 Yanwei Zhang1
Yanwei Zhang1 Wenrong Li2Xinming Xu2Mengwan Xie1Liping Tang1Peiyu Zheng1Nannan Song1Lijuan Yu1*Jiang Di1*
Wenrong Li2Xinming Xu2Mengwan Xie1Liping Tang1Peiyu Zheng1Nannan Song1Lijuan Yu1*Jiang Di1*- 1Key Laboratory of Evaluation and Utilization of Livestock and Poultry Resources (Sheep) of the Ministry of Agriculture and Rural Areas, Institute of Animal Sciences, Xinjiang Academy of Animal Sciences, Xinjiang, China
- 2Key Laboratory of Animal Biotechnology of Xinjiang, Institute of Biotechnology, Xinjiang Academy of Animal Sciences, Xinjiang, China
Introduction: The double-coated fleece is crucial for the adaptability and economic value of Hetian sheep, yet its underlying molecular mechanisms remain largely unexplored.
Methods: We integrated genome and transcriptome data from double-coated Hetian sheep and single-coated Chinese Merino sheep. Candidate genes associated with coat fleece type and environmental adaptation were identified using combined selective sweep and differential expression analyses. Subsequent analyses included Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment, protein-protein interaction (PPI) network construction, and machine learning-based screening.
Results: Selective sweep and differential expression analyses identified 101 and 106 candidate genes in Hetian sheep and Chinese Merino sheep, respectively. Enrichment analyses revealed these genes were primarily involved in pathways related to wool growth and energy metabolism. PPI network analysis and machine learning identified IRF2BP2 and EGFR as key functional genes associated with coat fleece type.
Discussion: This study enhances understanding of the genetic mechanisms governing double-coated fleece formation in Hetian sheep. The identification of key genes (IRF2BP2, EGFR) and the methodological approach provide valuable insights for developing machine learning-driven multi-omics selection models in sheep breeding.
1 Introduction
Hetian sheep is a unique local breed from Hetian, Xinjiang, China, known for its high-quality carpet wool. Hetian carpets, made from Hetian sheep wool, are famous worldwide. The breed was listed in the National List for the Protection of Livestock and Poultry Genetic Resources in 2006. Hetian sheep are adapted to the temperate continental desert climate, characterized by drought resistance, heat tolerance, ability to thrive on coarse feed, strong disease resistance, and exceptional adaptability to harsh environments (Han et al., 2023). One of the most distinctive features of Hetian sheep is their double-coated fleece, which includes coarse wool, fine wool, and heterotypical hair. These fibers differ in structure, with heterotypical hair being particularly notable for its interrupted medullary layers, a characteristic that contributes to the plush and durable texture of Hetian carpets (Chen et al., 2024; Shi et al., 2022a). This complex fleece structure raises important questions about the genetic mechanisms behind its formation.
Despite the recognized significance of Hetian sheep’s unique fleece, research on its double-coated fleece and heterotypical hair remains limited, primarily due to the complexity of the underlying genetic mechanisms (Wang et al., 2025; Shi et al., 2022b; Wang et al., 2021). Multiple genes interact in the development of double-coated fleece, and environmental factors, such as temperature variations and drought conditions, further complicate the genetic analysis. These challenges highlight the need for more focused research into the genetic basis of Hetian wool.
In recent years, multi-omic analysis methods have been increasingly applied in sheep breeding (Wang et al., 2023; Li et al., 2024; Xu et al., 2023). By integrating data across various omics levels, researchers can gain a more comprehensive understanding of the genetic and biological mechanisms underlying target traits, enhancing the precision and efficiency of breeding programs. For example, Banos et al. (2017) integrated genomic and transcriptomic data to identify 14 candidate genes related to innate immunity in Chios sheep. Zhao et al. (2021) combined transcriptomic and methylation datasets from Merino sheep skin to reveal differential expression profiles across four genotypes at six hair follicle developmental stages. They identified key transcripts involved in hair follicle development through regulatory network and gene co-expression analyses, and predicted that transcription factors (e.g., KLF4, LEF1, HOXC13, RBPJ, VDR, RARA, and STAT3) play stage-specific roles in hair follicle morphogenesis. In another study, Wang et al. (2020) demonstrated that the expression of certain hair follicle differentiation genes and transcription factors (TFs) in sheep was negatively correlated with DNA methylation levels, using integrated RNA-seq and WGBS analysis. These findings suggest that these genes and TFs may regulate hair morphogenesis by influencing the expression of related genes.
Machine learning has also gained traction in genetic breeding, particularly in genome-wide selection, gene network analysis, and multi-omics data interpretation (Tırınk et al., 2023; Hamadani et al., 2022; Shahinfar et al., 2019). For instance, Kirchner et al. (2004) employed the decision tree (DT) algorithm to classify and predict highly reproductive sows based on characteristics such as total litter size, live litter size, and healthy litter size, achieving promising prediction results. Piles et al. (2019) applied machine learning methods to molecular-level transcriptomic data to deeply explore candidate genes influencing pig feeding efficiency. Guo et al. (2024) utilized interpretable machine learning models alongside comparative transcriptomics to identify unique factors in sheep strongly associated with muscle growth. Additionally, Farhadi et al. (2023) investigated differences in fat deposition between fat-tailed and fine-tailed sheep breeds, using meta-analysis and machine learning techniques to identify three specific genes (POSTN, K35, and SETD4).
In this study, we employed a combination of multi-omics and machine learning techniques to investigate the genetic mechanisms underlying the formation of double-coated fleece. Our goal was to provide a scientific basis for precision breeding. Specifically, we selected Chinese Merino sheep (characterized by single-coated and homogeneous fleece) as a control and conducted selective sweep and differential expression analyses by integrating genomic and transcriptomic data. Additionally, we applied machine learning models, such as Neural Networks (NN), to analyze these multidimensional datasets. The aim of this approach was to uncover the regulatory networks associated with double-coated fleece traits in Hetian sheep, identify key functional genes, and explore the complex relationships between these traits and environmental adaptability.
2 Materials and methods
2.1 Ethics statement
All experimental protocols in this study (including the sample collection protocol), were approved by the Ethics Committee of Institute of Animal Husbandry, Xinjiang Academy of Animal Husbandry Sciences (China) under permission no. 2025001.
2.2 Animals and whole genome sequencing of pools
A total of 49 healthy two-year-old ewes of different coat fleece types were included in the study. Of these, 24 Hetian sheep (double-coated fleece, HT group) were from Tumuya village, Oytograk township, Yutian county, Xinjiang, and 25 Chinese Merino sheep (single-coated fleece, CM group) were from the Gongnaisi breeding sheep farm, Xinjiang (Figure 1).
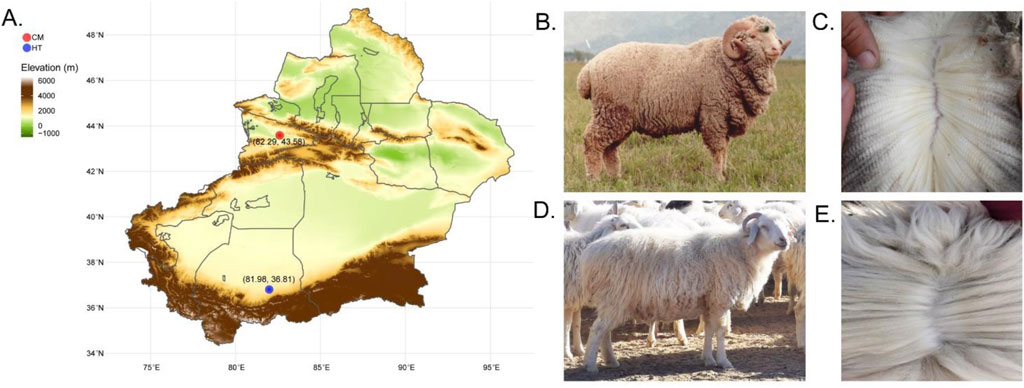
Figure 1. Geographical distribution of Hetian and Chinese Merino sheep, sampling locations and coat characteristics. (A) Geographic distribution and sampling locations. (B) Chinese merino sheep. (C) Single-coated fleece from Chinese Merino sheep. (D) Hetian sheep. (E) Double-coated fleece from Hetian sheep.
Genomic DNA was extracted from the ear tissue of each sheep using the QIAamp DNA Mini Kit (QIAGEN, 51304, Hilden, Germany). DNA from each group member was pooled in equimolar amounts (2 μg/sample) to construct the two pair-end sequencing libraries (insert sizes approximately ∼0.5 kb, with effective insert sequencing concentration >2 nM). Library construction and sequencing were performed on an Illumina HiSeq 2000TM platform supplied by Beijing Novogene Bioinformatics Technology Co.
2.3 Genome sequence processing, mapping and SNP analysis
Raw paired reads were preprocessed to remove adapters and low-quality sequences based on the following criteria: (a) reads with more than 50% of bases having a Qphred score ≤ 5; (b) reads containing ≥10% unidentified nucleotides (N); and (c) reads with >10 nucleotides aligning to adapter sequences. High-quality reads were then mapped to the sheep reference genome (ARS-UI_Ramb_v2.0) using BWA, and the resulting SAM files were cleaned with SAMtools (v1.21) (Danecek et al., 2021) to remove duplicates. Variants were further identified using GATK (v4.1.9.0) (McKenna et al., 2010) with HaplotypeCaller, CombineGVFs, and GenotypeGVFs. To annotate SNPs and classify mutations (nonsense, nonsynonymous, and synonymous), ANNOVAR (Wang et al., 2010) was employed. Additionally, loci were refined using VCFtools (Danecek et al., 2011) to exclude those with a minor allele frequency (MAF) < 5% or a genotype deletion rate >5%
2.4 Selective sweep analysis
Selective sweep analysis was conducted using CM as the reference and HT as the target population to identify genes associated with wool traits. To screen genomic regions, total SNP counts were calculated for different window sizes (10, 20, 30, 40, and 50 KB). When the window size exceeded 40 KB, the SNP count dropped below 20 (Supplementary Figure S1), indicating that a 40 KB window was optimal for the analysis.
The differentiation index (Fst) and nucleotide diversity (θπ) were calculated using VCFtools (v0.1.16) with parameters--fst-window-size 40,000 and--fst-window-step 20,000. These calculations divided the genome into intervals of 40 KB with a 20 KB step. Candidate genomic regions were identified based on the following criteria: regions with extreme θπ ratios (5% left-tailed or 95% right-tailed) and significantly high Fst values (top 5%). Overlapping areas of low θπ ratios and high Fst values were identified as selective regions for HT, while overlaps of high θπ ratios and high Fst values were identified for CM. Genes within these candidate regions were designated as candidate genes (CGs).
2.5 RNA extraction and library construction
Using a circular skin sampler with a radius of 0.44 mm, collect skin sample from the posterior edge of the left foreleg scapula and store it in a cryovial containing RNA protective solution. Total RNA was extracted from 20 skin samples (10 from HT and 10 from CM groups) using TRIzol reagent (Invitrogen, CA, United States). The quality of the RNA samples was assessed, with results showing OD260/280 ratios between 1.8 and 2.0, OD260/230 ratios above 2.0, and RIN values ranging from 7.0 to 8.5 for all samples, indicating they were suitable for further experiments.
Strand-specific libraries were constructed, with ribosomal RNA removed to enhance circRNA library preparation by eliminating linear RNA. The library was initially quantified using Qubit and diluted to 1 ng/μL. The insert size was checked using the Agilent 2100 Bioanalyzer, showing a size range of 250–300 bp, as expected. Once the insert size was confirmed, the effective concentration of the library was accurately measured by qPCR. The concentration exceeded 2 nM, ensuring the library met quality standards. The twenty libraries were pooled and sequenced on the Illumina platform at Novogene Bioinformatics Technology Co., Ltd.
2.6 RNA sequencing quality control and analysis
FastQC was used to assess the quality of the raw RNA sequencing reads. The data were then processed to remove impurities and obtain clean reads for further analysis. Clean data were aligned to the sheep reference genome (ARS-UI_Ramb_v2.0) using HISAT2 (Kim et al., 2019). The transcripts were assembled and quantified with StringTie (Pertea et al., 2015), and the expression level of genes or transcripts was measured by fragments per kilobase million (FPKM). Differentially expressed genes (DEGs) were identified using edgeR (Robinson et al., 2010). Genes were considered differentially expressed if they met the criteria of |log2(fold change)| > 0 and p < 0.05.
2.7 Identification of key functional genes and pathway enrichment
The intersections of candidate genes (CGs) and differentially expressed genes (DEGs) were taken to identify key functional genes (KFGs). These genes were then analyzed for pathway enrichment using KEGG analysis on Kobas 3.0 (Bu et al., 2021), with Ovis_aries selected as the background organism. The hypergeometric test/Fisher’s exact test was used for statistical analysis, and pathways with a p-value < 0.05 were considered significantly enriched. Plots were generated using the R package ggplot2 (Villanueva and Chen, 2019).
Additionally, DEGs were submitted to the STRING database (Szklarczyk et al., 2023) to construct a protein-protein interaction network. The interaction network was visually edited using Cytoscape software (Smoot et al., 2011).
2.8 Machine-learning screening for signature genes
Neural Network (NNET) algorithm is a computational model inspired by biological neural systems and is widely used in tasks such as pattern recognition, classification, and regression. This study employed a Multilayer Perceptron (MLP) neural network for biomarker discovery and classification modeling, utilizing gene expression level as predictors to identify characteristic genes distinguishing different populations (HT vs. CM). The analytical framework comprised four key phases: data preprocessing, feature selection, model training, and validation. All implementations were conducted in R (v4.2.2) using the caret (v6.0-94) Kuhn, 2008 and nnet (v7.3-19) packages.
2.8.1 Data preprocessing
The raw gene expression matrix was transposed to a sample × feature (gene) structure. An automated quantile-based criterion determined logarithmic transformation requirements: log2(x+1) transformation was applied to stabilize variance when either the 99th percentile exceeded 100 or the interquartile range surpassed 50 with a 25th percentile >0. In this study, the dynamic range of expression values spanned [QX1, QX6] (QX denotes specific quantile values), necessitating log2 transformation.
2.8.2 Dataset partitioning
Stratified sampling allocated 70% of samples to the training set and 30% to the independent test set, preserving class proportions (Case/Control = 1:1) through the createDataPartition function from the caret package. A fixed random seed (74,521) ensured reproducibility.
2.8.3 Feature selection
A two-stage feature screening strategy was implemented:
(1) Initial Screening: Boruta algorithm (100 iterations) evaluated global feature importance through statistical significance testing (p < 0.01) using shadow features.
(2) Refined Selection: A 10 × 10 repeated cross-validated neural network model was built on preselected features. Top 30 signature genes were identified via Variable Importance Measure (VIM) rankings computed through the connection weights algorithm, quantifying cumulative contributions of features to output nodes.
2.8.4 Model construction and validation
(1) Hyperparameter Optimization: Grid search tuned hidden layer neurons (size∈{5,10}) and weight decay coefficients (decay∈{0.01,0.1}), optimized by the receiver operating characteristic area under the curve (AUC).
(2) Training Protocol: Parallel computing acceleration (doParallel package(v1.0.17)) enabled 5 × 20 repeated cross-validation: The training set was randomly partitioned into 10 folds, with 20 independent validation cycles to mitigate sampling bias.
(3) Performance Evaluation: Independent test set metrics included: Accuracy, AUC, Sensitivity and specificity.
2.8.5 Statistical analysis
All visualizations (feature importance plots, ROC curves) were generated using ggplot2 (v3.4.2). Significance testing employed DeLong’s algorithm for ROC curve comparisons, with p < 0.05 considered statistically significant.
3 Results
3.1 Summary of genome and transcriptome sequencing
A total of 1,223.6 GB of clean data were obtained after genome resequencing, with an average of 24.97 GB per sample. The mapping rate across all samples ranged from 98.72% to 99.76%, and the average genome coverage depth (excluding N regions) ranged from 8.69X to 12.12X. Coverage of at least one base was above 97.67%, and coverage of at least four bases exceeded 91.54%. After mapping to the reference genome, SNP annotation was performed using ANNOVAR software, identifying 25,515,212 SNPs. The SNPs were located in intergenic (46.68%), intronic (40.74%), and exonic (1.59%) regions. The transition-to-transversion ratio (ts/tv) was 2.685, with 1816 exonic SNPs leading to stop-gain variations, 372 to stop-loss variations, and 154,692 to non-synonymous mutations.
For RNA-seq analysis, over 2.036 billion reads were analyzed, with 996 million from the HT group and 1.039 billion from the CM group. Each sample contained over 100 million reads, ranging from 84 million to 107 million. After trimming, approximately 29.4 million reads were removed, ensuring high data quality. Of the 2.006 billion clean reads, 1.036 billion (∼92.5%) were aligned to the genome, with 84.4% uniquely mapped and 8.1% mapped to multiple locations. The alignment rates for individual samples ranged from 89.2% to 94.7%. A summary of the RNA-seq datasets and mapping results is provided in Supplementary Table 4.
3.2 Screening of key functional genes
The distribution of the genetic differentiation statistic Fst across the genomes of Hetian sheep (HT) and Chinese Merino sheep (CM) is summarized in Figure 2A and Supplementary Table 5. SNP annotation (top 5% of Fst scores) using the ARS-UI_Ramb_v2.0 sheep genome identified a total of 3,533 genes. Notable overlaps were found between the Fst outliers and known QTLs associated with wool traits (e.g., KRT71, KRT74, IRF2BP2), reproductive traits (e.g., ASTN2, TSHR, GTF2A1), and production traits (e.g., PSAP, CDH23, UBE2B), among others (Supplementary Table 6).
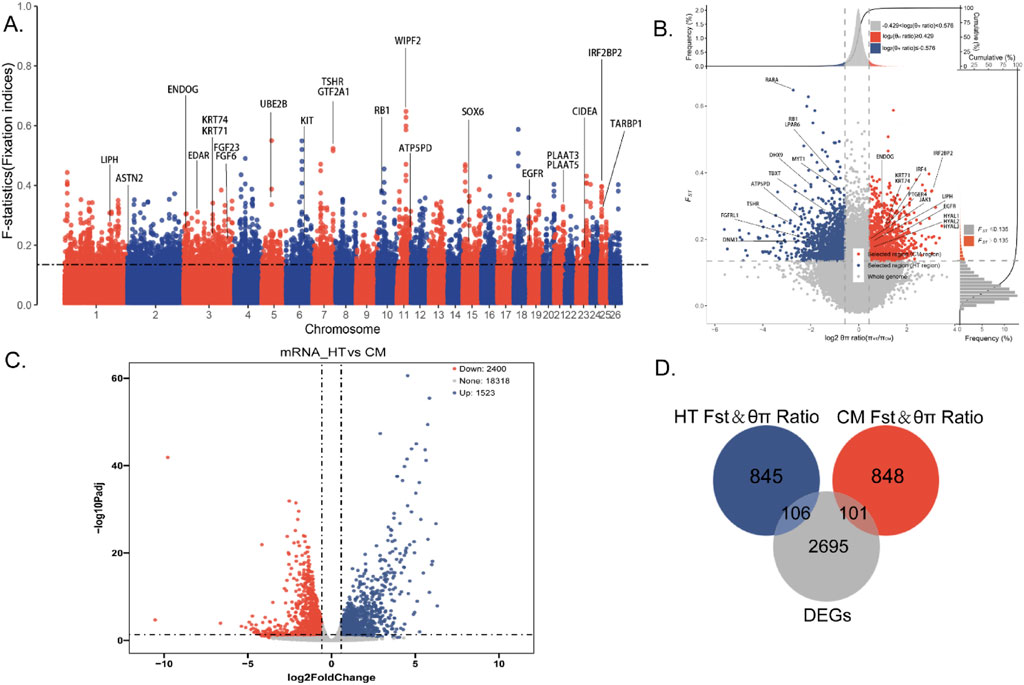
Figure 2. (A) Fst analysis of Manhattan (Fixation Indices: 0–0.05: genetic differentiation between populations is small and negligible. 0.05–0.15: Moderate genetic differentiation between populations. 0.15–0.25: Genetic differentiation between populations. >0.25: There is significant genetic differentiation between populations). (B) Fst and θπ ratio strategy selection signature analysis results are displayed (Note: The abscissa is the Log2π ratio, the ordinate is the Fst score, which corresponds to the frequency distribution map above and the frequency distribution map on the right, respectively, and the dot plot in the middle represents the corresponding Fst and Log2π ratio in different windows. The blue and redareas are the top 5% areas selected by Fst and Log2Pi ratio, blue represents Hetian sheep, and red represents Chinese merino sheep). (C) HT vs. CM mRNA gene level differential analysis results. (D) Venn diagram for Fst and θπ ratio and differential expression analysis (red: CM Fst and θπ ratio; blue: HT Fst and θπ ratio; grey: DEGs).
The combined selective sweep analysis of nucleotide diversity (θπ) and Fst yielded interesting results (Figure 2B; Supplementary Tables 7 and 8). The Fst and θπ ratio strategy revealed 949 genes in HT (Figure 2B, blue points) and 951 genes in CM (Figure 2B, red points). This analysis enabled complete differentiation of genes under selection pressure in both the HT and CM groups during growth.
RNA-seq data from the skin transcriptomes of 10 HT (experimental group) and 10 CM (control group) were compared, revealing a total of 2,902 differentially expressed genes (DEGs). Of these, 1,065 genes were upregulated and 1,837 were downregulated (Figure 2C; Supplementary Table 9).
A Venn diagram (Figure 2D) based on candidate genes from the Fst and θπ ratio and differential expression analysis identified 106 KFGs associated with single-coat fleece formation and 101 KFGs associated with double-coat fleece formation (Supplementary Table 10).
3.3 GO and KEGG enrichment analysis of KFGs
KFGs identified in the sheep genome and transcriptome using Fst and θπ ratio and differential expression analysis were further enriched for Gene Ontology (GO) and KEGG pathways.
For the double-coated fleece trait, 17 significant terms were identified (Figure 3A; Supplementary Table 11), including 16 KEGG pathways and 1 GO term (p < 0.05). Key pathways include Endocytosis (oas04144, PDGFRA, RABEP1, ARAP2, EPS15L1, RAB11FIP4), Toll-like receptor signaling (oas04620, MAP2K3, TICAM1, LY96), Cell cycle (oas04110, CCNB2, CDC6, RAD21), Cellular senescence (oas04218, MAP2K3, CCNB2, ITPR1), Pertussis (oas05133, LY96, TICAM1), Focal adhesion (oas04510, ITGA1, PDGFRA, FLNC), PD-L1 expression and PD-1 checkpoint pathway in cancer (oas05235, MAP2K3, TICAM1) and Proteoglycans in cancer (oas05205, FLNC, IQGAP1, ITPR1).
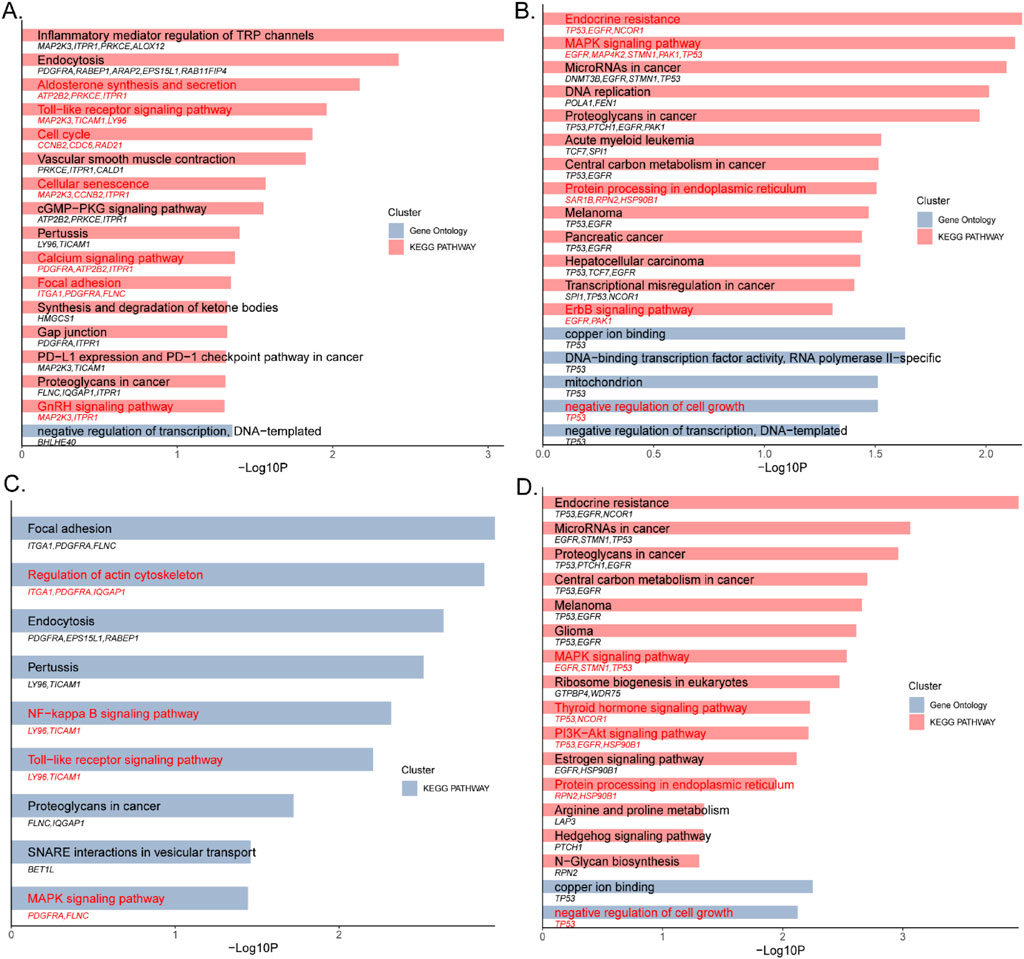
Figure 3. (A) The double-coated fleece trait (HT); (B) The single-coated fleece trait (CM); (C) The first interaction network of the double-coated fleece trait (HT); (D) The first interaction network of the single-coated fleece trait (CM).
For the single-coated fleece trait, 31 significant terms were identified (Figure 3B; Supplementary Table 12), including 25 KEGG pathways and 6 GO terms (p < 0.05). These included Endocrine resistance (oas01522, TP53, EGFR, NCOR1), MAPK signaling pathway (oas04010, EGFR, MAP4K2, STMN1, TP53), protein processing in the endoplasmic reticulum (oas04141, SAR1B, RPN2, HSP90B1), DNA replication (oas03030, POLA1, FEN1), ErbB signaling pathway (oas04012, EGFR, PAK1) and negative regulation of cell growth (GO: 0030308, TP53).
3.4 Protein protein interaction network of DEGs
To further investigate the relationships between KFGs, the selected KFGs were imported into the STRING database to construct the protein-protein interaction (PPI) network, and Cytoscape was used to visualize the network. The cytoHubba plugin (Chin et al., 2014) was applied for network topology and node centrality analysis, allowing the identification of hub genes and subnetworks through network algorithms.
The analysis revealed that 42 KFGs formed six distinct PPI networks associated with the double-coated fleece trait (Figure 4A; Supplementary Table 13). Key genes such as PDGFRA, RABEP1, EPS15L1, TICAM1, LY96, CCNB2, CDC6, RAD21, ITGA1, and FLNC were present in these networks. In the first reciprocal network, the KFGs were primarily enriched in pathways such as regulatory actin cytoskeleton, NF-kappa B signaling, Toll-like receptor signaling, and MAPK signaling (Figure 3C; Supplementary Table 14). Among these genes, there was an interaction relationship between the PDGFRA and EPS15L1, RABEP1 and EPS15L1, CCNB2 and CDC6, CCNB2 and RAD21, and ITGA1 and FLNC.
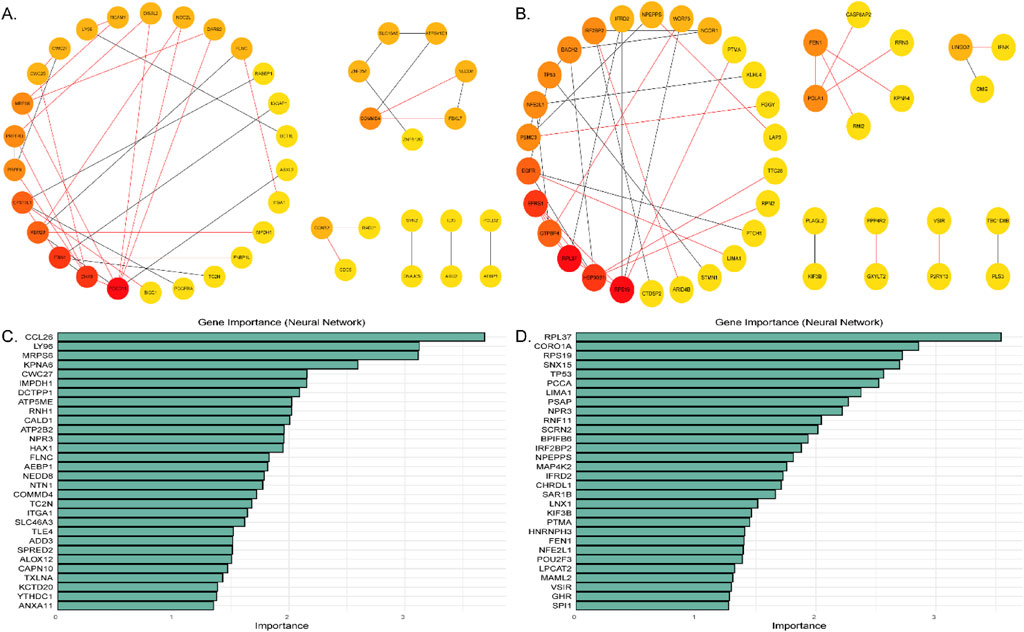
Figure 4. (A) Protein-protein interaction networks in the double-coated fleece trait. (B) Protein-protein interaction networks in the single-coated fleece trait. (C) Feature genes for double-coated fleece. (D) Feature genes for single-coated fleece. The abscissa coordinates show the importance of the features, sorted by the importance of the variables in descending order.
A total of 43 differentially expressed genes formed seven protein-protein interaction (PPI) networks for the single-coated fleece trait (Figure 4B; Supplementary Table 15). Key genes such as TP53, EGFR, NCOR1, STMN1, FEN1, PAK1, HSP90B1, and RPN2 were included in these networks. In the first reciprocal network, the KFGs were primarily enriched in pathways like MAPK signaling, thyroid hormone signaling, PI3K-Akt signaling, protein processing in the endoplasmic reticulum, and negative regulation of cell growth (Figure 3D; Supplementary Table 16). Among these genes, there was an interaction relationship between the TP53 and EGFR, EGFR and HSP90B1, EGFR and PTCH1, HSP90B1 and RPN2, and NCOR1 and IRF2BP2.
3.5 Machine learning analysis of key functional gene
Machine learning using the Neural Network (NNET) algorithm was applied to the 106 (single-coated fleece) and 101 (double-coated fleece) KFGs identified earlier, selecting the top 30 feature genes for each trait (Figures 4C, D). Among these, IRF2BP2, TP53, FEN1, ALOX12, LY96, FLNC, and ATP5ME were identified as highly important for wool growth traits and environmental adaptation. The results of transcriptome differential gene analysis also showed that the expression of these genes in HT and CM was extremely different (Figure 5). The performance of the constructed models was evaluated using tenfold cross-validation repeated ten times. Area Under the Curve (AUC) values were calculated based on Receiver Operating Characteristic (ROC) curves (Supplementary Figure S2). Detailed data used for the machine learning analysis is provided in Supplementary Tables 17 and 18.
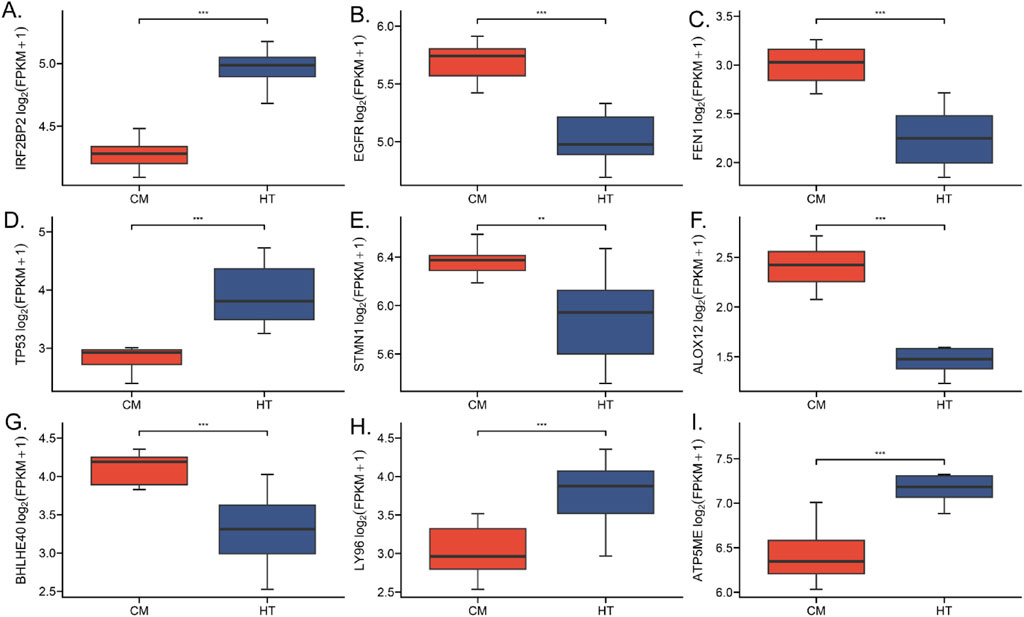
Figure 5. Expression levels of key genes associated with HT and CM coat types and wool growth traits. (A) IRF2BP2 gene. (B) EGFR gene. (C) FEN1 gene. (D) TP53 gene. (E) STMN1 gene. (F) ALOX12 gene. (G) BHLHE40 gene. (H) LY96 gene. (I) AT5ME gene.
4 Discussion
4.1 Enrichment analysis of KFGs in Chinese Merino and Hetian sheep
Enrichment analyses revealed that the KFGs enriched in Chinese Merino sheep were predominantly associated with pathways related to wool growth traits. In contrast, the KFGs enriched in Hetian sheep were linked not only to wool growth but also to adaptive traits. These differences reflect the distinct environments in which the two breeds have evolved. The Hetian sheep production region is characterized by a temperate continental desert climate with drought, high temperatures, and minimal rainfall. Such extreme climatic conditions, combined with long-term selective breeding, have resulted in the development of an exceptional local breed with remarkable drought and heat tolerance as well as high adaptability. On the other hand, the Chinese Merino sheep production region features a temperate continental climate with relatively mild ecological conditions. Decades of breeding efforts have focused on optimizing wool yield and fiber quality, particularly in terms of fineness, length, and crimp (Ciani et al., 2015; Wang et al., 2014).
In Chinese Merino sheep, 15 key functional genes (KFGs), including TP53, FEN1, EGFR, TCF7, HSP90B1, PTCH1, NCOR1, MAP4K2, STMN1, PAK1, DNMT3B, POLA1, SAR1B, RPN2, and SPI1, are involved in endocrine resistance, the MAPK signalling pathway, protein processing in the endoplasmic reticulum, the ErbB signalling pathway, and the negative regulation of cell growth, across 21 signalling pathways. These KFGs are likely to include potential regulators of wool growth in Chinese Merino sheep. Among these, the MAPK signalling pathway is well known for its close association with wool growth and development (Zhang et al., 2020; Öztürk et al., 2015; Sulayman et al., 2019; Lv et al., 2020). Endocrine resistance involves alterations in multiple signalling pathways and gene expression, with the ErbB pathway playing a key role in regulating cell proliferation and differentiation (Lu et al., 2021). Protein processing in the endoplasmic reticulum encompasses protein folding, modification, and transport (Potter and Nicchitta, 2002), processes that are critical for the proper formation of wool fibres (Yue et al., 2023). Additionally, the negative regulation of cell growth is essential for maintaining tissue homeostasis and preventing abnormal cell proliferation (Rousseau et al., 1996).
In Hetian sheep, 21 key functional genes (KFGs), including MAP2K3, ALOX12, ARAP2, ATP2B2, BHLHE40, CALD1, CCNB2, CDC6, EPS15L1, FLNC, HMGCS1, IQGAP1, ITGA1, ITPR1, LY96, PDGFRA, PRKCE, RAB11FIP4, RABEP1, RAD21, and TICAM1, are involved in aldosterone synthesis and secretion, the Toll-like receptor signalling pathway, the cell cycle, the calcium signalling pathway, focal adhesion, and the GnRH signalling pathway across 17 signalling pathways. These pathways are primarily associated with adaptation and wool growth in Hetian sheep. Aldosterone is an important corticosteroid that promotes sodium reabsorption and water retention, playing a critical role in the survival of Hetian sheep, which are adapted to saline soils (Bizzarri et al., 2016). The Toll-like receptor signalling pathway is vital to the immune system, with TLRs being central to inflammation, immune cell regulation, cell survival and proliferation, as well as adaptive immune responses by directing the differentiation of naïve T cells into effector T cells (Kulkarni et al., 2011; Kumar et al., 2024). The cell cycle pathway responds to external signals, such as growth factors, cytokines, and cell-to-cell contact, to regulate cell growth and division (Maddika et al., 2007). The GnRH signalling pathway is key in regulating the production and release of reproductive hormones, activating intracellular signalling cascades, and influencing both reproductive function and metabolic homeostasis (Kraus et al., 2001).
4.2 Genes associated with coat fleeced type in sheep
As early as 8,000 years ago, sheep were known to have a brown coat consisting of an outer layer of kemps (coarse hairs) that shed annually, along with a fine, woolly undercoat that also shed (Meadows et al., 2006). Over time, as food sources became more secure and sheep were domesticated, the characteristics of their wool evolved. It shifted from being coarse to fine, from short to long, and the coat fleece transitioned from double-layered to single-layered. In the present study, we identified two genes, IRF2BP2 and EGFR, that are associated with coat fleece type.
IRF2BP2 is involved in various cellular functions, including apoptosis, survival, and cell differentiation. It also plays a role in regulating the Hippo signaling pathway and acts as a tumor suppressor in hepatocellular carcinoma. Given the significant selection differences observed in the IRF2BP2 gene in our study, we propose that IRF2BP2 plays a pivotal role in the evolutionary transition from double-coated fleece to single-coated fleece. This is supported by previous studies, such as those by Demars et al. (2017); Lv et al. (2022); Sun et al. (2024), which identified mutations in IRF2BP2 that are linked to wool traits, further suggesting its role in fleece composition.
EGFR (epidermal growth factor receptor) is a member of the HER family, known for its crucial role in skin and hair development. Research has demonstrated that subcutaneous injection of EGF into neonatal mice can delay the development of hair follicles and the epidermis. In adult sheep, EGF injection not only inhibits hair fiber production and stimulates mitosis in basal epidermal cells, but also induces hair follicle degeneration. Further studies in mice have shown that EGFR deficiency leads to abnormal expression of LEF1, which causes differentiation disorders in medullary cells (Mak and Chan, 2003; Amberg et al., 2019). In Hetian sheep, the EGFR-LEF1 pathway may regulate the alternating appearance of myelinated and unmyelinated regions within certain hair follicle units, contributing to the distinctive structure of heterotypical hair.
4.3 Genes related to wool growth and adaptation
The TP53 gene encodes the p53 protein, a key regulator of cell division and cell death. It is involved in several signaling pathways directly related to wool growth, including the MAPK, Wnt, and PI3K-Akt pathways (Amberg et al., 2019; Chamcheu et al., 2019). Another critical gene, FEN1, encodes a flap endonuclease-1 enzyme that plays a central role in maintaining genome stability and replication. FEN1 interacts with various proteins required for genome stability. In highly proliferative tissues such as bone marrow, testis, and thymus, FEN1 expression is notably high. Studies of fusion gene expression in the epidermis (skin explants) of adult Fen1y/y mice revealed that proliferating keratin-forming cells were confined to the basal lamina and peri-follicular rondelles, suggesting that FEN1 is crucial for hair follicle cell proliferation and hair growth (Kleppa et al., 2012). Stathmin 1 (STMN1) is a ubiquitously expressed cytosolic phosphoprotein involved in integrating diverse intracellular signaling pathways that control cell proliferation, differentiation, and activity. Depletion of Stmn1 leads to increased apoptotic death, accelerated degenerative transformation, and premature inhibition of hair follicle proliferation, highlighting its critical role in the hair follicle cycle (Yang et al., 2022; Zhang et al., 2021; Bichsel et al., 2016). Achidonate 12-lipoxygenase (ALOX12) is downregulated in senescent hair follicles. Its inhibition can prevent the breakdown and conversion of arachidonic acid in hair follicles, thereby promoting stratum corneum maturation (Zheng et al., 2020). BHLHE40 Basic Helix-Loop-Helix Family Member E40 plays a pivotal role in cell differentiation. Knockdown of BHLHE40 impairs the ability of epidermal cells to regenerate hair follicles (Wang et al., 2024). LY96, also known as MD2, is a small glycoprotein expressed by macrophages and dendritic cells. It is functionally related to Toll-like receptor 4 (TLR4) and plays a significant role in innate immunity by participating in the Toll-like receptor signaling pathway (Cao and Yang, 2024). ATP5ME, a subunit of ATP synthase, is involved in cellular energy metabolism and thermogenesis (Zhuang et al., 2023).
5 Conclusion
In summary, our research offers valuable insights into the regulatory mechanisms underlying wool growth, coat fleece type, and adaptability in wool growth observed in Hetian sheep. By comparing genetic differences between Hetian and Chinese Merino sheep, we identified both novel and previously reported candidate genes through selective sweep analysis, transcriptome differences, and machine learning approaches. In the Chinese Merino sheep population, we identified candidate genes associated with wool growth (TP53, FEN1, and STMN1) and coat fleece type (IRF2BP2 and EGFR). In the Hetian sheep population, we identified key functional genes (KFGs) related to wool growth (ALOX12, BHLHE40, RICTOR, and PIP4K2A) and environmental adaptation (LY96 and ATP5ME). Further research is required to fully elucidate the complex interactions between these KFGs and to develop effective strategies for their application in sheep breeding programs.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal studies were approved by Experimental animal Ethics Committee, Institute of Animal Science, Xinjiang Academy of Animal Science. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
YZ: Conceptualization, Software, Writing – original draft. WL: Data curation, Supervision, Writing – review and editing. XX: Investigation, Resources, Supervision, Writing – review and editing. MX: Investigation, Writing – review and editing. LT: Investigation, Writing – review and editing. PZ: Investigation, Writing – review and editing. NS: Investigation, Writing – review and editing. LY: Resources, Writing – review and editing. JD: Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We would like to thank the National Key R&D Program of China (NO: 2021YFD1200901), the Key Laboratory of Xinjiang Autonomous Region (NO: 2023D04080), and the Tianshan Innovation Team Program (NO: 2022D14018) for funding this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1582244/full#supplementary-material
References
Amberg, N., Sotiropoulou, P. A., Heller, G., Lichtenberger, B. M., Holcmann, M., Camurdanoglu, B., et al. (2019). EGFR controls hair shaft differentiation in a p53-independent manner. iScience 15, 243–256. doi:10.1016/j.isci.2019.04.018
Banos, G., Bramis, G., Bush, S., Clark, E., McCulloch, M. E., Smith, J., et al. (2017). The genomic architecture of mastitis resistance in dairy sheep. BMC Genomics 18, 624–18. doi:10.1186/s12864-017-3982-1
Bichsel, K. J., Hammiller, B., Trempus, C. S., Li, Y., and Hansen, L. A. J. E. D. (2016). The epidermal growth factor receptor decreases S tathmin 1 and triggers catagen entry in the mouse. Exp. Dermatol. 25, 275–281. doi:10.1111/exd.12921
Bizzarri, C., Pedicelli, S., Cappa, M., and Cianfarani, S. J. H. (2016). Water balance and ‘salt wasting’ in the first year of life: the role of aldosterone-signaling defects. Horm. Res. Paediatr. 86, 143–153. doi:10.1159/000449057
Bu, D., Luo, H., Huo, P., Wang, Z., Zhang, S., He, Z., et al. (2021). KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325. doi:10.1093/nar/gkab447
Cao, J., and Yang, Z. J. C. (2024). FOXA2 inhibits the TLR4/NF-κB signaling pathway and alleviates inflammatory activation of macrophages in rheumatoid arthritis by repressing LY96 transcription. Cytokine 184, 156796. doi:10.1016/j.cyto.2024.156796
Chamcheu, J. C., Roy, T., Uddin, M. B., Banang-Mbeumi, S., Chamcheu, R.-C. N., Walker, A. L., et al. (2019). Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: a review of current status and future trends on natural and synthetic agents therapy. Cells 8, 803. doi:10.3390/cells8080803
Chen, X., Sun, S., and Sulaiman, Y. J. P. (2024). Analysis of differential expression of hair follicle tissue transcriptome in Hetian sheep undergoing different periodic changes. PeerJ 12, e18542. doi:10.7717/peerj.18542
Chin, C.-H., Chen, S.-H., Wu, H.-H., Ho, C.-W., Ko, M.-T., and Lin, C.-Y. J. B. (2014). cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11–S17. doi:10.1186/1752-0509-8-S4-S11
Ciani, E., Lasagna, E., D’andrea, M., Alloggio, I., Marroni, F., Ceccobelli, S., et al. (2015). Merino and merino-derived sheep breeds: a genome-wide intercontinental study. Genet. Sel. Evol. 47, 64. doi:10.1186/s12711-015-0139-z
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi:10.1093/bioinformatics/btr330
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10, giab008. doi:10.1093/gigascience/giab008
Demars, J., Cano, M., Drouilhet, L., Plisson-Petit, F., Bardou, P., Fabre, S., et al. (2017). Genome-wide identification of the mutation underlying fleece variation and discriminating ancestral hairy species from modern woolly sheep. Mol. Biol. Evol. 34, 1722–1729. doi:10.1093/molbev/msx114
Farhadi, S., Hasanpur, K., Ghias, J. S., Palangi, V., Maggiolino, A., and Landi, V. J. A. (2023). Comprehensive gene expression profiling analysis of adipose tissue in male individuals from fat-and thin-tailed sheep breeds. Animals 13, 3475. doi:10.3390/ani13223475
Guo, Y., Li, S., Na, R., Guo, L., Huo, C., Zhu, L., et al. (2024). Comparative transcriptome analysis of bovine, porcine, and sheep muscle using interpretable machine learning models. Animals 14, 2947. doi:10.3390/ani14202947
Hamadani, A., Ganai, N. A., Mudasir, S., Shanaz, S., Alam, S., and Hussain, I. J. S. R. (2022). Comparison of artificial intelligence algorithms and their ranking for the prediction of genetic merit in sheep. Sci. Rep. 12, 18726. doi:10.1038/s41598-022-23499-w
Han, Z., Zhou, W., Zhang, L., Wang, R., Liu, C., Bai, X., et al. (2023). Genetic diversity and runs of homozygosity analysis of Hetian sheep populations revealed by Illumina Ovine SNP50K BeadChip. Front. Ecol. Evol. 11. doi:10.3389/fevo.2023.1182966
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. J. N. b. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
Kirchner, K., Tölle, K.-H., and Krieter, J. (2004). The analysis of simulated sow herd datasets using decision tree technique. Computers and Electronics in Agriculture 42, 111–127. doi:10.1016/S0168-1699(03)00119-4
Kleppa, L., Mari, P.-O., Larsen, E., Lien, G. F., Godon, C., Theil, A. F., et al. (2012). Kinetics of endogenous mouse FEN1 in base excision repair. Nucleic Acids Res. 40, 9044–9059. doi:10.1093/nar/gks673
Kraus, S., Naor, Z., and Seger, R. J. A. (2001). Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch. Med. Res. 32, 499–509. doi:10.1016/s0188-4409(01)00331-9
Kuhn, M. (2008). Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26. doi:10.18637/jss.v028.i05
Kulkarni, R., Behboudi, S., and Sharif, S. J. C. (2011). Insights into the role of Toll-like receptors in modulation of T cell responses. Cell Tissue Res. 343, 141–152. doi:10.1007/s00441-010-1017-1
Kumar, R., Meena, A., Baraiya, T., Swarnkar, C., Misra, S., and Kumar, A. J. V. I. (2024). Expression of Toll-like receptors in Haemonchus Contortus resistant sheep: an innate immune parameter for host defense against gastrointestinal nematode infection. Immunopathology 275, 110813. doi:10.1016/j.vetimm.2024.110813
Li, C., Chen, B., Langda, S., Pu, P., Zhu, X., Zhou, S., et al. (2024). Multi-omic analyses shed light on the genetic control of high-altitude adaptation in sheep. Proteomics and Bioinforma. 22, qzae030. doi:10.1093/gpbjnl/qzae030
Lu, Q., Gao, Y., Fan, Z., Xiao, X., Chen, Y., Si, Y., et al. (2021). Amphiregulin promotes hair regeneration of skin-derived precursors via the PI3K and MAPK pathways. Cell Prolif. 54, e13106. doi:10.1111/cpr.13106
Lv, F.-H., Cao, Y.-H., Liu, G.-J., Luo, L.-Y., Lu, R., Liu, M.-J., et al. (2022). Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, and agronomically important loci. Mol. Biol. Evol. 39, msab353. doi:10.1093/molbev/msab353
Lv, X., Chen, W., Sun, W., Hussain, Z., Wang, S., and Wang, J. J. A. (2020). Analysis of lncRNAs expression profiles in hair follicle of Hu sheep lambskin. Animals 10, 1035. doi:10.3390/ani10061035
Maddika, S., Ande, S. R., Panigrahi, S., Paranjothy, T., Weglarczyk, K., Zuse, A., et al. (2007). Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist. Updat. 10, 13–29. doi:10.1016/j.drup.2007.01.003
Mak, K. K., and Chan, S. Y. J. (2003). Epidermal growth factor as a biologic switch in hair growth cycle. J. Biol. Chem. 278, 26120–26126. doi:10.1074/jbc.M212082200
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Meadows, J., Hanotte, O., Drögemüller, C., Calvo, J., Godfrey, R., Coltman, D., et al. (2006). Globally dispersed Y chromosomal haplotypes in wild and domestic sheep. Anim. Genet. 37, 444–453. doi:10.1111/j.1365-2052.2006.01496.x
Öztürk, Ö. A., Pakula, H., Chmielowiec, J., Qi, J., Stein, S., Lan, L., et al. (2015). Gab1 and Mapk signaling are essential in the hair cycle and hair follicle stem cell quiescence. Cell Rep. 13, 561–572. doi:10.1016/j.celrep.2015.09.015
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi:10.1038/nbt.3122
Piles, M., Fernandez-Lozano, C., Velasco-Galilea, M., González-Rodríguez, O., Sánchez, J. P., Torrallardona, D., et al. (2019). Machine learning applied to transcriptomic data to identify genes associated with feed efficiency in pigs. Genet. Sel. Evol. 51, 10–15. doi:10.1186/s12711-019-0453-y
Potter, M. D., and Nicchitta, C. V. (2002). Endoplasmic reticulum-bound ribosomes reside in stable association with the translocon following termination of protein synthesis. J. Biol. Chem. 277, 23314–23320. doi:10.1074/jbc.M202559200
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi:10.1093/bioinformatics/btp616
Rousseau, D., Gingras, A.-C., Pause, A., and Sonenberg, N. J. O. (1996). The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene 13, 2415–2420. doi:10.1016/S0014-5793(96)01321-X
Shahinfar, S., Kelman, K., and Kahn, L. J. (2019). Prediction of sheep carcass traits from early-life records using machine learning. Comput. Electron. Agric. 156, 159–177. doi:10.1016/j.compag.2018.11.021
Shi, R., Li, S., Liu, P., Guo, L., Gong, S., and Wan, Y. J. (2022a). Effects of testosterone on skin structure and factors related to androgen conversion and binding in Hetian sheep. Trop. Anim. Health Prod. 54, 218. doi:10.1007/s11250-022-03216-5
Shi, R., Li, S., Liu, P., Zhang, S., Wu, Z., Wu, T., et al. (2022b). Identification of key genes and signaling pathways related to Hetian sheep wool density by RNA-seq technology. PLoS One 17, e0265989. doi:10.1371/journal.pone.0265989
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. doi:10.1093/bioinformatics/btq675
Sulayman, A., Tian, K., Huang, X., Tian, Y., Xu, X., Fu, X., et al. (2019). Genome-wide identification and characterization of long non-coding RNAs expressed during sheep fetal and postnatal hair follicle development. Sci. Rep. 9, 8501. doi:10.1038/s41598-019-44600-w
Sun, X., Guo, J., Li, R., Zhang, H., Zhang, Y., Liu, G. E., et al. (2024). Whole-genome resequencing reveals genetic diversity and wool trait-related genes in liangshan semi-fine-wool sheep. Animals 14, 444. doi:10.3390/ani14030444
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., et al. (2023). The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. doi:10.1093/nar/gkac1000
Tırınk, C., Piwczyński, D., Kolenda, M., and Önder, H. J. A. (2023). Estimation of body weight based on biometric measurements by using random forest regression, support vector regression and CART algorithms. Animals (Basel) 13, 798. doi:10.3390/ani13050798
Villanueva, R. A. M., and Chen, Z. J. (2019). ggplot2: Elegant graphics for data analysis. Measurement: Interdisciplinary Research and Perspectives 17 (3), 160–167. doi:10.1080/15366367.2019.1565254
Wang, D., Jiang, J., Wang, M., Li, K., Liang, H., Wang, N., et al. (2024). Mitophagy promotes hair regeneration by activating glutathione metabolism. Research (Wash D C) 7, 0433. doi:10.34133/research.0433
Wang, H., Li, S., Wu, T., Wu, Z., and Guo, J. (2021). The effect of androgen on wool follicles and keratin production in Hetian sheep. 81, 526–536. doi:10.1590/1519-6984.224056
Wang, J., Fu, Y., Su, T., Wang, Y., Soladoye, O. P., Huang, Y., et al. (2023). A role of multi-omics technologies in sheep and goat meats: progress and way ahead. Foods 12, 4069. doi:10.3390/foods12224069
Wang, J., Lin, H., Wang, Q., Wu, Y., and Zhang, C. J. F. C. (2025). 4D DIA proteomic analysis of gender and age influences on meat quality and flavor in Hetian white sheep. Food Chem. 464, 141851. doi:10.1016/j.foodchem.2024.141851
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wang, S., Li, F., Liu, J., Zhang, Y., Zheng, Y., Ge, W., et al. (2020). Integrative analysis of methylome and transcriptome reveals the regulatory mechanisms of hair follicle morphogenesis in cashmere goat. Cells 9, 969. doi:10.3390/cells9040969
Wang, Z., Zhang, H., Yang, H., Wang, S., Rong, E., Pei, W., et al. (2014). Genome-wide association study for wool production traits in a Chinese Merino sheep population. PLoS One 9, e107101. doi:10.1371/journal.pone.0107101
Xu, Y.-X., Wang, B., Jing, J.-N., Ma, R., Luo, Y.-H., Li, X., et al. (2023). Whole-body adipose tissue multi-omic analyses in sheep reveal molecular mechanisms underlying local adaptation to extreme environments. Commun. Biol. 6, 159. doi:10.1038/s42003-023-04523-9
Yang, P., Zou, Z., and Gao, X. J. B. (2022). STMN1 promotes the proliferation and inhibits the apoptosis of acute myeloid leukemiacells by activating the PI3K/Akt pathway. Biocell 46, 207–218. doi:10.32604/biocell.2021.014728
Yue, L., Lu, Z., Guo, T., Liu, J., Yang, B., and Yuan, C. J. I. J. o. M. S. (2023). Proteome analysis of alpine merino sheep skin reveals new insights into the mechanisms involved in regulating wool fiber diameter. Int. J. Mol. Sci. 24, 15227. doi:10.3390/ijms242015227
Zhang, C., Li, Y., Qin, J., Yu, C., Ma, G., Chen, H., et al. (2021). TMT-based quantitative proteomic analysis reveals the effect of bone marrow derived mesenchymal stem cell on hair follicle regeneration. Front. Pharmacol. 12, 658040. doi:10.3389/fphar.2021.658040
Zhang, Y., Wu, K., Wang, L., Wang, Z., Han, W., Chen, D., et al. (2020). Comparative study on seasonal hair follicle cycling by analysis of the transcriptomes from cashmere and milk goats. Genomics 112, 332–345. doi:10.1016/j.ygeno.2019.02.013
Zhao, B., Luo, H., He, J., Huang, X., Chen, S., Fu, X., et al. (2021). Comprehensive transcriptome and methylome analysis delineates the biological basis of hair follicle development and wool-related traits in Merino sheep. BMC Biol. 19, 197–18. doi:10.1186/s12915-021-01127-9
Zheng, Z., Li, Y., Jin, G., Huang, T., Zou, M., and Duan, S. J. B. (2020). The biological role of arachidonic acid 12-lipoxygenase (ALOX12) in various human diseases. Biomed Pharmacother. 129, 110354. doi:10.1016/j.biopha.2020.110354
Keywords: Hetian sheep, Chinese merino sheep, coat fleece type, multi-omics, machine learning
Citation: Zhang Y, Li W, Xu X, Xie M, Tang L, Zheng P, Song N, Yu L and Di J (2025) Machine learning driven multi-omics analysis of the genetic mechanisms behind the double-coat fleece formation in Hetian sheep. Front. Genet. 16:1582244. doi: 10.3389/fgene.2025.1582244
Received: 24 February 2025; Accepted: 30 May 2025;
Published: 11 June 2025.
Edited by:
Zhe Zhang, South China Agricultural University, ChinaReviewed by:
Herman Revelo, Fundación Universitaria San Martín, ColombiaXiaolong Li, Lanzhou University, China
Copyright © 2025 Zhang, Li, Xu, Xie, Tang, Zheng, Song, Yu and Di. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Yu, eWxqeGluamlhbmcxMjBAc2luYS5jb20=; Jiang Di, ZGlqaWFuZzY5QDE2My5jb20=