 Maria Judit Molnar1,2*
Maria Judit Molnar1,2* Tamas Szlepak1,2
Tamas Szlepak1,2 Ildikó Csürke3Szendile Loth4Rita Káposzta5
Ildikó Csürke3Szendile Loth4Rita Káposzta5 Melinda Erdős6
Melinda Erdős6 Antal Dezsőfi4
Antal Dezsőfi4- 1Institute of Genomic Medicine and Rare Disorders, Semmelweis University, Budapest, Hungary
- 2ELKH-SE Multiomics Neurodegeneration Research Group, Eötvös Loránd Research Network, Budapest, Hungary
- 3Department of Pediatrics, Josa Andras County Hospital, Nyiregyhaza, Hungary
- 4Department of Pediatrics, Semmelweis University, Budapest, Hungary
- 5Department of Pediatrics, University of Debrecen, Debrecen, Hungary
- 6PID Clinical Unit and Laboratory, Department of Dermatology, Venereology, and Dermatooncology, Semmelweis University, Budapest, Hungary
Acid sphingomyelinase deficiency (ASMD) is an autosomal recessive disease caused by biallelic pathogenic variants in the sphingomyelin phosphodiesterase-1 (SMPD1) gene. Acid sphingomyelinase deficiency is characterized by a spectrum of disease and is broadly divided into three types (ASMD type A, ASMD type A/B, and ASMD type B). More than 220 disease-associated SMPD1 variants have been reported, and genotype/phenotype correlations are limited. Here we report the first description of all six diagnosed acid sphingomyelinase deficiency cases in Hungary. Nine SMPD1 variants are present in this cohort, including 3 SMPD1 variants (G247D, M384R, and F572L), which have only been described in Hungarian patients. All described variants are deemed to be pathogenic. Eight of the variants are missense, and one is a frameshift variant. The treatment of an ASMD type A/B patient in this cohort using hematopoietic stem cell transplantation is also detailed. This study may help to support diagnosis, patient genetic counseling, and management of acid sphingomyelinase deficiency.
1 Introduction
Acid sphingomyelinase deficiency (ASMD), which was historically known as Niemann-Pick disease, is a rare, progressive, debilitating, and potentially fatal disease (McGovern et al., 2017b). ASMD is an autosomal recessive lysosomal storage disease (LSD) caused by biallelic pathogenic variants in the sphingomyelin phosphodiesterase-1 (SMPD1) gene, which encodes acid sphingomyelinase (ASM). Reduced levels and/or activity of ASM results in the progressive accumulation of sphingomyelin and other lipids, notably within tissues with a high reticuloendothelial cell content, such as the spleen, liver, lung, lymph nodes, and bone marrow (McGovern et al., 2017b). To date, more than 220 pathogenic sequence variants of SMPD1 have been associated with ASMD (Hu et al., 2021). The numerous gene variants of SMPD1, in conjunction with other genetic and epigenetic factors, give rise to a wide range of disease symptoms and severity (McGovern et al., 2021).
ASMD is broadly divided into three types, which are characterized by their clinical manifestations, age of onset, and rapidity of disease progression (Schuchman and Desnick, 2017).
ASMD type A is an early-onset, rapidly progressing neurovisceral disorder leading to death at 2–3 years of age. These patients may exhibit symptoms including central nervous system involvement, hepatosplenomegaly, pulmonary symptoms, and a failure to thrive or reach developmental milestones (McGovern et al., 2006). ASMD type B has a slow, progressive course with limited or no neurologic involvement, and patients may survive until adolescence or adulthood. Hepatosplenomegaly is the predominant manifestation; other common symptoms include pulmonary involvement and liver dysfunction (McGovern et al., 2017a). ASMD type A/B is thought to be an intermediate clinical phenotype characterized by slow progressive, variable visceral disease and neurological degeneration (Pavlů-Pereira et al., 2005; Wasserstein et al., 2006).
Given the variety of clinical manifestations of ASMD, a wide range of clinical evaluations (including cardiac, neurological, metabolic, pulmonary, musculoskeletal, and gastrointestinal) may be undertaken before ASMD is suspected and diagnosed (McGovern et al., 2017b). The clinical diagnosis of ASMD may also be confounded by multiple symptoms that overlap with other LSDs. Consensus diagnostic guidelines for ASMD (McGovern et al., 2017b), therefore, recommend that diagnosis is made based on reduced ASM activity in whole blood, dried blood spots, fibroblasts, or leukocytes. The gold standard method of ASM estimation is tandem mass spectrometry, but fluorometric ASM activity assays are also widely used (McGovern et al., 2017b). When ASM deficiency, defined as a residual ASM less than 10% of control values (Wasserstein and Schuchman, 1993), is apparent, genetic sequencing is performed to obtain additional information and predict genotype/phenotype correlation if none has been established. Genetic sequencing may be independently diagnostic if 2 pathogenic variants are detected (McGovern et al., 2017b).
ASMD is a pan-ethnic disorder with a worldwide distribution (Simonaro et al., 2002). However, some SMPD1 variants have a higher incidence in particular geographic regions; for example, the p.R610del variant is prevalent within the Maghreb region of North Africa (Vanier et al., 1993). Detailed studies of the ASMD genotype and phenotype have been conducted in some European areas, such as in Czech and Slovak patients (Pavlů-Pereira et al., 2005). To date, a limited amount of information on Hungarian ASMD genotypes and phenotypes is available from a research study of three Hungarian ASMD families (Tóth et al., 2011).
Here we describe the genotype and phenotype of 6 Hungarian pediatric patients and the treatment of one with a hematopoietic stem cell transplant (HSCT).
2 Patients and methods
Patients diagnosed with ASMD in Hungary were identified with the involvement of physicians from Hungarian Rare Disease Expert Centers and lysosomal disease treatment centers. All patients described in this case series (Table 1) are Hungarian, patients 1-5 are Caucasian, and patient 6 is of Gypsy/Roma ethnicity. SMPD1 variants are described with reference to the GenBank (Accession Number) NM_000543.4 sequence. The impact analysis tool VarSome (Kopanos et al., 2019) and the Franklin online platform (https://franklin.genoox.com - Franklin by Genoox) were used for variant classification. ACMG (American College of Medical Genetics) classification were performed in all cases. The damaging rare variants were validated by Sanger sequencing in all cases. The trans position of the heterozygous rare variants were proven by the segregation analysis of the parents.
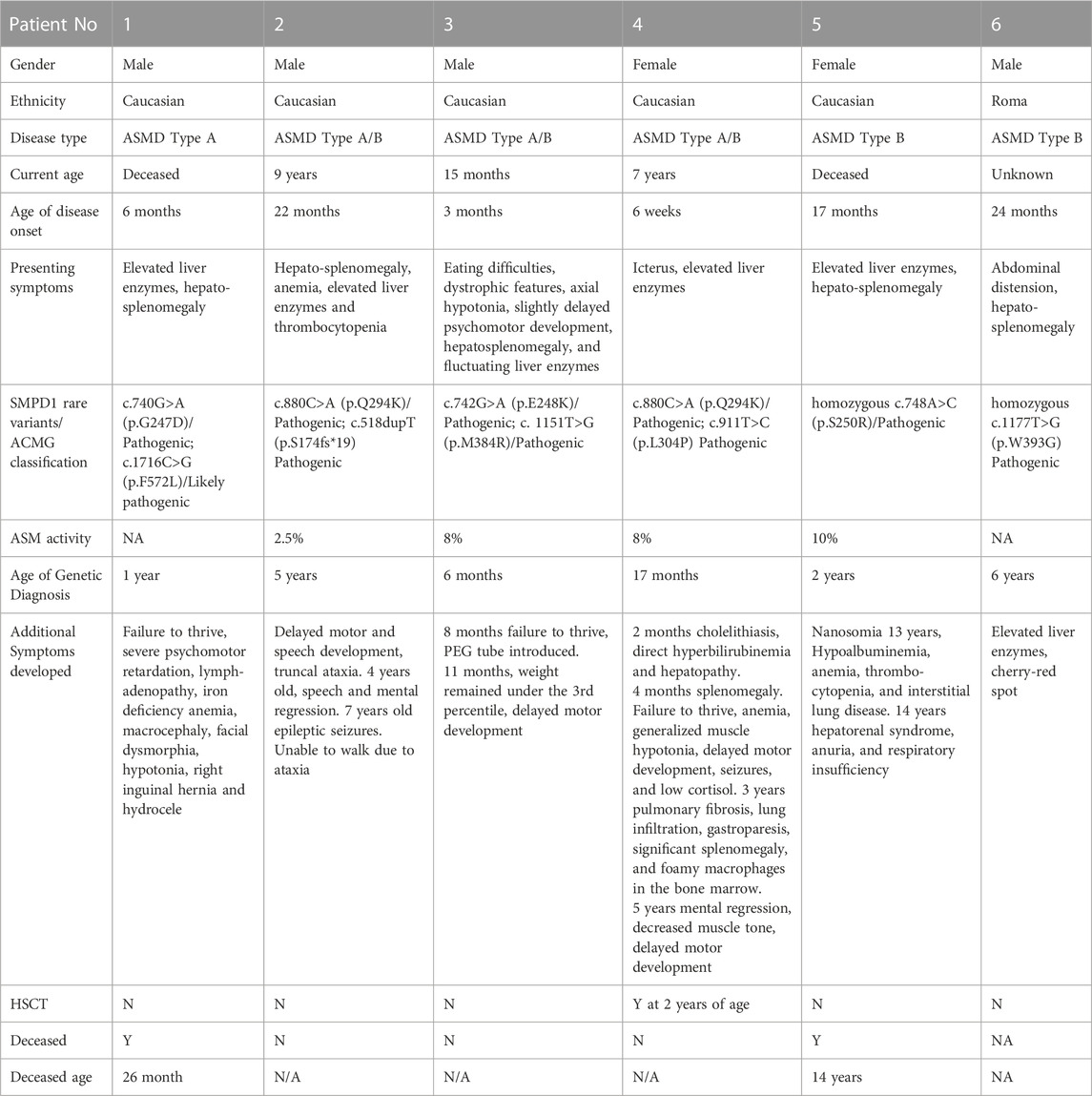
TABLE 1. Timeline, natural history, and genotype/phenotype of ASMD patients.
2.1 Case reports
All patients described in this case series (Table 1) are Hungarian, patients 1-5 are Caucasian, and patient 6 is of Romani person ethnicity. SMPD1 variants are described with reference to the GenBank (Accession Number) NM_000543.4 sequence. The impact analysis tool VarSome (Kopanos et al., 2019) and the Franklin online platform (https://franklin.genoox.com - Franklin by Genoox) were used for variant classification (Supplementary Table S1).
Patient 1 (a male deceased at 26 months) was diagnosed at 6 months of age with ASMD type A disease, which was characterized by hepatosplenomegaly and elevated liver enzymes. He was born at term with a birth weight of 3,020 g. His perinatal adaptation was normal; no prolonged jaundice was detected. He presented with hepatosplenomegaly at the age of 6 months. Laboratory tests showed elevated liver enzymes. By 4 months, he exhibited failure to thrive and delayed motor development. From the age of 5 months, he was treated for a right inguinal hernia and hydrocele. He received iron supplementation due to iron deficient anemia. By the age of 1 year, he only learned to roll over, but could not sit, stand or walk. He had hypotonia and he could not eat chunky food. He also developed macrocephaly and facial dysmorphia characterized by distant eyes and swollen eyelids, slight exophthalmos, wide nasal roots and deep-set ears. Biallelic SMPD1 c.740G>A (p.G247D), c.1716C>G (p.F572L) gene variants were identified at 1 year. During the second year of life, severe progression of the disease was observed, and rapid deterioration of organ functions led to death at 26 months of age.
2.1.1 Patients 2–4 had ASMD A/B phenotype
Patient 2 (a 9-year-old male) was 22 months old at disease onset and presented with hepatosplenomegaly, elevated liver enzymes, anemia, and thrombocytopenia. The patient was determined (at 5 years) to have an ASMD type A/B phenotype and to carry compound heterozygous c.880C>A (p.Q294K) and c.518dupT (p.S174fs*19) SMPD1 variants. Acid sphingomyelinase activity was decreased. Delayed motor and speech development and truncal ataxia were exhibited. At 4 years, speech and mental regression were noted, and epileptic seizures developed aged 7. The patient was unable to walk as a consequence of ataxia.
Patient 3 (a 15-month-old male) presented at 3 months with eating difficulties, dystrophic features, axial hypotonia, slightly delayed psychomotor development, hepatosplenomegaly, and elevated liver enzymes. Ferritin was 502 μg/L. C-triol (cholestane-3β,5α,6β-triol) was 109 ng/ml (normal range 0–40 ng/mL), 7-ketocholesterol: 395 ng/ml (normal range 0–85 ng/mL). Acid sphingomyelinase activity was decreased at 0.1 μmol/L/h (cut off > 1.2). He was determined (at 6 months) to have compound heterozygous c.742G>A (p.E248K) and c.1151T>G (p.M384R) SMPD1 variants. By 8 months, he exhibited a failure to thrive, and a PEG tube was introduced. At 11 months, his weight remained under the 3rd percentile, and he has delayed motor development. He is able to roll over in both directions but cannot crawl. He does not exhibit any pulmonary symptoms.
Patient 4 (a 7-year-old female) first exhibited symptoms of icterus and elevated liver enzymes at 6 weeks. She demonstrated cholelithiasis, direct hyperbilirubinemia and hepatopathy (at 2 months), and splenomegaly (at 4 months). Liver enzymes were elevated. Subsequently, failure to thrive, anemia, generalized muscle hypotonia, delayed motor development, seizures, and low cortisol were noted. Her acid sphingomyelinase activity was decreased at 0.1 μmol/L/h (cut off > 1.2). Genetic analysis (at 17 months of age) revealed biallelic c.880C>A (p.Q294K) and c.911T>C (p.L304P) SMPD1 variants. At 2 years of age, the patient received an allogeneic bone marrow transplant. However, donor chimerism rapidly decreased in the peripheral blood. Approximately 6 weeks after the initial procedure, an additional “mega-dose” haploidentical transplant with continual G-CSF was required. Three months later, a (CD 20 negative, mucosa-associated lymphoid tissue-type) post-transplant lymphoproliferative disorder developed, which was successfully treated with dexamethasone. A bone marrow biopsy confirmed normal thrombopoiesis; however, a low peripheral platelet count was noted, which may have been a consequence of hypersplenism. At 3 years of age, signs of disease progression were observed, including pulmonary fibrosis, lung infiltration, gastroparesis, significant splenomegaly, and foamy macrophages in the bone marrow. At 5 years, she is still PEG tube fed and demonstrates some mental regression, decreased muscle tone, and delayed motor development.
2.1.2 Patient 5–6 were classified as ASMD type B
Patient 5 (a female deceased at 14 years of age) was born at term with a birth weight of 2,750 g. Her perinatal adaptation was normal, no prolonged jaundice was detected. She presented with elevated liver enzymes and hepatosplenomegaly at 17 months. She also had nanosomia. Laboratory tests showed elevated liver enzymes. She had significantly decreased acid sphingomyelinase activity (0.24 μkat/kg protein, normal range: 2.3–3.9 μkat/kg protein). Genetic analysis (conducted at 2 years) revealed this patient was homozygous for the c.748A>C (p.S250R) SMPD1 variant. She had no neurological disease manifestations. Hypoalbuminemia, anemia, thrombocytopenia, and interstitial lung disease were noted at 13 years, and hepatorenal syndrome, anuria, and respiratory insufficiency developed by 14 years of age. The patient died aged 14 years due to severe hepatic dysfunction, hepatorenal syndrome, interstitial lung disease, and cardiac failure.
Patient 6 (male) displayed abdominal distension and hepatosplenomegaly at 24 months. He had no neurological disease manifestations at diagnosis. Liver enzymes were elevated, and a cherry-red spot was noted. This patient is Roma origin and is homozygous for the c.1177T>G (p.W393G) SMPD1 variant. A genetic diagnosis was established at 6 years of age. Limited further clinical information is available for this patient.
3 Discussion
Here we describe the genotype and phenotype of 6 Hungarian ASMD patients. Nine SMPD1 variants are present in this cohort. To date, 3 SMPD1 variants identified in this study (G247D, M384R, and F572L) have solely been described in Hungarian patients. The remaining SMPD1 variants have been previously documented in Bulgarian, Jewish, Chinese, Korean, Italian, Czech, and Slovakian patients (Levran et al., 1992; Ferlinz et al., 1995; Simonaro et al., 2002; Harzer et al., 2003; Sikora et al., 2003; Ricci et al., 2004; Pavlů-Pereira et al., 2005; Mihaylova et al., 2007; Cho et al., 2009; Gan-Or et al., 2013; Zampieri et al., 2016; Kaseniit et al., 2020; Hu et al., 2021). Eight of the variants are missense, which are deemed to be pathogenic, and one is a frameshift variant (Zampieri et al., 2016). Several of the missense variants present in this cohort of patients have been functionally evaluated (G247D, Q294K, L304P, W393G, and F572L), which demonstrated a range of residual activity from less than 5% up to 30% of wild-type ASM activity (Tóth et al., 2011; Zampieri et al., 2016). Frameshift variants are deleterious as they can produce mRNA, which is eliminated by nonsense-mediated decay or lead to the generation of truncated and potentially afunctional proteins (Zampieri et al., 2016). Frameshift variants generally result in limited or no ASM activity (McGovern et al., 2017b). However, the frameshift variant (p.S174fs*19) identified in this case series has not been further characterized.
Genetic testing revealed that the only patient diagnosed with ASMD type A (patient 1) carried p.G247D and p.F572L SMPD1 variants. This patient was previously reported by Tóth et al. (2011). Their functional in vitro analysis demonstrated that both variants (but notably p.G247D) decreased the half-life and catalytic activity of ASM, which may explain the severe and ultimately fatal ASMD disease trajectory in p.G247D, p.F572L patients (Tóth et al., 2011).
One of the ASMD type B patients described here (patient 6) is of Roma origin and homozygous for the p.W393G variant. A study of 20 Bulgarians with Roma ancestry showed that all the individuals were homozygous for the p.W393G variant and had an ASMD A/B phenotype. These patients demonstrated a wide spectrum of neurological involvement (Mihaylova et al., 2007). Functional studies of the resultant protein demonstrated limited ASM stability which may underpin the limited sphingomyelinase activity in these patients (Ferlinz et al., 1995). These data suggest that this variant is a Roma founder mutation.
Two patients in this case series carry the p.Q294K variant in heteroallelic form either alongside the p.L304P variant [ASMD type A/B (patient 4)]; or the p.S174fs*19 frameshift variant in the intermediate ASMD type A/B participant (patient 2). The p.L302P (also known as p.L304P) variant accounted for ∼24% of the mutant alleles in a group of Ashkenazi Jewish ancestry who had ASMD type A (Levran et al., 1992). Q294K is a common SMPD1 gene variant that is strongly associated (in either homoallelic or heteroallelic form) with neurological symptoms and disease progression (Pavlů-Pereira et al., 2005; Wasserstein et al., 2006). When p.Q294K is present in heteroallelic form, the phenotype is modified by the second SMPD1 variant (Pavlů-Pereira et al., 2005). A study in Czech and Slovak patients revealed that p.Q294K is one of the most common variants in Eastern Europe (Pavlů-Pereira et al., 2005). The p.Q294K variant was previously identified in the Hungarian population by anonymous retrospective testing of newborn screening cards for LSDs (Wittmann et al., 2012). However, this study was not designed to provide information on phenotype and disease trajectory or allow for patient follow-up (Wittmann et al., 2012).
An ASMD type A/B patient in this case series (patient 3) is heteroallelic for the p.E248K missense variant (Ricci et al., 2004), which is thought to be a functionally important variant as it resulted in charge changes (Ricci et al., 2004). p.E248K has been previously documented in Korean (Cho et al., 2009), Italian (Ricci et al., 2004), and Chinese (Hu et al., 2021) patients.
The Hungarian ASMD type B patient homozygous for p.S250R (patient 5) died aged 14. The p.S250R variant was also present in heteroallelic form (alongside p.Q294K) in a previously described 4-year-old patient who exhibited massive hepatosplenomegaly, lung infiltration but no neurological symptoms (Harzer et al., 2003). However, no follow-up information on patient longevity is provided (Harzer et al., 2003).
The most frequently reported worldwide SMPD1 variant is c.1829_1831del (p. R610del) (Zampieri et al., 2016). Interestingly it was not present in any of the Hungarian patients studied. However, Eastern European populations may have a lower incidence of the p.R610del variant, given that it was not identified in a cohort of patients from the Czech Republic (Pavlů-Pereira et al., 2005; Zampieri et al., 2016).
Until early 2022, when enzyme replacement therapy for ASMD was initially approved (Keam, 2022), no disease-specific treatments for ASMD were available. Limited reports document the successful use of bone marrow or stem cell transplantation in ASMD (Victor et al., 2003; Shah et al., 2005). Despite the use of 2 allogeneic stem cell transplants in patient 4, the (p.Q294K; p.L304P) ASMD type A/B patient described, disease progression has continued.
In conclusion, this case report represents the most comprehensive description of ASMD in Hungary to date. Nine SMPD1 variants are present in this cohort (Supplementary Table S1), including 3 SMPD1 variants (p.G247D, p.M384R, and p.F572L), which have solely been described in Hungarian patients. In patients who present with rare or novel SMPD1 variants, genotyping has limited prognostic value. Therefore, the analysis of possible correlations between the genotype and ASMD disease course can inform clinical management and help to develop novel or personalized therapies.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Hungarian Scientific and Research Ethical Committee (No. 44599–2). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
Patient workup and collation of clinical data: AD, ME, IC, RK, and SL. Genetic analysis: TSZ, MM, and ME. Manuscript drafting: MM, TSZ, AD, ME, and IC. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by the following grants: TKP2021-NVA-15, TKP2021-EGA-25, OTKA 139010.
Acknowledgments
The authors respectfully acknowledge the patients whose cases were presented in this manuscript. The Institute of Genomic Medicine and Rare Disorders is a member of ERN Rare Neurological Disorders and Neuromuscular Disorders. Writing support was provided by Julia Jenkins, Ph.D., of GK Pharmacomm Ltd., and funded via an unrestricted grant from Sanofi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1158108/full#supplementary-material
References
Cho, Y.-U., Chae, J. D., Lee, W. M., Woo, J. J., Lee, H. B., Gong, S. J., et al. (2009). A case of a Korean adult affected by type B niemann-pick disease: Secondary sea-blue histiocytosis and molecular characterization. Korean J. Lab. Med. 29, 97–103. doi:10.3343/kjlm.2009.29.2.97
Ferlinz, K., Hurwitz, R., Weiler, M., Suzuki, K., Sandhoff, K., and Vanier, M. T. (1995). Molecular analysis of the acid sphingomyelinase deficiency in a family with an intermediate form of Niemann-Pick disease. Am. J. Hum. Genet. 56, 1343–1349.
Gan-Or, Z., Ozelius, L. J., Bar-Shira, A., Saunders-Pullman, R., Mirelman, A., Kornreich, R., et al. (2013). The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80, 1606–1610. doi:10.1212/WNL.0b013e31828f180e
Harzer, K., Rolfs, A., Bauer, P., Zschiesche, M., Mengel, E., Backes, J., et al. (2003). Niemann-pick disease type A and B are clinically but also enzymatically heterogeneous: Pitfall in the laboratory diagnosis of sphingomyelinase deficiency associated with the mutation Q292 K. Neuropediatrics 34, 301–306. doi:10.1055/s-2003-44668
Hu, J., Maegawa, G. H. B., Zhan, X., Gao, X., Wang, Y., Xu, F., et al. (2021). Clinical, biochemical, and genotype-phenotype correlations of 118 patients with Niemann-Pick disease Types A/B. Hum. Mutat. 42, 614–625. doi:10.1002/humu.24192
Kaseniit, K. E., Haque, I. S., Goldberg, J. D., Shulman, L. P., and Muzzey, D. (2020). Genetic ancestry analysis on >93,000 individuals undergoing expanded carrier screening reveals limitations of ethnicity-based medical guidelines. Genet. Med. 22, 1694–1702. doi:10.1038/s41436-020-0869-3
Keam, S. J. (2022). Olipudase alfa: First approval. Drugs 82, 941–947. doi:10.1007/s40265-022-01727-x
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980. doi:10.1093/bioinformatics/bty897
Levran, O., Desnick, R. J., and Schuchman, E. H. (1992). Identification and expression of a common missense mutation (L302P) in the acid sphingomyelinase gene of Ashkenazi Jewish type A Niemann-Pick disease patients. Blood 80, 2081–2087. doi:10.1182/blood.v80.8.2081.bloodjournal8082081
McGovern, M. M., Aron, A., Brodie, S. E., Desnick, R. J., and Wasserstein, M. P. (2006). Natural history of type A niemann-pick disease: Possible endpoints for therapeutic trials. Neurology 66, 228–232. doi:10.1212/01.wnl.0000194208.08904.0c
McGovern, M. M., Avetisyan, R., Sanson, B.-J., and Lidove, O. (2017a). Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 12, 41. doi:10.1186/s13023-017-0572-x
McGovern, M. M., Dionisi-Vici, C., Giugliani, R., Hwu, P., Lidove, O., Lukacs, Z., et al. (2017b). Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet. Med. 19, 967–974. doi:10.1038/gim.2017.7
McGovern, M. M., Wasserstein, M. P., Bembi, B., Giugliani, R., Mengel, K. E., Vanier, M. T., et al. (2021). Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: Eleven years of observation. Orphanet J. Rare Dis. 16, 212. doi:10.1186/s13023-021-01842-0
Mihaylova, V., Hantke, J., Sinigerska, I., Cherninkova, S., Raicheva, M., Bouwer, S., et al. (2007). Highly variable neural involvement in sphingomyelinase-deficient Niemann-Pick disease caused by an ancestral Gypsy mutation. Brain 130, 1050–1061. doi:10.1093/brain/awm026
Pavlů-Pereira, H., Asfaw, B., Poupctová, H., Ledvinová, J., Sikora, J., Vanier, M. T., et al. (2005). Acid sphingomyelinase deficiency. Phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J. Inherit. Metab. Dis. 28, 203–227. doi:10.1007/s10545-005-5671-5
Ricci, V., Stroppiano, M., Corsolini, F., Di Rocco, M., Parenti, G., Regis, S., et al. (2004). Screening of 25 Italian patients with Niemann-Pick a reveals fourteen new mutations, one common and thirteen private, in SMPD1. Hum. Mutat. 24, 105. doi:10.1002/humu.9258
Schuchman, E. H., and Desnick, R. J. (2017). Types A and B niemann-pick disease. Mol. Genet. Metab. 120, 27–33. doi:10.1016/j.ymgme.2016.12.008
Shah, A. J., Kapoor, N., Crooks, G. M., Parkman, R., Weinberg, K. I., Wilson, K., et al. (2005). Successful hematopoietic stem cell transplantation for Niemann-Pick disease type B. Pediatrics 116, 1022–1025. doi:10.1542/peds.2005-0867
Sikora, J., Pavlu-Pereira, H., Elleder, M., Roelofs, H., and Wevers, R. A. (2003). Seven novel acid sphingomyelinase gene mutations in Niemann-Pick type A and B patients. Ann. Hum. Genet. 67, 63–70. doi:10.1046/j.1469-1809.2003.00009.x
Simonaro, C. M., Desnick, R. J., McGovern, M. M., Wasserstein, M. P., and Schuchman, E. H. (2002). The demographics and distribution of type B niemann-pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 71, 1413–1419. doi:10.1086/345074
Tóth, B., Erdős, M., Székely, A., Ritli, L., Bagossi, P., Sümegi, J., et al. (2011). Molecular genetic characterization of novel sphingomyelin phosphodiesterase 1 mutations causing niemann–pick disease. JIMD Rep. 3, 125–129. doi:10.1007/8904_2011_80
Vanier, M. T., Ferlinz, K., Rousson, R., Duthel, S., Louisot, P., Sandhoff, K., et al. (1993). Deletion of arginine (608) in acid sphingomyelinase is the prevalent mutation among Niemann-Pick disease type B patients from northern Africa. Hum. Genet. 92, 325–330. doi:10.1007/BF01247328
Victor, S., Coulter, J. B. S., Besley, G. T. N., Ellis, I., Desnick, R. J., Schuchman, E. H., et al. (2003). Niemann-pick disease: Sixteen-year follow-up of allogeneic bone marrow transplantation in a type B variant. J. Inherit. Metab. Dis. 26, 775–785. doi:10.1023/B:BOLI.0000009950.81514.c8
Wasserstein, M., Aron, A., Brodie, S., Simonaro, C., Desnick, R., and Mcgovern, M. (2006). Acid sphingomyelinase deficiency: Prevalence and characterization of an intermediate phenotype of NIEMANN-PICK disease. J. Pediatr. 149, 554–559. doi:10.1016/j.jpeds.2006.06.034
Wasserstein, M. P., and Schuchman, E. H. (1993). “Acid sphingomyelinase deficiency,” in GeneReviews®, , eds. M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Beanet al. (Seattle, WA: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1370/ [Accessed January 4, 2023].
Wittmann, J., Karg, E., Turi, S., Legnini, E., Wittmann, G., Giese, A.-K., et al. (2012). Newborn screening for lysosomal storage disorders in Hungary. JIMD Rep. 6, 117–125. doi:10.1007/8904_2012_130
Keywords: ASMD, acid sphingomyelinase deficiency type A/B, intermediate-type acid sphingomyelinase deficiency, Niemann-Pick disease type A/B, SMPD1
Citation: Molnar MJ, Szlepak T, Csürke I, Loth S, Káposzta R, Erdős M and Dezsőfi A (2023) Case report: The spectrum of SMPD1 pathogenic variants in Hungary. Front. Genet. 14:1158108. doi: 10.3389/fgene.2023.1158108
Received: 03 February 2023; Accepted: 24 May 2023;
Published: 06 June 2023.
Edited by:
Sunita Bijarnia-Mahay, Sir Ganga Ram Hospital, IndiaReviewed by:
May Christine Malicdan, National Institutes of Health (NIH), United StatesJie-Yuan Jin, Central South University, China
Copyright © 2023 Molnar, Szlepak, Csürke, Loth, Káposzta, Erdős and Dezsőfi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Judit Molnar, bW9sbmFyLm1hcmlhanVkaXRAbWVkLnNlbW1lbHdlaXMtdW5pdi5odQ==