 Yanan Gao1,2†
Yanan Gao1,2† Lamei Yuan1,2,3†
Lamei Yuan1,2,3† Jinzhong Yuan4
Jinzhong Yuan4 Yan Yang5
Yan Yang5 Jiangang Wang1
Jiangang Wang1 Yong Chen6
Yong Chen6 Hao Zhang4
Hao Zhang4 Yinze Ai2
Yinze Ai2 Hao Deng1,2,3*
Hao Deng1,2,3*- 1Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Center for Experimental Medicine, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Disease Genome Research Center, Central South University, Changsha, China
- 4Department of Nephrology, The Third Xiangya Hospital, Central South University, Changsha, China
- 5Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 6National Health Committee Key Laboratory of Birth Defects for Research and Prevention, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
Background: Benign familial hematuria and Alport syndrome are common causes of familial hematuria among children and young adults, which are attributable to variants in the collagen type IV alpha chain genes, COL4A3, COL4A4, or COL4A5. The study was conducted to identify the underlying genetic causes in patients with familial hematuria.
Methods: Two unrelated Han-Chinese pedigrees with familial hematuria were recruited for this study. Whole exome sequencing was combined with in silico analysis to identify potential genetic variants, followed by variant confirmation by Sanger sequencing. Reverse transcription, PCR, and Sanger sequencing were performed to evaluate the effect of the detected splicing variant on mRNA splicing.
Results: A novel heterozygous splicing c.595-1G>A variant and a known heterozygous c.1715G>C variant in the collagen type IV alpha 4 chain gene (COL4A4) were identified and confirmed in patients of pedigree 1 and pedigree 2, respectively. Complementary DNA analysis indicated this splicing variant could abolish the canonical splice acceptor site and cause a single nucleotide deletion of exon 10, which was predicted to produce a truncated protein.
Conclusions: The two COL4A4 variants, c.595-1G>A variant and c.1715G>C (p.Gly572Ala) variant, were identified as the genetic etiologies of two families with familial hematuria, respectively. Our study broadened the variant spectrum of the COL4A4 gene and explained the possible pathogenesis, which will benefit clinical management and genetic counseling.
Introduction
The familial hematuria of glomerular origin is usually due to a group of genetically and phenotypically heterogeneous disorders, such as benign familial hematuria (BFH), Alport syndrome (AS), glomerulopathy with fibronectin deposits (GFND), and C3 glomerulonephritis (C3GN) (Deltas et al., 2013). The common causes of familial hematuria among children and young adults are BFH and AS (Kashtan, 2009; Gale, 2013). BFH (OMIM 141200) is usually a non-progressive autosomal dominant renal disorder with a penetrance of 70% (Blumenthal et al., 1988; Lemmink et al., 1996; Savige et al., 2013), which is mainly characterized by persistent or recurrent glomerular hematuria, diffuse thinning of the glomerular basement membrane (GBM), and normal renal function (Slajpah et al., 2007). BFH is usually referred to as thin basement membrane nephropathy (TBMN) (Marcocci et al., 2009; Deltas et al., 2013; Gale, 2013), but TBMN is no longer considered benign as some can develop into end-stage renal disease (ESRD) in adults (Dische et al., 1985; Naylor et al., 2021). The frequency of this condition is estimated to be at least 1.0% in the worldwide population (Buzza et al., 2001; Chan and Gale, 2015), 5.2% in a population with microscopical examination and morphometry of GBM (Dische et al., 1990), and 15.5%–26% in children with hematuria and renal biopsy (Piqueras et al., 1998; Roth et al., 2001). It seems to be more prevalent in females than in males (Buzza et al., 2001; Savige et al., 2003). BFH is often attributable to heterozygous variants in the collagen type IV alpha 3 chain gene (COL4A3) or the collagen type IV alpha 4 chain gene (COL4A4), arranging head to head on chromosome 2 (Boye et al., 1998; Tazón Vega et al., 2003). AS most often manifests hematuria, proteinuria, and progressive renal failure, sometimes associated with sensorineural hearing loss and ocular abnormalities (Hudson et al., 2003), and the renal pathology is characterized by irregular thickening, thinning, and lamellation of GBM (Savige et al., 2013; Kamiyoshi et al., 2016). The COL4A3/COL4A4 heterozygous variants were also reported to cause autosomal dominant Alport syndrome (ADAS, OMIM 104200) (Boye et al., 1998), while homozygous or compound heterozygous variants were responsible for the autosomal recessive Alport syndrome (ARAS) (Longo et al., 2002; Voskarides et al., 2007). The heterozygous carriers of the ARAS family can present manifestations like BFH (Heidet et al., 2001; Savige et al., 2003). X-linked AS (XLAS), the most common form, accounts for about 85% of AS patients, which is ascribed to variants in the collagen type IV alpha 5 chain gene (COL4A5) (Slajpah et al., 2007; Nabais Sá et al., 2015). Compared with ARAS and XLAS, ADAS is relatively milder and slowly progressive with rare extrarenal manifestations (Kamiyoshi et al., 2016). Given the heterotrimeric association of type IV collagen α3/α4/α5 chain in the GBM and the overlapping clinical symptoms, a spectrum ranging from totally asymptomatic or isolated microscopic hematuria to proteinuria, up to chronic renal failure (CRF) and ESRD (Nabais Sá et al., 2015; Furlano et al., 2021; Kashtan, 2021), it is rational to consider BFH and AS as an entity and to term them collectively as collagen IV-related nephropathies (Tazón Vega et al., 2003; Pieri et al., 2014). BFH is considered to be the mildest end in the continuous spectrum, and the most severe end is represented by ARAS (Lemmink et al., 1996; Nieuwhof et al., 1997; Xu et al., 2016). However, BFH can resemble early AS, especially ADAS, in initial clinical presentation and the electron-microscopic features of the GBM, rendering the exact diagnosis notoriously difficult (Badenas et al., 2002; Gross et al., 2003; Voskarides et al., 2007). A complete investigation of family history, careful clinical evaluation, long-term specialist follow-up, and available renal biopsy analysis are vital in avoiding misdiagnosis or missed diagnosis (Kashtan, 2021). This study aimed to identify the genetic etiologies of familial hematuria in two unrelated Han-Chinese families and to elucidate possible morbigenous mechanisms.
Materials and methods
Subjects and clinical evaluations
Members of two Han-Chinese pedigrees were recruited from the Third Xiangya Hospital of Central South University, China (Figure 1). Detailed audiological examinations and ophthalmologic assessments were performed, including pure-tone audiometry, acoustic immittance, otoacoustic emissions, auditory brainstem response, slit-lamp evaluation, fundus examination, and optical coherence tomography. Peripheral blood samples were collected from four patients and four asymptomatic family members of two pedigrees, together with available clinical information. Written informed consents were obtained from all participating individuals. The entire study complied with the Declaration of Helsinki guidelines and was approved by the Institutional Review Board of the Third Xiangya Hospital of Central South University, China.
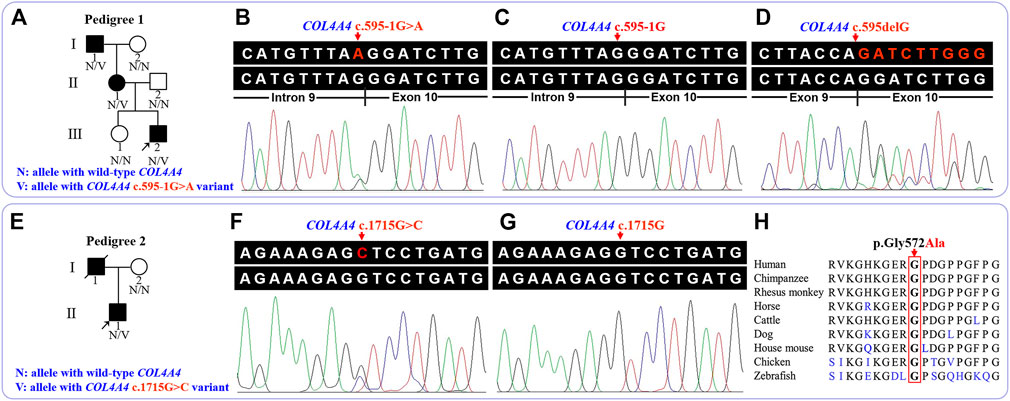
FIGURE 1. Pedigree and sequence analysis of the two unrelated Han-Chinese families with familial hematuria. (A) Pedigree 1. Squares and circles represent males and females, respectively. The fully shaded symbol indicates the affected individual. Arrow indicates the proband. (B) The genomic sequence with heterozygous COL4A4 c.595-1G>A variant (III:2 in pedigree 1). (C) The wild-type COL4A4 genomic sequence (II:2 in pedigree 1). (D) Analysis of complementary DNA revealed a deletion of the first base of exon 10 (III:2 in pedigree 1). (E) Pedigree 2. Arrow indicates the proband. The slash indicates deceased individual. (F) The genomic sequence with heterozygous COL4A4 c.1715G>C variant (II:1 in pedigree 2). (G) The wild-type COL4A4 genomic sequence (I:2 in pedigree 2). (H) Conservation analysis of the COL4A4 p.Gly572 residue. COL4A4, the collagen type IV alpha 4 chain gene.
DNA extraction, exome capture, and sequencing
Whole exome sequencing (WES) was conducted on the proband and their parents of pedigree 1, and the proband of pedigree 2. Genomic DNA (gDNA) of participating individuals was extracted from peripheral venous blood lymphocytes following the manufacturer’s protocol (Huang et al., 2021; Xiong et al., 2021). The qualified gDNA was fragmented randomly and then processed for end-repairing, A-tailing, and adaptor ligation. These adapter-ligated fragments were amplified by polymerase chain reactions (PCR) followed by purification and exome array hybridization. After further enrichment and purification, the captured libraries were sequenced on the high-throughput sequencing platforms: Illumina NextSeq500 (pedigree 1) and DNBSEQ (pedigree 2).
Variant analysis and Sanger sequencing
After filtering the raw data from the sequencing platform, data processing including sequence alignment, variant calling, annotation, and analysis was performed according to previously described criteria (Wu et al., 2016; Wu et al., 2019), as detailed in the Supplementary Methods.
The potential pathogenic variants identified in patients were further tested in participating individuals via Sanger sequencing. The following are the sequences of primers: 5′-GCTGGTGGCTGTGATTTCTT-3′ and 5′-CACCTGTGTCTGACCCAAAA-3′ for detecting the potential variant in the pedigree 1, 5′-CCAACCCAGAATCAAGGTCA-3′ and 5′-TCCTGGATCCCCTTTTTCTC-3′ for detecting potential variant in pedigree 2.
RNA isolation, complementary DNA synthesis, and PCR analysis
To analyze the effect of identified splicing variant, total RNA was extracted from the peripheral blood lymphocytes of the patients (I:1, II:1, and III:2) in pedigree 1 using the TRIzol reagent (Invitrogen, Carlsbad, CA, United States). The complementary DNA (cDNA) was synthesized by reverse transcription using the First Strand cDNA Synthesis Kit (Toyobo, Japan), and further amplified by PCR using the specifically designed primers: 5′-TGGGGAAAAGGGAGAAAAAG-3′ and 5′-ACCTTGCTGACCAACCTCAC-3′. PCR products were checked by agarose gel electrophoresis and then analyzed on the ABI 3730XL DNA sequencer (ABI, Carlsbad, CA, United States).
Conservative analysis and variant evaluation
The protein sequence alignment among nine species was completed by the NCBI Basic Local Alignment Search Tool (BLAST, https://blast.ncbi.nlm.nih.gov/BlastAlign.cgi). The American College of Medical Genetics and Genomics (ACMG) guidelines for interpreting sequence variants were utilized to classify the identified variants (Richards et al., 2015; Savige et al., 2021).
Quantification of nonsense-mediated decay by Sanger sequencing
Nonsense-mediated decay (NMD) was determined by sequencing analysis of COL4A4 cDNA from patients I:1, II:1, and III:2 of pedigree 1, and by comparing the areas under the peaks (AUP) of wild-type (WT) and mutant (MT) alleles. The AUP of the WT and MT specific-alleles were quantified by the ImageJ software v1.53a (NIH, United States), and the ratio of AUP of MT over corresponding WT was then calculated. Statistical analysis was performed using the Microsoft Excel 2016 software (Microsoft, Inc.) and GraphPad Prism v9.4.0 (GraphPad Software, Inc.) using Student’s t test. p<.05 was considered significant.
Results
Clinical data
The proband (III:2) of pedigree 1, a 15-year-old boy, was accidentally found to have microscopic hematuria during the physical examination for enrollment 3 years ago. The dysmorphic erythrocytes in the urinary sediment observed by phase-contrast microscopy supported the diagnosis of glomerular hematuria. Further investigation found that his mother (II:1) also had long-term microscopic hematuria, occasionally accompanied by trace proteinuria by routine tests. His 71-year-old grandfather (I:1) had microscopic hematuria persisting for an uncertain time. All three patients exhibit no signs of impairment in blood pressure or renal function. Audiological and ophthalmologic assessments of the proband and his mother demonstrated no signs of sensorineural hearing loss or ocular lesions.
The proband (II:1) of pedigree 2, a 30-year-old male, displayed microscopic hematuria and a mildly elevated serum creatinine level on routine physical examination. His serum creatinine fluctuated between 110 and 123 μmol/L (reference interval: 59–104 μmol/L) during 3 years of follow-up. Renal biopsy was performed in the Sichuan Provincial People’s Hospital half a year ago. Light microscopy showed segmental mild hyperplasia of mesangial cells and matrix. Immunofluorescence staining of IgM, IgA, C3, κ, and λ was positive, showing a massive pattern of deposition within the mesangium. On electron microscopy, there is segmental mild mesangial expansion with massive electron-dense deposits and segmental irregular thickening and thinning of the GBM (Figure 2). The audiological and ophthalmologic evaluations revealed no abnormalities. Clinical data of these patients were summarized in Table 1.
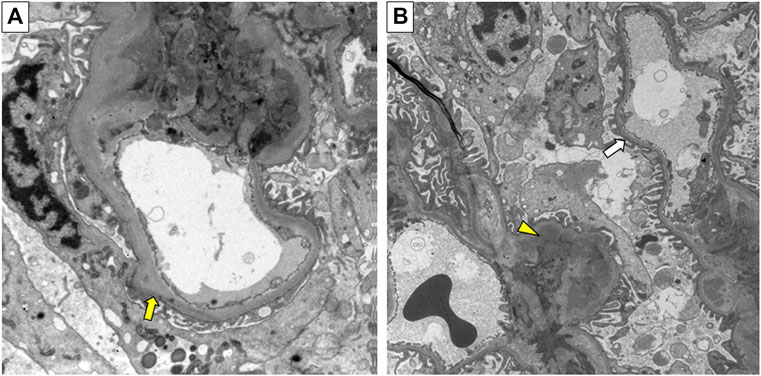
FIGURE 2. Electron micrograph of renal biopsy specimen from the proband (II:1) in pedigree 2. (A) Irregular thickening of the glomerular basement membrane (GBM) (yellow arrow). (B) Irregular thinning of the GBM (white arrow), and electron-dense deposits (yellow triangle). Original magnification 6500×.
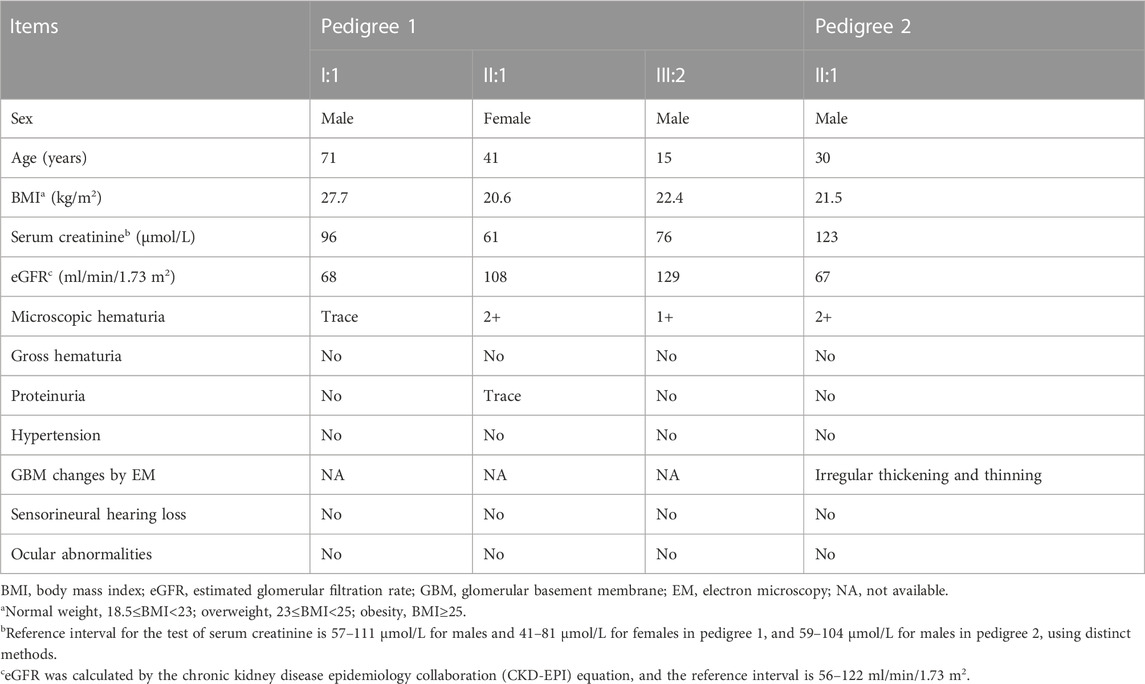
TABLE 1. Clinical data of patients with the COL4A4 gene variant.
WES and identification of pathogenic variants
The detailed WES data of four individuals are available in Supplementary Table 1. After database and in silico analysis screening, a novel heterozygous c.595-1G>A variant in the intron 9 of the COL4A4 gene (NG_011592.1, NM_000092.5) was suggested as the potential pathogenic factor of patients in pedigree 1, and a known heterozygous c.1715G>C (p.Gly572Ala) variant in the exon 24 of the COL4A4 gene was considered to be the potential disease-related variant of the proband in pedigree 2. The results of in silico analysis are presented in Table 2. After Sanger sequencing, the heterozygous splicing variant, c.595-1G>A, was identified in the patients I:1, II:1, and III:2 of pedigree 1, and the heterozygous missense variant c.1715G>C was confirmed in the proband II:1 of pedigree 2. These two variants were absent in the enrolled asymptomatic family members. For the reverse transcription PCR products, agarose gel electrophoresis and Sanger sequencing revealed no exon skipping or intron retention. Sanger sequencing indicated that the COL4A4 c.599-1G>A substitution can abolish the intron 9 canonical splice acceptor site and introduce a new splice site, resulting in the loss of the first base of exon 10 during splicing, which was predicted to cause a frame shift and premature termination of translation (c.595delG, p.Gly199Aspfs*20). The COL4A4 glycine at position 572 (p.Gly572) is highly conserved across species (Figure 1). According to ACMG guidelines, the two variants were classified as “pathogenic”.
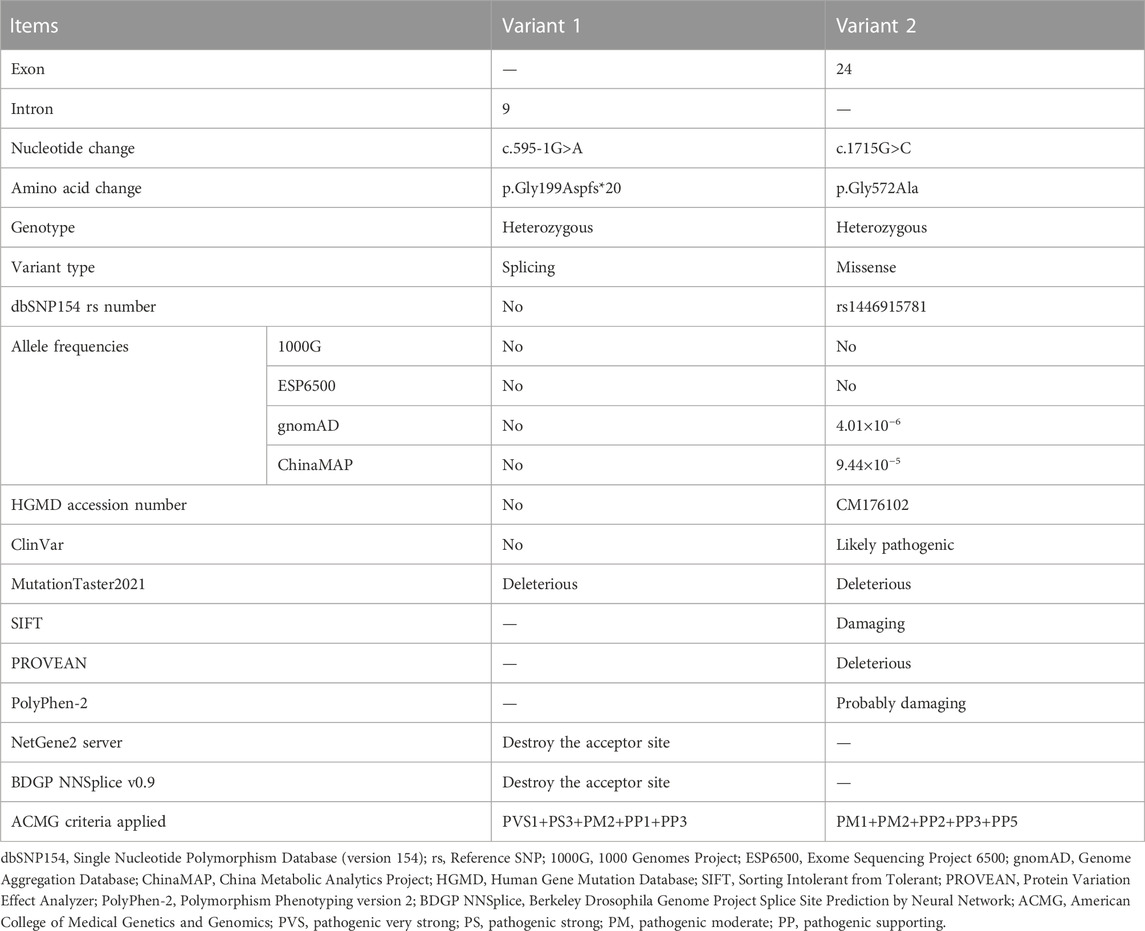
TABLE 2. Identification of the COL4A4 gene variants in two pedigrees with familial hematuria.
Quantification of NMD
The differences between the AUP of the WT and MT alleles are statistically significant (p<.0001). The relative expression of the MT allele is approximately 50% lower than the WT allele by comparing the AUP of specific alleles in the cDNA sequencing chromatogram of these three patients (I:1, II:1, and III:2) in pedigree 1 (Figure 3).
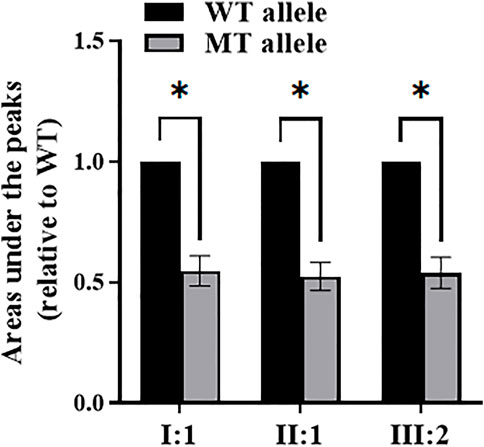
FIGURE 3. The areas under the peaks of wild-type (WT) and mutant (MT) alleles by sequencing analysis of COL4A4 cDNA from patients I:1, II:1, and III:2 of pedigree 1. *Statistically significant differences (Student’s t test, p<.0001).
Discussion
The COL4A4 gene (OMIM 120131), located at 2q36.3, contains 48 exons encoding the α4 chain of type IV collagen (Boye et al., 1998). Each chain contains an N-terminal 7S domain, a long, central, triple-helical collagenous domain of Gly-Xaa-Yaa repeats frequently interrupted by short non-collagenous regions, and a C-terminal non-collagenous (NC1) domain (Figure 4) (Boutaud et al., 2000; LeBleu et al., 2010). The six highly homologous chains of type IV collagen, namely α1(IV) to α6(IV), were encoded by six genes (COL4A1 to COL4A6) (Hudson et al., 2003; Cicuéndez et al., 2021). In mammals, the existence of six distinct α(IV) chains could only assemble into three different trimers, namely α1α1α2(IV), α3α4α5(IV), and α5α5α6(IV) (LeBleu et al., 2010). During fetal development, collagen α1α1α2(IV) is ubiquitously distributed in all basement membranes (BMs) (Sand et al., 2013), while in the mature GBM and BMs of cochlea and eye, α3α4α5(IV) is the major component (Hudson et al., 2003; Suleiman et al., 2013; Savige et al., 2015). The pathogenic variants in any one of the three genes (COL4A3, COL4A4, and COL4A5) may interfere with the developmental switch, resulting in the persistent expression or compensatory increase of fewer cross-linked and more protease-susceptible α1α1α2(IV), which leads to BFH or AS (Suleiman et al., 2013; Clark et al., 2016; Xu et al., 2016). Currently, more than 400 COL4A4 gene variants have been recorded in the Human Gene Mutation Database (HGMD), of which at least 29 and 65 variants are associated with BFH and ADAS, respectively, and three variants can cause both disorders. These different variants are widely distributed in the COL4A4 gene without hot spots described, and the most frequent variants are glycine substitutions (46/97) (Figure 4). The frequency of nonsense, splicing, and frameshift variants in ADAS seems to be higher than that in BFH (44.12% vs. 37.50%), consistent with that these variants usually cause more severe phenotypes (Shang et al., 2019).
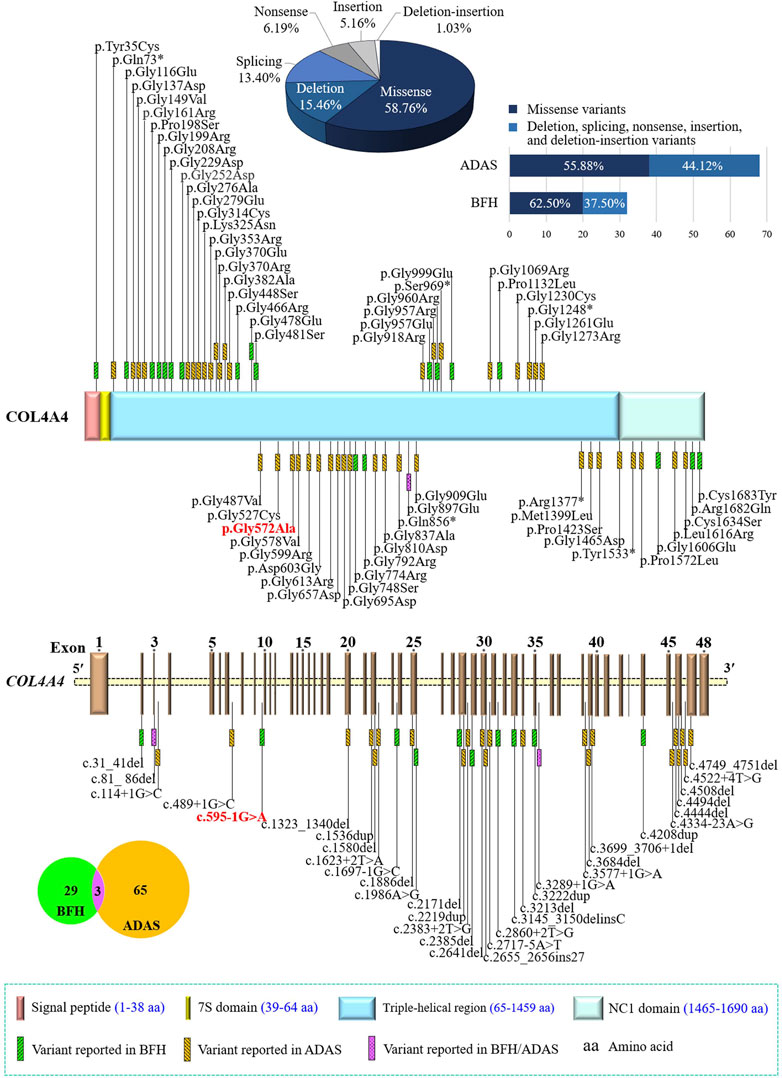
FIGURE 4. Statistics of the COL4A4 gene variants related to benign familial hematuria (BFH) or autosomal dominant Alport syndrome (ADAS). Collagen type IV alpha 4 chain contains an N-terminal 7S domain, a long, central triple-helical region, and a C-terminal non-collagenous (NC1) domain. Variants identified in this study were indicated in bold and red. COL4A4, the collagen type IV alpha 4 chain gene.
In this study, two variants in the COL4A4 gene were identified in two unrelated Chinese pedigrees with familial hematuria. The heterozygous c.595-1G>A variant is completely co-segregated with the disorder phenotype within pedigree 1. The heterozygous c.1715G>C transversion detected in the proband of pedigree 2 has been previously reported in compound heterozygosity with variant c.-23T>G in a Chinese AS male patient, and both his sons with heterozygous c.-23T>G variant only presented isolated hematuria (Liu et al., 2017), supporting that the identified c.1715G>C variant may be pathogenic and seems to exert a pathogenic effect to result in a more severe renal phenotype. The absence of these two variants in the asymptomatic family members, the prediction of its deleterious effect by in silico programs, cDNA analysis, protein sequence alignment, and ACMG criteria evaluation suggested that they were likely disease-causing variants in these two pedigrees.
The splicing variant c.595-1G>A may induce NMD of the altered COL4A4 mRNA or produce a truncated loss-of-function α4(IV) chain, consistent with similar studies (Schwarze et al., 2001; Davidson et al., 2007; Korstanje et al., 2014; Hashikami et al., 2018). The missense variant c.1715G>C (p.Gly572Ala) occurred in the first position of critical Gly-Xaa-Yaa repeats in the collagenous domain. The highly conserved glycine residue is the smallest amino acid that can fit tightly into the center of the triple helix structure of the α(IV) chain and is crucial to the helix formation (Deltas et al., 2013; Murray et al., 2014). Hence, both variants in the COL4A4 gene may disrupt the normal synthesis of the α4(IV) chain and cause defective synthesis, assembly, deposition, or function of the α3α4α5(IV) network (Korstanje et al., 2014; Kashtan et al., 2018), interfering natural development process and leading to the abnormal GBMs (Clark et al., 2016; Naylor et al., 2021). Additionally, the poor association of abnormal α4(IV) with the α3(IV) and α5(IV) chain may leave more misfolded protein in the endoplasmic reticulum (ER), disrupt podocyte’s special secretory capacity, and lead to GBM defects (Pieri et al., 2014). These defective GBMs may partially allow the escape of erythrocytes into Bowman’s space, thereby triggering the onset of hematuria (Yoshikawa et al., 1984).
In pedigree 1, isolated microscopic hematuria, normal renal function, and absence of other signs related to AS in the proband, his mother, and grandfather, are more suggestive of a heterozygous COL4A4-related BFH than AS. In pedigree 2, given that the proband presented microscopic hematuria, mildly elevated serum creatinine level, and irregular thickening and thinning of the GBM, without signs of sensorineural hearing loss or ocular lesions, a diagnosis of early ADAS cannot be ruled out. The IgA deposits within the mesangium corresponding to IgA nephropathy could be due to the predisposition of mesangial IgA deposition in defective GBMs caused by COL4A4 variant (Savige and Harraka, 2021). Usually, from clinical phenotype and renal biopsy, the coexistence of AS and IgA nephropathy is attributed to two independent causes. However, some variants in the COL4A3, COL4A4, or COL4A5 gene seem to increase the susceptibility to IgA nephropathy, which is supported by IgA deposits occasionally observed in the AS patients and the COL4A3, COL4A4, or COL4A5 gene variants reported in a minority of IgA nephropathy patients (Kamiyoshi et al., 2016; Li et al., 2020; Cambier et al., 2021). Based on the identified responsible variant, COL4A4-associated nephropathies, a sub-classification of the conditions in the two pedigrees, is strongly recommended. In this way, these similar or overlapping clinical manifestations could be attributed to a single disease spectrum rather than several distinct disease statuses. It would, therefore, be beneficial to risk assessment, disease management, and genetic counseling of diseased individuals.
Taken together, a novel c.595-1G>A variant and a known c.1715G>C (p.Gly572Ala) variant of the COL4A4 gene were identified in two Chinese families with familial hematuria, respectively. WES has been proven to be a powerful tool for uncovering the genetic etiologies of heterogeneous disorders and identifying at-risk individuals, which will assist in early clinical diagnosis and accurate disease classification. Currently, there is a lack of evidence-based treatment for persistent microscopic hematuria (Xu et al., 2016), but it is of paramount importance to regularly monitor proteinuria development, renal function, and blood pressure every 1-2 years (Savige et al., 2003). Our findings broadened the variant spectrum of the COL4A4 gene and may assist in reproductive risk counseling of these two families and improved medical care.
Data availability statement
The datasets presented in this article are not readily available because the data information is in a controlled state due to the national legislation, specifically the Ministry of Science and Technology of the People’s Republic of China. Data of this project can be accessed after an approval application by the China National GeneBank DataBase (CNGBdb). Please refer to CNGBdb: https://db.cngb.org/, or email: Q05HQmRiQGNuZ2Iub3Jn for detailed application guidance. The project accession code CNP0003636 should be included in the application.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board (IRB) of the Third Xiangya Hospital of Central South University (Changsha, China). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
YG, LY, and HD conceptualized and designed this study. JY, YY, JW, YC, HZ, and YA collected the patient samples and performed clinical data analysis. YG, LY, and HD performed the experiments and data analysis, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81873686 and 81800219), Natural Science Foundation of Hunan Province (Grant Nos. 2020JJ3057 and 2020JJ4830), the Hunan Provincial Innovation Foundation for Postgraduate (Grant No. CX20210378), Province-level College Students’ Innovative Training Plan Program (Grant No. S2022105330781), the Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (Grant No. YX202109), and Distinguished Professor of the Lotus Scholars Award Program of Hunan Province, China.
Acknowledgments
We extend our greatest gratitude to all the participating individuals and investigators for their contributions to this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1064491/full#supplementary-material
References
Badenas, C., Praga, M., Tazón, B., Heidet, L., Arrondel, C., Armengol, A., et al. (2002). Mutations in the COL4A4 and COL4A3 genes cause familial benign hematuria. J. Am. Soc. Nephrol. 13, 1248–1254. doi:10.1681/ASN.V1351248
Blumenthal, S. S., Fritsche, C., and Lemann, J. (1988). Establishing the diagnosis of benign familial hematuria. The importance of examining the urine sediment of family members. JAMA 259, 2263–2266. doi:10.1001/jama.1988.03720150039033
Boutaud, A., Borza, D. B., Bondar, O., Gunwar, S., Netzer, K. O., Singh, N., et al. (2000). Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 275, 30716–30724. doi:10.1074/jbc.M004569200
Boye, E., Mollet, G., Forestier, L., Cohen-Solal, L., Heidet, L., Cochat, P., et al. (1998). Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am. J. Hum. Genet. 63, 1329–1340. doi:10.1086/302106
Buzza, M., Wilson, D., and Savige, J. (2001). Segregation of hematuria in thin basement membrane disease with haplotypes at the loci for Alport syndrome. Kidney Int. 59, 1670–1676. doi:10.1046/j.1523-1755.2001.0590051670.x
Cambier, A., Robert, T., Hogan, J., Rabant, M., Peuchmaur, M., Boyer, O., et al. (2021). Rare collagenous heterozygote variants in children with IgA nephropathy. Kidney Int. Rep. 6, 1326–1335. doi:10.1016/j.ekir.2021.02.022
Chan, M. M., and Gale, D. P. (2015). Isolated microscopic haematuria of glomerular origin: Clinical significance and diagnosis in the 21st century. Clin. Med. 15, 576–580. doi:10.7861/clinmedicine.15-6-576
Cicuéndez, M., Casarrubios, L., Feito, M. J., Madarieta, I., Garcia-Urkia, N., Murua, O., et al. (2021). Effects of human and porcine adipose extracellular matrices decellularized by enzymatic or chemical methods on macrophage polarization and immunocompetence. Int. J. Mol. Sci. 22, 3847. doi:10.3390/ijms22083847
Clark, S. D., Nabity, M. B., Cianciolo, R. E., Dufek, B., and Cosgrove, D. (2016). X-linked Alport dogs demonstrate mesangial filopodial invasion of the capillary tuft as an early event in glomerular damage. PLoS One 11, e0168343. doi:10.1371/journal.pone.0168343
Davidson, A. G., Bell, R. J., Lees, G. E., Kashtan, C. E., Davidson, G. S., and Murphy, K. E. (2007). Genetic cause of autosomal recessive hereditary nephropathy in the English Cocker Spaniel. J. Vet. Intern. Med. 21, 394–401. doi:10.1892/0891-6640(2007)21[394:gcoarh]2.0.co;2
Deltas, C., Pierides, A., and Voskarides, K. (2013). Molecular genetics of familial hematuric diseases. Nephrol. Dial. Transpl. 28, 2946–2960. doi:10.1093/ndt/gft253
Dische, F. E., Anderson, V. E., Keane, S. J., Taube, D., Bewick, M., and Parsons, V. (1990). Incidence of thin membrane nephropathy: Morphometric investigation of a population sample. J. Clin. Pathol. 43, 457–460. doi:10.1136/jcp.43.6.457
Dische, F. E., Weston, M. J., and Parsons, V. (1985). Abnormally thin glomerular basement membranes associated with hematuria, proteinuria or renal failure in adults. Am. J. Nephrol. 5, 103–109. doi:10.1159/000166914
Furlano, M., Martínez, V., Pybus, M., Arce, Y., Crespí, J., Venegas, M. D. P., et al. (2021). Clinical and genetic features of autosomal dominant Alport syndrome: A cohort study. Am. J. Kidney Dis. 78, 560–570.e1. doi:10.1053/j.ajkd.2021.02.326
Gale, D. P. (2013). How benign is hematuria? Using genetics to predict prognosis. Pediatr. Nephrol. 28, 1183–1193. doi:10.1007/s00467-012-2399-y
Gross, O., Netzer, K. O., Lambrecht, R., Seibold, S., and Weber, M. (2003). Novel COL4A4 splice defect and in-frame deletion in a large consanguine family as a genetic link between benign familial haematuria and autosomal Alport syndrome. Nephrol. Dial. Transpl. 18, 1122–1127. doi:10.1093/ndt/gfg157
Hashikami, K., Asahina, M., Nozu, K., Iijima, K., Nagata, M., and Takeyama, M. (2018). Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem. Biophys. Rep. 17, 81–86. doi:10.1016/j.bbrep.2018.12.003
Heidet, L., Arrondel, C., Forestier, L., Cohen-Solal, L., Mollet, G., Gutierrez, B., et al. (2001). Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J. Am. Soc. Nephrol. 12, 97–106. doi:10.1681/ASN.V12197
Huang, Y., Yuan, L., Cao, Y., Tang, R., Xu, H., Tang, Z., et al. (2021). Novel compound heterozygous mutations in the CHST6 gene cause macular corneal dystrophy in a Han Chinese family. Ann. Transl. Med. 9, 622. doi:10.21037/atm-20-7178
Hudson, B. G., Tryggvason, K., Sundaramoorthy, M., and Neilson, E. G. (2003). Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N. Engl. J. Med. 348, 2543–2556. doi:10.1056/NEJMra022296
Kamiyoshi, N., Nozu, K., Fu, X. J., Morisada, N., Nozu, Y., Ye, M. J., et al. (2016). Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin. J. Am. Soc. Nephrol. 11, 1441–1449. doi:10.2215/CJN.01000116
Kashtan, C. E. (2021). Alport syndrome: Achieving early diagnosis and treatment. Am. J. Kidney Dis. 77, 272–279. doi:10.1053/j.ajkd.2020.03.026
Kashtan, C. E., Ding, J., Garosi, G., Heidet, L., Massella, L., Nakanishi, K., et al. (2018). Alport syndrome: A unified classification of genetic disorders of collagen IV α345: A position paper of the Alport syndrome classification working group. Kidney Int. 93, 1045–1051. doi:10.1016/j.kint.2017.12.018
Kashtan, C. E. (2009). Familial hematuria. Pediatr. Nephrol. 24, 1951–1958. doi:10.1007/s00467-007-0622-z
Korstanje, R., Caputo, C. R., Doty, R. A., Cook, S. A., Bronson, R. T., Davisson, M. T., et al. (2014). A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen α3α4α5(IV) trimers. Kidney Int. 85, 1461–1468. doi:10.1038/ki.2013.493
LeBleu, V., Sund, M., Sugimoto, H., Birrane, G., Kanasaki, K., Finan, E., et al. (2010). Identification of the NC1 domain of α3 chain as critical for α3α4α5 type IV collagen network assembly. J. Biol. Chem. 285, 41874–41885. doi:10.1074/jbc.M110.149534
Lemmink, H. H., Nillesen, W. N., Mochizuki, T., Schröder, C. H., Brunner, H. G., van Oost, B. A., et al. (1996). Benign familial hematuria due to mutation of the type IV collagen alpha4 gene. J. Clin. Invest. 98, 1114–1118. doi:10.1172/JCI118893
Li, Y., Groopman, E. E., D'Agati, V., Prakash, S., Zhang, J., Mizerska-Wasiak, M., et al. (2020). Type IV collagen mutations in familial IgA nephropathy. Kidney Int. Rep. 5, 1075–1078. doi:10.1016/j.ekir.2020.04.011
Liu, J. H., Wei, X. X., Li, A., Cui, Y. X., Xia, X. Y., Qin, W. S., et al. (2017). Novel mutations in COL4A3, COL4A4, and COL4A5 in Chinese patients with Alport syndrome. PLoS One 12, e0177685. doi:10.1371/journal.pone.0177685
Longo, I., Porcedda, P., Mari, F., Giachino, D., Meloni, I., Deplano, C., et al. (2002). COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 61, 1947–1956. doi:10.1046/j.1523-1755.2002.00379.x
Marcocci, E., Uliana, V., Bruttini, M., Artuso, R., Silengo, M. C., Zerial, M., et al. (2009). Autosomal dominant Alport syndrome: Molecular analysis of the COL4A4 gene and clinical outcome. Nephrol. Dial. Transpl. 24, 1464–1471. doi:10.1093/ndt/gfn681
Murray, L. S., Lu, Y., Taggart, A., Van Regemorter, N., Vilain, C., Abramowicz, M., et al. (2014). Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum. Mol. Genet. 23, 283–292. doi:10.1093/hmg/ddt418
Nabais Sá, M. J., Storey, H., Flinter, F., Nagel, M., Sampaio, S., Castro, R., et al. (2015). Collagen type IV-related nephropathies in Portugal: Pathogenic COL4A3 and COL4A4 mutations and clinical characterization of 25 families. Clin. Genet. 88, 456–461. doi:10.1111/cge.12521
Naylor, R. W., Morais, M. R. P. T., and Lennon, R. (2021). Complexities of the glomerular basement membrane. Nat. Rev. Nephrol. 17, 112–127. doi:10.1038/s41581-020-0329-y
Nieuwhof, C. M., de Heer, F., de Leeuw, P., and van Breda Vriesman, P. J. (1997). Thin GBM nephropathy: Premature glomerular obsolescence is associated with hypertension and late onset renal failure. Kidney Int. 51, 1596–1601. doi:10.1038/ki.1997.219
Pieri, M., Stefanou, C., Zaravinos, A., Erguler, K., Stylianou, K., Lapathitis, G., et al. (2014). Evidence for activation of the unfolded protein response in collagen IV nephropathies. J. Am. Soc. Nephrol. 25, 260–275. doi:10.1681/ASN.2012121217
Piqueras, A. I., White, R. H., Raafat, F., Moghal, N., and Milford, D. V. (1998). Renal biopsy diagnosis in children presenting with haematuria. Pediatr. Nephrol. 12, 386–391. doi:10.1007/s004670050471
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Roth, K. S., Amaker, B. H., and Chan, J. C. (2001). Pediatric hematuria and thin basement membrane nephropathy: What is it and what does it mean? Clin. Pediatr. 40, 607–613. doi:10.1177/000992280104001105
Sand, J. M., Larsen, L., Hogaboam, C., Martinez, F., Han, M., Røssel Larsen, M., et al. (2013). MMP mediated degradation of type IV collagen alpha 1 and alpha 3 chains reflects basement membrane remodeling in experimental and clinical fibrosis--validation of two novel biomarker assays. PLoS One 8, e84934. doi:10.1371/journal.pone.0084934
Savige, J., Gregory, M., Gross, O., Kashtan, C., Ding, J., and Flinter, F. (2013). Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol. 24, 364–375. doi:10.1681/ASN.2012020148
Savige, J., and Harraka, P. (2021). Pathogenic variants in the genes affected in Alport syndrome (COL4A3-COL4A5) and their association with other kidney conditions: A review. Am. J. Kidney Dis. 78, 857–864. doi:10.1053/j.ajkd.2021.04.017
Savige, J., Rana, K., Tonna, S., Buzza, M., Dagher, H., and Wang, Y. Y. (2003). Thin basement membrane nephropathy. Kidney Int. 64, 1169–1178. doi:10.1046/j.1523-1755.2003.00234.x
Savige, J., Sheth, S., Leys, A., Nicholson, A., Mack, H. G., and Colville, D. (2015). Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 10, 703–709. doi:10.2215/CJN.10581014
Savige, J., Storey, H., Watson, E., Hertz, J. M., Deltas, C., Renieri, A., et al. (2021). Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 29, 1186–1197. doi:10.1038/s41431-021-00858-1
Schwarze, U., Schievink, W. I., Petty, E., Jaff, M. R., Babovic-Vuksanovic, D., Cherry, K. J., et al. (2001). Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 69, 989–1001. doi:10.1086/324123
Shang, S., Peng, F., Wang, T., Wu, X., Li, P., Li, Q., et al. (2019). Genotype-phenotype correlation and prognostic impact in Chinese patients with Alport Syndrome. Mol. Genet. Genomic Med. 7, e00741. doi:10.1002/mgg3.741
Slajpah, M., Gorinsek, B., Berginc, G., Vizjak, A., Ferluga, D., Hvala, A., et al. (2007). Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. 71, 1287–1295. doi:10.1038/sj.ki.5002221
Suleiman, H., Zhang, L., Roth, R., Heuser, J. E., Miner, J. H., Shaw, A. S., et al. (2013). Nanoscale protein architecture of the kidney glomerular basement membrane. Elife 2, e01149. doi:10.7554/eLife.01149
Tazón Vega, B., Badenas, C., Ars, E., Lens, X., Milà, M., Darnell, A., et al. (2003). Autosomal recessive Alport's syndrome and benign familial hematuria are collagen type IV diseases. Am. J. Kidney Dis. 42, 952–959. doi:10.1016/j.ajkd.2003.08.002
Voskarides, K., Damianou, L., Neocleous, V., Zouvani, I., Christodoulidou, S., Hadjiconstantinou, V., et al. (2007). COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Am. Soc. Nephrol. 18, 3004–3016. doi:10.1681/ASN.2007040444
Wu, Y., Guo, Y., Yuan, J., Xu, H., Chen, Y., Zhang, H., et al. (2019). A COL4A5 missense variant in a Han-Chinese family with X-linked Alport syndrome. Curr. Mol. Med. 19, 758–765. doi:10.2174/1566524019666190906144214
Wu, Y., Hu, P., Xu, H., Yuan, J., Yuan, L., Xiong, W., et al. (2016). A novel heterozygous COL4A4 missense mutation in a Chinese family with focal segmental glomerulosclerosis. J. Cell Mol. Med. 20, 2328–2332. doi:10.1111/jcmm.12924
Xiong, Y., Xia, H., Yuan, L., Deng, S., Ding, Z., and Deng, H. (2021). Identification of compound heterozygous DNAH11 variants in a Han-Chinese family with primary ciliary dyskinesia. J. Cell Mol. Med. 25, 9028–9037. doi:10.1111/jcmm.16866
Xu, Y., Guo, M., Dong, H., Jiang, W., Ma, R., Liu, S., et al. (2016). A novel COL4A4 mutation identified in a Chinese family with thin basement membrane nephropathy. Sci. Rep. 6, 20244. doi:10.1038/srep20244
Keywords: benign familial hematuria, Alport syndrome, collagen type IV alpha 4 chain gene, whole exome sequencing, splicing variant
Citation: Gao Y, Yuan L, Yuan J, Yang Y, Wang J, Chen Y, Zhang H, Ai Y and Deng H (2023) Identification of COL4A4 variants in Chinese patients with familial hematuria. Front. Genet. 13:1064491. doi: 10.3389/fgene.2022.1064491
Received: 08 October 2022; Accepted: 15 December 2022;
Published: 09 January 2023.
Edited by:
Cagri Gulec, Istanbul University, TürkiyeReviewed by:
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, IranJunjiang Fu, Southwest Medical University, China
Huaping Xie, Hunan Normal University, China
Zheng Liu, Guilin Medical University, China
Copyright © 2023 Gao, Yuan, Yuan, Yang, Wang, Chen, Zhang, Ai and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Deng, aGRlbmcwMDhAMTYzLmNvbQ==
†These authors have contributed equally to this work and share the first authorship