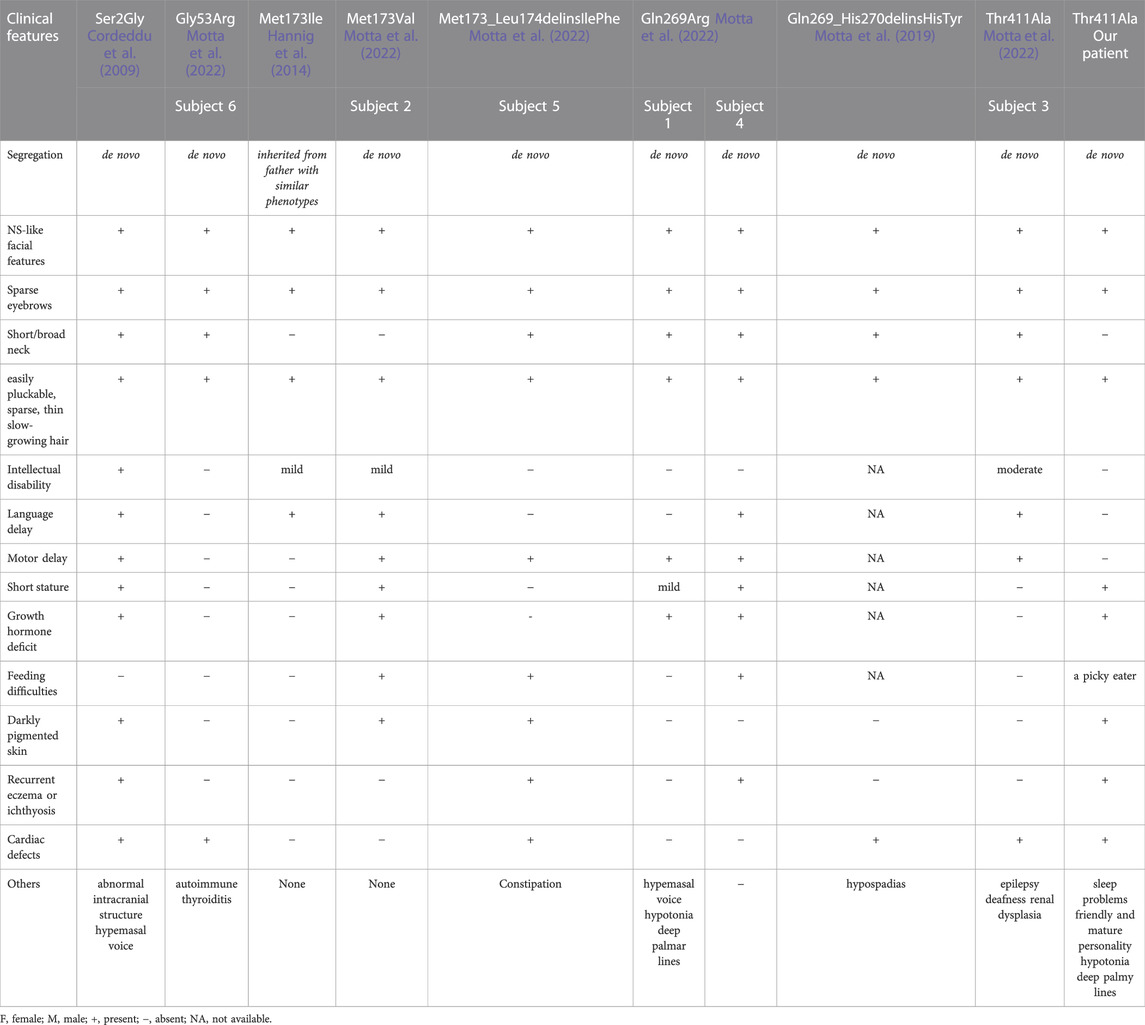
TABLE 1. Overview of variants and phenotypes observed in patients with NS/LAH.
Remarkably, in contrast to our patient with severe short stature, the de novo SHOC2 variant (Thr411Ala) was previously identified in Subject 3 in the recently published literature, who had normal height (Motta et al., 2022). Both individuals presented with facial features typical of NS. However, Subject 3 displayed severe phenotypes. Subject 3 showed significant speech delay, moderate intellectual disabilities and epilepsy, whereas our patient had normal neurodevelopment. Furthermore, Subject 3 suffered from bilateral sensorineural deafness requiring hearing aids and unilateral renal dysplasia due to ureteral reflux and requiring left nephrectomy (Motta et al., 2022). These phenotypes are not consistent with those of NS, and our patient did not show these features. Our patient’s milder phenotypes seem to be more consistent with those caused by pathogenic variants other than p.Ser2Gly. Therefore, it is possible that there may be additional pathogenic variants or copy number variants (CNVs) contributing to these differential phenotypes observed for Subject 3, as the variant analysis for this subject was detected using the RASopathy gene panel instead of whole-exome sequencing. This significant discrepancy needs to be further investigated. It is worth mentioning that our patient displayed slight sleep problems and a friendly and relatively mature personality, which have not been depicted in previously reported NS/LAH individuals. Thus, the present case enriches the clinical features of NS/LAH.
In conclusion, the SHOC2 Thr411Ala variant identified here further confirmed the pathogenic nature of the variant. Next, we systematically reviewed the clinical phenotypes of NS/LAH individuals. The novel phenotypes, slight sleep problems and a friendly and relatively mature personality, were observed in our patient. These findings will enrich our knowledge of the clinical characteristics, clinical management and genetic counseling of NS/LAH, which needs to be further explored.
Data availability statement
The data analyzed in this study is subject to the following licenses/restrictions: The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author. Requests to access these datasets should be directed to haimingyuan, haimingyuan@sina.cn.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Dongguan Maternal and Child Healthcare Hospital. Written informed consent to participate in this study was provided by the participantsandapos; legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’; legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
QW drafted the first versions of the manuscript. HY performed the design of the study, the clinical evaluation and data analysis, and manuscript polishing. YF performed data entry, the experiments, and data analysis. SC made the clinical evaluation and collected clinical information of the patient in detail. All authors read and approved the final manuscript.
Funding
This study was financed by the Dongguan Social Development Projects (No. 202050715007852 and 202050715007220). The funding body participated in the design, experimental operation and result interpretation of the project.
Acknowledgments
We would like to express our sincere gratitude to our patient and her parents for their cooperation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1040124/full#supplementary-material
References
Baldassarre, G., Mussa, A., Banaudi, E., Rossi, C., Tartaglia, M., Silengo, M., et al. (2014). Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation. Am. J. Med. Genet. A 164A (12), 3120–3125. doi:10.1002/ajmg.a.36697
Capalbo, D., Melis, D., De Martino, L., Palamaro, L., Riccomagno, S., Bona, G., et al. (2012a). Noonan-like syndrome with loose anagen hair associated with growth hormone insensitivity and atypical neurological manifestations. Am. J. Med. Genet. A 158A (4), 856–860. doi:10.1002/ajmg.a.35234
Capalbo, D., Scala, M. G., Melis, D., Minopoli, G., Improda, N., Palamaro, L., et al. (2012b). Clinical Heterogeneity in two patients with Noonan-like Syndrome associated with the same SHOC2 mutation. Ital. J. Pediatr. 38, 48. doi:10.1186/1824-7288-38-48
Cordeddu, V., Di Schiavi, E., Pennacchio, L. A., Ma'ayan, A., Sarkozy, A., Fodale, V., et al. (2009). Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet. 41 (9), 1022–1026. doi:10.1038/ng.425
Gripp, K. W., Zand, D. J., Demmer, L., Anderson, C. E., Dobyns, W. B., Zackai, E. H., et al. (2013). Expanding the SHOC2 mutation associated phenotype of noonan syndrome with loose anagen hair: Structural brain anomalies and myelofibrosis. Am. J. Med. Genet. A 161A (10), 2420–2430. doi:10.1002/ajmg.a.36098
Hannig, V., Jeoung, M., Jang, E. R., Phillips, J. A., and Galperin, E. (2014). A novel SHOC2 variant in rasopathy. Hum. Mutat. 35 (11), 1290–1294. doi:10.1002/humu.22634
Hoban, R., Roberts, A. E., Demmer, L., Jethva, R., and Shephard, B. (2012). Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am. J. Med. Genet. A 158A, 1411–1413. doi:10.1002/ajmg.a.35318
Li, X., Yao, R., Tan, X., Li, N., Ding, Y., Li, J., et al. (2019). Molecular and phenotypic spectrum of Noonan syndrome in Chinese patients. Clin. Genet. 96 (4), 290–299. doi:10.1111/cge.13588
Lores, J., Ramirez-Montano, D., Prada, C. E., Ramírez-Montaño, D., Nastasi-Catanese, J. A., and Pachajoa, H. (2020). Clinical and molecular analysis of 26 individuals with Noonan syndrome in a reference institution in Colombia. Am. J. Med. Genet. C Semin. Med. Genet. 184 (4), 1042–1051. doi:10.1002/ajmg.c.31869
Matsunaga-Udagawa, R., Fujita, Y., Yoshiki, S., Terai, K., Kamioka, Y., Kiyokawa, E., et al. (2010). The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J. Biol. Chem. 285 (10), 7818–7826. doi:10.1074/jbc.M109.053975
Mazzanti, L., Tamburrino, F., Scarano, E., Perri, A., Vestrucci, B., Guidetti, M., et al. (2013). GH Therapy and first final height data in Noonan-like syndrome with loose anagen hair (Mazzanti syndrome). Am. J. Med. Genet. A 161A (11), 2756–2761. doi:10.1002/ajmg.a.36255
Motta, M., Giancotti, A., Mastromoro, G., Chandramouli, B., Pinna, V., Pantaleoni, F., et al. (2019). Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum. Mutat. 40 (8), 1046–1056. doi:10.1002/humu.23767
Motta, M., Solman, M., Bonnard, A. A., Kuechler, A., Pantaleoni, F., Priolo, M., et al. (2022). Expanding the molecular spectrum of pathogenic SHOC2 variants underlying Mazzanti syndrome. Hum. Mol. Genet. 31, 2766–2778. doi:10.1093/hmg/ddac071
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. and ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rodriguez-Viciana, P., Oses-Prieto, J., Burlingame, A., Fried, M., and McCormick, F. (2006). A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell 22 (2), 217–230. doi:10.1016/j.molcel.2006.03.027
Takasawa, K., Takishima, S., Morioka, C., Nishioka, M., Ohashi, H., Aoki, Y., et al. (2015). Improved growth velocity of a patient with Noonan-like syndrome with loose anagen hair (NS/LAH) without growth hormone deficiency by low-dose growth hormone therapy. Am. J. Med. Genet. A 167A (10), 2425–2429. doi:10.1002/ajmg.a.37191
Keywords: SHOC2, Noonan syndrome-like with loose anagen hair (NS/LAH), short stature, growth hormone (GH), growth hormone deficiency (GHD)
Citation: Wang Q, Cheng S, Fu Y and Yuan H (2022) Case report: A de novo RASopathy-causing SHOC2 variant in a Chinese girl with noonan syndrome-like with loose anagen hair. Front. Genet. 13:1040124. doi: 10.3389/fgene.2022.1040124
Received: 08 September 2022; Accepted: 28 November 2022;
Published: 09 December 2022.
Edited by:
Anna Papadopoulou, University General Hospital Attikon, GreeceReviewed by:
Juan Caballero, European Bioinformatics Institute (EMBL-EBI), United KingdomFeliciano J. Ramos, School of Medicine, University of Zaragoza, Spain
Copyright © 2022 Wang, Cheng, Fu and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiming Yuan, aGFpbWluZ3l1YW5Ac2luYS5jbg==
†These authors have contributed equally to this work