 Ying Su1,2
Ying Su1,2 Chun-Qiong Ran1,2
Chun-Qiong Ran1,2 Zhe-Long Liu1,2
Zhe-Long Liu1,2 Yan Yang1,2
Yan Yang1,2 Gang Yuan1,2Shu-Hong Hu1,2
Gang Yuan1,2Shu-Hong Hu1,2 Xue-Feng Yu1,2
Xue-Feng Yu1,2 Wen-Tao He1,2*
Wen-Tao He1,2*- 1Division of Endocrinology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Branch of National Clinical Research Center for Metabolic Diseases, Wuhan, China
Background: Stickler syndrome (SS) is a group of hereditary collagenopathies caused by a variety of collagen and non-collagen genes. Affected patients have characteristic manifestations involving ophthalmic, articular, craniofacial and auditory disorders. SS is classified into several subtypes according to clinical and molecular features. Type 3 SS is an ultra-rare disease, known as non-ocular SS or otospondylomegaepiphyseal dysplasia (OSMED) with only a few pathogenic COL11A2 variants reported to date.
Case presentation: A 29-year-old Chinese male was referred to our hospital for hearing loss and multiple joint pain. He presented a phenotype highly suggestive of OSMED, including progressive sensorineural deafness, spondyloepiphyseal dysplasia with large epiphyses, platyspondyly, degenerative osteoarthritis, and sunken nasal bridge. We detected compound heterozygous mutations in COL11A2, both of which were predicted to be splicing mutations. One is synonymous mutation c.3774C>T (p.Gly1258Gly) supposed to be a splice site mutation, the other is a novel intron mutation c.4750 + 5 G>A, which is a highly conservative site across several species. We also present a review of the current known pathogenic mutation spectrum of COL11A2 in patients with type 3 SS.
Conclusion: Both synonymous extonic and intronic variants are easily overlooked by whole-exome sequencing. For patients with clinical manifestations suspected of SS syndrome, next-generation whole-genome sequencing is necessary for precision diagnosis and genetic counseling.
Introduction
Stickler syndrome (SS) is a group of multisystem collagenopathies characterized by ophthalmic, auditory, craniofacial, articular and skeletal abnormalities. The first case of “hereditary progressive arthro-ophthalmopathy” was reported by Stickler et al dates back to 1965 (Stickler et al., 1965). SS is clinically and molecularly heterogeneous, which makes recognition and precise diagnosis of the disease difficult. Currently, pathogenic variants in six collagen-type genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2 and COL9A3) and at least five non-collagen genes (LRP2, GZF1, BMP4, PLOD3 and LOXL3) have been demonstrated to be involved in the pathogenesis of SS (Schrauwen et al., 2014; Alzahrani et al., 2015; Chan et al., 2019; Ewans et al., 2019; Nixon et al., 2019). The abnormal extracellular matrix modelling in affected tissues caused by genetic mutations leads to characteristic manifestations in patients with SS. To date, it has been delineated that more than 40 different collagen or collagen-related genes are responsible for synthesizing and assembling at least 29 types of collagens (Carter and Raggio, 2009). Nevertheless, the most causative genetic alterations in SS are associated with abnormal production or assembly of fibrillar collagens II, IX and XI, which are enriched in the vitreous humor as well as hyaline and elastic cartilage (Ihanamaki et al., 2004).
Type 1 SS (OMIM #108300), accounting for 80%–90% of cases, is featured by a high risk for blindness resulting from retinal detachment and membranous vitreous changes. The haploinsufficiency of COL2A1, the encoding gene for collagen type II alpha one chain, is responsible for autosomal dominant type 1 SS (Roche and Cresswell, 1991).
Type 2 SS (OMIM #604841), representing the remaining 10%–20% of cases, is usually caused by monoallelic mutations in COL11A1, which encodes the collagen type XI alpha one chain (Soh et al., 2022). However, biallelic pathogenic variants of COL11A1 have also been reported occasionally (Nixon et al., 2022). Type 2 SS is characterized by a beaded vitreous phenotype, different from type 1 SS.
Type 3 SS (OMIM #184840) is a rare form, previously known as non-ocular SS, or otospondylomegaepiphyseal dysplasia (OSMED) (OMIM #184840, #215150), or Weissenbacher Zweymüller syndrome. The first detailed description of this syndrome was made by pediatricians, Dr. Weissenbacher and Zweymüller, in 1964 (Weissenbacher and Zweymueller, 1964). The affected patient was a normal-sized newborn with several signs of chondrodysplasia, including a markedly snub nose, coronal vertebral clefts, and rhizomelic shortness of limbs with dumb-bell-shaped femora (Weissenbacher and Zweymueller, 1964; Pihlajamaa et al., 1998). The patient did not have ocular signs such as myopia and vitreoretinal degeneration. The follow-up results of the original patient showed that he had developed sensorineural deafness at 5 years old, and spondyloepiphyseal dysplasia at about 13 years old (Giedion et al., 1982; Pihlajamaa et al., 1998). After summarizing the common features of the four affected patients, Giedion et al. had postulated the nomenclature, OSMED, to characterize this condition better (Giedion et al., 1982). The pathogenic gene responsible for OSMED was identified by directed sequencing of two candidate genes (COL2A1 and COL11A2) expressed in cartilage (Pihlajamaa et al., 1998). Finally, COL11A2 mutation has been demonstrated to be the etiology of OSMED. Both autosomal dominant and recessive inheritable patterns of COL11A2 mutation have been documented to be involved in the pathogenesis of type 3 SS (Vikkula et al., 1995; Melkoniemi et al., 2000), depending on the mutation sites.
The uncommon causative genes for SS include coding genes for collagen IX protein, namely, COL9A1, COL9A2 and COL9A3, together with non-collagen genes mentioned previously.
In this report, we describe a 29-year-old Chinese male with non-ocular OSMED-like manifestations. Whole-genome sequencing of DNA from the peripheral blood has uncovered a compound heterozygous mutation of COL11A2. We also review the currently known pathogenic mutations of COL11A2 for type 3 SS.
Case description
A 29-year-old Chinese male was admitted to our hospital for recurrent pain in the low back, knee and ankle joints, which had troubled him for 2 years. He was from a non-consanguineous family. He had been born with normal weight and height. During his infancy, he was noted to have enlarged interphalangeal joints and O-shaped legs. Non-progressive hearing impairment occurred since childhood, so his language development was retarded. His mental development seemed normal. No visual signs or symptoms had been observed. No similar symptoms occurred in his family members. On admission, he was 168 cm in height and 54.7 kg in weight (body mass index: 19.3 kg/m2). Mild abnormal body proportions were observed, with a shorter upper part compared to the lower part (80 cm vs. 88 cm). In addition, his arm span was shorter than his height (160 cm vs. 168 cm). The patient had a snub nose, a high arched palate, micrognathia, and bilateral clinodactyly of the finger and toe joints. The ophthalmologic evaluation showed normal lens, vitreous, fundus and visual acuity. According to an audiological examination, the patient has congenital bilateral sensorineural hearing loss. His middle and inner ear space appeared unremarkable with a magnetic resonance imaging (MRI) scan. Routine laboratory examinations were all negative, including erythrocyte sedimentation rate, complete blood cell count, antinuclear antibody, rheumatoid factor, thyroid hormone profiles and glucose metabolism. Body mass density (BMD) of the lumber spine and femur showed normal BMD and osteoporosis, respectively (Z-score: L1-4: −0.3, −0.5, 0, −0.2; femur: −2.5). The procollagen type 1 N-terminal prope-peptide (P1NP) was 83.2 ng/mL (9.06–76.24 ng/mL), and the carboxy-terminal cross-linked telopeptide of type 1 collagen (CTX) was 0.54 ng/mL (0.043–0.783 ng/mL). A radiographic examination revealed moderate to severe osteoarthritis in his metacarpophalangeal joints, wrists, knees and ankles. He also had various degrees of flattening of the vertebral bodies due to compression fracture (Figures 1A, B). Given the involvement of systematic chondroskeleton in the proband, we advised the performance of genetic diagnosis. After the consent of the patient and his family members, we employed high-throughput sequencing of the whole-genome of the proband’s peripheral blood. It revealed that the proband harbored compound heterozygous mutations in COL11A2. One is c.3774 C > T and the other is c.4750 + 5G > A (Figures 2A, B). The two mutations were confirmed by Sanger’s sequencing both in the proband and his parents. COL11A2 c.3774 C > T is located in a consensus sequence of 5′ splice site (Figure 3). Base on the American College of Medical Genetics and Genomics (ACMG) guidelines, the variation is considered to be uncertain significance. Therefore, we investigated the influence of this synonymous mutation on splicing process in silico (NetGene2, NNSPLICE0.9) (Table 1). These analyses suggested that c.3774 C > T may result in the aberrant 5′ splice site. He was treated with bisphosphonates, calcium and Vitamin D to alleviate osteoporosis. The pains in multiple joints were treated with nonsteroidal anti-inflammatory drugs or gabapentin intermittently. Primers used to amplify the exons of COL11A2 were shown in Supplementary Table S1.
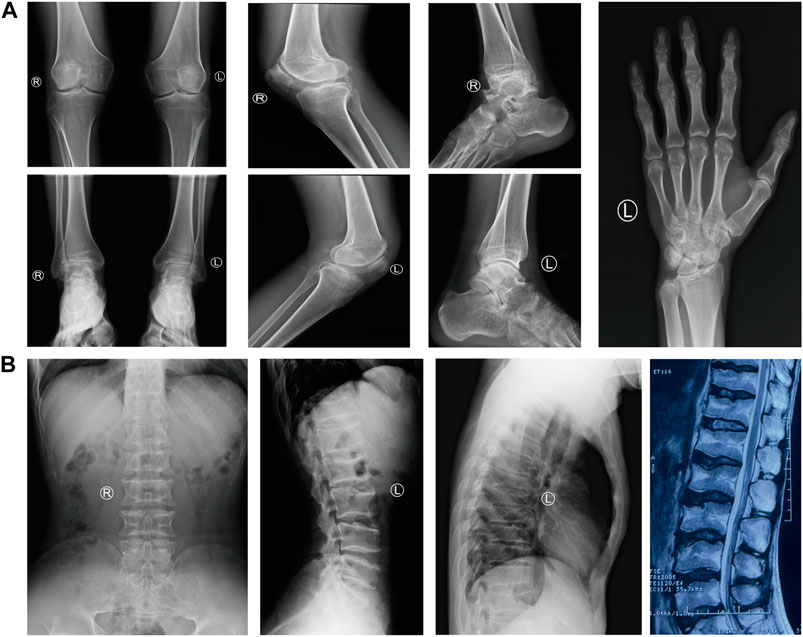
FIGURE 1. The radiological findings of osteoarthritis in the proband. (A) Radiographs of the knees, ankles, left hand and foot, showing the diffuse joint-space-narrowing. (B) Radiographs and MRI of the spine, showing platyspondyly.
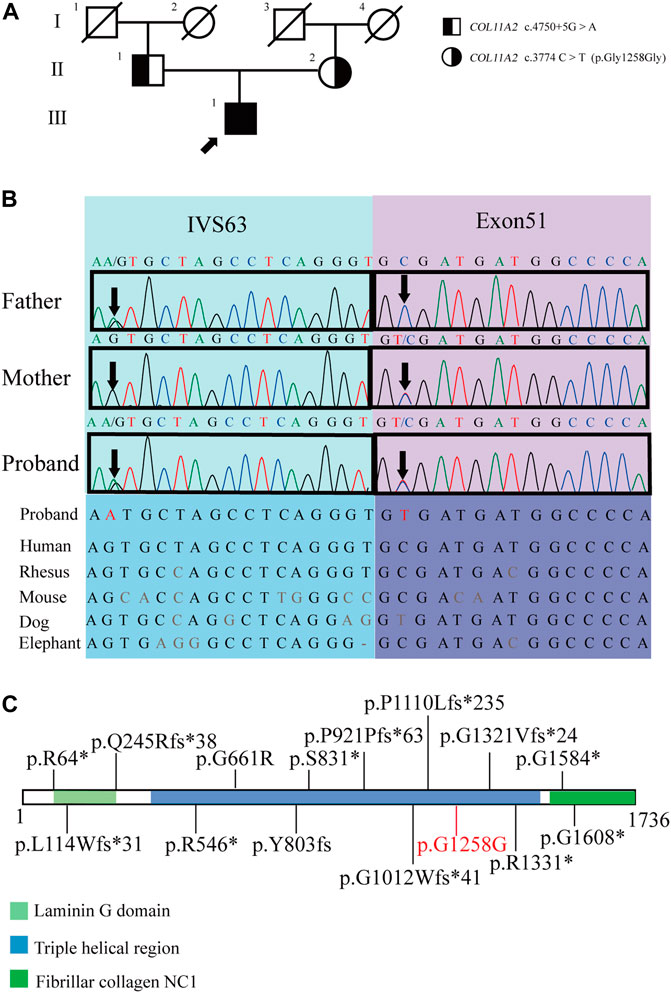
FIGURE 2. Pedigrees of the proband and the genetic characteristics of the pedigree. (A) Pedigrees of the proband (arrow indicates proband). (B) The Sanger sequencing of the family and the evolutionary conservation of the cluster across multiple species. (C) COL11A2 variants associated with type 3 SS, variants were collected from ClinVar and previously reported (variants written in black), the pathogenic variant in the proband is marked in red (the suspected splicing mutation COL11A2 c.4750+5G > A is not shown) (https://legacy.uniprot.org/uniprot/P13942).
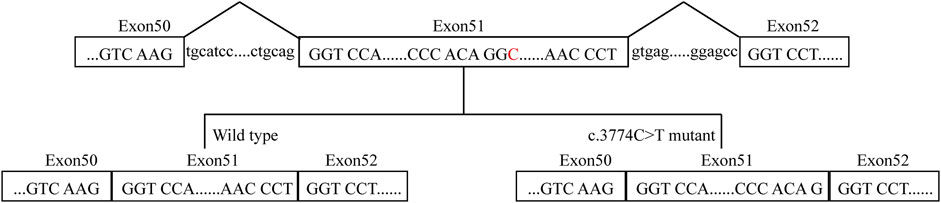
FIGURE 3. A proposed model for the role of the COL11A2 c.3774C>T mutation. The red C represents the mutation site.
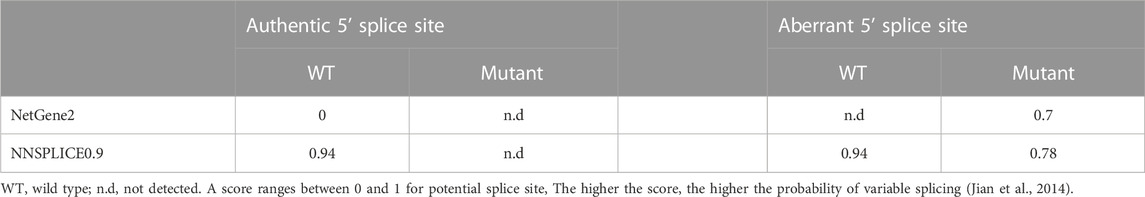
TABLE 1. In silico splicing simulation of COL11A2 c.3774 C > T.
Discussion
In this study, the patient was diagnosed with autosomal recessive type 3 SS based on clinical manifestations, imaging findings and genetic sequencing. As an uncommon cause of SS, type 3 SS is also known as non-ocular SS or OSMED. For the patient, one of the compound heterozygous mutations has been discovered in similar patients (c.3774C>T (p.Gly1258Gly) (Acke et al., 2014), and the other is an intron mutation site that locates in a highly-conservative site across species. Both mutations are supposed to cause abnormal splicing mutations which affect the function of type XI collagen.
Type XI collagen is a quantitatively minor component of collagen in cartilage, which co-polymerizes with type II and type IX collagen to form a heterotrimer. Type XI collagen is mainly composed of three types of chains, namely, α1 (XI), α2 (XI), and α3 (XI). COL11A2 is the coding gene for the α2 (XI) chain (Bruckner and van der Rest, 1994). Hence, defects in COL11A2 will only affect osteoarthritis with the normal ocular phenotype (Snead et al., 2011).
The mutation sites of the patient are both located in the triple helix region of collagen (Figure 2C), which is the sequence repetition of the amino acid triad -Gly-X-Y-. The majority of -X-Y- consists of prolines and hydroxyprolines that are essential for the formation and stabilization of the triple helix. The mutation sites in this region may lead to changes in the structure of the triple helix and corresponding functional changes (Snead et al., 2011; Chakchouk et al., 2015).
COL11A2 gene mutations cause different clinical manifestations depending on mutation types. Missense mutations in the helix domain may destroy the stability of the helix structure. When mutations occur at the carboxyl end, namely, the starting point of the collagen helical trimer, it will result in the most serious clinical phenotype (Chen et al., 2005). It has long been recognized that synomymous mutation has no functional impact. However, growing studies have shown that synonymous mutations affect DNA, RNA, and protein-based features (Vuoristo et al., 2004; Hunt et al., 2014; Zeng et al., 2021). The COL11A2 c.3774 C > T is close to the marginal zone of exons and may cause splicing abnormalities. The other mutation site, c.4750 + 5G>A, is an intron mutation that has not previously been reported. According to the ACMG guidelines, the variation is considered uncertain significance. Currently, it cannot be ruled out that deep intron mutations could result in disease (Snead et al., 2022). Therefore, we investigated the influence of c.3774 C > T on the COL11A2 mRNA splicing process in silico (Table 1), which resulted in aberrant splicing via the aberrant 5′ splice site (Figure 3). The proband’s parents both are asymptomatic with a heterozygous mutation.
Based on the characteristics of the vitreous, a taxonomy of the SS subgroups has developed over the past 20 years. It has been shown that this taxonomy is a reliable strategy for both diagnosing SS and directing molecular study (Nixon et al., 2022; Soh et al., 2022). Type 3 SS is a non-ocular type of SS, manifesting as prelingual development of sensorineural hearing loss, growth of the epiphyses with generalized shortening of the limbs, and vertebral body dysplasia. Distinguishing facial characteristics include an upturned nose, a low nasal bridge, and midface hypoplasia. The condition appears radiographically to impact numerous joints. The upper and lower extremity’s long bones are frequently seen to be shorter, with extensive epiphyses and metaphyseal flare that give them a dumbbell form. The coronal clefts in the spine’s vertebral bodies indicate faulty ossification, making the vertebral bodies platyspondyly. Despite having shorter stature as children, most patients grow to be average height. Patients typically experience joint discomfort due to prevalent arthritic changes. Clinically, the possibility of type 3 SS should be considered in patients with early sensorineural deafness, skeletal abnormalities, and imaging alterations. Genetic detection should be carried out as soon as possible to determine the cause.
At present, there is no efficient treatment for SS. Individualized treatment seems appropriate according to the different symptoms and signs. Early-stage surgical repair of skeletal abnormalities in children with type 3 SS has been reported in the past, and the short-term outcome is favorable. However, as age increases and the disease symptoms worsen, the surgical intervention becomes more challenging (Williams et al., 2020).
There are several limitations to our case report. First of all, we did not perform any additional functional testing. It still needs to be established whether the synonymous mutation in this patient leads to alternative splicing. Secondly, due to the paucity of clinical awareness of the disease, the patient had spent a lot of time on disease diagnosis. SS should be considered when patients exhibit typical deafness, bone abnormalities, and other features. In the end, although the patient has type 3 SS, there is currently no effective treatment. However, the validation of the causative gene is beneficial for genetic counseling.
In conclusion, we report a rare case with autosomal recessive type 3 SS. Both intronic and synonymous mutations are easily missed by conventional whole-exome sequencing. Next-generation whole-genome sequencing, in silico prediction, and possibly supplementary functional analysis are warranted for the accurate diagnosis of these patients.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Ethical Committee approval number: TJ-IRB20221309). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YS: manuscript writing, collection, and assembly of data. C-QR, Z-LL: Gene sequencing and genetic counseling. YY, GY, S-HH, X-FY, and W-TH: manuscript editing and approval of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by China Diabetes Excellence Study (2018-N-01) of China International Medical Exchange Foundation.
Acknowledgments
We are grateful to the patient and his family members.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1154087/full#supplementary-material
Abbreviations
SS, Stickler syndrome; COL11A2, collagen alpha-2 (XI) chain; OSMED, otospondylomegaepiphyseal dysplasia; COL2A1, collagen alpha-1(II) chain; COL11A1, collagen alpha-1 (XI) chain; COL9A1, collagen alpha-1(IX) chain; COL9A2, collagen alpha-2(IX) chain; COL9A3, collagen alpha-3(IX); LRP2, low-density lipoprotein receptor-related protein 2; GZF1, GDNF-inducible zinc finger protein 1; BMP4, bone morphogenetic protein 4; PLOD3, multifunctional procollagen lysine hydroxylase and glycosyltransferase LH3; LOXL3, lysyl oxidase homolog 3; MRI, magnetic resonance imaging; BMD, body mass density; P1NP, procollagen type 1 N-terminal prope-peptide; CTX, carboxy-terminal cross-linked telopeptide of type 1 collagen.
References
Acke, F. R., Malfait, F., Vanakker, O. M., Steyaert, W., De Leeneer, K., Mortier, G., et al. (2014). Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by targeted NGS and exome sequencing. Mol. Genet. Metabolism 1133, 230–235. doi:10.1016/j.ymgme.2014.09.001
Alzahrani, F., Al Hazzaa, S. A., Tayeb, H., and Alkuraya, F. S. (2015). LOXL3, encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Hum. Genet. 1344, 451–453. doi:10.1007/s00439-015-1531-z
Bruckner, P., and van der Rest, M. (1994). Structure and function of cartilage collagens. Microsc. Res. Tech. 285, 378–384. doi:10.1002/jemt.1070280504
Carter, E. M., and Raggio, C. L. (2009). Genetic and orthopedic aspects of collagen disorders. Curr. Opin. Pediatr. 211, 46–54. doi:10.1097/mop.0b013e32832185c5
Chakchouk, I., Grati, M., Bademci, G., Bensaid, M., Ma, Q., Chakroun, A., et al. (2015). Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53. Mol. Genet. Genomics 2904, 1327–1334. doi:10.1007/s00438-015-0995-9
Chan, T. K., Alkaabi, M. K., ElBarky, A. M., and El-Hattab, A. W. (2019). LOXL3 novel mutation causing a rare form of autosomal recessive Stickler syndrome. Clin. Genet. 952, 325–328. doi:10.1111/cge.13465
Chen, W., Kahrizi, K., Meyer, N. C., Riazalhosseini, Y., Van Camp, G., Najmabadi, H., et al. (2005). Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J. Med. Genet. 4210, e61. doi:10.1136/jmg.2005.032615
Ewans, L. J., Colley, A., Gaston-Massuet, C., Gualtieri, A., Cowley, M. J., McCabe, M. J., et al. (2019). Pathogenic variants in PLOD3 result in a Stickler syndrome-like connective tissue disorder with vascular complications. J. Med. Genet. 569, 629–638. doi:10.1136/jmedgenet-2019-106019
Giedion, A., Brandner, M., Lecannellier, J., Muhar, U., Prader, A., Sulzer, J., et al. (1982). Oto-spondylo-megaepiphyseal dysplasia (OSMED). Helv. Paediatr. Acta 374, 361–380.
Hunt, R. C., Simhadri, V. L., Iandoli, M., Sauna, Z. E., and Kimchi-Sarfaty, C. (2014). Exposing synonymous mutations. Trends Genet. 307, 308–321. doi:10.1016/j.tig.2014.04.006
Ihanamaki, T., Pelliniemi, L. J., and Vuorio, E. (2004). Collagens and collagen-related matrix components in the human and mouse eye. Prog. Retin Eye Res. 234, 403–434. doi:10.1016/j.preteyeres.2004.04.002
Jian, X., Boerwinkle, E., and Liu, X. (2014). In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 4222, 13534–13544. doi:10.1093/nar/gku1206
Melkoniemi, M., Brunner, H. G., Manouvrier, S., Hennekam, R., Superti-Furga, A., Kaariainen, H., et al. (2000). Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am. J. Hum. Genet. 662, 368–377. doi:10.1086/302750
Nixon, T. R. W., Richards, A. J., Martin, H., Alexander, P., and Snead, M. P. (2022). Autosomal recessive stickler syndrome. Genes (Basel) 137, 1135. doi:10.3390/genes13071135
Nixon, T. R. W., Richards, A., Towns, L. K., Fuller, G., Abbs, S., Alexander, P., et al. (2019). Bone morphogenetic protein 4 (BMP4) loss-of-function variant associated with autosomal dominant Stickler syndrome and renal dysplasia. Eur. J. Hum. Genet. 273, 369–377. doi:10.1038/s41431-018-0316-y
Pihlajamaa, T., Prockop, D. J., Faber, J., Winterpacht, A., Zabel, B., Giedion, A., et al. (1998). Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am. J. Med. Genet. 802, 115–120. doi:10.1002/(sici)1096-8628(19981102)80:2<115:aid-ajmg5>3.0.co;2-o
Roche, P. A., and Cresswell, P. (1991). Proteolysis of the class II-associated invariant chain generates a peptide binding site in intracellular HLA-DR molecules. Proc. Natl. Acad. Sci. U. S. A. 88, 3150–3154. doi:10.1073/pnas.88.8.3150
Schrauwen, I., Sommen, M., Claes, C., Pinner, J., Flaherty, M., Collins, F., et al. (2014). Broadening the phenotype of LRP2 mutations: A new mutation in LRP2 causes a predominantly ocular phenotype suggestive of stickler syndrome. Clin. Genet. 863, 282–286. doi:10.1111/cge.12265
Snead, M. P., McNinch, A. M., Poulson, A. V., Bearcroft, P., Silverman, B., Gomersall, P., et al. (2011). Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (Lond) 2511, 1389–1400. doi:10.1038/eye.2011.201
Snead, M. P., Richards, A. J., McNinch, A. M., Alexander, P., Martin, H., Nixon, T. R. W., et al. (2022). Stickler syndrome - lessons from a national cohort. Eye (Lond). 3610, 1966–1972. doi:10.1038/s41433-021-01776-8
Soh, Z., Richards, A. J., McNinch, A., Alexander, P., Martin, H., and Snead, M. P. (2022). Dominant stickler syndrome. Genes (Basel) 136, 1089. doi:10.3390/genes13061089
Stickler, G. B., Belau, P. G., Farrell, F. J., Jones, J. D., Pugh, D. G., Steinberg, A. G., et al. (1965). Hereditary progressive arthro-ophthalmopathy. Mayo Clin. Proc. 40, 433–455.
Vikkula, M., Mariman, E. C., Lui, V. C., Zhidkova, N. I., Tiller, G. E., Goldring, M. B., et al. (1995). Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 803, 431–437. doi:10.1016/0092-8674(95)90493-x
Vuoristo, M. M., Pappas, J. G., Jansen, V., and Ala-Kokko, L. (2004). A stop codon mutation in COL11A2 induces exon skipping and leads to non-ocular Stickler syndrome. Am. J. Med. Genet. A 130A2, 160–164. doi:10.1002/ajmg.a.30111
Weissenbacher, G., and Zweymueller, E. (1964). Simultaneous occurrance of the pierre robin syndrome and fetal chondrodysplasia. Monatsschr Kinderheilkd 112, 315–317.
Williams, B. R., Calhoun, A., Holton, K. J., Kelly, B. J., and Sembrano, J. N. (2020). Otospondylomegaepiphyseal dysplasia: A case report of clinical and radiographic findings. JBJS Case Connect. 104, e20 00140. doi:10.2106/JBJS.CC.20.00140
Keywords: Col11a2, splicing mutation, case report, type 3 Stickler syndrome, genetic counseling
Citation: Su Y, Ran C-Q, Liu Z-L, Yang Y, Yuan G, Hu S-H, Yu X-F and He W-T (2023) Case report: Autosomal recessive type 3 Stickler syndrome caused by compound heterozygous mutations in COL11A2. Front. Genet. 14:1154087. doi: 10.3389/fgene.2023.1154087
Received: 30 January 2023; Accepted: 25 May 2023;
Published: 06 June 2023.
Edited by:
Mahmood Rasool, King Abdulaziz University, Saudi ArabiaReviewed by:
Sajjad Karim, King Abdulaziz University, Saudi ArabiaAyca Kocaaga, Eskisehir City Hospital, Türkiye
Copyright © 2023 Su, Ran, Liu, Yang, Yuan, Hu, Yu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Tao He, d3RoZUB0amgudGptdS5lZHUuY24=