 Andrey Eremin1*
Andrey Eremin1* Alexander Sergeev1,2
Alexander Sergeev1,2 Arthur Kopylov1,2
Arthur Kopylov1,2 Vladimir Rodin1Daniil Malyshev1Tatiana Panova1Igor Polyakov1
Vladimir Rodin1Daniil Malyshev1Tatiana Panova1Igor Polyakov1 Maria Zvereva1
Maria Zvereva1- 1Lomonosov Moscow State University, Moscow, Russia
- 2Institute of Biomedical Chemistry, Moscow, Russia
Background: Ogataea parapolymorpha DL-1 is a versatile thermotolerant organism with numerous applications in biotechnology, particularly in the production of recombinant proteins and the study of methanol metabolism and peroxisome functions. This study presents a comprehensive genome and methylome analysis of Ogataea parapolymorpha DL-1 using long-read sequencing technology. The research builds upon previous short-read sequencing efforts, revealing enhancements in genome assembly and epigenomic insights.
Methods: We used long-read sequencing technology to achieve a telomere-to-telomere (T2T) genome assembly of Ogataea parapolymorpha DL-1. High-quality reads were obtained and assembled de novo, followed by polishing to enhance accuracy. The genome was analyzed to identify coding genes, telomeric motifs, rRNA genes, and methylation patterns, including the detection of 5mC and 6 mA modifications. Epigenetic features were further assessed and validated through liquid chromatography-mass spectrometry.
Results: Key findings include the absence of 5 mC DNA modification and the presence of 6 mA in the genome, unusual telomere regulation mechanism based on the addition of non-telomeric dT and the introduction of long-read enhanced telomere-to-telomere assembly.
Conclusion: This work provides deeper insights into the yeast’s genome organization and methylation patterns, contributing to the understanding of its genetics and therefore potential biotechnological applications.
Introduction
Ogataea parapolymorpha DL-1 is a methylotrophic yeast with distinctive features and broad applications in both biotechnology and basic research. Known for its capacity to utilize methanol as a sole carbon source, this organism has been extensively studied for its roles in methanol metabolism, peroxisome biogenesis, and related functions. It also serves as a robust cell factory for recombinant protein production and is a promising candidate for metabolic engineering, particularly in high-temperature ethanol production. One of the most intriguing aspects of Ogataea parapolymorpha DL-1 is its genome organization, particularly in subtelomeric regions, which are enriched with genes involved in diverse metabolic processes. These include transporters for metal ions, amino acids, and carbohydrates, as well as genes associated with redox processes and NADPH regeneration. The dynamic nature of these subtelomeric regions suggests a genetic mechanism for rapid adaptation to environmental changes and stressors (Liebal et al., 2021; Xie et al., 2024).
Transcriptomic analyses reveal that Ogataea parapolymorpha DL-1 undergoes dramatic gene expression changes when grown on methanol compared to glucose (Ravin et al., 2013). These changes affect various cellular processes, including metabolism, stress responses, and mitochondrial respiratory function. Key upregulated genes include those encoding enzymes for methanol utilization, proteins required for peroxisome assembly and maintenance, and components of the antioxidant defense system. Additionally, phenotype microarray analyses have provided insights into the metabolic versatility of this yeast, highlighting its ability to utilize a wide range of carbon substrates and adapt to diverse growth conditions (Manfrão-Netto et al., 2019).
One of the significant advantages of Ogataea parapolymorpha DL-1 in biotechnological applications is its highly efficient methanol-inducible promoters, which drive enhanced gene expression in the presence of methanol. Moreover, its derepression in glycerol-containing media offers a competitive advantage over other methylotrophic yeasts, such as Pichia pastoris. Another noteworthy trait is its thermotolerance, which facilitates industrial processes by minimizing microbial contamination, reducing cooling costs, and enabling advanced techniques such as Simultaneous Saccharification and Fermentation (SSF) (Ravin et al., 2013).
Telomerase, a ribonucleoprotein enzyme, maintains genomic stability by synthesizing telomeric DNA using an RNA template. In Ogataea parapolymorpha DL-1, telomerase uniquely incorporates a non-templated dT nucleotide at telomere ends, contributing to telomere length regulation. This process highlights a distinct telomere maintenance mechanism that differs from other yeasts. Structural analyses of Ogataea parapolymorpha DL-1 telomerase RNA reveal conserved elements with unique adaptations, emphasizing the enzyme’s evolutionary divergence and functional versatility under thermotolerant conditions (Smekalova ElenaM. et al., 2013).
The yeast has also been a valuable model for telomere biology. Previously referred to as Hansenula polymorpha DL-1, strain ATCC 26012 was reclassified as Ogataea parapolymorpha DL-1 in 2010 (Suh and Zhou, 2010). Its reference genome, assembled in earlier studies, provided few insights into its genomic architecture, subtelomeric regions and repetitive elements (Ravin et al., 2013; Smekalova ElenaM. et al., 2013).
Although previous short-read assembly of Ogataea parapolymorpha DL-1 was of appropriate quality, it remained incomplete, especially in repetitive and subtelomeric regions critical for functional and structural genomics. We resequenced the genome using advanced long-read sequencing technology to achieve a comprehensive, telomere-to-telomere assembly. T2T assembly would provide a reliable reference for genomic and epigenomic studies and also enable a deeper understanding of telomere biology, genome stability, accurate mapping of DNA methylation states, and serve as a superior benchmark for ongoing comparative genomic and biotechnological research.
DNA methylation is an important epigenetic mechanism influencing gene expression, genome stability, and adaptation (Mattei et al., 2022). In yeasts, 5-methylcytosine (5 mC) has been reported at varying levels, with some species displaying barely detectable amounts of this modification (Tang et al., 2012). Understanding which methylation systems are present or absent helps clarify how these organisms regulate transcription, respond to stress, or maintain genomic integrity. In this study, we specifically investigate 5mC, 5hmC and 6 mA in Ogataea parapolymorpha DL-1 to determine the exact levels of those modifications.
Methods
Ogataea parapolymorpha DL-1 genomic DNA extraction
Ogataea parapolymorpha DL-1 yeast was grown in 2 mL of YPD medium (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) dextrose) at 200 rpm, 37°C overnight. The cells were pelleted by centrifugation at 12,100
Library preparation
PromethION sequencing libraries were prepared using 1
PromethION flow cell preparation and DNA sequencing
The sequencing mix was prepared with 35
Raw sequencing data processing
Dorado (Oxford Nanopore Technologies, 0.9.0+9dc15a8, model ZG5hX3IxMC40LjFfZTguMl80MDBicHNfc3VwQHY0LjMuMA==), was used for basecalling and demultiplexing. Porechop (v0.2.4) (Bonenfant et al., 2022) was used for adapter trimming. FastQC (v0.12.1) (FastQC, 2024) was used to assess the quality of reads. Fastp (v0.24.0) (Chen et al., 2018) was used to filter out low quality reads with a threshold of Q35.
De novo genome assembly and polishing
Long-read genome assembly was performed using miniasm (v0.3) (Li, 2016), a fast and lightweight assembler that constructs contigs based on read overlaps without performing error correction. Filtered sequencing reads were used to identify overlaps using minimap2 (v2.24-r1122) (Li, 2018) with the “ava-ont” preset for all-vs-all read alignment. The resulting overlap file in PAF format was then used as input for miniasm using default parameters for assembly. The assembly graph was generated in GFA format and subsequently converted to FASTA using a custom awk script. Since miniasm does not perform consensus error correction, the assembled sequences were further polished using medaka (v2.0.1, model r1041_e82_400bps_sup_v4.3.0, Oxford Nanopore Technologies), a neural network-based tool specifically designed for Oxford Nanopore data. Raw reads were aligned to the draft assembly using minimap2, and medaka_consensus was run with default parameters. The final polished assembly was used for downstream analysis.
Quality evaluation and genomic features annotation
Quality of De novo assembled genome was evaluated using two reference free quality assessment tools Inspector (v1.3) (Chen et al., 2021) and Merqury (v1.3) (Rhie et al., 2020). To generate a k-mer database for reference-free quality assessment using Merqury, we used Meryl tool (v.1.3, part of Merqury) to count 21-mers from high-accuracy short-read sequencing data. Coding genes were annotated with AUGUSTUS (v3.5.0) (Stanke and Augustus, 2005). rRNA genes were found with rRNAFinder (v1.1.1) (Dong, 2019). Comparison with previous assembly was carried out by the use of MUMmer (v4.0.1) (Marçais et al., 2018). Assembly to assembly alignment was visualized with CIRCOS (v0.52) (Krzywinski et al., 2009). Tandem repeats were annotated with RPTRF (v1.0) (Behboudi et al., 2023).
Methylation analysis
5mC, 5hmC and 6 mA modification data was called from raw pod5 data using dorado basecaller (Oxford Nanopore Technologies, version 0.9.0+9dc15a8, models ZG5hX3IxMC40LjFfZTguMl80MDBicHNfc3VwQHY0LjMuMF81bUNfNWhtQ0B2MQ== and ZG5hX3IxMC40LjFfZTguMl80MDBicHNfc3VwQHY0LjMuMF82bUFAdjI=), bedmethyl files were obtained using modkit pileup (Oxford Nanopore Technologies, version 0.4.1). The threshold for counting modifications was frequency >0.5 and coverage >10X.
To assess the consistency of the modification calling results, we took a ‘hac’ model with all of the versions available (hac_v4.3.0_6 mA_v1, hac_v4.3.0_6 mA_v2, hac_v5.0.0_6 mA_v1, hac_v5.0.0_6 mA_v2, hac_v5.0.0_6 mA_v3, hac_v4.3.0_5 mC_5hmC_v1, hac_v5.0.0_5 mC_5hmC_v2, hac_v5.0.0_5 mC_5hmC_v3. Supplementary Table S2; Supplementary Table S3; Supplementary Table S4).
Methylation motifs were searched using MEME (v5.5.7) (Bailey and Elkan, 1994). To extract a batch of sequences for MEME we searched for coordinates of modified bases with high frequency >0.5 and coverage >10X, then added 10 bp upstream and downstream those coordinates resulting in a bed file of 21 bp-long sequences. Fasta file was created using bedtools (v2.31.1) (Quinlan and Hall, 2010).
To validate the absence of 5 mC in our primary study organism, we performed a parallel sequencing analysis of the Escherichia coli genome as a positive control.
Methylation density over chromosomes was calculated as follows:
Methylation density per 1 Kbp in the specific chromosome was calculated as follows:
Search for putative methyltransferases in Ogatea parapolymorpha DL-1 genome
To find putative methyltransferases we created a reference database of protein sequences fasta files using UniProtKB (The UniProt Consortium, 2016) with the query: ““DNA methyltransferase” AND ((N6-adenine) OR (cytosine-5)) NOT (RNA OR tRNA)”. It yielded 121,392 results. We used DIAMOND (Buchfink et al., 2021) (v 2.1.11) to create a database out of UniProtKB’s search results. We then took protein sequences predicted by AUGUSTUS and searched them over a constructed database using diamond blastp in sensitive mode with default parameters. Protein sequences from top 5 search results were subjected to online BLAST (Altschul et al., 1990) search to NCBI’s non-redundant (NR) protein database.
LC-SRM analysis and instrumentation
Samples were analyzed on an Infinity II 1290 UPLC system (Agilent, Santa Clara, CA, United States) hyphenated with a G6490A triple quadrupole mass spectrometer (Agilent, Santa Clara, CA, United States). The UPLC system was equipped with an Acquity HSS T3 column (150
The mass spectrometer was equipped with a Jet Stream ionization source and operated in a positive ionization mode with a drying gas (nitrogen) temperature of 260 °C at a flow rate of 13 L/min and sheath gas (nitrogen) temperature of 320 °C at a flow rate of 7 L/min. The capillary voltage was set to 3200 V and the nozzle voltage was set to 600 V with a high pressure RF adjusted to 120 V and low pressure RF to 55 V. Spectra were acquired in SRM (selected reactions monitoring) mode for the duty cycle time of 238.5 milliseconds (4.19 cycles per s). Nucleotides were detected by transitions using an optimized set of collision energy, cell accelerating voltage, and dwell time parameters (Table 1). Reference standards: 2′-Deoxyadenosine, 2′-deoxycytidine, 2′-deoxyguanosine (Sigma, Germany), 5-(methyl-d3)-2′-deoxycytidine (Santa Cruz Biotechnology, TX, United States).
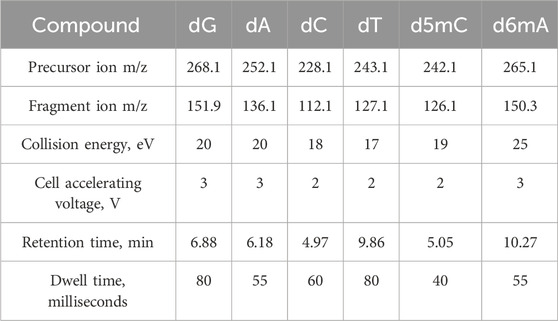
Table 1. SRM parameters optimized for acquisition of nucleotides.
External standard (ESTD) was adulterated by combing of nucleotide standards and diluted in 0.1% formic acid to 100 pg/
Dot blot 6 mA analysis
Genomic DNA from Ogataea parapolymorpha DL-1 was isolated and treated with Proteinase K (overnight, 40°C in Q5 buffer) to remove residual proteins, followed by heat inactivation at 95°C and purification using magnetic beads (GentaPure kit). DNA was denatured by incubation with 2 M NaOH and 100 mM EDTA at 95°C for 10 min, chilled on ice, and neutralized with 1 M Tris-HCl (pH 7.5).
Denatured DNA (3
DNA from Raji cells and Escherichia coli served as positive controls for 6mA, while unmethylated PCR products were used as a negative control (Supplementary Figure S2).
3′-telomere extension
To investigate the addition of non-telomeric nucleotides at telomeric ends, we processed BAM files containing reads aligned to defined telomeric regions across all chromosomes. Quality trimming of reads was applied conservatively (bases with quality scores below Q30 were removed) to eliminate ambiguous or low-confidence base calls at the read termini. Quality trimming at Q30 ensures that the terminal nucleotide retained in each read is confidently sequenced and reliably represents the true biological end of the read, rather than a sequencing artifact or low-confidence call. Bases removed during trimming were of low confidence and, therefore, likely erroneous; bases remaining after trimming are thus of high-quality, robust indicators of actual genomic termini and suitable for accurate biological interpretation. Subsequently, we isolated the last 200 base pairs from the trimmed reads and specifically quantified occurrences at the ends of trimmed reads of thymine (T), as well as adenine (A) as its complementary base. By calculating the percentage of trimmed reads ending with these nucleotides, we confidently assessed the prevalence of non-templated nucleotide additions at telomere ends, ensuring that detected terminal nucleotides represent genuine genomic termini rather than sequencing artifacts.
Results
The resulting total amount of reads after fastp filtration with a quality threshold of Q35 (>80% of bases with Q35) was 285,134 with an average read length of 3,402 bp (Table 2). The threshold for quality was chosen as Q35 in order to avoid hybrid assembly with its obligation of obtaining high-quality short reads. After aligning reads to a previously reported assembly (PRJNA60503), the resulting coverage of Ogataea parapolymorpha DL-1 was 102X, and the coverage of the positive control E. coli was 112X. There were no plasmids assembled de novo from the reads, only seven chromosomes and one circular mito genome.
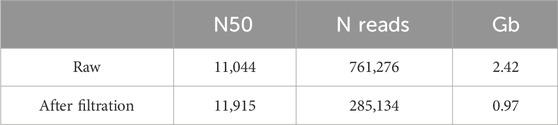
Table 2. Descriptive statistics of sequencing data before and after filtration.
Long-read genomic assembly reveals an extension in the length of every chromosome compared to previous data. (Table 3).
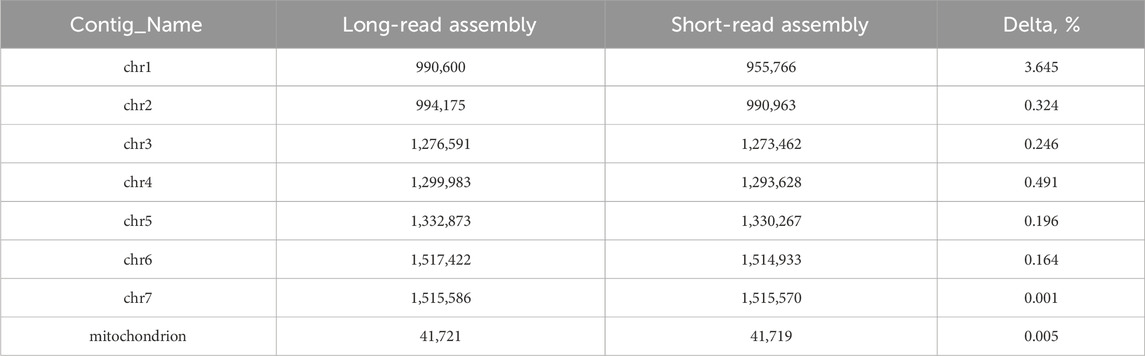
Table 3. Comparison of long-read and short-read assemblies with the corresponding delta percentages.
As shown in Table 4, the QV scores calculated with Mercury and Inspector for the long-read-based assembly of Ogataea parapolymorpha DL-1 were consistently higher than those for the short-read-based draft. This confirms that the long-read assembly provides a more accurate representation of the genome. For additional validation, we evaluated an E. coli K-12 BW25113 delta-MutM reference assembly as a control. Both tools reported high QV scores for E. coli, consistent with its well-characterized, high-quality reference, further supporting the reliability of the evaluation metrics.
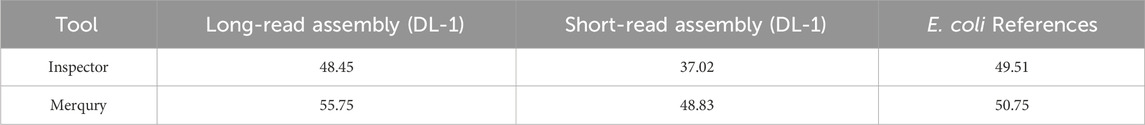
Table 4. Comparison of QV scores reported by Inspector and Merqury.
Our long-read based assembly achieves a telomere-to-telomere (T2T) completeness, a significant improvement over the previous assembly that only identified incomplete parts of telomeric sequences for chromosomes 4 and 7. Our analysis has confirmed the presence of the GGTGGCGG telomere motif (Table 5). Furthermore, the telomeres in our assembly are considerably longer, by 2–3 times, than those noted in the earlier assembly for chromosomes 4 and 7. In contrast to this incomplete representation, our gapless T2T assembly provides a seamless genomic map (Figure 3). The breakpoints identified in the assembly-to-assembly alignment coincided precisely with the gaps previously reported in chromosomes 1 and 4 of the earlier assembly.
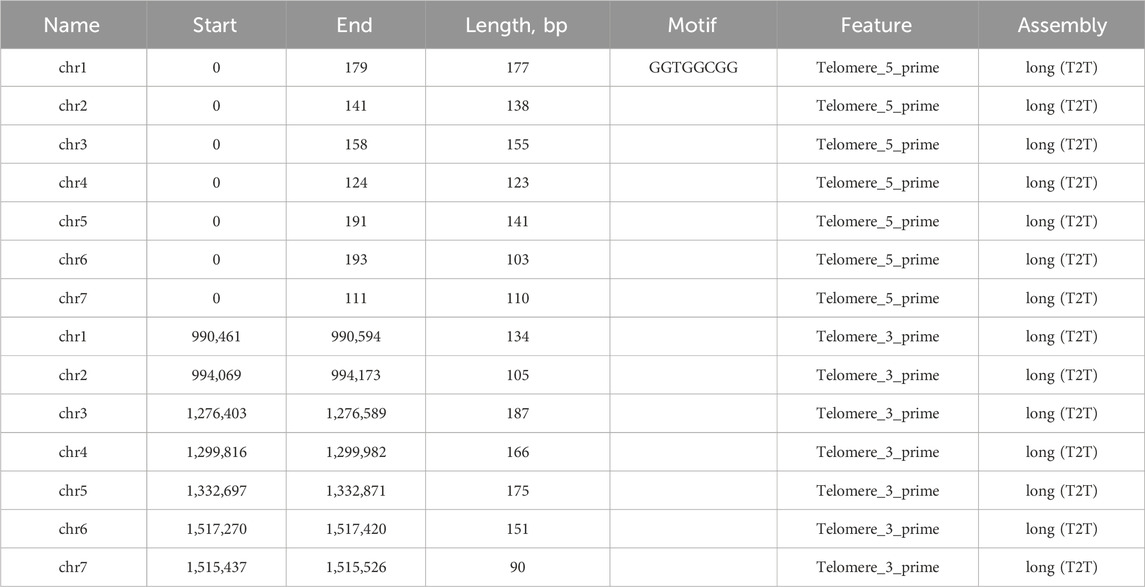
Table 5. Telomeric sequences found in the long read and short read assemblies.
Through the application of “Inspector” (Chen et al., 2021) a tool for reference-free assembly quality assessment, we detected an erroneous expansion within chromosome 7 of up to 1,000 bp in the previous short-read assembly, highlighting the superior resolution of long-read sequencing in identifying and correcting assembly errors (Figure 1).
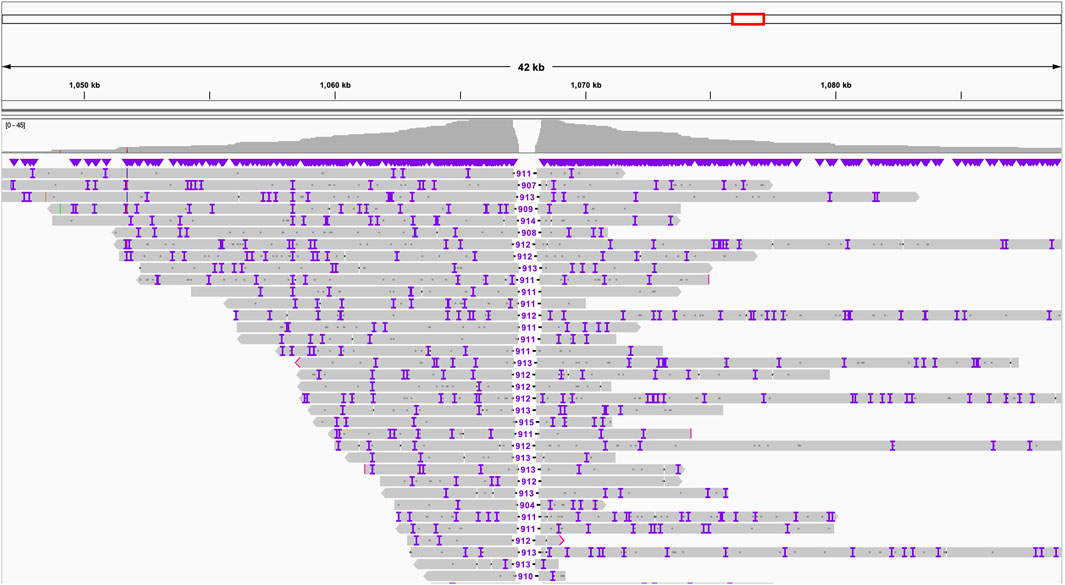
Figure 1. Expansion chr7:1,067,235-1,068,147 in the old assembly revealed by Inspector using long-read data.
Examination of rRNA gene encoding regions has showed the presence of five copies of each of the four types of rRNA genes (Table 6), in contrast to the single copy of three genes found in the previous assembly.
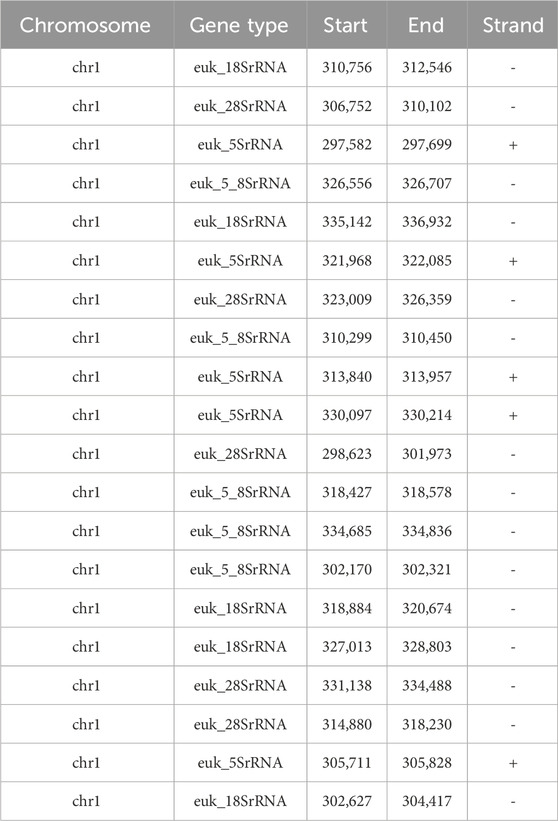
Table 6. rRNA genes coordinates in the newly assembled genome.
Analysis of the telomeric regions across various chromosomes of Ogataea parapolymorpha DL-1 revealed a significant predominance of thymine (T) nucleotides at the 3′ ends of the telomeres, supporting the hypothesis of non-telomeric dT addition by telomerase (Table 7 “A_end” - number of Adenines at the 3′end, “T_end” -- number of Thymidines respectively, “perc_A” - percentage of 3′end aligned reads containing dA at the end. N_total_reads - total reads, aligned to the 3′end).
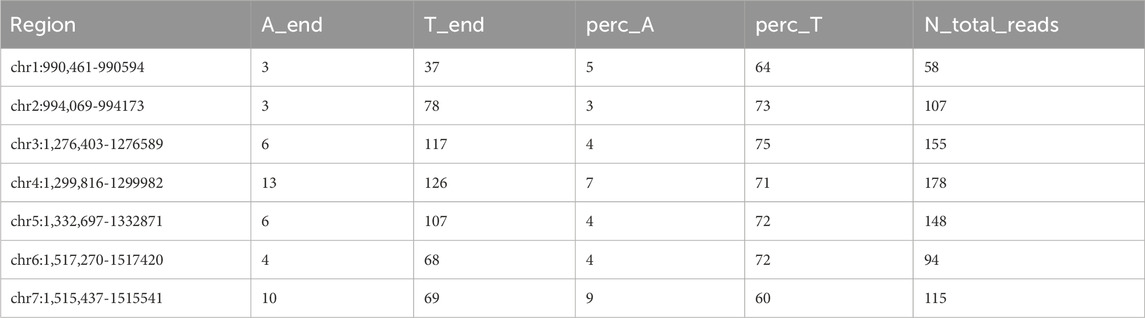
Table 7. Count of nontelomeric dT addition derived from reads aligned to the 3′end of the chromosomes.
The respective counts of basecalled modifications for Ogataea parapolymorpha DL-1 and positive control Escherichia coli are presented in the (Table 8). Based on modification calling data, the mitochondrial genome, as well as the other chromosomes, had the 6 mA modifications and did not have the 5 mC.
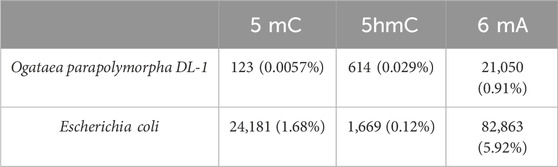
Table 8. Comparative analysis of total number modifications (5mC, 5hmC and 6 mA) found with dorado. Percentage in brackets shows the part from all of the A or C in the genome.
We explored the little amount of 5 mC data that was basecalled. We looked at the rDNA locus (Figure 2) to check whether it contains 5 mC or not, as it was previously reported to be present in other yeast species (Sandesh Pai et al., 2022). Our data showed no 5 mC in the rDNA locus of Ogataea parapolymorpha DL-1.
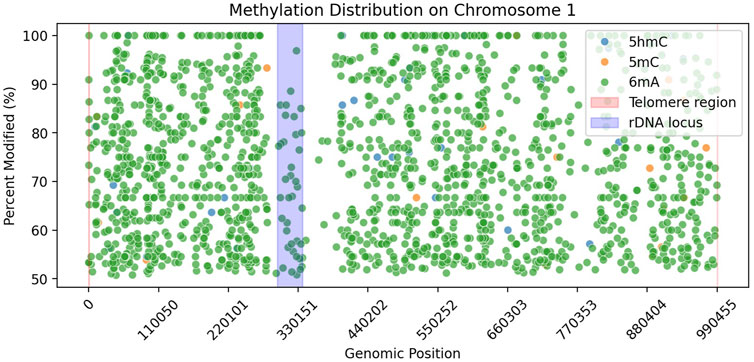
Figure 2. Methylation distribution along Chromosome 1 of Ogataea parapolymorpha DL-1. Scatter plot shows genomic positions versus percent modified sites for three DNA modifications: 5hmC (blue), 5 mC (orange), and 6 mA (green), filtered by >50% modification and >10
To validate the absence of 5mC, genomic DNA samples isolated from the Ogataea parapolymorpha DL-1 yeast were hydrolyzed to the nucleosides, and the content of 5 mC was determined using UPLC/MS-MS. 5mC was not detected in the hydrolyzed samples, confirming the results obtained from nanopore sequencing data (Supplementary Table S1).
The presence of 6 mA in Ogataea parapolymorpha DL-1 genomic DNA was confirmed by dot blot analysis (Supplementary Figure S2).
Sequence motif TCCACCA was found in the regions

Table 9. Modifications analysis. Motifs found with MEME in regions
Methylation density analysis showed a uniform distribution across the chromosomes. Density per 1 Kbp also was consistent across the chromosomes showing approximately 2 6 mA modifications per kilobase (Table 10 Figure 3).
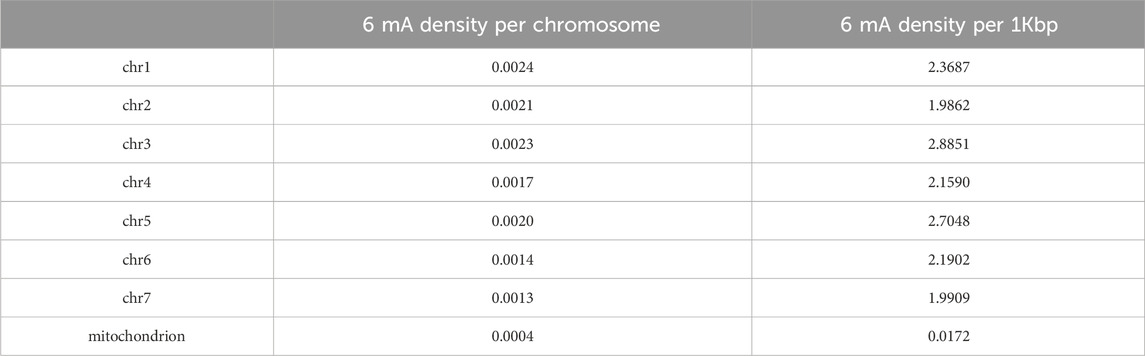
Table 10. 6 mA Density per chromosome and per 1Kbp for different chromosomes calculated using Equation 1 and Equation 2.
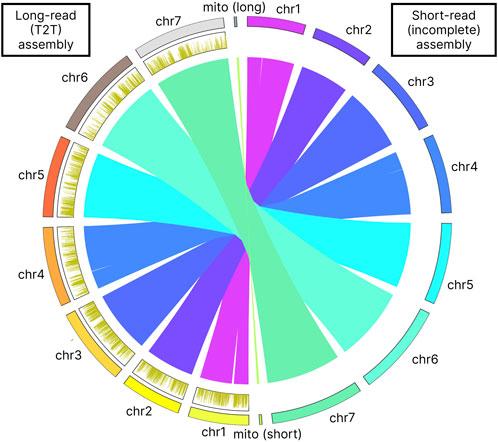
Figure 3. Assembly-to-assembly alignment visualization. The figure also shows the 6 mA methylation profile on the left side of the figure as distribution plot. 6 mA density plots were computed in 1 kb bins.
Search for putative methyltransferases in Ogatea parapolymorpha DL-1 genome with diamond blastp reported 279 partial alignments. No homologous methyltransferases were found using this method.
Discussion
Long read based enhanced WGS assembly
As we produced a huge amount of high quality sequencing data, we were able to use only long reads for assembly and do not produce short reads for the hybrid approach. For samples with an average read length of 8 Kbp or more, a coverage depth of 30X is sufficient (Khrenova et al., 2022). For samples with shorter reads, averaging around 2 Kbp, a higher coverage depth of 50X is required. Our reads with N50 more than 11 Kbp and coverage depth more than 100X allowed for high quality genomic assembly. We also filtered data to ensure highest quality. Our analysis of Ogataea parapolymorpha DL-1 has culminated in the acquisition of extended chromosome lengths compared to the preceding short-read assembly. Specifically, chromosome 1 exhibited the most significant extension, growing from 955,766 base pairs in the short-read assembly to 990,600 base pairs in the long-read assembly, marking a delta of 3.645%. Similarly, chromosome 4 saw a noteworthy lengthening, from 1,293,628 to 1,299,983 base pairs, reflecting a 0.491% increase. What is more, those two chromosomes were the only ones that could not be assembled gaplessly in the previous study.
Both tools Inspector and Merqury were used to evaluate the consensus quality (QV) of the genome assemblies in a reference-free manner. These tools rely on distinct principles and provide complementary insights. Inspector evaluates assembly quality by aligning long reads back to the assembly and identifying small-scale and structural errors directly from read-to-contig alignments. It is particularly well-suited for identifying localized misassemblies and quantifying structural integrity, including inversions, collapses, and haplotype switches. Merqury, on the other hand, assesses base-level consensus accuracy through k-mer spectra analysis. It calculates consensus Quality Value (QV), completeness, and haplotype phasing based on the presence or absence of k-mers in the assembly compared to the read set. In our study, both tools showed broadly consistent results (Table 4) but differed in absolute QV values, which can be attributed to their underlying methodologies.
We also analyzed the rDNA locus within chromosome 1. The rDNA locus in most yeast species, including Ogataea polymorpha DL-1, consists of tens or hundreds of tandemly repeated units, typically spanning hundreds of kilobases to over a megabase in length (Kobayashi and Sasaki, 2017). Genome assemblies generally represent this array as a collapsed segment due to the repetitive nature of the region. In our study, we identified five tandemly repeated units of the rDNA locus in our assembly, including complete copies of the 18S, 28S, 5S, and 5.8S rRNA genes. However, this does not reflect the total number of rDNA repeats in Ogataea polymorpha DL-1, as determining the full array size would require extremely long sequencing reads (approximately 1 Mb or longer) that traverse the entire rDNA array. A comparative analysis between the two assemblies of chromosome 1 revealed a substantial deletion of 32,415 base pairs in the previous short-read assembly. This deleted region corresponds to the rDNA locus and includes tandemly repeated units of the 18S, 28S, 5S, and 5.8S rRNA genes. This specific nature of genomic sequence is the most probable reason why the previous short-read based assembly had a gap in this region. Unfortunately, its not clear why the chromosome 4 and 7 had misassembly. We examined those regions for tandem repeats using RPTRF and did not find any repeats spanning longer regions than short reads are able to cover.
Using the Inspector tool, we uncovered another misassembled region in previously reported draft - a significant expansion in chromosome 7 (Figure 1). This expansion, spanning 912 base pairs (from position 1,067,235 to 1,068,147), represents a notable deviation from the newly established long-read assembly. Though it is not clear why did the previous effort this region.
Our analyses also confirmed the presence of the previously identified telomeric motif (GGTGGCGG) at the 3′ ends. This confirmation across all chromosome ends, coupled with the discovery of the 5′ CCACCCCG motif, enhances our understanding of the chromosomal termini in this species. The detailed data, including coordinates and lengths of these telomeric regions, are provided in the accompanying table. In contrast to the previous short-read assembly of Ogataea parapolymorpha DL-1, our long-read sequencing approach successfully closed the gaps, resulting in a complete and continuous genome. Specifically, the previous assembly exhibited gaps in chromosomes 1 and 4, at positions chr1:297,310–297409, chr1:305,147-305246, and chr4:366,734-370733.
3′-telomere extension
In the study (Smekalova EM. et al., 2013) of telomerase activity within Ogataea parapolymorpha DL-1, a remarkable discovery was made regarding the addition of a non-telomeric dT nucleotide by the telomerase enzyme. Unlike the conventional understanding of telomerase function, which primarily focuses on the elongation of telomeres through the addition of specific telomeric repeats to counteract telomere shortening, Ogataea parapolymorpha DL-1 exhibits a unique mechanism of telomere length regulation in budding yeasts (Malyavko et al., 2014). The telomerase RNA from this thermotolerant yeast species, identified as HpTER, extends beyond merely synthesizing telomeric sequences. It actively incorporates a dT nucleotide beyond the anticipated boundary of the RNA template in vitro, a modification not part of the telomeric repeat itself. Subsequent sequencing of chromosomal ends carried out by the authors (Smekalova EM. et al., 2013) validated the presence of this dT nucleotide as a terminal element at the 3′ end of telomeres with independent methodological confirmation of the presence of an additional nontelomeric nucleotide at the 3′-end of the chromosomes (Malyavko et al., 2016). Mutational analysis of HpTER template region elucidated that this unconventional addition of non telomeric nucleotide plays a crucial role in limiting telomere length within Ogataea parapolymorpha DL-1 and one of natural mechanism of ‘chromosome capping’ for telomere length regulation (Wellinger, 2010).
In our study we have confirmed this finding using long read sequencing technology. This phenomenon underscores a novel layer of complexity in telomere length regulation, revealing that telomerase can extend its functional repertoire beyond traditional repeat addition. This adaptation suggests a species-specific regulatory mechanism for telomere maintenance, potentially contributing to the genomic stability of Ogataea parapolymorpha DL-1 and highlighting the diversity of telomerase activity across different organisms.
DNA methylation anomaly: Absence of 5 mC
A paramount discovery is the absence of 5-methylcytosine (5 mC), which was supported by the UPLC data. This suggests an evolutionary trajectory where the organism has possibly discarded the methylation system typically essential for other species, yet maintains normal physiological functions. The dorado modification calling model is prone to producing errors, nevertheless, in Table 8 we see that Ogataea parapolymorpha DL-1 doesn’t have the cytosine modification if we compare it to E. coli which does have it. We also observe that the count of 5hmC for Ogataea parapolymorpha DL-1 exceeds the count of 5mC, which is impossible because 5hmC is produced from 5 mC. It should also be mentioned that different modification calling tools and different versions of models for them are prone to producing different results. For example, it was reported that the open-source 5 mC analysis tool DeepMod 2 (Umair Ahsan et al., 2024) demonstrated greater accuracy in detecting canonical cytosines compared to Oxford Nanopore’s Dorado in study (Sergeev et al., 2025). In our study, we have shown the inconsistency of modification calling results by comparing different models for dorado basecaller (Supplementary Table S2; Supplementary Table S3; Supplementary Table S4). This result clearly shows that the modification data obtained from nanopore sequencer must be confirmed by orthogonal methods such as LC-SRM or dot blot used in our study.
Positive control E. coli in contrast, shows that the number of 5 mC exceeds 5hmC greatly. This suggests that the minimal amount of modified cytosines basecalled for Ogataea parapolymorpha DL-1 is attributable to errors in the basecalling model. The phenomenon of DNA methylation, a crucial epigenetic mechanism for regulating gene expression, exhibits remarkable variability across other living organisms like the fungal kingdom, primarily due to the differential presence of DNA methyltransferases (DNMTs) (Nai et al., 2021). In certain fungal species, the complete absence of DNMTs shows the lack of DNA methylation, highlighting a direct link between the availability of these enzymes and the existence of methylation processes within the genome. This absence indicates that some fungi might employ alternative epigenetic strategies for gene regulation, diverging significantly from the methylation-dependent pathways observed in other eukaryotes. There are other reported cases in which 6 mA is present and 5 mC is not present in the genome (Lieberman Greer et al., 2015), (Lizarraga et al., 2020).
In our study, we observed an absence of detectable 5 mC in Ogataea parapolymorpha DL-1, consistent with previous findings suggesting that DNA methylation in budding yeast species can vary significantly, often presenting at low or undetectable levels. For instance, low-level methylation (0.014%–0.364%) has been reported in various budding yeast species including Saccharomyces cerevisiae (Tang et al., 2012), often only detectable by highly sensitive analytical methods such as GC/MS. In contrast, our analysis utilizing nanopore sequencing complemented by ultra-sensitive UPLC-MS/MS did not detect 5 mC modifications. Additionally, the few nanopore-based predictions of 5 mC we identified (Table 8) were closely scrutinized and attributed to algorithmic false positives due to their extremely low frequency and lack of reproducibility by orthogonal methods. Furthermore, the genomic distribution of these few computationally predicted 5 mC sites did not align with previously reported methylation hotspots such as the rDNA locus (Figure 2) observed in Saccharomyces cerevisiae (Sandesh Pai et al., 2022).
Given these observations, we conclude that the absence of 5 mC in Ogataea parapolymorpha DL-1 is authentic rather than a limitation of analytical sensitivity, and it may reflect evolutionary divergence in epigenetic regulatory mechanisms among closely related yeast species.
Possible methyltransferase motif
In our study, we identified a potential methyltransferase-associated motif, TCCACCA (Table 9), within the Ogataea parapolymorpha DL-1 genome. Interestingly, 6 mA is not located within the site. Instead, the modified base could be found up to 4 base pairs upstream of this site.
It is known from other organisms that methylation motifs do not always directly coincide with the modified bases (Wang et al., 2019). Rather, these motifs commonly serve as binding sites recognized by methyltransferases or associated proteins, thereby influencing methylation states at adjacent nucleotides. For instance, in bacteria such as Escherichia coli, the known DNA methyltransferase Dam recognizes and methylates adenine residues within a specific GATC motif, directly methylating adenines within that precise context (Kahramanoglou et al., 2012). However, in contrast, studies in eukaryotes, such as Tetrahymena thermophila and Chlamydomonas reinhardtii, have shown that 6 mA methyltransferases may recognize extended sequence contexts or distal motifs rather than direct, contiguous recognition sequences (Cheng et al., 2019).
To our knowledge, no data currently exist on this specific TCCACCA motif or similar motifs in Ogataea parapolymorpha DL-1 or other closely related yeast species. Additionally, no characterized methyltransferase in yeast has been linked to the recognition of this sequence.
Conclusion
Our comprehensive investigation into the Ogataea parapolymorpha DL-1 genome through long-read sequencing technology has advanced the understanding of this organism’s genomic architecture and methylation patterns. By achieving a telomere-to-telomere assembly, we have closed previous gaps and also extended the known lengths of chromosomes, offering a complete and continuous genomic map. This study has also uncovered crucial genomic regions previously unresolved, including a significant 32,415 bp deletion containing multiple rRNA gene copies, enhancing our insight into the genomic organization and its functional implications in rRNA processing. The absence of 5-methylcytosine across the genome suggests unique methylation dynamics and an evolutionary divergence from typical eukaryotic methylation patterns. These findings underscore the power of long-read sequencing to reveal detailed and accurate genomic features that are often obscured by short-read sequencing technologies. This work enriches our genomic knowledge of Ogataea parapolymorpha DL-1 and contributes to the broader understanding of genomic structure and stability in eukaryotes, paving the way for future studies on genomic and epigenetic diversity.
Data availability statement
The Genomic assembly as well as the sequencing reads generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA1091144.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
AE: Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review and editing. AS: Data curation, Methodology, Resources, Writing – original draft, Writing – review and editing. AK: Resources, Writing – original draft, Writing – review and editing. VR: Methodology, Validation, Writing – review and editing. DM: Validation, Writing – review and editing. TP: Data curation, Writing – review and editing. IP: Software, Writing – review and editing. MZ: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research has been conducted in the Interdisciplinary Scientific and Educational School of Moscow University <Molecular Technologies of the Living Systems and Synthetic Biology> for yeast genome sequencing and analysis. This work was financed by the Ministry of Science and Higher Education of the Russian Federation within the framework of Agreement 075-15-2024-643.
Acknowledgments
We are grateful to Mikhail Agafonov for providing the strain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1574332/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi:10.1016/S0022-2836(05)80360-2
Bailey, T. L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Behboudi, R., Nouri-Baygi, M., and Naghibzadeh, M. (2023). RPTRF: a rapid perfect tandem repeat finder tool for DNA sequences. Biosystems 226 (apr), 104869. doi:10.1016/j.biosystems.2023.104869
Bonenfant, Q., Noé, L., and Touzet, H. (2022). Porechop_ABI: discovering unknown adapters in Oxford Nanopore Technology sequencing reads for downstream trimming. Bioinforma. Adv. 3 (1), vbac085. doi:10.1093/bioadv/vbac085
Buchfink, B., Reuter, K., and Drost, H.-G. (2021). Sensitive protein alignments at tree-of-life scale using diamond. Nat. Methods 18 (4), 366–368. doi:10.1038/s41592-021-01101-x
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), pages=i884–i890. doi:10.1093/bioinformatics/bty560
Chen, Yu, Zhang, Y., Wang, A. Y., Gao, M., and Chong, Z. (2021). Accurate long-read de novo assembly evaluation with Inspector. Genome Biol. 22 (1), 312. doi:10.1186/s13059-021-02527-4
Cheng, T., Wang, Y., Huang, J., Chen, X., Zhao, X., Gao, S., et al. (2019). Our recent progress in epigenetic research using the model ciliate, tetrahymena thermophila. Mar. Life Sci. and Technol. 1 (1), 4–14. doi:10.1007/s42995-019-00015-0
Dong, X. (2019). rRNAFinder. Available online at: https://github.com/xiaoli-dong/rRNAFinder.
FastQC, A. (2024). Quality control tool for high throughput sequence data. on Mon. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed April 22, 2024).
Kahramanoglou, C., Prieto, A. I., Khedkar, S., Haase, B., Gupta, A., Benes, V., et al. (2012). Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat. Commun. 3 (1), 886. doi:10.1038/ncomms1878
Khrenova, M. G., Panova, T. V., Rodin, V. A., Kryakvin, M. A., Lukyanov, D. A., Osterman, I. A., et al. (2022). Nanopore sequencing for de novo bacterial genome assembly and search for single-nucleotide polymorphism. Int. J. Mol. Sci. 23 (15), 8569. doi:10.3390/ijms23158569
Kobayashi, T., and Sasaki, M. (2017). Ribosomal dna stability is supported by many ‘buffer genes’—introduction to the yeast rdna stability database. FEMS Yeast Res. 17 (1). doi:10.1093/femsyr/fox001
Krzywinski, M., Schein, J., Birol, İ., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), pages=1639–1645. doi:10.1101/gr.092759.109
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32 (14), pages=2103–2110. doi:10.1093/bioinformatics/btw152
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi:10.1093/bioinformatics/bty191
Liebal, U. W., Fabry, B. A., Ravikrishnan, A., Schedel, C. V. L., Schmitz, S., Blank, L. M., et al. (2021). Genome-scale model reconstruction of the methylotrophic yeast Ogataea polymorpha. BMC Biotechnol. 21 (1), 23. doi:10.1186/s12896-021-00675-w
Lieberman Greer, E., Andres Blanco, M., Gu, L., Sendinc, E., Liu, J., Aristizábal-Corrales, D., et al. (2015). DNA methylation on N6-adenine in C. elegans. Cell. 161 (4), may, pages=868–878. doi:10.1016/j.cell.2015.04.005
Lizarraga, A., Klapholz O’Brown, Z., Boulias, K., Roach, L., Greer, E. L., Johnson, P. J., et al. (2020). Adenine DNA methylation, 3D genome organization, and gene expression in the parasite Trichomonas vaginalis. Proc. Natl. Acad. Sci. 117 (23), 13033–13043. doi:10.1073/pnas.1917286117
Malyavko, A. N., Parfenova, Y. Y., Zvereva, M. I., and Dontsova, O. A. (2014). Telomere length regulation in budding yeasts. FEBS Lett. 588 (15), pages=2530–2536. doi:10.1016/j.febslet.2014.05.049
Malyavko, A. N., Petrova, O. A., Zvereva, M. I., and Dontsova, O. A. (2016). Designing a system to test for the presence of any nontelomeric nucleotide at the 3′-chromosomal end. Mosc. Univ. Chem. Bull. 71 (1), 45–47. doi:10.3103/s0027131416010107
Manfrão-Netto, J. H. C., Gomes, A. M. V., and Parachin, N. S. (2019). Advances in using Hansenula polymorpha as chassis for recombinant protein production. Front. Bioeng. Biotechnol. 7, 94. doi:10.3389/fbioe.2019.00094
Marçais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: a fast and versatile genome alignment system. PLOS Comput. Biol. 14 (1), e1005944. doi:10.1371/journal.pcbi.1005944
Mattei, A. L., Bailly, N., and Meissner, A. (2022). Dna methylation: a historical perspective. Trends Genet. 38 (7), 676–707. doi:10.1016/j.tig.2022.03.010
Nai, Y.-S., Huang, Y.-C., Yen, M.-R., and Chen, P.-Y. (2021). Diversity of fungal DNA methyltransferases and their association with DNA methylation patterns. Front. Microbiol. 11, 616922. doi:10.3389/fmicb.2020.616922
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 (6), jan, pages=841–842. doi:10.1093/bioinformatics/btq033
Ravin, N. V., Eldarov, M. A., Kadnikov, V. V., Beletsky, A. V., Schneider, J., Mardanova, E. S., et al. (2013). Genome sequence and analysis of methylotrophic yeast Hansenula polymorpha DL1. BMC Genomics 14 (1), 837. doi:10.1186/1471-2164-14-837
Rhie, A., Walenz, B. P., Koren, S., and Phillippy, A. M. (2020). Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21 (1), 245. doi:10.1186/s13059-020-02134-9
Sandesh Pai, S., Ranjan, S., Mathew, A. R., Anindya, R., and Meur, G. (2022). Analysis of the long-read sequencing data using computational tools confirms the presence of 5-methylcytosine in the saccharomyces cerevisiae genome. Access Microbiol. 4 (6), acmi000363. doi:10.1099/acmi.0.000363
Sergeev, A. V., Malyshev, D. P., Genatullina, A. I., Pavlova, G. V., Gromova, E. S., and Zvereva, M. I. (2025). Single-molecule nanopore sequencing of the cpg island from the promoter of o6-methylguanine-dna methyltransferase provides insights into the mechanism of de novo methylation of g/c-rich regions. Epigenomes 9 (1), 4. doi:10.3390/epigenomes9010004
Smekalova, E. M., Malyavko, A. N., Zvereva, M. I., Mardanov, A. V., Ravin, N. V., Skryabin, K. G., et al. (2013a). Specific features of telomerase RNA from Hansenula polymorpha. RNA 19 (11), 1563–1574. pages=1563–1574. doi:10.1261/rna.038612.113
Smekalova, E. M., Malyavko, A. N., Zvereva, M. I., Mardanov, A. V., Ravin, N. V., Skryabin, K. G., et al. (2013b). Specific features of telomerase RNA from Hansenula polymorpha. RNA 19, 1563–1574. doi:10.1261/rna.038612.113
Stanke, M., and Augustus, B. M. (2005). A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, pages=W465–W467. doi:10.1093/nar/gkl200
Suh, S.-O., and Zhou, J. J. (2010). Methylotrophic yeasts near Ogataea (Hansenula) polymorpha: a proposal of Ogataea angusta comb. nov. and Candida parapolymorpha sp. nov. FEMS Yeast Res. 10, 631–638. doi:10.1111/j.1567-1364.2010.00634.x
Tang, Y., Gao, X.-D., Wang, Y., Yuan, B.-F., and Feng, Y.-Qi (2012). Widespread existence of cytosine methylation in yeast dna measured by gas chromatography/mass spectrometry. Anal. Chem. 84 (16), 7249–7255. doi:10.1021/ac301727c
The UniProt Consortium (2016). Uniprot: the universal protein knowledgebase. Nucleic Acids Res. 45 (D1), D158–D169. doi:10.1093/nar/gkw1099
Umair Ahsan, M., Gouru, A., Chan, J., Zhou, W., and Wang, K. (2024). A signal processing and deep learning framework for methylation detection using oxford nanopore sequencing. Nat. Commun. 15 (1), 1448. doi:10.1038/s41467-024-45778-y
Wang, M., Zhang, K., Ngo, Vu, Liu, C., Fan, S., Whitaker, J. W., et al. (2019). Identification of DNA motifs that regulate DNA methylation. Nucleic Acids Res. 47 (13), 6753–6768. doi:10.1093/nar/gkz483
Wellinger, R. J. (2010). When the caps fall off: responses to telomere uncapping in yeast. FEBS Lett. 584 (17), 3734–3740. doi:10.1016/j.febslet.2010.06.031
Keywords: genomics, long-read sequencing, methylomics, Ogataea parapolymorpha DL-1, T2T-assembly
Citation: Eremin A, Sergeev A, Kopylov A, Rodin V, Malyshev D, Panova T, Polyakov I and Zvereva M (2025) Long-read sequencing reveals absence of 5mC in Ogataea parapolymorpha DL-1 genome and introduces telomere-to-telomere assembly. Front. Genet. 16:1574332. doi: 10.3389/fgene.2025.1574332
Received: 10 February 2025; Accepted: 22 April 2025;
Published: 09 May 2025.
Edited by:
Nunzio D'Agostino, University of Naples Federico II, ItalyReviewed by:
Sara Hanson, Colorado College, United StatesCarlos Lax Molina, University of Murcia, Spain
Copyright © 2025 Eremin, Sergeev, Kopylov, Rodin, Malyshev, Panova, Polyakov and Zvereva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrey Eremin, ZXJlbWluYWFAbXkubXN1LnJ1