 Yan Zhao
Yan Zhao Juntao Hu
Juntao Hu Jiajiao Wu3
Jiajiao Wu3 Zhihong Li
Zhihong Li- 1Key Laboratory of Surveillance and Management for Plant Quarantine Pests, Ministry of Agriculture and Rural Affairs, College of Plant Protection, China Agricultural University, Beijing, China
- 2Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institute of Biodiversity Science, Center of Evolutionary Biology, School of Life Sciences, Fudan University, Shanghai, China
- 3Technology Center of Guangzhou Customs, Guangzhou, China
Introduction: While it has been suggested that histone modifications can facilitate animal responses to rapidly changing environments, few studies have profiled whole-genome histone modification patterns in invasive species, leaving the regulatory landscape of histone modifications in invasive species unclear.
Methods: Here, we screen genome-wide patterns of two important histone modifications, trimethylated Histone H3 Lysine 4 (H3K4me3) and trimethylated Histone H3 Lysine 27 (H3K27me3), in adult thorax muscles of a notorious invasive pest, the Oriental fruit fly Bactrocera dorsalis (Hendel) (Diptera: Tephritidae), using Chromatin Immunoprecipitation with high-throughput sequencing (ChIP-seq).
Results: We identified promoters featured by the occupancy of H3K4me3, H3K27me3 or bivalent histone modifications that were respectively annotated with unique genes key to muscle development and structure maintenance. In addition, we found H3K27me3 occupied the entire body of genes, where the average enrichment was almost constant. Transcriptomic analysis indicated that H3K4me3 is associated with active gene transcription, and H3K27me3 is mostly associated with transcriptional repression. Importantly, we identified genes and putative motifs modified by distinct histone modification patterns that may possibly regulate flight activity.
Discussion: These findings provide the first evidence of histone modification signature in B. dorsalis, and will be useful for future studies of epigenetic signature in other invasive insect species.
Introduction
Understanding the mechanisms underlying animal responses to environmental change is essential to predict the shifts in animal distribution range (Parmesan, 2006; Merilä and Hendry, 2014; González-Tokman et al., 2020; Aguirre-Liguori et al., 2021). While a large number of studies have demonstrated the important role of genetic mechanism in animal responses to environmental stressors, such as temperature (Dixon et al., 2015; Cao et al., 2021; Chen and Narum, 2021), salinity (Berg et al., 2015; Garcia-Elfring et al., 2021; Rautsaw et al., 2021), oxygen (Yu et al., 2016; Yang et al., 2021; Zhang et al., 2021), and precipitation (Dudaniec et al., 2018; Gheyas et al., 2021; Wiener et al., 2021), a surge of recent evidence suggests that non-genetic mechanism, especially epigenetic modifications, can also contribute to responses to changing environment in animal populations with little genetic variation (Bossdorf et al., 2008; Richards and Pigliucci, 2020; Stajic and Jansen, 2021). Unlike genetic variation, epigenetic variation can directly interact with ambient environments, and induce possibly heritable gene expression changes without altering underlying gene sequence, thus providing a faster route for animals adapting to novel environments (Bossdorf et al., 2008; Feil and Fraga, 2012; Hu and Barrett, 2017). Epigenetic variation may therefore be an important mechanism underlying phenotypic variation and adaptive evolution within the context of limited genetic variation (Jaenisch and Bird, 2003; Verhoeven et al., 2016).
To date, the most extensively studied epigenetic mechanism is DNA methylation, which has been found involved in phenotypic responses to a number of environmental cues in animal populations [e.g., salinity (Artemov et al., 2017; Metzger and Schulte, 2018; Heckwolf et al., 2020), temperature (Marsh and Pasqualone, 2014; Paredes et al., 2016; Metzger and Schulte, 2017; Sheldon et al., 2020), photoperiod (Stevenson and Prendergast, 2013; Stevenson, 2018; Lindner et al., 2021), chemical pollutants (Baccarelli and Bollati, 2009; Devóz et al., 2017; Sanchez et al., 2017; Barouki et al., 2018)]. However, other epigenetic modifications underlying phenotypic plasticity have been less characterized in animals (Hu and Barrett, 2017; Jeremias et al., 2018; Vogt, 2021). The research bias toward DNA methylation also holds true in insect studies (Mukherjee et al., 2015; Maleszka, 2016; Glastad et al., 2019). With the exception for a few studies (Dai et al., 2018; Jones et al., 2018), almost all insect studies have focused on the relationship between DNA methylation variation and phenotypic change in model and non-invasive insects such as bees (Lyko et al., 2010; Foret et al., 2012; Yagound et al., 2020), ants (Bonasio et al., 2012; Libbrecht et al., 2016; Oldroyd and Yagound, 2021), aphids (Walsh et al., 2010; Mathers et al., 2019), and mealybug (Bain et al., 2021). Because it has been suggested that invasive species are more phenotypically plastic than non-invasive species in growth, morphological, and physiological traits (Davidson et al., 2011; Kelly, 2019; Bates et al., 2020), it is possible that the epigenetic patterns underlying plasticity are different between invasive and non-invasive insects. However, without proper profiling of regulatory landscape and functions, the role of epigenetic modifications other than DNA methylation in regulating phenotypes of invasive insects remains unclear.
In addition to DNA methylation, post-translational modifications of histones (hPTMs) including lysine acetylation, methylation, phosphorylation, sumoylation, and ubiquitinylation, are important epigenetic modifications of transcriptional regulation through altering chromatin structure (Kouzarides, 2007; Bonasio et al., 2010; Zhou et al., 2011). Genome-wide epigenomic profiles of specific histone modification and their transcriptional regulation roles have been previously reported in Drosophila (Kharchenko et al., 2011; Nègre et al., 2011). In particular, trimethylated Histone H3 Lysine 4 (H3K4me3) is usually found around the transcription start sites (TSSs) of genes, and targets active promoters (Bernstein et al., 2005; Mikkelsen et al., 2007; Benayoun et al., 2014), while trimethylation of Histone H3 Lysine 27 (H3K27me3) is usually located in intergenic regions, and acts as a repressive epigenetic marker to silence gene transcription (Grossniklaus and Paro, 2014; Aranda et al., 2015; Conway et al., 2015). In addition, the two marks can co-localize as bivalent domains to maintain gene expression levels at poised state (Bernstein et al., 2006; Vastenhouw and Schier, 2012). Studies of hPTMs in insects have been largely limited to the model species Drosophila, with hPTMs found involving in regulating phenotypic behaviors or changes such as circadian rhythm (Bu et al., 2020; Gong et al., 2021), aging (Siebold et al., 2010; Ma et al., 2018), and adaptation to stressful environments (Sharma et al., 2017; Wang et al., 2021). The few hPTMs studies in non-model insect species have focused on phenotypic change in non-invasive insects, such as nutritionally affected polyphenisms in honey bees and ants (Simola et al., 2013; Glastad et al., 2015; Simola et al., 2016; Wojciechowski et al., 2018), pupal diapause in response to photoperiod in the flesh fly and oil-collecting bee (Reynolds et al., 2016; Santos et al., 2018), and morphological development influenced by nutritional conditions in the flour beetle (Ozawa et al., 2016). Taken together, it is evident that hPTMs can obviously regulate important physiological processes and behavioral activities of insects. However, the role of histone modifications in invasive insects are still unknown.
To provide histone modification landscape of invasive insect species, we used Bactrocera dorsalis (Hendel) (Diptera: Tephritidae) (Figure 1), an notorious invasive species that has caused significant loss to global agriculture due to its polyphagia, high fecundity, superior mobility, high insecticide resistance, and strong environmental stress resistance (Santos-Rosa et al., 2002; Ekesi et al., 2007; Han et al., 2011; Jin et al., 2011; Hu et al., 2014; Magagula et al., 2015; Pieterse et al., 2017; Gu et al., 2019). After its first detection in 1912 in southern China, Bactrcocera dorsalis is now widely distributed in China with weak population genetic structure (Qin et al., 2018). Besides, it has strong flight capacity, and they can fly long distance or even fly to a high altitude and then disperse via moving air currents, which is prerequisite for long distance migration (Steiner, 1957; Chen et al., 2007). As its strong flight capacity is important for entry, establishment and spreading during invasiveness (Roques et al., 2009; Guo et al., 2018), it provides an excellent insect model to study the regulatory role of histone modification on insect flight activity. We used Chromatin Immunoprecipitation followed by high-throughput sequencing (ChIP-seq) (Furey, 2012; Park et al., 2017), which has been proven useful to identify transcription factor binding sites (Schmidt et al., 2010; Stefflova et al., 2013), conserved regulatory regions (Tena et al., 2014), and the activity of tissue-specific regulatory elements (Visel et al., 2009; Blow et al., 2010; Attanasio et al., 2013), to profile two histone modifications, H3K4me3 and H3K27me3, in thorax muscles, a key tissue to insect flight ability (Maughan and Vigoreaux, 1999; Josephson, 2006; Roques et al., 2009; Guo et al., 2018). We then correlated the histone modification profiles with levels of genome-wide gene expression obtained by RNA-seq, which allowed us to link enrichment or depletion of active and repressive histone modifications to their target genes. In this study we investigate two specific questions: a) What are the general regulatory landscapes of the two histone modifications in the flight muscles of this invasive insect? b) Is there functional relevance of the two histone modifications to flight activity? Answering the two questions will help us better understand the histone modification signature of the flight muscles of B. dorsalis, and serve as the baseline for future studies of mapping histone modification of other invasive insects.
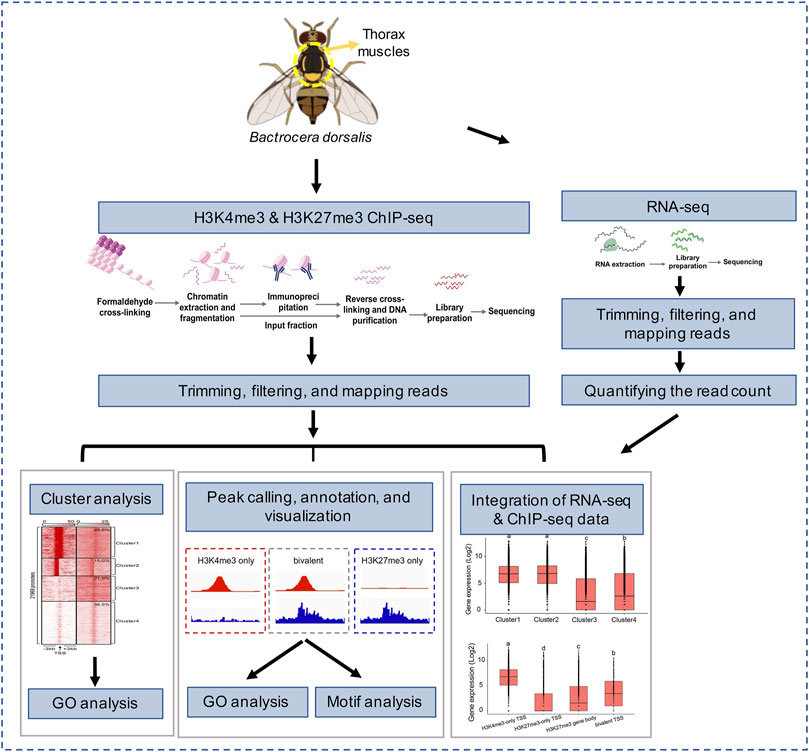
FIGURE 1. Overview of experimental design and data analysis.
Materials and methods
Insect source and husbandry
We collected B. dorsalis larvae individuals from the southern edge of its distribution in China (Danzhou, Hainan Province; 19°30′N, 108°50′E), within the initial entry region of this species in China, which could provide a baseline and more comprehensive genome-wide regulatory landscape of hPTMs in B. dorsalis. Then we reared the larvae for two generations in a common condition in lab as described in Li et al. (2012) to remove environmental effects from previous generations (Sanford and Kelly, 2011; Somero, 2012). Briefly, we distributed 500 eggs to the surface of an artificial diet (Supplementary Table S1) until larvae hatching. After hatching and emergence from pupae, we maintained all adult flies at a density of 200 individuals per rearing cage (45 × 45 × 50 cm), air temperature of 25°C ± 0.5°C, humidity of 70% ± 5%, and a photoperiod of 14 L: 10 D in an artificial climate cabinet (PXZ-430B, Ningbo Jiangnan Instrument Factory, China). We fed adult flies with 7.5 g sucrose and 2.5 g peptone (AOBOX, China) every 2 days. To control for developmental stage and sex, equal number of sexually matured, 15-day-old male and female adults were randomly selected, snap frozen in microcentrifuge tubes, and stored at −80°C until extraction of genetic material.
Tissue of choice
The choice of tissue used for genome-wide mapping of histone modification can influence the interpretation of modification landscape (Bonn et al., 2012). Because the strong invasion ability of B. dorsalis is partially due to its remarkable flight capacity that significantly increases when reaching sexual maturity, we selected thorax muscles from sexually matured adults (Liang et al., 2001; Josephson, 2006), which is likely to increases the likelihood of identifying histone modification peaks and associated genes with functions relevant to flight activity.
Chromatin immunoprecipitation
To identify the regulatory landscape of histone modification, we performed ChIP experiments using two independent biological replicates for both H3K4me3 and H3K27me3, with each replicate consisting of mixed 25 males and 25 females. We used two replicates by following the recommendation of ENCODE Consortium, because more than two replicates do not significantly improve site discovery (Rozowsky et al., 2009). To prepare ChIP libraries, we followed the protocol as described in Nagalingam et al. (2018) with some minor modifications. Briefly, we first dissected and pooled thorax muscle tissue (hereafter referred to as “muscles”) from 50 adults in cold 1X phosphate-buffered saline (PBS, P1022, Solarbio, China) mixed with 1X protease inhibitor (A8260, Solarbio, China). After thorough disruption and filtration through cell strainer, chromatin released from muscles was cross-linked in 200 μL of 1% fresh formaldehyde (HUSH, China) and cold PBS at room temperature for 15 min. We stopped the reaction by adding 1 M glycine (MG0167, MesGenBiotech, China) to a final concentration of 0.2M, and incubating in a rotator mixer at room temperature for an additional 10 min. The fixed tissues were washed three times using cold PBS, and homogenized by centrifuging at 4°C in a speed of 700g for 5 min. To lyse cells and retrieve single cell suspension, we resuspended pellets in 1 mL of animal cell nuclear lysate (20–163, Millipore, US) on ice for 30 min. The fragmentation of chromatin DNA was achieved by transferring 100 μL of the lysate into a sonication tube, and sonicating by Bioruptor® UCD-200 to the length of 200–600 bp (Supplementary Figure S1A). After running electrophoresis on agarose gel to check sonication results, we diluted the supernatant containing sheared chromatin DNA with 900 μL ChIP dilution buffer (20–153, Millipore, US) containing 4.5 μL protease inhibitors, and mixed with 60 μL of Protein G Agarose (16–201D, Millipore, US) for 1 h at 4°C in a rotator mixer. After pellet agarose by brief centrifugation at a speed of 5000g for 1 min, we collected precleared chromatin from the remaining supernatant.
To set up ChIP reactions, we used anti-H3K4me3 (ab8580, Abcam) and anti-H3K27me3 (ab6002, Abcam) after searching antibodies from the Antibody Validation Database (http://compbio.med.harvard.edu/antibodies), and assessed their quality using Western blot. We performed Western blot following Marcon et al. (2015) with some minor modifications. Briefly, we first extracted histones, boiled them in sample buffer, ran stacking gel of 4.5% acrylamide in 10% polyacrylamide to electrophorese, and transferred the proteins from gel to PVDF membrane (Immobilon-P Transfer membrane, Millipore). Following incubation with primary and secondary antibodies, we performed chemiluminescence detection with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, US) following the manufacturer’s instructions (Supplementary Figure S1B). We then separately mixed precleared chromatin with one of the two antibodies (5 ug of antibody per reaction) following manufacture’s instruction, and incubated the mix overnight on a rotator mixer at 4°C. We added 60 μL Protein G Agarose to each reaction, and incubated all reactions on a rotator mixer at 4°C for 1 h to collect the antibody/antigen/DNA complex. Beads were then sequentially washed once with Low Salt Immune Complex Wash Buffer (20–154, Millipore, US), High Salt Immune Complex Wash Buffer (20–155, Millipore, US), LiCl Immune Complex Wash Buffer (20–156, Millipore, US), and twice with TE Buffer (20–157, Millipore, US). DNA was then eluted from the beads by incubation in 200 μL elution buffer (10 μL 20% SDS, 20 μL 1 M NaHCO3 and 170 μL ddH2O) for 15 min at room temperature. We then added 8 μL 5 M NaCl to all samples, followed by incubation at 65°C overnight to reverse the DNA-protein crosslinks. Then, we incubated the samples for 30 min at 37°C with 1 μL RNase A (R6513, Merck, Germany), followed by mixing with 4 μL 0.5 M EDTA (E8030, Solarbio, China), 8 μL 1 M Tris-HCl (T1020, Solarbio, China) and 1 μL proteinase K (4333793, ThermoFisher, US) and incubating at 45°C for 2 h. Finally, DNA was purified using Bioline DNeasy minikit following manufacturer’s protocol. The final concentration and amount of DNA used for ChIP-seq library preparation were shown in Supplementary Figure S1C.
Library preparation and sequencing
ChIP-seq libraries were prepared, and sequenced by following the ENCODE guidelines (Landt et al., 2012) at Wuhan IGENEBOOK Biotechnology Co., Ltd. DNA fragments ranging from 200 to 300 bp were selected using SPRI beads, and amplified by 15 PCR cycles following end repairing and adapter ligation. The quality of libraries was checked on Bioanalyzer 2100 (Agilent) and Qubit fluorometer (Invitrogen, Carlsbad, CA, United States). All ChIP-seq libraries were sequenced in one lane (150-bp pair-end reads) of Illumina HiSeq 2000 platform.
Quality control and read mapping
To remove adapter contamination, low-quality bases, and bases artificially introduced during library construction, we filtered ChIP-seq reads following the steps described in Zhou et al. (2011) with some modifications. We examined read quality of each sample using FASTQC v0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and trimmed reads using Trim Galore! v0.6.6 (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with default parameters. We used Bowtie2 v2.4.2 (Langmead and Salzberg, 2012) to align trimmed reads for each replicate to the genome (NCBI assembly accession ASM78921v2) with default parameters. We only included reads that uniquely mapped to the reference genome in downstream analysis. We used SAMtools v1.3.1 (Li et al., 2009) to sort mapped reads based on reference coordinates, and filtered unmapped, non-uniquely mapped and discordantly paired reads by setting ‘view -F 3852’ and ‘view -b -f 2’, and removed reads with low mapping quality (MAPQ <30). PCR duplicated reads were removed by using MarkDuplicates function in Picard v2.25.1 (http://broadinstitute.github.io/picard/). We checked the reproducibility between biological replicates by calculating the Pearson correlation coefficient (PCC) and Spearman’s correlation coefficient (SCC) for genome-wide read distribution, using the average signals in individual 500-bp non-overlapping genomic windows with “multiBamSummary” and “plotCorrelation” functions in deepTools v3.5.1 (Bardet et al., 2012; Ramirez et al., 2014). We used Integrative Genome Viewer (IGV) v2.9.4 (Thorvaldsdottir et al., 2013) for visualization of enrichment profiles. Read counts were normalized to counts per million mapped reads (CPM) in 10-bp windows with the extendReads to 200, and converted to bigWig format using the “bamCoverage” function in deepTools.
Evaluating ChIP-seq data
We used a number of indicators to evaluate the quality of the ChIP-seq data, following the Encyclopedia of DNA elements Consortium (ENCODE) guideline (Robertson et al., 2007; Landt et al., 2012). We considered a library to be highly complex when its non-redundant fraction (NRF) was at least 0.8, PCR Bottlenecking Coefficients (PBC) 1 was higher than 0.5 and PBC2 was higher than 1. In addition to peak calling, we separately quantified enrichment by conducting a strand cross-correlation analysis to assess the degree of immunoprecipitated fragment clustering in ChIP-seq experiments. We considered a library with good clustering when its normalized strand coefficient (NSC) was higher than 1.05, and its relative strand correlation (RSC) was higher than 0.8. Finally, we used fraction of reads in peaks (FRiP), which evaluates the number and strength of peaks obtained for each ChIP replicate, to measure the signal-to-noise ratio (S/N). We considered an immunoprecipitation reaction successful when its FRiP was at least 0.01.
Cluster analysis
To explore the enrichment of aligned ChIP-seq reads for H3K4me3 and H3K27me3 in the 3-kb windows around the transcription starts sites (TSSs) (hereafter referred to as ‘TSS regions’), we first extracted reads and reference coordinates (position of 21,968 TSSs, assumed to be the 5′-end of all annotated transcripts), using the ‘getPromoter’ function implemented in the R package ChIPseeker v1.26.2 (Yu et al., 2015). Windows of negative strand transcripts were inverted so that they have the same orientation as the positive strand transcripts. Following Kratochwil and Meyer (2015), we performed clustering normalization using K-means linear, and distinguished between four categories of clusters: 1) Cluster 1, which are clusters featured by dual peaks of H3K4me3 around TSSs, with one peak highly occupied (mean density ≥200) and shifted to the 3′-end of TSSs (<200bp) and the other peak moderately occupied (20 ≤ mean density <200) and shifted to the 5’ -end of TSSs (<400bp), and very low or no (0 ≤ mean density <10) H3K27me3 occupancy around TSSs, 2) Cluster 2, which are clusters featured by one H3K4me3 peak highly occupied and shifted to the 5′ end of TSSs (<200 bp), and very low or no H3K27me3 occupancy around TSSs, 3) Cluster 3, which are clusters featured by low (10 ≤ mean density <20) H3K4me3 and H3K27me3 occupancy around TSSs, and 4) Cluster 4, which are clusters of featured by one H3K4me3 peak of low occupancy and very low or no H3K27me3 occupancy around TSSs. Finally, we annotated genes with their TSSs within the 3 kb distance to Cluster 1-3, using the ‘annotatePeak’ function implemented in the ChIPseeker for functional analysis. We visualized genomic distribution of clusters in heatmaps, using Seqminer v1.3.4 (Ye et al., 2011).
Peak calling and genomic context analysis
To perform peak calling for each replicate, we used ‘ChIP-seq’ function, implemented in MACS2 v2.1.1 (Feng et al., 2012), with input group as control. For narrow peak such as H3K4me3, we determined reproducible peaks between replicates using irreproducible discovery rate (IDR) (Babu et al., 2011; Li et al., 2011). We used the ‘callpeak’ module with following parameters: -g 4.53e8 --keep-dup all, and a narrow peak cut-off of p-value <0.01. We then used the ‘idr’ module to measure the reproducibility of replicates, and ranked the peaks by their p-values with an IDR threshold of 0.01. Following the ENCODE ChIP-seq guidelines (Landt et al., 2012), peaks that passed the IDR threshold by comparing true replicates were retained for downstream analysis. For broad peak such as H3K27me3, we identified peaks by using the ‘callpeak’ module with following parameters: ‘--broad -g 4.53e8 --broad-cutoff 0.1 --keep-dup all’. We then identified the overlapping peaks between both replicates using the ‘findOverlapsOfPeaks’ function in the R package ChIPpeakAnno v3.10.2 (Zhu et al., 2010; Zhu, 2013). We used BEDtools v2.30.0 (Quinlan and Hall, 2010) intersect commands to identify the bivalent domains, which consist of nucleosomes containing both H3K4me3 and H3K27me3 simultaneously, with at least a 100-bp overlap for the estimation of one nucleosome overlap (Ku et al., 2021). We then mapped the H3K4me3 and H3K27me3 peaks to TSS and gene body regions [defined as the genomic region from the TSS to the transcription termination site (TTS) (Young et al., 2011)], respectively, to generate tagMatrix using the ‘getTagMatrix’ function in ChIPseeker, and visualized average profiles of the peaks binding to TSS regions (i.e., 5′ to 3′) and gene body regions, respectively.
To identify the positions of H3K4me3 and H3K27me3 peaks within genomic features, we annotated identified peaks to the B. dorsalis genome, using ‘annotatePeak’ function in ChIPseeker with default parameters. We gave the precedence to promoter >5′untranslated region (UTR) > 3′UTR > exon > intron > downstream > intergenic regions when features overlapped, and defined downstream regions as downstream 300 bp from the gene end. We then compared the distributions of peaks to null distributions of individual genomic features in the reference genome using G test.
Effect on histone modification on gene expression
To demonstrate the regulatory role of the two histone modifications in gene expression, we constructed RNA-seq libraries for the same thorax muscle tissues as described above. We extracted total RNA using TRIzol reagent kit (15596–018, Invitrogen, United States) following the manufacturer’s protocol. The quality and concentration of extracted RNA was evaluated using Nanodrop 8000 spectrophotometer (Thermo Fisher, United States) and Agilent 2100 Bioanalyzer (Agilent Technologies, United States). RNA-seq libraries were prepared by Novogene Bioinformatics Technology Co. Ltd. (Tianjin, China) using NEBNext® UltraTM Directional RNA Library Prep Kit (E7420, NEB, United States), and sequenced on the Illumina NovaSeq 6000 platform (Novogene) with 150-bp paired-end reads.
To quantify gene expression, we used Trim Galore! to remove adapter sequences and low-quality reads. The remaining quality-filtered reads were assessed with FASTQC and mapped to the B. dorsalis genome (NCBI Assembly accession ASM78921v2) using HISAT2 v2.1.0 (Kim et al., 2015), with the following parameters: --new-summary -k 1 --rna-strandness RF. We removed PCR duplicated reads using the MarkDuplicates function implemented in Picard. We used HTSeq-Count v0.12.4 (Anders et al., 2015) to output read counts for each gene with parameters “-f bam -s reverse -r name -t exon -union”. Finally, we compared the average expression levels (log2 of RPKM values) among genes 1) associated with Cluster 1–4, 2) with their TSSs enriched by H3K4me3, H3K27me3 or bivalent domains, and 3) with their gene bodies enriched by H3K27me3 using one-way analysis of variance (ANOVA).
Gene annotation and gene ontology analysis
To perform the functional analysis of peaks, we identified genes with TSS regions enriched by H3K4me3, H3K27me3, or both peaks, using the ‘annotatePeak’ function in ChIPseeker. In addition, because H3K27me3 is also featured with broad domains across gene bodies, we thus also annotated genes with their gene bodies enriched with H3K27me3 peaks.
For gene ontology (GO) enrichment analysis, we first complied a GO term list for all protein-coding genes in the B. dorsalis genome, using eggNOG-mapper (Huerta-Cepas et al., 2017) to search for orthologous genes in the eggNOG database. The list was then used as input to the universal enrichment protocol, using the R package clusterProfiler v4.0.5 (Yu et al., 2012) and q-value cut-off of 0.001 to determine enrichment significance.
Motif analysis
Because it has been suggested that motif analysis can facilitate the discovery of unanticipated sequence signals such as transcription factor binding sites associated with histone modifications (Bailey et al., 2013; Ruiz et al., 2019; Chen et al., 2020), we performed de novo motif analysis for H3K4me3, H3K27me3 and bivalent domains. We first extracted a list of sequences that are 50 bp upstream and 50 bp downstream from the summits of the top 500 peaks overlapping with TSS regions (Niu et al., 2018). We then used the list as input to search for conserved sequence motifs, using motif elicitation (MEME)-ChIP (Bailey et al., 2009) software with default parameters. We used TOMTOM (Khan et al., 2018) with default settings to match discovered motifs to the JASPAR database (Fornes et al., 2020). Finally, we used FIMO (Hertz and Stormo, 1999; Grant et al., 2011) to map motif prediction, and visualize genomic locations of the motifs in gene list as described above.
Potential regulatory role of histone modifications in flight activity
As flight activity is closely related to wing development, and long-distance migration is an important and widespread flight activity, we acquired a list of previously documented genes involved in wing development of B. dorsalis and migration-associated genes from recent papers (Supplementary Table S2), and compared them to peak-annotated genes identified above to investigate the possible functional relevance of histone modification to flight activity (Jones et al., 2015; Guo et al., 2018; Doyle et al., 2022). Specifically, Guo et al. (2018) profiled transcriptomes of B. dorsalis, and identified a group of key genes with functions relevant to wing development, which showed highest expression level in the pupal stage and poised state in other stages, through comparative transcriptome analyses. Jones et al. (2015) used comparative transcriptomics of flight phenotypes to determine a suite of expressed candidate genes associated with flight activity in the cotton bollworm, Helicoverpa armigera, including odorant binding proteins, flight muscle structure, fatty acid synthesis, etc. Doyle et al. (2022) undertook a genome-wide transcriptomic comparison of actively migrating marmalade hoverfly, Episyrphus balteatus and found the features of the migrant phenotype have arisen by the integration and modification of pathways such as insulin signalling for diapause and longevity, JAK/SAT for immunity, and those leading to octopamine production and fuelling to boost flight capabilities. Specifically, upregulated genes associated with migration include genes related to metabolic processes, sensory functions, octopamine synthesis, neuropeptide hormones, muscle function, and immunity genes, and the downregulated genes include genes involved in insulin and TGF-β signalling, hormonal regulation, and multiple ankyrin repeats single KH domain (mask). We then compared the proportion of peak-annotated genes in each gene category to the proportion of peak-annotated genes in the reference genome using G test.
Results
Mapping statistics and quality check of ChIP-seq data in B. dorsalis
After alignment, 55.4%–55.7% of H3K4me3 reads, 40.2%–40.3% of H3K27me3 reads, and 37.3% of input reads were unambiguously mapped to the reference genome, with high PCC and SCC values between replicates, suggesting high experimental reproducibility (Supplementary Table S3; Supplementary Figure S3A). While 2.2% of H3K4me3 reads, 2.4%–2.6% of H3K27me3 reads, and 3.4% of input reads were filtered due to nonspecific matches, 42.1%–42.4% of H3K4me3 reads, 57.2%–57.3% of H3K27me3 reads, and 59.3% of input reads did not map at all. After further filtering of PCR duplicates, unmapped, non-uniquely mapped, improperly paired, and low mapping quality reads, 36.1%–36.3% of H3K4me3 reads, 17.4%–17.8% of H3K27me3 reads, and 19.6% of input reads were retained for downstream analysis (Supplementary Figure S2).
When analyzing uniquely mapped reads, all replicate and input samples passed the PBC1 and PBC2 quality threshold, while four of five samples failed to meet the NRF cut-offs (Supplementary Figure S4A), suggesting low library complexity, similar to the recent studies in other non-model species (Kingsley et al., 2019). However, when checking the NSC and RSC values, all samples met the ENCODE standard, and had larger fragment-length peaks compared to read-length peaks (Supplementary Figure S4B), suggesting high immunoenrichment quality despite lower library complexity. When checking FRiP value, both H3K4me3 replicate samples produced a high proportion of aligned reads within peaks, with the FRiP scores ranging from 0.611 to 0.612 (Supplementary Figure S4C), and the called regions ranging from 7,695 to 7,814 (Supplementary Figure S4D). In contrast, both H3K27me3 replicate sample had lower FRiP value of 0.295–0.319 and 14,929–16,481 called regions (Supplementary Figures S4C, S4D). In summary, we found that ChIP-seq can generally be adapted to the non-model invasive species B. dorsalis, and provide sufficient data for regulatory landscape analysis.
Cluster analysis
We present the results obtained by running the analysis on Replicate 1 (H3K4me3_1 and H3K27me3_1) here. Results concerning Replicate 2 (H3K4me3_2 and H3K27me3_2) were similar (Supplementary Figure S6). Based on K-means cluster analysis, we found 30.6%, 15.2%, 17.8%, and 36.4% of TSSs falling into Cluster 1-4, respectively (Figures 2A, B). Therefore, we assumed that 45.8% of the genes were in either active or poised (Cluster 1 and Cluster 2) state, while 54.2% of the genes were in either weakly expressed (Cluster 3) or inactive (Cluster 4) state. Indeed, the gene expression patterns involved in each cluster have supported it (Cluster 1: 6.4 ± 0.02, Cluster 2: 6.4 ± 0.03, Cluster 3: 3.0 ± 0.04, Cluster 4: 3.6 ± 0.03; the gene expression were calculated based on the log2 of RPKM values), with the average expression levels of genes associated with Cluster 1 and Cluster 2 were significantly higher than those in Cluster 3 and Cluster 4 (ANOVA test, p < 0.05, Figure 2D).
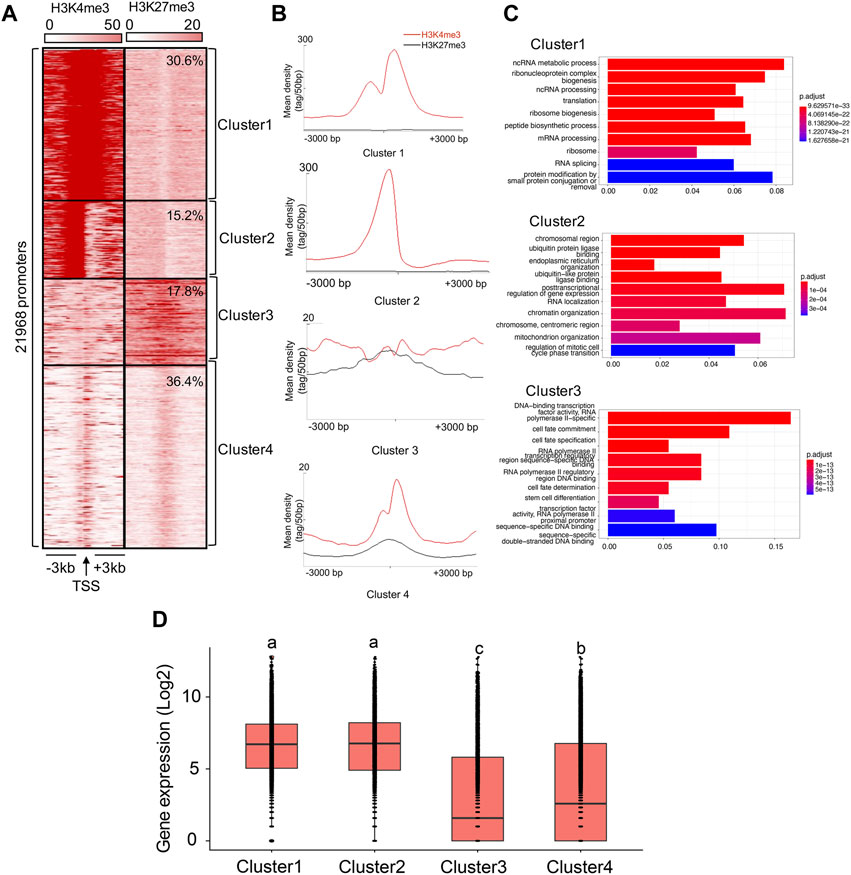
FIGURE 2. Cluster analysis of H3K4me3 and H3K27me3 in the B. dorsalis genome. (A) Heatmap and (B) density profile of the K-means clustering of mapped read position for H3K4me3 and H3K27me3 around TSS regions (±3 kb) in the B. dorsalis genome. (C) Top 10 over-represented GO terms of genes associated with Cluster 1–3. Corrected p-values are indicated by color, the warmer the color, the smaller the p-values. The vertical axis represents the GO term names and the horizontal axis represents gene ratio. (D) Boxplots showing the mean level of gene expression for genes with the TSSs within the 3 kb distance to Cluster 1–4. The gene expression levels were calculated based on the log2 of RPKM values. The differences in gene expression levels among clusters were evaluated using one-way analysis of variance (ANOVA).
H3K4me3 and H3K27me3 peak analyses
In total, we identified 7,226 and 11,417 peaks for H3K4me3 and H3K27me3, respectively (Supplementary Table S4), with the two replicates displaying high reproducibility and low noise interfering (Supplementary Figures S3, S5). 95.0% (6,865 out of 7,226) of H3K4me3 peaks and 45.9% (5,206 out of 11,417) of H3K27me3 peaks overlapped with 45.6% and 11.2% of genes annotated in B. dorsalis genome, respectively, suggesting that these two histone marks were widely involved in regulatory activities in this species. When analyzing the length of peaks, 45.3% of the H3K4me3 peaks had a length shorter than 1 kb, and 44.9% of peaks had a length ranging between 1 kb and 2 kb. As for H3K27me3 peaks, the length ranged from 251bp to 47,936 bp, with the majority of peaks (51.7%) having length longer than 1 kb. We extracted 5,541 and 843 genes with TSS regions annotated with H3K4me3-only and H3K27me3-only peaks, respectively, and we also obtained 295 bivalent domains, in which 197 bivalent domains were overlapped with 139 gene TSS regions (Figure 3A; Supplementary Table S5). Due to the key regulatory role of H3K27me3 locating within gene bodies, we also identified 1,398 genes with their gene bodies overlapped with H3K27me3 peaks. We then assessed the relationship between different histone modification profiles and gene expression. We found significant differences in average gene expression levels between H3K4me3-only TSS (log2 of RPKM values: 6.6 ± 0.02), H3K27me3-only TSS (2.1 ± 0.06), H3K27me3 gene body (2.7 ± 0.05), bivalent TSS (3.8 ± 0.2), and genes without hPTMs (3.3 ± 0.04), with the highest average expression levels were H3K4me3-only TSS, followed by bivalent TSS, genes without hPTMs and H3K27me3 gene body, and the lowest average expression levels were H3K27me3-only TSS (ANOVA test, p < 0.05, Figure 3C). However, genes with TSSs enriched by bivalent domains were not in the posited state, defined by Gaertner et al. (2012) as those with transcript levels below 10 RPKM as determined by RNA-seq, which was consistent with that in Drosophila melanogaster.
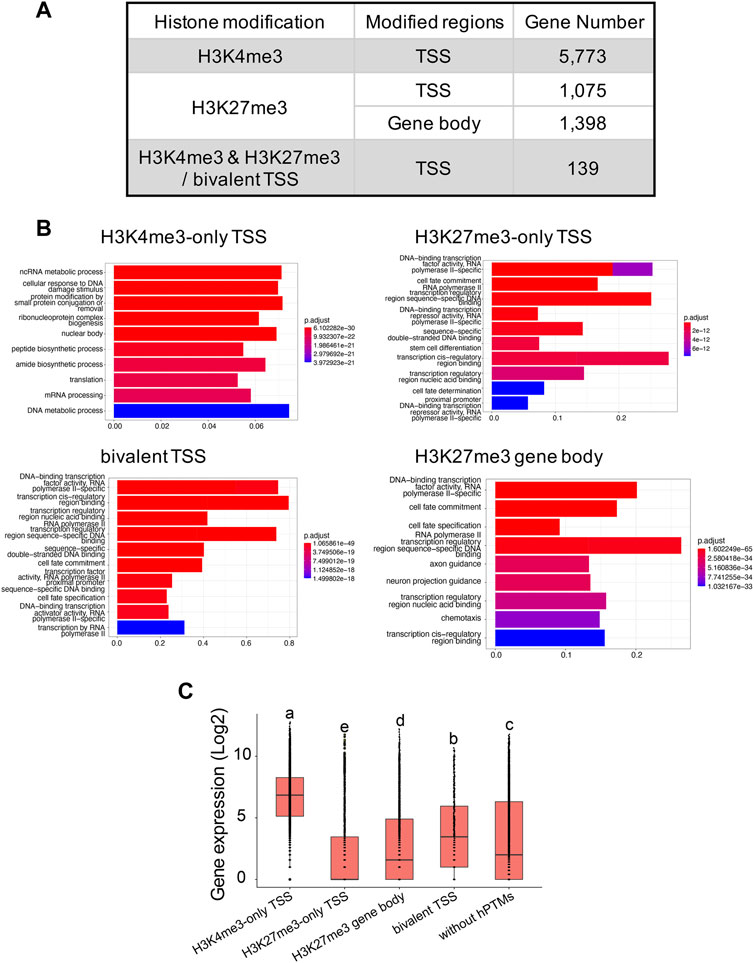
FIGURE 3. H3K4me3 and H3K27me3 peak analyses in the B. genome. (A) Summary of the genes with H3K4me3-only, H3K27me3-only and bivalent domains at the TSSs, and genes with H3K27me3 at the gene body regions in B. dorsalis. (B) Top 10 over-represented GO terms in genes with H3K4me3-only, H3K27me3-only and bivalent domains at the TSSs, and genes with H3K27me3 at the gene body regions in B. dorsalis. Corrected p-values are indicated by color, the warmer the color, the smaller the p-values. The vertical axis represents the GO term names and the horizontal axis represents gene ratio. (C) Boxplots showing the mean level of gene expression for genes with TSSs enriched by H3K4me3, H3K27me3, or both peaks and genes with gene bodies enriched by H3K27me3 and genes that are not modified by hPTMs. The gene expression levels were calculated based on the log2 of RPKM values. The differences in gene expression levels among different modifications were evaluated using one-way analysis of variance (ANOVA).
When examining the average histone modification peak profiles, we found a mutually exclusive pattern between the two marks where H3K4me3 peaks showed a strong dual peak pattern with a central notch around the TSSs (Figure 4A). In contrast, H3K27me3 tended to occupy broader regions, with the average enrichment was almost constant throughout gene bodies (Figure 4B). When analyzing the genomic features of H3K4me3 peaks, we found a significant enrichment of peaks in genic regions (G = 38.5, df = 1, p = 5.4 × 10−10), and promoter regions (within 1 kb upstream and 1 kb downstream to TSSs) (G = 419.4, df = 1, p < 2.2 × 10−16) when compared to the null distribution built on genome GFF annotation file (Figure 4C; Supplementary Table S5). While as for H3K27me3 peaks, only 8.0% of the peaks overlapped with the promoter regions (within 1 kb upstream and 1 kb downstream to TSSs) (G = 1.2, df = 1, p = 0.3) and more than half of the peaks located within intergenic regions (G = 28.3, df = 1, p = 1.0 × 10−7) (Figure 4D; Supplementary Table S5). In summary, the peak annotation results showed contrasting distribution patterns between H3K4me3 and H3K27me3, suggesting possibly distinct regulatory roles between the two modifications (Benayoun et al., 2014; Aranda et al., 2015; Conway et al., 2015).
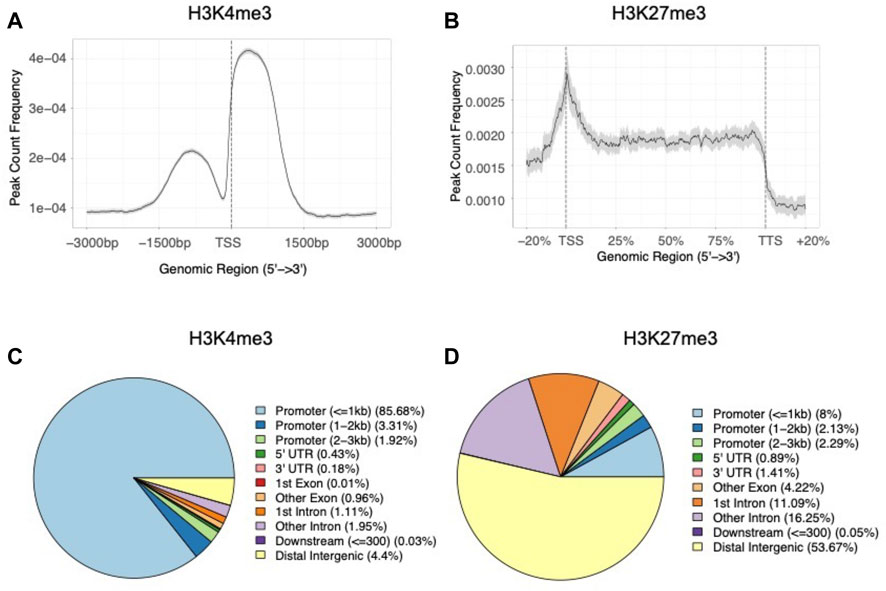
FIGURE 4. Distribution patterns of (A) average H3K4me3 peaks around TSS regions (±3 kb) and (B) average H3K27me3 peaks in gene body regions of the B. dorsalis genome. The proportion of genomic features of (C) H3K4me3 peaks and (D) H3K27me3 peaks compared with genomic features.
Functional analysis of clusters and peaks
We found a number of GO terms significantly enriched within each Cluster (Figure 2C; Supplementary Table S6), with genes involved in metabolic, biosynthetic processes, cell cycle, gene expression and chromatin organization significantly overrepresented (q-value <0.001) in Cluster 1 (503 GO terms) and Cluster 2 (37 GO terms). In contrast, genes annotated with Cluster 3 (229 GO terms) displayed different GO term enrichment, with functions related to cell fate, tissue development and morphogenesis, negative regulation of transcription by RNA polymerase II, and imaginal disc development significantly enriched (q-value <0.001). Genes associated to H3K4me3 vs. H3K27me3 also differed in the number and functions of GO terms (Figure 3B; Supplementary Table S6). Genes with TSSs uniquely modified by H3K4me3 significantly enriched in 622 GO terms (q-value <0.001) related to GO:0034660 (ncRNA metabolic process), GO:0006974 (cellular response to DNA damage stimulus), and GO:0006412 (translation), which were totally different from the GO terms associated with genes with TSSs uniquely modified by H3K27me3, which were enriched for GO:0001227 (DNA-binding transcription repressor activity, RNA polymerase II-specific), GO:0000976 (transcription cis-regulatory region binding) and GO:0021953 (central nervous system neuron differentiation). In contrast, we found 159 GO terms shared between the genes with TSSs (161 GO terms) and gene bodies (735 GO terms) modified by H3K27me3. Genes with bivalent protomers significantly enriched in 381 GO terms related to cell fate, transcription cis-regulatory, tissue development and morphogenesis, and imaginal disc development, with 104 GO terms overlapped with GO terms associated with genes uniquely modified by H3K27me3 at TSS regions and gene bodies, and only two GO terms, GO:0003712 (transcription coregulator activity) and GO:0003682 (chromatin binding), overlapped with genes uniquely modified by H3K4me3 at TSS regions.
Motif analysis
We detected one, three and one significantly enriched motifs in the TSS regions of genes uniquely modified by H3K4me3, H3K27me3, and bivalent domains, respectively (Figures 5A–C; Supplementary Table S7). The significantly enriched motif for H3K4me3 was ‘GCTGCT’ (E-value = 1.0 × 10−20, frequency = 401), similar to the motif of odd (E-value = 1.3 × 10−2) identified in the JASPAR2018_CORE_insects database. The most enriched motif for H3K27me3 was ‘ACAT’ (E-value = 8.4 × 10−176, frequency = 73,968), which is similar to the motif of Cf2 (E-value = 3.0 × 10−2) in the database. The second most enriched motif for H3K27me3 was a TGT-rich motif (E-value = 1.5 × 10−37, frequency = 69,886). The third most significantly enriched motif was an AA-rich motif (E-value = 5.8 × 10−15, frequency = 4,139). The significantly enriched motif for bivalent domains was ‘GTTGTT’ (E-value = 7.3 × 10−21, frequency = 74), which is similar to the known motif of Bgb:run (E-value = 5.5 × 10−3).
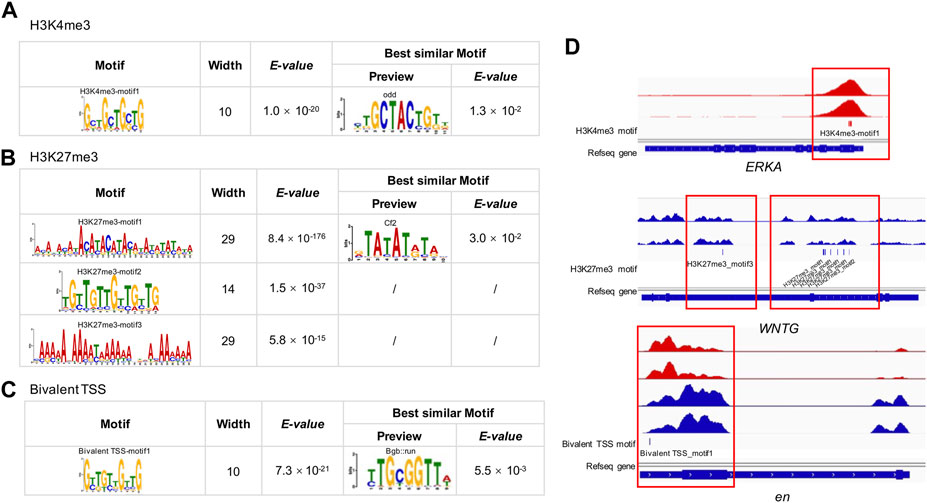
FIGURE 5. MEME-CHIP analysis of H3K4me3, H3K27me3 and bivalent domains motif in the B. dorsalis genome. Top consensus sequences identified by de novo motif discovery for (A) H3K4me3, (B) H3K27me3 and (C) bivalent TSSs, and the best similar known motifs previously described in Drosophila. (D) Examples of the locations of putative motifs within genes related to flight activity. See Supplementary Table S7 for motif position information for all genes related to flight activity.
Potential regulatory role of histone modifications in flight activity
To explore the possible relevance of histone modification to flight activity, we analyzed the overlap between genes annotated with peaks and previously documented genes involved in wing development and migratory flight activity. We found 20 genes overlapped between peak-annotated genes and genes involved in the development of insect wings (G = 4.2, df = 1, p = 4 × 10−2, Supplementary Table S2), for example, epidermal growth factor receptor (EGFR), wingless (wg), cubitus interruptyus (ci), decapentaplegic (dpp), hedgehog (hh), including nine genes modified by H3K4me3, eight genes modified by H3K27me3, and three genes with bivalent promoters. Besides, we found 268 genes overlapped between peak-annotated genes and genes involved in migratory flight activity (Supplementary Table S2), including 55 metabolism related genes (G = 4.1, df = 1, p = 4 × 10−2), 14 genes related to environmental sensing, timing and navigation (G = 3.0, df = 1, p = 8 × 10−2), 15 genes involved in insulin signaling pathways (G = 12.3, df = 1, p = 5 × 10−4), 11 juvenile hormone (JH) and ecdysone related genes (G = 3.0, df = 1, p = 8 × 10−2), 5 genes associated with octopamine synthesis (G = 207.46, df = 1, p < 2.2 × 10−16), 10 genes associated with neuropeptide hormones (G = 3.3, df = 1, p = 8 × 10−7), 73 muscle function related genes (G = 20.4, df = 1, p = 6.3 × 10−6), 16 genes involved in transforming growth factor β signalling (G = 27.2, df = 1, p = 1.9 × 10−7), and 69 genes involved in JAK/STAT pathway and stress and immunity (G = 1.1, df = 1, p = 0.3). In total, the two hPTMs showed significant enrichment in 60% of the gene categories in Supplementary Table S2 when compared to expected by chance. As H3K4me3 and H3K27me3 are generally associated with actively transcribed genes and repressed genes, respectively (Benayoun et al., 2014; Aranda et al., 2015), we further found that 76.1% of H3K4me3-modified genes overlapped with upregulated genes, and 60.0% of H3K27me3-modified genes overlapped with downregulated genes involved in Supplementary Table S2. The widespread modification of genes relevant to wing development and migratory flight activity suggest a potentially key role of histone modifications in flight activity. When analyzing the relevance of motifs to wing development and metabolism, we found that 38.7% (29 out of 75) of the genes in the list of insect wings and muscle energy supply contained one or more putative DNA motifs (Figure 5D; Supplementary Table S7), suggesting the potential roles of these motifs in regulating insect flight activity.
Discussion
The role of histone modification in regulating phenotypic responses to environmental change in model and non-invasive species has received increased attention in recent years (Simola et al., 2016; Kacsoh et al., 2017; Wojciechowski et al., 2018; Gong et al., 2021). However, the regulatory landscape and potential functional relevance of histone modification in invasive species remain poorly understood (Jeremias et al., 2018; Vogt, 2021). We used a quantitative, single-base-resolution technique (ChIP-seq) to profile genome-wide patterns of H3K4me3 and H3K27me3, which are two key histone modifications involved in gene regulation and phenotypic change, from thorax muscle tissue of a highly invasive insect, B. dorsalis. Despite a generally distinct genomic locations and functions between the two histone modifications, we identified a small number of overlapping regions possibly due to promoter bivalency. The combined analysis of ChIP-seq and RNA-seq data supports that H3K4me3 coincides with actively transcribed genes and H3K27me3 is associated with repressed genes that show low levels of transcription. Importantly, we found genes that are previously identified to be associated with wing development and migratory flight activity were largely modified by H3K4me3 and H3K27me3 in B. dorsalis (Supplementary Table S2), providing a support for the possible role of histone modification in flight activity. Finally, we identified one, three and one putative motifs for H3K4me3, H3K27me3 and bivalent TSSs modified regions, respectively that were also likely to be important for flight activity. Overall, our study provides the first genome-wide investigation of histone modifications in an invasive insect pest, and identifies genomic regions modified by histone markers that may be relevant to rapid range expansion of invasive insect species.
Application of ChIP-seq on the non-model B. dorsalis
In this study, we present a protocol for preparing and analyzing H3K4me3 and H3K27me3 ChIP-seq data on a non-model invasive species, providing a powerful opportunity to examine the role of an under-explored epigenetic mechanism in insect that is likely to regulate invasiveness. The efficacy of our protocol was evaluated by a number of lines of evidence: 1) 40.2%–55.7% of reads were uniquely mapped to the reference genome, which is similar to the results from recent studies in other insects, for example, mosquito (whole body, 53.7%–58.2%) (Gómez-Díaz et al., 2014) and ant (brain, 17.2%–23.0%) (Simola et al., 2013). 2) Based on the uniquely mapped reads, we identified 7,226 H3K4me3 and 11,417 H3K27me3 peaks. While the number of H3K4me3 and H3K27me3 peaks identified in our study were smaller than the number of peaks found in D. melanogaster, mosquito (Gómez-Díaz et al., 2014; Ruiz et al., 2019) and silkworm (Cheng et al., 2018), and the genome size of B. dorsalis (468.7 Mb) is larger than D. melanogaster (138.9 Mb), mosquito (250.4 Mb) and silkworm (429.0 Mb), it could be due to the quality of the available genome annotation. 3) We found different distribution patterns between the two histone modifications, where H3K27me3 was more likely to be located within the intergenic regions, and H3K4me3 predominantly enriched in the genetic regions, in consistent with previous studies in yeast, plants, worms, flies and mammals (Bernstein et al., 2005; Liu et al., 2005; Zhang et al., 2009; Benayoun et al., 2014). 4) The combined analysis of ChIP-seq and RNA-seq data supports a relationship between transcription levels and profiles of histone modifications, with H3K4me3 coincides with actively transcribed genes, H3K27me3 is associated with repressed genes that show low levels of transcription, and the average expression level of genes not modified by hPTMs is lower than the average expression level of genes modified by H3K4me3 but higher than the genes modified by H3K27me3. 5) Gene annotation and ontology analysis of the two histone modifications showed the assumed expression of genes and an enrichment of GO terms expected to be found in muscle tissue. Taken together, the distribution patterns and functional annotations of H3K4me3 and H3K27me3 produced by our ChIP-seq protocol were comparable with the known results of histone modifications from other animal species, indicating strong reliability of our ChIP-seq data, and offering a proof-of-concept validation of our approach.
Functional analyses of H3K4me3, H3K27me3, and bivalent modifications in B. dorsalis
Our functional analyses showed that H3K4me3 modifications were enriched around the TSSs and 5′end of genes, while H3K27me3 modifications were found in the entire body of genes, which are consistent with the distinct distribution patterns between these two histone modifications observed in Drosophila and mammals (Boyer et al., 2006; Lee et al., 2006; Eissenberg et al., 2007; Schuettengruber et al., 2009; Mohan et al., 2011), suggesting a conserved regulatory role of histone modifications across animal taxa. In addition, we found a clear difference between genes and functions annotated with the two histone modifications. The peaks of H3K4me3 overlapped with genes both encoding proteins relevant to general molecular regulation progress and physiologic processes, and specific functions in muscles such as energy metabolism process, suggesting an involvement of H3K4me3 in regulating both house-keeping genes essential to all tissues (Pan et al., 2007) and signaling pathways active in muscles (Brisson et al., 2007; Cao and Jiang, 2017). In contrast, genes annotated with H3K27me3 peaks were more likely to repress genes that are not expected to highly expressed in muscles, for example, Oct-TyrR, Nlg2, and Nlg4 are genes associated with nervous system-related processes that were modified by H3K27me3, which was also found weakly expressed in adult Drosophila muscle tissue (Zhang et al., 2018). Taken together, these findings indicate that H3K4me3 likely regulates active transcription, while H3K27me3 is associated with transcriptional repression in B. dorsalis.
Despite a general mutually exclusive pattern of the genomic distributions and regulatory roles between H3K4me3 and H3K27me3, we also found bivalent TSS regions marked by both modifications. However, in contrast to previous finding that bivalent marks were widely distributed in the genomes of mammals and fish (Pan et al., 2007; Zhao et al., 2007; Vastenhouw et al., 2010; Sachs et al., 2013; Zhu et al., 2019; Blanco et al., 2020), and genes with bivalent promoters considered poised for expression (Mikkelsen et al., 2007; Voigt et al., 2013; Harikumar and Meshorer, 2015), we only identified 2.1% (139 out of 6,616) of all TSSs as bivalent TSSs in B. dorsalis, and the genes with bivalent TSSs were not in poised state (Gaertner et al., 2012). This is consistent with a recent findings in Drosophila that both marks were not significant coexistence in TSS regions (Schuettengruber et al., 2007; Schuettengruber et al., 2009; Gan et al., 2010; Schwartz and Cavalli, 2017), suggesting that bivalent domains might be restricted to certain organisms (Voigt et al., 2013). Such species-specific patterns have also been observed in DNA methylation, where vertebrate genomes are usually highly methylated while insect genomes are typically sparsely methylated (Roberts and Gavery, 2012). Because it is often suites of epigenetic mechanisms that act in concert to influence biological functions in animals, our finding suggests that the distribution and regulatory roles of bivalent modifications are phylogenetically unique.
Motif analysis of H3K4me3 and H3K27me3 in B. dorsalis
To further characterize the functional role of the two histone modifications that cannot be explored in the regular peak calling analysis, we performed motif analysis, and identified a number of motifs for both marks and the bivalent domains within TSS regions, consistent with the binding preferences of known motifs important for Drosophila development (Fornes et al., 2020). For example, the ‘GCTGCT’ motifs, identified as the principal motifs of the H3K4me3 peak binding, was highly homologous to the odd motif in Drosophila (Nüsslein-Volhard and Wieschaus, 1980). odd encodes a zinc finger protein that represses other segmentation genes in the early Drosophila embryo (Ward and Coulter, 2000), and plays a key role in wg signalling (Cadigan et al., 1994; Beaven and Denholm, 2018). We also identified an important motif for H3K27me3 peaks, ‘ACAT’, which is homologous to Cf2 (Chorion factor 2) in Drosophila. Cf2 is a key regulator during embryo muscle formation, and participates in the regulation of the final size and number of nuclei present in skeletal, visceral and cardiac muscles (Bagni et al., 2002; Arredondo et al., 2017). Due to their highly functional relevance, this collection of motifs may be important for facilitating muscle development and structure maintenance, and warrant future studies to provide a more complete understanding of the functional link between muscle functions and the underlying motifs.
Epigenetic regulation of flight activity in B. dorsalis
The strong flight ability along with its underlying physiological, morphological and behavioral mechanisms facilitating its expansion to new environments consist of a key component of invasiveness in B. dorsalis (Liang et al., 2001; Froerer et al., 2010). When comparing the overlap between a list of previously documented genes involved in wing development and migratory flight activity of B. dorsalis and peak-annotated genes, we found that a large number of these genes were modified by histone modifications in B. dorsalis, suggesting a possibly regulatory role of histone modification in flight activity. For example, we identified a number of overlapped genes with functions relevant to wing development, including EGFR, wg, ci, dpp, hh, omb, and ubx signaling pathways that have been found especially important in wing vein morphogenesis and structural basis for long-distance flight of insects (Strigini and Cohen, 2000; Roch et al., 2002; Zecca and Struhl, 2002; Brisson et al., 2010; Shen et al., 2010; Paul et al., 2013; Quah et al., 2015; Tomoyasu, 2017; Liu et al., 2020). Insect flight is the most energy demanding activity known in the animal species (Arrese and Soulages, 2010). In this study, we identified overlapped genes with functions relevant to muscle energy metabolism, for example, genes identified with roles in lipid metabolism, such as long-chain fatty-acid CoA ligase, desaturase related genes (Parisi et al., 2013; Jeffries et al., 2014), and carbohydrate metabolism genes, such as genes involved in trehalose transporter (Tret), glucosidase, maltase, hexokinase, glycogen phosphorylase (GlyP), and glycerol-3-phosphate dehydrogenase (Gpdh) (Barnes and Laurie-Ahlberg, 1986; Eanes et al., 2006; Kanamori et al., 2010; Inomata et al., 2019). Besides, we also identified overlapped genes related to PI3K signaling pathways that participate in redox reactions and the metabolism of lipids and carbohydrates (Nässel et al., 2013; Mattila and Hietakangas, 2017). Muscle assembly is focused in the thorax of flies and is primary associated with flight. We also identified a suite of overlapped genes associated with flight muscle structure, such as genes involved in lamins, Actin, sallimus (sls), flightin, and collagen (Hiromi and Hotta, 1985; Reedy et al., 2000; Bullard et al., 2005; Uchino et al., 2013). In addition, we annotated various genes involved in hormonal regulation, for example, insulin signaling pathway that regulates nutrient delivery and redistribution, which have been suggested playing a key role in wing formation and flight muscle development (Grönke and Partridge, 2010; Lin et al., 2016). Particularly, we found genes with bivalent TSSs that were relevant to JH signaling pathways, which has been suggested affecting flight, energy metabolism, and reproduction in insects (Flatt et al., 2005; Wang et al., 2012; Guo et al., 2018; Jones et al., 2018). In summary, our results suggest that histone modifications possibly play an important role in regulating genes relevant to flight activity in B. dorsalis (Roques et al., 2009; Guo et al., 2018; Jiang et al., 2022), which could provide the basis information for studies of epigenetic signatures of the flight activity of other invasive insects in the future study.
Potential caveats
Our study has some limitations that should be noted. First, unlike model species such as human, mouse, worm and fly, there is no blacklist in non-model organisms that contains the regions of high signal that presumably represent unannotated repeats in the genome (Consortium, 2012; Boyle et al., 2014; Yue et al., 2014). Therefore, our results were inevitably influenced by the adverse effect of significant enrichment of signal from amplification of noise (Carroll et al., 2014). Second, tissue and developmental stage can influence the genome-wide histone modification landscape (Bonn et al., 2012; Jambhekar et al., 2019). We only used muscle tissue from sexually matured adult stage of B. dorsalis, preventing the generalization of our results to other tissues or developmental stage that may also be important to flight, especially migratory flight activity. Sampling during the initial stage of ovarian development, when the migratory flight behavior of migratory insect often occurs (Xiao et al., 2013), maybe give more information in the future study. Third, the flight activities of insects are underpinned by large modifications in gene regulation (Doyle et al., 2022). We only profiled two histone modifications that are typically found in promoters and coding regions, while a number of different histone marks, such as H3K27ac, H3K9me1 and H3K36me1 are known to be enriched in non-coding elements and are also functionally important (Zhou et al., 2011). In the future, considering multiple hPTMs and link them with specific flight capacity phenotype will further reveal the regulatory role of histone modifications in insect flight activities. In addition, it is important to compare the pattern of hPTMs in invasive vs. non-invasive species, which could provide more information on epigenetic regulation of invasiveness. The closely related B. minax, which has much more restricted host use and is less invasive compared to B. dorsalis could have been a relevant species for comparison in the future study. Thus, using ChIP-seq to profile additional histone modifications in more tissues and developmental stages will be helpful to provide a more comprehensive view of the regulatory landscape of histone modification and epigenetic architecture in B. dorsalis.
Conclusion
Consistent with previous studies of model and endemic species, here we present the first genome-wide regulatory landscape of H3K4me3 and H3K27me3 histone modifications in the invasive B. dorsalis. We validated the efficacy of our ChIP-seq protocol in B. dorsalis, and found generally contrasting distributions of the two histone marks across the genome, with H3K4me3 mainly located downstream of the TSSs of genes, H3K27me3 tended to occupy broader regions covering the entire body of genes, and a small number of bivalent TSS regions modified by both histone modifications. Transcriptomic analysis supported a link between various histone modification profiles and transcription levels in B. dorsalis, with H3K4me3 associated with active gene transcription, and H3K27me3 is mostly associated with transcriptional repression. We also identified distinct GO enrichment patterns between the two histone modifications that are likely to reflect the distinct roles of H3K4me3 vs. H3K27me3 in regulating tissue development and structure maintenance. Importantly, we identified modified regions annotated with genes that overlapped with previously documented genes involving in wing development and migratory flight activity in B. dorsalis, suggesting a potential role of histone modifications in flight activity. Our work adds to the few studies using non-model, invasive insect species to test for the utilization of ChIP-seq to profile histone modification. Our results suggest that H3K4me3 and H3K27me3 could play an important role in facilitating phenotypic change during insect development as well as range expansion for invasive species.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/sra/PRJNA911513.
Author contributions
YZ, JH, and ZL conceived the study. YZ and JW sampled and reared insects, and collected tissue. YZ performed experiments, generated and analyzed sequencing data. YZ and JH wrote the manuscript with input from ZL
Funding
This work was supported by National Natural Science Foundation of China (32172394) to ZL, and National Natural Science Foundation of China (32170417) and Shanghai Sailing Program (2021YF1403200) to JH.
Acknowledgments
The authors would like to thank Prof. Juan Du and Prof. Zhichuang Lv for helping with experimental design.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1108104/full#supplementary-material
References
Aguirre-Liguori, J. A., Ramírez-Barahona, S., and Gaut, B. S. (2021). The evolutionary genomics of species’ responses to climate change. Nat. Ecol. Evol. 5 (10), 1350–1360. doi:10.1038/s41559-021-01526-9
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31 (2), 166–169. doi:10.1093/bioinformatics/btu638
Aranda, S., Mas, G., and Di Croce, L. (2015). Regulation of gene transcription by Polycomb proteins. Sci. Adv. 1 (11), e1500737. doi:10.1126/sciadv.1500737
Arredondo, J. J., Vivar, J., Laine-Menéndez, S., Martínez-Morentin, L., and Cervera, M. (2017). CF2 transcription factor is involved in the regulation of Mef2 RNA levels, nuclei number and muscle fiber size. PLoS One 12 (6), e0179194. doi:10.1371/journal.pone.0179194
Arrese, E. L., and Soulages, J. L. (2010). Insect fat body: Energy, metabolism, and regulation. Annu. Rev. Entomology 55, 207–225. doi:10.1146/annurev-ento-112408-085356
Artemov, A. V., Mugue, N. S., Rastorguev, S. M., Zhenilo, S., Mazur, A. M., Tsygankova, S. V., et al. (2017). Genome-wide DNA methylation profiling reveals epigenetic adaptation of stickleback to marine and freshwater conditions. Mol. Biol. Evol. 34 (9), 2203–2213. doi:10.1093/molbev/msx156
Attanasio, C., Nord, A. S., Zhu, Y., Blow, M. J., Li, Z., Liberton, D. K., et al. (2013). Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342 (6157), 1241006. doi:10.1126/science.1241006
Babu, M., Gagarinova, A., and Emili, A. (2011). Array-based synthetic genetic screens to map bacterial pathways and functional networks in Escherichia coli. Methods Mol. Biol. 781, 99–126. doi:10.1007/978-1-61779-276-2_7
Baccarelli, A., and Bollati, V. (2009). Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 21 (2), 243–251. doi:10.1097/mop.0b013e32832925cc
Bagni, C., Bray, S., Gogos, J. A., Kafatos, F. C., and Hsu, T. (2002). The Drosophila zinc finger transcription factor CF2 is a myogenic marker downstream of MEF2 during muscle development. Mech. Dev. 117 (1-2), 265–268. doi:10.1016/s0925-4773(02)00176-4
Bailey, T., Krajewski, P., Ladunga, I., Lefebvre, C., Li, Q., Liu, T., et al. (2013). Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput. Biol. 9 (11), e1003326. doi:10.1371/journal.pcbi.1003326
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). Meme SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37 (2), W202–W208. doi:10.1093/nar/gkp335
Bain, S. A., Marshall, H., de la Filia, A. G., Laetsch, D. R., Husnik, F., and Ross, L. (2021). Sex-specific expression and DNA methylation in a species with extreme sexual dimorphism and paternal genome elimination. Mol. Ecol. 30 (22), 5687–5703. doi:10.1111/mec.15842
Bardet, A. F., He, Q., Zeitlinger, J., and Stark, A. (2012). A computational pipeline for comparative ChIP-seq analyses. Nat. Protoc. 7 (1), 45–61. doi:10.1038/nprot.2011.420
Barnes, P. T., and Laurie-Ahlberg, C. C. (1986). Genetic variability of flight metabolism in Drosophila melanogaster. III. Effects of GPDH allozymes and environmental temperature on power output. Genetics 112 (2), 267–294. doi:10.1093/genetics/112.2.267
Barouki, R., Melén, E., Herceg, Z., Beckers, J., Chen, J., Karagas, M., et al. (2018). Epigenetics as a mechanism linking developmental exposures to long-term toxicity. Environ. Int. 114, 77–86. doi:10.1016/j.envint.2018.02.014
Bates, O. K., Ollier, S., and Bertelsmeier, C. (2020). Smaller climatic niche shifts in invasive than non-invasive alien ant species. Nat. Commun. 11 (1), 5213–5218. doi:10.1038/s41467-020-19031-1
Beaven, R., and Denholm, B. (2018). Release and spread of Wingless is required to pattern the proximo-distal axis of Drosophila renal tubules. Elife 7, e35373. doi:10.7554/eLife.35373
Benayoun, B. A., Pollina, E. A., Ucar, D., Mahmoudi, S., Karra, K., Wong, E. D., et al. (2014). H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158 (3), 673–688. doi:10.1016/j.cell.2014.06.027
Berg, P. R., Jentoft, S., Star, B., Ring, K. H., Knutsen, H., Lien, S., et al. (2015). Adaptation to low salinity promotes genomic divergence in Atlantic cod (Gadus morhua L.). Genome Biol. Evol. 7 (6), 1644–1663. doi:10.1093/gbe/evv093
Bernstein, B. E., Kamal, M., Lindblad-Toh, K., Bekiranov, S., Bailey, D. K., Huebert, D. J., et al. (2005). Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120 (2), 169–181. doi:10.1016/j.cell.2005.01.001
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 (2), 315–326. doi:10.1016/j.cell.2006.02.041
Blanco, E., González-Ramírez, M., Alcaine-Colet, A., Aranda, S., and Di Croce, L. (2020). The bivalent genome: Characterization, structure, and regulation. Trends Genet. 36 (2), 118–131. doi:10.1016/j.tig.2019.11.004
Blow, M. J., McCulley, D. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2010). ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42 (9), 806–810. doi:10.1038/ng.650
Bonasio, R., Li, Q., Lian, J., Mutti, N. S., Jin, L., Zhao, H., et al. (2012). Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 22 (19), 1755–1764. doi:10.1016/j.cub.2012.07.042
Bonasio, R., Tu, S., and Reinberg, D. (2010). Molecular signals of epigenetic states. Science 330 (6004), 612–616. doi:10.1126/science.1191078
Bonn, S., Zinzen, R. P., Girardot, C., Gustafson, E. H., Perez-Gonzalez, A., Delhomme, N., et al. (2012). Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 44 (2), 148–156. doi:10.1038/ng.1064
Bossdorf, O., Richards, C. L., and Pigliucci, M. (2008). Epigenetics for ecologists. Ecol. Lett. 11 (2), 106–115. doi:10.1111/j.1461-0248.2007.01130.x
Boyer, L. A., Plath, K., Zeitlinger, J., Brambrink, T., Medeiros, L. A., Lee, T. I., et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441 (7091), 349–353. doi:10.1038/nature04733
Boyle, A. P., Araya, C. L., Brdlik, C., Cayting, P., Cheng, C., Cheng, Y., et al. (2014). Comparative analysis of regulatory information and circuits across distant species. Nature 512 (7515), 453–456. doi:10.1038/nature13668
Brisson, J. A., Davis, G. K., and Stern, D. L. (2007). Common genome-wide patterns of transcript accumulation underlying the wing polyphenism and polymorphism in the pea aphid (Acyrthosiphon pisum). Evol. Dev. 9 (4), 338–346. doi:10.1111/j.1525-142X.2007.00170.x
Brisson, J. A., Ishikawa, A., and Miura, T. (2010). Wing development genes of the pea aphid and differential gene expression between winged and unwinged morphs. Insect Mol. Biol. 19 (2), 63–73. doi:10.1111/j.1365-2583.2009.00935.x
Bu, B., Chen, L., Zheng, L., He, W., and Zhang, L. (2020). Nipped-A regulates the Drosophila circadian clock via histone deubiquitination. EMBO J. 39 (1), e101259. doi:10.15252/embj.2018101259
Bullard, B., Burkart, C., Labeit, S., and Leonard, K. (2005). The function of elastic proteins in the oscillatory contraction of insect flight muscle. J. Muscle Res. Cell Motil. 26 (6), 479–485. doi:10.1007/s10974-005-9032-7
Cadigan, K. M., Grossniklaus, U., and Gehring, W. J. (1994). Localized expression of sloppy paired protein maintains the polarity of Drosophila parasegments. Genes. and Dev. 8 (8), 899–913. doi:10.1101/gad.8.8.899
Cao, L. J., Li, B. Y., Chen, J. C., Zhu, J. Y., Hoffmann, A. A., and Wei, S. J. (2021). Local climate adaptation and gene flow in the native range of two co-occurring fruit moths with contrasting invasiveness. Mol. Ecol. 30 (17), 4204–4219. doi:10.1111/mec.16055
Cao, X., and Jiang, H. (2017). An analysis of 67 RNA-seq datasets from various tissues at different stages of a model insect, Manduca sexta. BMC Genomics 18 (1), 796. doi:10.1186/s12864-017-4147-y
Carroll, T. S., Liang, Z., Salama, R., Stark, R., and de Santiago, I. (2014). Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 5, 75. doi:10.3389/fgene.2014.00075
Chen, P., Ye, H., and Mu, Q. (2007). Migration and dispersal of the oriental fruit fly, Bactrocera dorsalis in regions of Nujiang River based on fluorescence mark. Acta Ecol. Sin. 27 (6), 2468–2476.
Chen, Y., Lyu, R., Rong, B., Zheng, Y., Lin, Z., Dai, R., et al. (2020). Refined spatial temporal epigenomic profiling reveals intrinsic connection between PRDM9-mediated H3K4me3 and the fate of double-stranded breaks. Cell Res. 30 (3), 256–268. doi:10.1038/s41422-020-0281-1
Chen, Z., and Narum, S. R. (2021). Whole genome resequencing reveals genomic regions associated with thermal adaptation in redband trout. Mol. Ecol. 30 (1), 162–174. doi:10.1111/mec.15717
Cheng, D., Cheng, T., Yang, X., Zhang, Q., Fu, J., Feng, T., et al. (2018). The genome-wide transcriptional regulatory landscape of ecdysone in the silkworm. Epigenetics Chromatin 11 (1), 48–13. doi:10.1186/s13072-018-0216-y
Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489 (7414), 57–74. doi:10.1038/nature11247
Conway, E., Healy, E., and Bracken, A. P. (2015). PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 37, 42–48. doi:10.1016/j.ceb.2015.10.003
Dai, T. M., Lü, Z. C., Wang, Y. S., Liu, W. X., Hong, X. Y., and Wan, F. H. (2018). Molecular characterizations of DNA methyltransferase 3 and its roles in temperature tolerance in the whitefly, Bemisia tabaci Mediterranean. Insect Mol. Biol. 27 (1), 123–132. doi:10.1111/imb.12354
Davidson, A. M., Jennions, M., and Nicotra, A. B. (2011). Do invasive species show higher phenotypic plasticity than native species and, if so, is it adaptive? A meta-analysis. Ecol. Lett. 14 (4), 419–431. doi:10.1111/j.1461-0248.2011.01596.x
Devóz, P. P., Gomes, W. R., De Araújo, M. L., Ribeiro, D. L., Pedron, T., Greggi Antunes, L. M., et al. (2017). Lead (Pb) exposure induces disturbances in epigenetic status in workers exposed to this metal. J. Toxicol. Environ. Health, Part A 80 (19-21), 1098–1105. doi:10.1080/15287394.2017.1357364
Dixon, G. B., Davies, S. W., Aglyamova, G. V., Meyer, E., Bay, L. K., and Matz, M. V. (2015). CORAL REEFS. Genomic determinants of coral heat tolerance across latitudes. Science 348 (6242), 1460–1462. doi:10.1126/science.1261224
Doyle, T., Jimenez-Guri, E., Hawkes, W. L., Massy, R., Mantica, F., Permanyer, J., et al. (2022). Genome-wide transcriptomic changes reveal the genetic pathways involved in insect migration. Mol. Ecol. 31 (16), 4332–4350. doi:10.1111/mec.16588
Dudaniec, R. Y., Yong, C. J., Lancaster, L. T., Svensson, E. I., and Hansson, B. (2018). Signatures of local adaptation along environmental gradients in a range-expanding damselfly (Ischnura elegans). Mol. Ecol. 27 (11), 2576–2593. doi:10.1111/mec.14709
Eanes, W. F., Merritt, T. J., Flowers, J. M., Kumagai, S., Sezgin, E., and Zhu, C. T. (2006). Flux control and excess capacity in the enzymes of glycolysis and their relationship to flight metabolism in Drosophila melanogaster. Proc. Natl. Acad. Sci. 103 (51), 19413–19418. doi:10.1073/pnas.0607095104
Eissenberg, J. C., Lee, M. G., Schneider, J., Ilvarsonn, A., Shiekhattar, R., and Shilatifard, A. (2007). The trithorax-group gene in Drosophila little imaginal discs encodes a trimethylated histone H3 Lys4 demethylase. Nat. Struct. Mol. Biol. 14 (4), 344–346. doi:10.1038/nsmb1217
Ekesi, S., Nderitu, P. W., and Chang, C. L. (2007). Adaption to and small-scale rearing of invasive fruit fly Bactrocera invadens (Diptera: Tephritidae) on artificial diet. Ann. Entomological Soc. Am. 100 (4), 562–567. doi:10.1603/0013-8746(2007)100[562:atasro]2.0.co;2
Feil, R., and Fraga, M. F. (2012). Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 13 (2), 97–109. doi:10.1038/nrg3142
Feng, J., Liu, T., Qin, B., Zhang, Y., and Liu, X. S. (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7 (9), 1728–1740. doi:10.1038/nprot.2012.101
Flatt, T., Tu, M. P., and Tatar, M. (2005). Hormonal pleiotropy and the juvenile hormone regulation of Drosophila development and life history. Bioessays 27 (10), 999–1010. doi:10.1002/bies.20290
Foret, S., Kucharski, R., Pellegrini, M., Feng, S., Jacobsen, S. E., Robinson, G. E., et al. (2012). DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl. Acad. Sci. 109 (13), 4968–4973. doi:10.1073/pnas.1202392109
Fornes, O., Castro-Mondragon, J. A., Khan, A., Van der Lee, R., Zhang, X., Richmond, P. A., et al. (2020). Jaspar 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48 (D1), D87–D92. doi:10.1093/nar/gkz1001
Froerer, K. M., Peck, S. L., McQuate, G. T., Vargas, R. I., Jang, E. B., and McInnis, D. O. (2010). Long-distance movement of Bactrocera dorsalis (Diptera: Tephritidae) in puna, Hawaii: How far can they go? Am. Entomologist 56, 88–95. doi:10.1093/ae/56.2.88
Furey, T. S. (2012). ChIP-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 13 (12), 840–852. doi:10.1038/nrg3306
Gaertner, B., Johnston, J., Chen, K., Wallaschek, N., Paulson, A., Garruss, A. S., et al. (2012). Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep. 2 (6), 1670–1683. doi:10.1016/j.celrep.2012.11.024
Gan, Q., Schones, D. E., Eun, S. H., Wei, G., Cui, K., Zhao, K., et al. (2010). Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol. 11 (4), R42–R17. doi:10.1186/gb-2010-11-4-r42
Garcia-Elfring, A., Paccard, A., Thurman, T. J., Wasserman, B. A., Palkovacs, E. P., Hendry, A. P., et al. (2021). Using seasonal genomic changes to understand historical adaptation to new environments: Parallel selection on stickleback in highly-variable estuaries. Mol. Ecol. 30 (9), 2054–2064. doi:10.1111/mec.15879
Gheyas, A. A., Vallejo-Trujillo, A., Kebede, A., Lozano-Jaramillo, M., Dessie, T., Smith, J., et al. (2021). Integrated environmental and genomic analysis reveals the drivers of local adaptation in African indigenous chickens. Mol. Biol. Evol. 38 (10), 4268–4285. doi:10.1093/molbev/msab156
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. D. (2019). Epigenetics in insects: Genome regulation and the generation of phenotypic diversity. Annu. Rev. Entomology 64, 185–203. doi:10.1146/annurev-ento-011118-111914
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. (2015). DNA methylation and chromatin organization in insects: Insights from the ant Camponotus floridanus. Genome Biol. Evol. 7 (4), 931–942. doi:10.1093/gbe/evv039
Gómez-Díaz, E., Rivero, A., Chandre, F., and Corces, V. G. (2014). Insights into the epigenomic landscape of the human malaria vector Anopheles gambiae. Front. Genet. 5, 277. doi:10.3389/fgene.2014.00277
Gong, N. N., Dilley, L. C., Williams, C. E., Moscato, E. H., Szuperak, M., Wang, Q., et al. (2021). The chromatin remodeler ISWI acts during Drosophila development to regulate adult sleep. Sci. Adv. 7 (8), eabe2597. doi:10.1126/sciadv.abe2597
González-Tokman, D., Córdoba-Aguilar, A., Dáttilo, W., Lira-Noriega, A., Sánchez-Guillén, R. A., and Villalobos, F. (2020). Insect responses to heat: Physiological mechanisms, evolution and ecological implications in a warming world. Biol. Rev. 95 (3), 802–821. doi:10.1111/brv.12588
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). FIMO: Scanning for occurrences of a given motif. Bioinformatics 27 (7), 1017–1018. doi:10.1093/bioinformatics/btr064
Grönke, S., and Partridge, L. (2010). “The functions of insulin-like peptides in insects,” in IGFs: Local repair and survival factors throughout life span (Berlin, Heidelberg: Springer), 105–124.
Grossniklaus, U., and Paro, R. (2014). Transcriptional silencing by polycomb-group proteins. Cold Spring Harb. Perspect. Biol. 6 (11), a019331. doi:10.1101/cshperspect.a019331
Gu, X., Zhao, Y., Su, Y., Wu, J., Wang, Z., Hu, J., et al. (2019). A transcriptional and functional analysis of heat hardening in two invasive fruit fly species, Bactrocera dorsalis and Bactrocera correcta. Evol. Appl. 12 (6), 1147–1163. doi:10.1111/eva.12793
Guo, S., Zhao, Z., Liu, L., Li, Z., and Shen, J. (2018). Comparative transcriptome analyses uncover key candidate genes mediating flight capacity in Bactrocera dorsalis (Hendel) and Bactrocera correcta (Bezzi)(Diptera: Tephritidae). Int. J. Mol. Sci. 19 (2), 396. doi:10.3390/ijms19020396
Han, P., Wang, X., Niu, C. Y., Dong, Y. C., Zhu, J. Q., and Desneux, N. (2011). Population dynamics, phenology, and overwintering of Bactrocera dorsalis (Diptera: Tephritidae) in Hubei Province, China. J. Pest Sci. 84 (3), 289–295. doi:10.1007/s10340-011-0363-4
Harikumar, A., and Meshorer, E. (2015). Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 16 (12), 1609–1619. doi:10.15252/embr.201541011
Heckwolf, M. J., Meyer, B. S., Häsler, R., Höppner, M. P., Eizaguirre, C., and Reusch, T. B. H. (2020). Two different epigenetic information channels in wild three-spined sticklebacks are involved in salinity adaptation. Sci. Adv. 6 (12), eaaz1138. doi:10.1126/sciadv.aaz1138
Hertz, G. Z., and Stormo, G. D. (1999). Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinforma. Oxf. Engl. 15 (7), 563–577. doi:10.1093/bioinformatics/15.7.563
Hiromi, Y., and Hotta, Y. (1985). Actin gene mutations in Drosophila; heat shock activation in the indirect flight muscles. EMBO J. 4 (7), 1681–1687. doi:10.1002/j.1460-2075.1985.tb03837.x
Hu, J. T., and Barrett, R. D. H. (2017). Epigenetics in natural animal populations. J. Evol. Biol. 30 (9), 1612–1632. doi:10.1111/jeb.13130
Hu, J. T., Chen, B., and Li, Z. H. (2014). Thermal plasticity is related to the hardening response of heat shock protein expression in two Bactrocera fruit flies. J. Insect Physiology 67, 105–113. doi:10.1016/j.jinsphys.2014.06.009
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34 (8), 2115–2122. doi:10.1093/molbev/msx148
Inomata, N., Takahasi, K. R., and Koga, N. (2019). Association between duplicated maltase genes and the transcriptional regulation for the carbohydrate changes in Drosophila melanogaster. Gene 686, 141–145. doi:10.1016/j.gene.2018.11.007
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 33, 245–254. doi:10.1038/ng1089
Jambhekar, A., Dhall, A., and Shi, Y. (2019). Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20 (10), 625–641. doi:10.1038/s41580-019-0151-1
Jeffries, K. A., Dempsey, D. R., Behari, A. L., Anderson, R. L., and Merkler, D. J. (2014). Drosophila melanogaster as a model system to study long-chain fatty acid amide metabolism. FEBS Lett. 588 (9), 1596–1602. doi:10.1016/j.febslet.2014.02.051
Jeremias, G., Barbosa, J., Marques, S. M., Asselman, J., Gonçalves, F. J., and Pereira, J. L. (2018). Synthesizing the role of epigenetics in the response and adaptation of species to climate change in freshwater ecosystems. Mol. Ecol. 27 (13), 2790–2806. doi:10.1111/mec.14727
Jiang, F., Liang, L., Wang, J., and Zhu, S. (2022). Chromosome-level genome assembly of Bactrocera dorsalis reveals its adaptation and invasion mechanisms. Commun. Biol. 5 (1), 25–11. doi:10.1038/s42003-021-02966-6
Jin, T., Zeng, L., Lin, Y., Lu, Y., and Liang, G. (2011). Insecticide resistance of the oriental fruit fly, Bactrocera dorsalis (Hendel)(Diptera: Tephritidae), in mainland China. Pest Manag. Sci. 67 (3), 370–376. doi:10.1002/ps.2076
Jones, C. M., Lim, K. S., Chapman, J. W., and Bass, C. (2018). Genome-wide characterization of DNA methylation in an invasive lepidopteran pest, the cotton bollworm Helicoverpa armigera. G3 Genes., Genomes, Genet. 8 (3), 779–787. doi:10.1534/g3.117.1112
Jones, C. M., Papanicolaou, A., Mironidis, G. K., Vontas, J., Yang, Y., Lim, K. S., et al. (2015). Genomewide transcriptional signatures of migratory flight activity in a globally invasive insect pest. Mol. Ecol. 24 (19), 4901–4911. doi:10.1111/mec.13362
Josephson, R. K. (2006). “Comparative physiology of insect flight muscle,” in Nature’s versatile engine: Insect flight muscle inside and out (Berlin, Heidelberg: Springer), 34–43.
Kacsoh, B. Z., Greene, C. S., and Bosco, G. (2017). Machine learning analysis identifies Drosophila grunge/atrophin as an important learning and memory gene required for memory retention and social learning. G3 Genes., Genomes, Genet. 7 (11), 3705–3718. doi:10.1534/g3.117.300172
Kanamori, Y., Saito, A., Hagiwara-Komoda, Y., Tanaka, D., Mitsumasu, K., Kikuta, S., et al. (2010). The trehalose transporter 1 gene sequence is conserved in insects and encodes proteins with different kinetic properties involved in trehalose import into peripheral tissues. Insect Biochem. Mol. Biol. 40 (1), 30–37. doi:10.1016/j.ibmb.2009.12.006
Kelly, M. (2019). Adaptation to climate change through genetic accommodation and assimilation of plastic phenotypes. Philosophical Trans. R. Soc. B 374 (1768), 20180176. doi:10.1098/rstb.2018.0176
Khan, A., Fornes, O., Stigliani, A., Gheorghe, M., Castro-Mondragon, J. A., Van Der Lee, R., et al. (2018). Jaspar 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46 (D1), D1284–D266. doi:10.1093/nar/gkx1188
Kharchenko, P. V., Alekseyenko, A. A., Schwartz, Y. B., Minoda, A., Riddle, N. C., Ernst, J., et al. (2011). Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471 (7339), 480–485. doi:10.1038/nature09725
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Kingsley, N., Kern, C., Creppe, C., Hales, E. N., Zhou, H., Kalbfleisch, T., et al. (2019). Functionally annotating regulatory elements in the equine genome using histone mark ChIP-Seq. Genes. 11 (1), 3. doi:10.3390/genes11010003
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128 (4), 693–705. doi:10.1016/j.cell.2007.02.005
Kratochwil, C. F., and Meyer, A. (2015). Mapping active promoters by ChIP-seq profiling of H3K4me3 in cichlid fish-a first step to uncover cis-regulatory elements in ecological model teleosts. Mol. Ecol. Resour. 15 (4), 761–771. doi:10.1111/1755-0998.12350
Landt, S. G., Marinov, G. K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22 (9), 1813–1831. doi:10.1101/gr.136184.111
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi:10.1038/nmeth.1923
Lee, T. I., Jenner, R. G., Boyer, L. A., Guenther, M. G., Levine, S. S., Kumar, R. M., et al. (2006). Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125 (2), 301–313. doi:10.1016/j.cell.2006.02.043
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, Q., Brown, J. B., Huang, H., and Bickel, P. J. (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Statistics 5 (3), 1752–1779. doi:10.1214/11-aoas466
Li, Y., Wu, Y., Chen, H., Wu, J., and Li, Z. (2012). Population structure and colonization of Bactrocera dorsalis (Diptera: Tephritidae) in China, inferred from mtDNA COI sequences. J. Appl. Entomology 136 (4), 241–251. doi:10.1111/j.1439-0418.2011.01636.x
Liang, F., Wu, J. J., and Liang, G. Q. (2001). The first report of the test on the flight ability of oriental fruit fly. Acta Agric. Univ. Jiangxiensis 23 (2), 259–260.
Libbrecht, R., Oxley, P. R., Keller, L., and Kronauer, D. J. C. (2016). Robust DNA methylation in the clonal raider ant brain. Curr. Biol. 26 (3), 391–395. doi:10.1016/j.cub.2015.12.040
Lin, X., Yao, Y., Wang, B., Emlen, D. J., and Lavine, L. C. (2016). Ecological trade-offs between migration and reproduction are mediated by the nutrition-sensitive insulin-signaling pathway. Int. J. Biol. Sci. 12 (5), 607–616. doi:10.7150/ijbs.14802
Lindner, M., Laine, V. N., Verhagen, I., Viitaniemi, H. M., Visser, M. E., van Oers, K., et al. (2021). Rapid changes in DNA methylation associated with the initiation of reproduction in a small songbird. Mol. Ecol. 30 (15), 3645–3659. doi:10.1111/mec.15803
Liu, C. L., Kaplan, T., Kim, M., Buratowski, S., Schreiber, S. L., Friedman, N., et al. (2005). Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 3 (10), e328. doi:10.1371/journal.pbio.0030328
Liu, F., Li, X., Zhao, M., Guo, M., Han, K., Dong, X., et al. (2020). Ultrabithorax is a key regulator for the dimorphism of wings, a main cause for the outbreak of planthoppers in rice. Natl. Sci. Rev. 7 (7), 1181–1189. doi:10.1093/nsr/nwaa061
Lyko, F., Foret, S., Kucharski, R., Wolf, S., Falckenhayn, C., and Maleszka, R. (2010). The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 8 (11), e1000506. doi:10.1371/journal.pbio.1000506
Ma, Z., Wang, H., Cai, Y., Wang, H., Niu, K., Wu, X., et al. (2018). Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. Elife 7, e35368. doi:10.7554/eLife.35368
Magagula, C. N., Cugala, D. C., Monadjem, A., and Dlamini, W. M. (2015). Predicted regional and national distribution of Bactrocera dorsalis(syn.B. invadens) (Diptera: Tephritidae) in southern Africa and implications for its management. Afr. Entomol. 23 (2), 427–437. doi:10.4001/003.023.0220
Maleszka, R. (2016). Epigenetic code and insect behavioural plasticity. Curr. Ipinion Insect Sci. 15, 45–52. doi:10.1016/j.cois.2016.03.003
Marcon, E., Jain, H., Bhattacharya, A., Guo, H., Phanse, S., Pu, S., et al. (2015). Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. methods 12 (8), 725–731. doi:10.1038/nmeth.3472
Marsh, A. G., and Pasqualone, A. A. (2014). DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiology 5, 173. doi:10.3389/fphys.2014.00173
Mathers, T. C., Mugford, S. T., Percival-Alwyn, L., Chen, Y., Kaithakottil, G., Swarbreck, D., et al. (2019). Sex-specific changes in the aphid DNA methylation landscape. Mol. Ecol. 28 (18), 4228–4241. doi:10.1111/mec.15216
Mattila, J., and Hietakangas, V. (2017). Regulation of carbohydrate energy metabolism in Drosophila melanogaster. Genetics 207 (4), 1231–1253. doi:10.1534/genetics.117.199885
Maughan, D. W., and Vigoreaux, J. O. (1999). An integrated view of insect flight muscle: Genes, motor molecules, and motion. Physiology 14 (3), 87–92. doi:10.1152/physiologyonline.1999.14.3.87
Merilä, J., and Hendry, A. P. (2014). Climate change, adaptation, and phenotypic plasticity: The problem and the evidence. Evol. Appl. 7 (1), 1–14. doi:10.1111/eva.12137
Metzger, D. C., and Schulte, P. M. (2017). Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc. R. Soc. B Biol. Sci. 284 (1864), 20171667. doi:10.1098/rspb.2017.1667
Metzger, D. C., and Schulte, P. M. (2018). The DNA methylation landscape of stickleback reveals patterns of sex chromosome evolution and effects of environmental salinity. Genome Biol. Evol. 10 (3), 775–785. doi:10.1093/gbe/evy034
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 (7153), 553–560. doi:10.1038/nature06008
Mohan, M., Herz, H. M., Smith, E. R., Zhang, Y., Jackson, J., Washburn, M. P., et al. (2011). The COMPASS family of H3K4 methylases in Drosophila. Mol. Cell. Biol. 31 (21), 4310–4318. doi:10.1128/MCB.06092-11
Mukherjee, K., Twyman, R. M., and Vilcinskas, A. (2015). Insects as models to study the epigenetic basis of disease. Prog. Biophysics Mol. Biol. 118 (1-2), 69–78. doi:10.1016/j.pbiomolbio.2015.02.009
Nagalingam, K., Lorenc, M. T., Manoli, S., Cameron, S. L., Clarke, A. R., and Dudley, K. J. (2018). Chromatin immunoprecipitation (ChIP) method for non-model fruit flies (Diptera: Tephritidae) and evidence of histone modifications. PLoS One 13 (3), e0194420. doi:10.1371/journal.pone.0194420
Nässel, D. R., Kubrak, O. A., Liu, Y., Luo, J., and Lushchak, O. V. (2013). Factors that regulate insulin producing cells and their output in Drosophila. Front. Physiology 4, 252. doi:10.3389/fphys.2013.00252
Nègre, N., Brown, C. D., Ma, L., Bristow, C. A., Miller, S. W., Wagner, U., et al. (2011). A cis-regulatory map of the Drosophila genome. Nature 471 (7339), 527–531. doi:10.1038/nature09990
Niu, B., Coslo, D. M., Bataille, A. R., Albert, I., Pugh, B. F., and Omiecinski, C. J. (2018). In vivo genome-wide binding interactions of mouse and human constitutive androstane receptors reveal novel gene targets. Nucleic acids Res. 46 (16), 8385–8403. doi:10.1093/nar/gky692
Nüsslein-Volhard, C., and Wieschaus, E. (1980). Mutations affecting segment number and polarity in Drosophila. Drosoph. Nat. 287 (5785), 795–801. doi:10.1038/287795a0
Oldroyd, B. P., and Yagound, B. (2021). The role of epigenetics, particularly DNA methylation, in the evolution of caste in insect societies. Philosophical Trans. R. Soc. B 376 (1826), 20200115. doi:10.1098/rstb.2020.0115
Ozawa, T., Mizuhara, T., Arata, M., Shimada, M., Niimi, T., Okada, K., et al. (2016). Histone deacetylases control module-specific phenotypic plasticity in beetle weapons. Proc. Natl. Acad. Sci. 113 (52), 15042–15047. doi:10.1073/pnas.1615688114
Pan, G., Tian, S., Nie, J., Yang, C., Ruotti, V., Wei, H., et al. (2007). Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1 (3), 299–312. doi:10.1016/j.stem.2007.08.003
Paredes, U., Radersma, R., Cannell, N., While, G. M., and Uller, T. (2016). Low incubation temperature induces DNA hypomethylation in lizard brains. J. Exp. Zoology Part A Ecol. Genet. Physiology 325 (6), 390–395. doi:10.1002/jez.2024
Parisi, F., Riccardo, S., Zola, S., Lora, C., Grifoni, D., Brown, L. M., et al. (2013). dMyc expression in the fat body affects DILP2 release and increases the expression of the fat desaturase Desat1 resulting in organismal growth. Dev. Biol. 379 (1), 64–75. doi:10.1016/j.ydbio.2013.04.008
Park, S. J., Kim, J. H., Yoon, B. H., and Kim, S. Y. (2017). A ChIP-seq data analysis pipeline based on bioconductor packages. Genomics and Inf. 15 (1), 11–18. doi:10.5808/GI.2017.15.1.11
Parmesan, C. (2006). Ecological and evolutionary responses to recent climate change. Annu. Rev. Ecol. Evol. Syst. 37, 637–669. doi:10.1146/annurev.ecolsys.37.091305.110100
Paul, L., Wang, S. H., Manivannan, S. N., Bonanno, L., Lewis, S., Austin, C. L., et al. (2013). Dpp-induced Egfr signaling triggers postembryonic wing development in Drosophila. Proc. Natl. Acad. Sci. 110 (13), 5058–5063. doi:10.1073/pnas.1217538110
Pieterse, W., Terblanche, J. S., and Addison, P. (2017). Do thermal tolerances and rapid thermal responses contribute to the invasion potential of Bactrocera dorsalis (Diptera: Tephritidae)? J. Insect Physiology 98, 1–6. doi:10.1016/j.jinsphys.2016.11.004
Qin, Y. J., Krosch, M. N., Schutze, M. K., Zhang, Y., Wang, X. x., Prabhakar, C. S., et al. (2018). Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evol. Appl. 11 (10), 1990–2003. doi:10.1111/eva.12701
Quah, S., Hui, J. H., and Holland, P. W. (2015). A burst of miRNA innovation in the early evolution of butterflies and moths. Mol. Biol. Evol. 32 (5), 1161–1174. doi:10.1093/molbev/msv004
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A., and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42 (W1), W187–W191. doi:10.1093/nar/gku365
Rautsaw, R. M., Schramer, T. D., Acuña, R., Arick, L. N., DiMeo, M., Mercier, K. P., et al. (2021). Genomic adaptations to salinity resist gene flow in the evolution of floridian watersnakes. Mol. Biol. Evol. 38 (3), 745–760. doi:10.1093/molbev/msaa266
Reedy, M. C., Bullard, B., and Vigoreaux, J. O. (2000). Flightin is essential for thick filament assembly and sarcomere stability in Drosophila flight muscles. J. Cell Biol. 151 (7), 1483–1500. doi:10.1083/jcb.151.7.1483
Reynolds, J., Bautista-Jimenez, R., and Denlinger, D. (2016). Changes in histone acetylation as potential mediators of pupal diapause in the flesh fly, Sarcophaga bullata. Insect Biochem. Mol. Biol. 76, 29–37. doi:10.1016/j.ibmb.2016.06.012
Richards, C. L., and Pigliucci, M. (2020). Epigenetic inheritance. A decade into the extended evolutionary synthesis. Paradigmi 38, 463–494. doi:10.30460/99624
Roberts, S. B., and Gavery, M. R. (2012). Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiology 2, 116. doi:10.3389/fphys.2011.00116
Robertson, G., Hirst, M., Bainbridge, M., Bilenky, M., Zhao, Y., Zeng, T., et al. (2007). Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4 (8), 651–657. doi:10.1038/nmeth1068
Roch, F., Jiménez, G., and Casanova, J. (2002). EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129 (4), 993–1002. doi:10.1242/dev.129.4.993
Roques, A., Rabitsch, W., Rasplus, J. Y., Lopez-Vaamonde, C., Nentwig, W., and Kenis, M. (2009). “Alien terrestrial invertebrates of Europe,” in Handbook of alien species in Europe (Berlin, Heidelberg: Springer), 63–79.
Rozowsky, J., Euskirchen, G., Auerbach, R. K., Zhang, Z. D., Gibson, T., Bjornson, R., et al. (2009). PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 27 (1), 66–75. doi:10.1038/nbt.1518
Ruiz, J. L., Yerbanga, R. S., Lefèvre, T., Ouedraogo, J. B., Corces, V. G., and Gómez-Díaz, E. (2019). Chromatin changes in Anopheles gambiae induced by Plasmodium falciparum infection. Epigenetics Chromatin 12 (1), 5–18. doi:10.1186/s13072-018-0250-9
Sachs, M., Onodera, C., Blaschke, K., Ebata, K. T., Song, J. S., and Ramalho-Santos, M. (2013). Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 3 (6), 1777–1784. doi:10.1016/j.celrep.2013.04.032
Sanchez, O. F., Lee, J., Yu King Hing, N., Kim, S. E., Freeman, J. L., and Yuan, C. (2017). Lead (Pb) exposure reduces global DNA methylation level by non-competitive inhibition and alteration of dnmt expression. Metallomics 9 (2), 149–160. doi:10.1039/c6mt00198j
Sanford, E., and Kelly, M. W. (2011). Local adaptation in marine invertebrates. Annu. Rev. Mar. Sci. 3, 509–535. doi:10.1146/annurev-marine-120709-142756
Santos, P. K. F., de Souza Araujo, N., Françoso, E., Zuntini, A. R., and Arias, M. C. (2018). Diapause in a tropical oil-collecting bee: Molecular basis unveiled by RNA-seq. BMC genomics 19 (1), 305–311. doi:10.1186/s12864-018-4694-x
Santos-Rosa, H., Schneider, R., Bannister, A. J., Sherriff, J., Bernstein, B. E., Emre, N. C., et al. (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419 (6905), 407–411. doi:10.1038/nature01080
Schmidt, D., Wilson, M. D., Ballester, B., Schwalie, P. C., Brown, G. D., Marshall, A., et al. (2010). Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328 (5981), 1036–1040. doi:10.1126/science.1186176
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B., and Cavalli, G. (2007). Genome regulation by polycomb and trithorax proteins. Cell 128 (4), 735–745. doi:10.1016/j.cell.2007.02.009
Schuettengruber, B., Ganapathi, M., Leblanc, B., Portoso, M., Jaschek, R., Tolhuis, B., et al. (2009). Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol. 7 (1), e1000013. doi:10.1371/journal.pbio.1000013
Schwartz, Y. B., and Cavalli, G. (2017). Three-dimensional genome organization and function in Drosophila. Genetics 205 (1), 5–24. doi:10.1534/genetics.115.185132
Sharma, V., Kohli, S., and Brahmachari, V. (2017). Correlation between desiccation stress response and epigenetic modifications of genes in Drosophila melanogaster: An example of environment-epigenome interaction. Biochim. Biophys. Acta Gene Regul. Mech. 1860 (10), 1058–1068. doi:10.1016/j.bbagrm.2017.08.001
Sheldon, E. L., Schrey, A. W., Hurley, L. L., and Griffith, S. C. (2020). Dynamic changes in DNA methylation during postnatal development in zebra finches Taeniopygia guttata exposed to different temperatures. J. Avian Biol. 51 (5), jav.02294. doi:10.1111/jav.02294
Shen, J., Dahmann, C., and Pflugfelder, G. O. (2010). Spatial discontinuity of optomotor-blind expression in the Drosophila wing imaginal disc disrupts epithelial architecture and promotes cell sorting. BMC Dev. Biol. 10 (1), 23–12. doi:10.1186/1471-213X-10-23
Siebold, A. P., Banerjee, R., Tie, F., Kiss, D. L., Moskowitz, J., and Harte, P. J. (2010). Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. 107 (1), 169–174. doi:10.1073/pnas.0907739107
Simola, D. F., Graham, R. J., Brady, C. M., Enzmann, B. L., Desplan, C., Ray, A., et al. (2016). Epigenetic (re) programming of caste-specific behavior in the ant Camponotus floridanus. Science 351 (6268), aac6633. doi:10.1126/science.aac6633
Simola, D. F., Ye, C., Mutti, N. S., Dolezal, K., Bonasio, R., Liebig, J., et al. (2013). A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res. 23 (3), 486–496. doi:10.1101/gr.148361.112
Somero, G. N. (2012). The physiology of global change: Linking patterns to mechanisms. Annu. Rev. Mar. Sci. 4, 39–61. doi:10.1146/annurev-marine-120710-100935
Stajic, D., and Jansen, L. E. (2021). Empirical evidence for epigenetic inheritance driving evolutionary adaptation. Philosophical Trans. R. Soc. B 376 (1826), 20200121. doi:10.1098/rstb.2020.0121
Stefflova, K., Thybert, D., Wilson, M. D., Streeter, I., Aleksic, J., Karagianni, P., et al. (2013). Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell 154 (3), 530–540. doi:10.1016/j.cell.2013.07.007
Steiner, L. F. (1957). Field evaluation of oriental fruit fly insecticides in Hawaii. J. Econ. Entomology 50 (1), 16–24. doi:10.1093/jee/50.1.16
Stevenson, T. J. (2018). Epigenetic regulation of biological rhythms: An evolutionary ancient molecular timer. Trends Genet. 34 (2), 90–100. doi:10.1016/j.tig.2017.11.003
Stevenson, T. J., and Prendergast, B. J. (2013). Reversible DNA methylation regulates seasonal photoperiodic time measurement. Proc. Natl. Acad. Sci. 110 (41), 16651–16656. doi:10.1073/pnas.1310643110
Strigini, M., and Cohen, S. M. (2000). Wingless gradient formation in the Drosophila wing. Curr. Biol. 10 (6), 293–300. doi:10.1016/s0960-9822(00)00378-x
Tena, J. J., Gonzalez-Aguilera, C., Fernandez-Minan, A., Vazquez-Marin, J., Parra-Acero, H., Cross, J. W., et al. (2014). Comparative epigenomics in distantly related teleost species identifies conserved cis-regulatory nodes active during the vertebrate phylotypic period. Genome Res. 24 (7), 1075–1085. doi:10.1101/gr.163915.113
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinforma. 14 (2), 178–192. doi:10.1093/bib/bbs017
Tomoyasu, Y. (2017). Ultrabithorax and the evolution of insect forewing/hindwing differentiation. Curr. Opin. Insect Sci. 19, 8–15. doi:10.1016/j.cois.2016.10.007
Uchino, R., Nonaka, Y.-k., Horigome, T., Sugiyama, S., and Furukawa, K. (2013). Loss of Drosophila A-type lamin C initially causes tendon abnormality including disintegration of cytoskeleton and nuclear lamina in muscular defects. Dev. Biol. 373 (1), 216–227. doi:10.1016/j.ydbio.2012.08.001
Vastenhouw, N. L., and Schier, A. F. (2012). Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 24 (3), 374–386. doi:10.1016/j.ceb.2012.03.009
Vastenhouw, N. L., Zhang, Y., Woods, I. G., Imam, F., Regev, A., Liu, X. S., et al. (2010). Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464 (7290), 922–926. doi:10.1038/nature08866
Verhoeven, K. J. F., vonHoldt, B. M., and Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Mol. Ecol. 25, 1631–1638. doi:10.1111/mec.13617
Visel, A., Blow, M. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457 (7231), 854–858. doi:10.1038/nature07730
Vogt, G. (2021). Epigenetic variation in animal populations: Sources, extent, phenotypic implications, and ecological and evolutionary relevance. J. Biosci. 46 (1), 24–47. doi:10.1007/s12038-021-00138-6
Voigt, P., Tee, W. W., and Reinberg, D. (2013). A double take on bivalent promoters. Genes. and Dev. 27 (12), 1318–1338. doi:10.1101/gad.219626.113
Walsh, T. K., Brisson, J. A., Robertson, H. M., Gordon, K., Jaubert-Possamai, S., Tagu, D., et al. (2010). A functional DNA methylation system in the pea aphid, Acyrthosiphon pisum. Insect Mol. Biol. 19 (2), 215–228. doi:10.1111/j.1365-2583.2009.00974.x
Wang, Y., Brent, C. S., Fennern, E., and Amdam, G. V. (2012). Gustatory perception and fat body energy metabolism are jointly affected by vitellogenin and juvenile hormone in honey bees. PLoS Genet. 8 (6), e1002779. doi:10.1371/journal.pgen.1002779
Wang, Y., Ferveur, J. F., and Moussian, B. (2021). Eco-genetics of desiccation resistance in Drosophila. Biol. Rev. 96 (4), 1421–1440. doi:10.1111/brv.12709
Ward, E. J., and Coulter, D. E. (2000). odd-skipped is expressed in multiple tissues during Drosophila embryogenesis. Mech. Dev. 96 (2), 233–236. doi:10.1016/s0925-4773(00)00389-0
Wiener, P., Robert, C., Ahbara, A., Salavati, M., Abebe, A., Kebede, A., et al. (2021). Whole-genome sequence data suggest environmental adaptation of Ethiopian sheep populations. Genome Biol. Evol. 13 (3), evab014. doi:10.1093/gbe/evab014
Wojciechowski, M., Lowe, R., Maleszka, J., Conn, D., Maleszka, R., and Hurd, P. J. (2018). Phenotypically distinct female castes in honey bees are defined by alternative chromatin states during larval development. Genome Res. 28 (10), 1532–1542. doi:10.1101/gr.236497.118
Xiao, J. J., Jin, Z. F., Xu, H. X., and Li, R. Z. (2013). Overview of long-distance migration of rice planthopper. Chin. Agric. Sci. Bull. 29, 147–152. doi:10.11924/j.issn.1000-6850.2012-1881
Yagound, B., Remnant, E. J., Buchmann, G., and Oldroyd, B. P. (2020). Intergenerational transfer of DNA methylation marks in the honey bee. Proc. Natl. Acad. Sci. 117 (51), 32519–32527. doi:10.1073/pnas.2017094117
Yang, L., Wang, Y., Sun, N., Chen, J., and He, S. (2021). Genomic and functional evidence reveals convergent evolution in fishes on the Tibetan Plateau. Mol. Ecol. 30 (22), 5752–5764. doi:10.1111/mec.16171
Ye, T., Krebs, A. R., Choukrallah, M. A., Keime, C., Plewniak, F., Davidson, I., et al. (2011). seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 39 (6), e35. doi:10.1093/nar/gkq1287
Young, M. D., Willson, T. A., Wakefield, M. J., Trounson, E., Hilton, D. J., Blewitt, M. E., et al. (2011). ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 39 (17), 7415–7427. doi:10.1093/nar/gkr416
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287. doi:10.1089/omi.2011.0118
Yu, G., Wang, L. G., and He, Q. Y. (2015). ChIPseeker: An R/bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31 (14), 2382–2383. doi:10.1093/bioinformatics/btv145
Yu, L., Wang, G. D., Ruan, J., Chen, Y. B., Yang, C. P., Cao, X., et al. (2016). Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat. Genet. 48 (8), 947–952. doi:10.1038/ng.3615
Yue, F., Cheng, Y., Breschi, A., Vierstra, J., Wu, W., Ryba, T., et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515 (7527), 355–364. doi:10.1038/nature13992
Zecca, M., and Struhl, G. (2002). Subdivision of the Drosophila wing imaginal disc by EGFR-mediated signaling. Development 129 (6), 1357–1368. doi:10.1242/dev.129.6.1357
Zhang, S., Ratliff, E. P., Molina, B., El-Mecharrafie, N., Mastroianni, J., Kotzebue, R. W., et al. (2018). Aging and intermittent fasting impact on transcriptional regulation and physiological responses of adult drosophila neuronal and muscle tissues. Int. J. Mol. Sci. 19 (4), 1140. doi:10.3390/ijms19041140
Zhang, T., Chen, J., Zhang, J., Guo, Y. T., Zhou, X., Li, M. W., et al. (2021). Phenotypic and genomic adaptations to the extremely high elevation in plateau zokor (Myospalax baileyi). Mol. Ecol. 30 (22), 5765–5779. doi:10.1111/mec.16174
Zhang, X., Bernatavichute, Y. V., Cokus, S., Pellegrini, M., and Jacobsen, S. E. (2009). Genome-wide analysis of mono-di-and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 10 (6), R62–R14. doi:10.1186/gb-2009-10-6-r62
Zhao, X. D., Han, X., Chew, J. L., Liu, J., Chiu, K. P., Choo, A., et al. (2007). Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1 (3), 286–298. doi:10.1016/j.stem.2007.08.004
Zhou, V. W., Goren, A., and Bernstein, B. E. (2011). Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12 (1), 7–18. doi:10.1038/nrg2905
Zhu, L. J., Gazin, C., Lawson, N. D., Lin, S. M., Lapointe, D. S., Green, M. R., et al. (2010). ChIPpeakAnno: A bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinforma. 11 (1), 237. doi:10.1186/1471-2105-11-237
Zhu, L. J. (2013). “Integrative analysis of ChIP-chip and ChIP-seq dataset,” in Tiling arrays (Berlin, Heidelberg: Springer), 105–124.
Keywords: epigenetics, histone modification, chromatin immunoprecipitation high throughput sequencing, Bactrocera dorsalis, flight activity
Citation: Zhao Y, Hu J, Wu J and Li Z (2023) ChIP-seq profiling of H3K4me3 and H3K27me3 in an invasive insect, Bactrocera dorsalis. Front. Genet. 14:1108104. doi: 10.3389/fgene.2023.1108104
Received: 25 November 2022; Accepted: 10 February 2023;
Published: 23 February 2023.
Edited by:
Seth Frietze, University of Vermont Cancer Center, United StatesReviewed by:
Sara Helms Cahan, University of Vermont, United StatesWan Qiang Qian, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2023 Zhao, Hu, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihong Li, bGl6aEBjYXUuZWR1LmNu
†These authors have contributed equally to this work