 Xinpeng Guo
Xinpeng Guo Junrong Guo
Junrong Guo Xijing Liu
Xijing Liu Ting Hu
Ting Hu- 1Department of Medical Genetics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 2West China School of Medicine, Sichuan University, Chengdu, Sichuan, China
- 3Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, Sichuan, China
Schizophrenia is a complex neuropsychiatric disorder closely associated with genetic factors. Copy number variations (CNVs) play a key role in the genetic etiology of schizophrenia, with the distal 1q21.1 microdeletion identified as a rare CNV that serves as a significant genetic risk factor for the disorder. This microdeletion is found in 0.2%–0.6% of individuals with schizophrenia and is associated with an eightfold increased risk of developing the condition. The distal 1q21.1 region contains several schizophrenia risk genes, including PRKAB2, BCL9, CHD1L, GJA5, and GJA8. This review focuses on the roles of these five genes in brain function and explores their potential pathophysiological mechanisms in schizophrenia. By synthesizing current evidence, this review aims to deepen the understanding of schizophrenia by outlining its genetic architecture and molecular mechanisms, thereby providing a comprehensive framework for exploring disease pathogenesis.
1 Introduction
Schizophrenia is a severe mental disorder characterized primarily by hallucinations, delusions, cognitive impairments, and disorganized speech and behavior (Birnbaum and Weinberger, 2017). It is a multifactorial disease with a high hereditary component, with an estimated heritability of approximately 80% (Legge et al., 2021). Schizophrenia not only affects the diagnosed individual but also presents significant challenges to families and society, affecting about 1% of the global population (Birnbaum and Weinberger, 2017; Smeland and Andreassen, 2022). Despite extensive studies, its pathogenesis remains unclear. However, recent advances in genetic research have highlighted the crucial role of genetic factors in the risk of schizophrenia. In the 1980s (Sherrington et al., 1988), first suggested a potential link between schizophrenia and a specific chromosome (chromosome 5) through familial linkage studies. The advent of genome-wide association studies (GWAS) by the early 21st century marked significant progress, enabling the identification of numerous susceptibility loci associated with the disorder (O'Donovan et al., 2008; Iyegbe and O'Reilly, 2022; Biological insights from 108 schizophrenia, 2014). These findings underscore that schizophrenia, better conceptualized as a syndrome or a spectrum rather than a single disease entity, is associated with multiple genetic loci, reflecting its biological and clinical heterogeneity.
In recent years, emerging evidence has highlighted that CNVs contribute to the increased risk of schizophrenia (Rare chromosomal deletions and duplications, 2008; Bergen et al., 2019). Numerous epidemiological studies have determined that CNVs are closely associated with the genetic etiology of schizophrenia (Mollon et al., 2023; Rees et al., 2011; Rees et al., 2016). Among the known genetic alterations in schizophrenia, CNVs are one of the most common pathogenic changes. Marshall et al. (2017) conducted a genome-wide study with 41,321 participants, including 21,094 schizophrenia cases and 20,227 controls. The study identified a significant increase in CNV burden in schizophrenia, particularly at the 1q21.1 region (OR = 1.11, P = 5.7 × 10−15), even after excluding previously identified risk loci. The 1q21.1 region harbors several genes involved in synaptic function and neurodevelopment. The increased CNV burden in this region may disrupt the normal function of these genes, affecting neurodevelopment and synaptic function, thereby elevating the risk of schizophrenia. Among rare CNVs, the distal 1q21.1 microdeletion has been identified as a significant genetic risk factor for schizophrenia, occurring in 0.2%–0.6% of schizophrenia cases (Marshall et al., 2017; Levinson et al., 2011; Guo et al., 1993). Furthermore, individuals with 1q21.1 microdeletions have an eightfold increased risk of schizophrenia, further highlighting the significant association between distal deletions at 1q21.1 and schizophrenia (Rare chromosomal deletions and duplications, 2008; Stefansson et al., 2008). This review summarizes the evidence linking the distal 1q21.1 microdeletion to schizophrenia and outlines the mechanisms of action of five genes (PRKAB2, CHD1L, GJA5, GJA8, BCL9) in the nervous system and their potential relationship with schizophrenia.
2 Chromosomal structure and genetic mechanisms of the 1q21.1
Chromosomal region 1q21.1 is a structurally complex genomic locus, characterized by abundance of low-copy repeats, which makes it highly prone to non-allelic homologous recombination (Malhotra and Sebat, 2012). This susceptibility leads to recurrent chromosomal deletions and duplications. The region harbors four primary segmental duplication clusters, termed breakpoints (BP1-BP4), which can be classified into two distinct regions: the proximal (BP2-BP3) and distal (BP3-BP4) regions (Brunetti-Pierri et al., 2008; Rosenfeld et al., 2012). The distal 1q21.1 microdeletion refers to a deletion of approximately 0.8 Mb between BP3 and BP4 (located at approximately 147.1–147.9 Mb, hg38). This region contains at least seven unique protein-coding genes, including PRKAB2, CHD1L, FMO5, BCL9, ACP6, GJA5, and GJA8 (Edwards et al., 2021) (Figure 1). According to data from the UK Biobank (https://www.ukbiobank.ac.uk), the prevalence of distal 1q21.1 microdeletions in general population is approximately 0.027% (Kendall et al., 2017). Among individuals with developmental delay, intellectual disability, and/or congenital malformations, the detection rate of distal 1q21.1 microdeletions is approximately 0.2% (Guo et al., 1993). This microdeletion is usually inherited in an autosomal dominant manner, with inheritance possible from either parent. Carriers may exhibit either normal or abnormal phenotypes. Additionally, approximately 18%–35% of cases are de novo (Guo et al., 1993). The clinical presentation of 1q21.1 distal microdeletions shows considerable variability, with incomplete penetrance (Edwards et al., 2021; Upadhyai et al., 2020). Affected individuals may be asymptomatic or may present with a range of neurodevelopmental disorders, including developmental delay, autism spectrum disorder (ASD), schizophrenia, and attention-deficit hyperactivity disorder (Rare chromosomal deletions and duplications, 2008; Guo et al., 1993; Mefford et al., 2008; Busè et al., 2017; Bucan et al., 2009; Gudmundsson et al., 2019; Bernier et al., 2016). Furthermore, this microdeletion has been associated with other features such as cataracts, microcephaly, congenital heart defects, ocular anomalies, seizures, and renal abnormalities (Guo et al., 1993; Brunetti-Pierri et al., 2008; Mefford et al., 2008; Busè et al., 2017; Bernier et al., 2016; Christiansen et al., 2004; Costain et al., 2016; Weber et al., 2011).
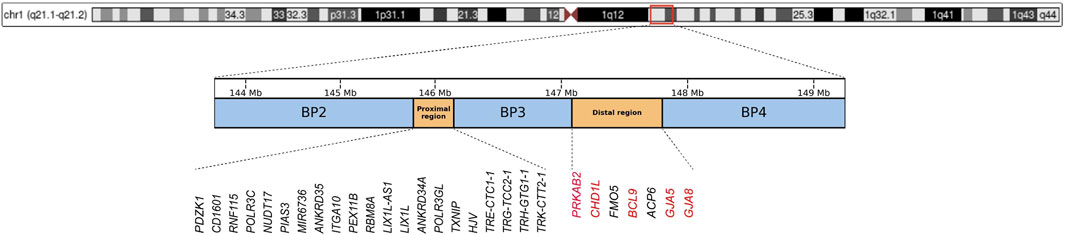
Figure 1. Chromosomal structure of the 1q21.1-1q21.2 region (hg38), with blue boxes representing the three breakpoint regions (BP2, BP3, and BP4), and orange boxes indicating the Proximal and Distal regions. Genes within the Proximal and Distal regions are annotated below, with schizophrenia-associated risk genes in the Distal region highlighted in red.
3 Impact of distal 1q21.1 microdeletion on brain structural development
Sønderby et al. (2021) conducted a large-scale neuroimaging meta-analysis, revealing dose-dependent effects on brain structures in carriers of distal 1q21.1 microdeletions, which were accompanied by cognitive deficits. The study demonstrated that distal 1q21.1 microdeletions exert a positive dose-dependent effect on intracranial volume and total cortical surface area, particularly in the frontal and cingulate cortices, while negatively affecting the caudate nucleus and hippocampus volume. These findings link distal 1q21.1 microdeletions to structural brain changes, suggesting that this genetic variation may increase the risk of neuropsychiatric disorders by affecting structural changes during brain development. Szecówka et al. (2023) further demonstrated that individuals at high risk for schizophrenia exhibited reduced subcortical volumes, including the hippocampus and thalamus, supporting the hypothesis that volumetric changes in brain structures are associated with psychiatric disorder risk. Notably, alterations in key brain regions, particularly the hippocampus, may play a crucial role in schizophrenia pathogenesis. The hippocampus, a critical region for memory and emotional regulation, has been shown to exhibit significant structural changes in individuals with schizophrenia, including reduced hippocampal volume, which is associated with cognitive deficits and emotional dysregulation (Peterson et al., 2023; Sasabayashi et al., 2021; Wegrzyn et al., 2022). Consistent with previous study, van (van Erp et al., 2016) reported significant reductions in subcortical structure volumes in patients carrying schizophrenia-associated CNVs, including 1q21.1 microdeletions/microduplications and 22q11.2 deletion syndrome, compared to CNV carriers without schizophrenia. These findings suggest that distal 1q21.1 microdeletions not only disrupt normal brain development but also contribute to abnormal volumetric changes in specific brain regions, further emphasizing the role of distal 1q21.1 microdeletions in the risk of neuropsychiatric disorders. Additionally, Boen et al. (2024) identified region-specific effects beyond overall brain measurement differences in distal 1q21.1 microdeletions. Collectively, these findings indicate that distal 1q21.1 microdeletions contribute to schizophrenia by altering early brain structure development, thereby supporting the hypothesis that the distal 1q21.1 microdeletion is significant genetic risk factors for schizophrenia.
Induced pluripotent stem cell (iPSC) models have been utilized to examine the impact of distal 1q21.1 microdeletions on neuronal proliferation, differentiation, maturation, and synaptic function, revealing disruptions in early neurodevelopment, potentially associated with an increase in lower-layer cortical neurons. These findings were corroborated in mouse models carrying the 1q21.1 deletion (Chapman et al., 2022). In particular, (Nielsen et al., 2017) demonstrated that distal 1q21.1 microdeletions contribute to schizophrenia pathophysiology by affecting dopaminergic signaling in the Df (h1q21)/+ mouse model. The Df (h1q21)/+ mice exhibited several schizophrenia-like behaviors, including hyperactivity in response to amphetamine, impaired prepulse inhibition (PPI), and altered dopamine receptor sensitivity, which are consistent with the positive symptoms seen in human schizophrenia. Further, Gordon et al. (2021) found a general downregulation of mitochondrial-related gene expression in neurons from 1q21.1 microdeletion mice, consistent with transcriptomic alterations observed in postmortem brains of individuals with schizophrenia and ASD, suggesting neuronal energy dysfunction plays a crucial role in schizophrenia pathogenesis. Overall, the results from iPSC models, murine models, and transcriptomic analyses highlight the critical role of distal 1q21.1 microdeletions in disrupting neuronal development, synaptic function, and energy metabolism, contributing to the pathophysiology of neurodevelopmental disorders involving schizophrenia. These findings further support distal 1q21.1 microdeletions as genetic risk factors for schizophrenia and related neurodevelopmental conditions.
4 Risk genes within distal 1q21.1 region involving in schizophrenia
4.1 PRKAB2
PRKAB2 (OMIM: 602741) encodes the β2 subunit of the AMP-activated protein kinase (AMPK) complex, a highly conserved heterotrimeric kinase critical for cellular energy metabolism and acting as an essential energy sensor within cells (Ronnett et al., 2009; Hardie et al., 2012; Hardie, 2007; Zhou et al., 2022). Specifically, in the nervous system, AMPK regulates energy metabolism, provides neuroprotection, modulates neuronal conduction, and participates in neural development, all of which are vital for maintaining normal neuronal function (Brunetti-Pierri et al., 2008; Muraleedharan and Dasgupta, 2022; Bobela et al., 2017; Shah et al., 2017; Yang et al., 2022; Belforte et al., 2021). Moreover, the functional significance of AMPK in the central nervous system is further supported by its suppressive effect on the mTOR signaling pathway, a key regulatory axis involved in synaptic plasticity, learning processes, and memory formation (Harvard et al., 2011; Wang and Jia, 2023). AMPK regulates glucose metabolism in the brain by modulating glucose uptake, glycolysis, and glycogen metabolism, thereby maintaining energy homeostasis and proper neuronal function (Muraleedharan and Dasgupta, 2022; Dienel, 2019; Muraleedharan et al., 2020). Bioenergetic dysfunction, including aberrant insulin signaling and impaired glucose homeostasis, has been implicated in schizophrenia pathophysiology, contributing to core clinical symptoms (Henkel et al., 2022; Bryll et al., 2020; Agarwal et al., 2020). In this context, evidence shows that a single copy deletion of PRKAB2 reduces the expression of AMPK-β2, thereby impairing AMPK activation. Conversely, duplication of PRKAB2 does not adversely affect AMPK activity (Harvard et al., 2011). Further, Nagy et al. (2018) have demonstrated that a deficiency of PRKAB2 and AMPK complex activity in the nervous system of a fruit fly model leads to decreased learning ability and severe sleep disturbances. These findings are particularly relevant because cognitive impairments and changes in sleep patterns are common symptoms of schizophrenia. Thus, PRKAB2 may increase the risk of developing this condition. Furthermore, deficiency in AMPK can shorten lifespan and cause abnormalities in neuronal dendritic structures, a phenotype also associated with schizophrenia (Nagy et al., 2018). Interestingly, Ng et al. (2012) discovered that activation of AMPK effectively alleviates dopaminergic signaling deficits and mitochondrial dysfunction in Drosophila models of Parkinson’s disease. However, this protective effect is lost when AMPK is inactivated. In a comprehensive approach, (Wagh et al., 2021) through integrated analysis of various studies, identified 227 differentially expressed genes (DEGs) between schizophrenia patients and controls, demonstrating that PRKAB2 significantly appears among these DEGs, showing genetic and epigenetic changes associated with schizophrenia. Additionally, it indicates that polymorphisms in PRKAB2 are associated with weight gain in patients with schizophrenia or affective disorders treated with antipsychotic drugs (chlorpromazine or olanzapine) (Souza et al., 2012). Based on these findings, PRKAB2 plays a pivotal role in energy metabolism and the maintenance of normal function within the nervous system. Its genetic deficiency may impair AMPK activation, thereby disrupting cerebral energy metabolism and ultimately contributing to cognitive deficits and sleep disturbances, which are common symptoms associated with schizophrenia. Therefore, the haploinsufficiency of PRKAB2 may be closely linked to the onset and progression of schizophrenia.
4.2 BCL9
BCL9 (OMIM: 602597), located at 1q21.1, is a schizophrenia susceptibility gene that encodes a nuclear retention factor for β-catenin. It plays a crucial role in the Wnt signaling pathway, a key regulatory mechanism in neural development that modulates the proliferation, migration, and differentiation of neural stem cells. Additionally, this pathway is essential for maintaining neuroplasticity and promoting neurogenesis (de la Roche et al., 2008; Yoon and Mao, 2021; Adachi et al., 2004; Hoseth et al., 2018; Kuwabara et al., 2009; Zechner et al., 2003). Wnt signaling is activated during neural development and is crucial for synaptic plasticity in the adult brain (Jensen et al., 2012; He et al., 2018). The neurodevelopmental hypothesis of schizophrenia implicates disturbances in this pathway in the pathogenesis of schizophrenia (Inestrosa et al., 2012; Panaccione et al., 2013; Okerlund and Cheyette, 2011). A GWAS covering the Han Chinese population, including 5,772 controls and 4,187 schizophrenia patients, revealed multiple single nucleotide polymorphisms (SNPs) located in the BCL9 gene that are significantly associated with schizophrenia (Li et al., 2011). Additionally, another GWAS analyzing data from 1,774 European and American schizophrenia patients and 2,726 controls identified three SNPs in the BCL9 strongly linked to negative symptoms of schizophrenia. Among these, rs583583 showed the most significant association, further suggesting a potential role of BCL9 in schizophrenia susceptibility (Xu et al., 2013). However, a study conducted by Kimura et al. (2015) examined the association between the rs583583 polymorphism and the manifestation of negative symptoms in Japanese individuals diagnosed with schizophrenia. Their results indicate that BCL9 is unlikely to harbor a common genetic variant that contributes to the increased risk of schizophrenia in the Japanese population. Additional GWAS and meta-analyses provide further evidence supporting these findings, suggesting that BCL9 is implicated in the negative symptoms of schizophrenia and designating it as one of the most prominent high-risk candidate genes for the disorder. Notably, other components of Wnt signaling have also been found to be associated with schizophrenia and other mental illnesses, further emphasizing the pathway’s significant role in schizophrenia (Yoon and Mao, 2021). Disruptions during early neurodevelopment have been closely associated with the underlying pathological mechanisms of schizophrenia (Birnbaum and Weinberger, 2017; Cheng et al., 2022; Ahmad et al., 2018; Weinberger, 2017). Wnt proteins are essential components of critical signaling pathways that regulate fetal brain development, contributing to angiogenesis, neurogenesis, cell survival, synaptic development, and neuronal extension, thereby playing indispensable roles in these processes. β-catenin, a key component of the classical Wnt pathway, whose mutations have been identified as a common cause of intellectual disability, highlights its importance in the development of the neural system (Yoon and Mao, 2021). Schizophrenia is also marked by aberrant mRNA expression of Wnt-related genes, weakening typical β-catenin-dependent signaling and enhancing atypical Wnt signaling. Schizophrenia features abnormal expression of Wnt-related genes and abnormal levels of plasma proteins, suggesting that drugs targeting the Wnt pathway may play a role in the treatment of severe mental disorders (Hoseth et al., 2018; Freyberg et al., 2010). BCL9 has emerged as a key genetic contributor to schizophrenia susceptibility through its regulation of the Wnt pathway. Moreover, specific SNPs within the BCL9 gene have been significantly associated with the negative symptoms of schizophrenia, further supporting its potential as a risk candidate gene.
4.3 CHD1L
CHD1L (OMIM: 613039) (Chromodomain helicase/ATPase DNA binding protein 1-like gene) encodes the CHD1L protein, a multifunctional factor involved in chromatin remodeling processes essential for DNA replication, transcription regulation, repair, and recombination. In addition, it exerts a crucial role in various cellular biological processes, including cell differentiation and development (Xiong et al., 2021; Wang et al., 2021; Jiang et al., 2015; Ahel et al., 2009). CHD1L exhibits the highest expression in the brain, where it is pivotal for nervous system development (Brockschmidt et al., 2012). A study on human embryonic stem cells (hESC) demonstrated that the overexpression of CHD1L upregulates the expression of ectodermal genes, particularly the key regulatory gene PAX6, which is critical for neural development. Such upregulation promotes the differentiation of hESCs into neuroepithelia. Conversely, the knockout of CHD1L significantly impairs this differentiation process (Dou et al., 2017). Neuroepithelial cells, as progenitor cells in the early nervous system, differentiate into various types of neurons and glial cells, forming the complex structure of the brain and nervous system (Merkle and Alvarez-Buylla, 2006; Eze et al., 2021). Pahlevan et al. (2024) reported that knockdown of CHD1L in human iPSC-derived neurons, along with its knockout in zebrafish, disrupt normal neuronal development and impair neural function. The importance of CHD1L in early nervous system development is further underscored by findings suggesting its central role in normal neuronal differentiation and function. Furthermore, studies indicate that disturbances in neuronal differentiation could be linked to the development of schizophrenia (Notaras et al., 2022; Robicsek et al., 2013; Robicsek et al., 2018). CHD1L participates in DNA damage response through interactions with multiple repair-associated factors, thereby promoting genomic stability and supporting the fidelity of DNA repair pathways (Xiong et al., 2021; Wang et al., 2021; Bulut-Karslioglu et al., 2021). Moreover, studies suggest that polymorphisms in DNA repair genes are potential etiological factors for mental disorders, and impaired DNA repair functions may increase the risk of schizophrenia (Odemis et al., 2016; Yang et al., 2017; Benes, 2011). Currently, the hypothesis that genetic damage to DNA and/or repair genes is linked to the pathogenesis of schizophrenia is supported by gradually accumulating evidence. However, the causal relationship between DNA damage/repair and schizophrenia remains controversial, highlighting that our current understanding of DNA damage and repair mechanisms in these disorders is still evolving (Markkanen et al., 2016). The relationship between CHD1L and schizophrenia likely involves its role in both neuronal differentiation and DNA repair. While the precise causal connection between CHD1L and schizophrenia remains unclear, growing evidence suggests that genetic variations or functional alterations in CHD1L may contribute to the pathogenesis of schizophrenia by impairing neurodevelopment and compromising genomic stability, thereby elevating the risk of the disorder.
4.4 GJA5, GJA8
The connexins (Cxs) encoded by GJA5 (OMIM: 121013) and GJA8 (OMIM: 600897) are a class of transmembrane proteins that are essential components of gap junctions, essential for intercellular communication. The Cx40 protein encoded by GJA5 is primarily expressed in the cardiac conduction system and atria, where it facilitates electrical coupling of atrial myocytes. Similarly, the Cx50 protein, encoded by GJA8, is mainly expressed in the lens epithelium, where it plays a crucial role in maintaining the transparency of the human lens (Bai, 2014; Ceroni et al., 2019; Brodie et al., 2019; Dong et al., 2024). In addition, GJA5 and GJA8 are also expressed in the nervous system, where Cx40 and Cx50 participate in neuronal gap junctions, regulating neuronal excitability and synaptic plasticity in the central nervous system. Connexin-mediated gap junctions facilitate electrotonic coupling within chemically homogeneous GABAergic networks, allowing rapid ion exchange and promoting synchronized neuronal firing, thereby contributing to fast and spatially distributed inhibition of neural excitability (Lapato and Tiwari-Woodruff, 2018; Decrock et al., 2015; Rash et al., 2001). Moreover, the loss of astroglia gap junction functionality may lead to severe cognitive impairments in patients with schizophrenia (Mitterauer, 2009; Mitterauer, 2011). Importantly, GJA5 and GJA8 are considered candidate genes for schizophrenia, largely due to their involvement in neural signaling and their location within the 1q21 chromosomal region, which is consistently linked to the disorder. Studies using cumulative scoring have found that GJA5 and GJA8 are genes frequently disrupted by CNVs in schizophrenia. Integrating accumulated priority data with known schizophrenia susceptibility genes, further analysis has identified GJA8 as a promising candidate gene for schizophrenia, supported by two independent pieces of evidence (from CNVs and genetic association or linkage studies) (Luo et al., 2014). In a paired case-control sample from Toronto, Canada, the Cx50 rs989192-rs4950495 haplotype was found to be associated with schizophrenia, a finding that was replicated in a Portuguese family study. Finally, analyses of alleles, genotypes, and haplotypes have not found an association between Cx40 and schizophrenia, highlighting that it is Cx50, not Cx40, that may play a role in the genetic susceptibility to schizophrenia (Ni et al., 2007). Overall, the exact mechanisms by which GJA5 and GJA8 contribute to schizophrenia remain unclear. However, based on existing evidence, it is hypothesized that these genes, which encode Cxs, play a critical role in intercellular communication and membrane junctions. Haploinsufficiency of GJA5 and GJA8 may disrupt gap junction assembly or electrical coupling, thereby affecting the transmission of information across neural circuits. This disruption of neural communication could contribute to the pathogenesis of schizophrenia by impairing the function of various neural networks.
5 Concluding remarks and future perspectives
Schizophrenia is a complex and multifactorial neurodevelopmental disorder, involving genetic factors. The distal 1q21.1 microdeletion is a major genetic risk factor for schizophrenia, with studies showing an eightfold increased risk in affected individuals. This review discusses the potential roles of genes within the 1q21.1 region, particularly PRKAB2, BCL9, CHD1L, and GJA5/GJA8, and their possible molecular mechanisms in the pathogenesis of schizophrenia. Supplementary Table S3 summarizes the identified risk genes and their biological roles in disease-related alterations.
The pathogenesis of schizophrenia may involve the synergistic action of multiple genes, such as PRKAB2, BCL9, CHD1L, GJA5, and GJA8. For example, mutations in PRKAB2 may disrupt neuronal energy metabolism, leading to imbalances in neurotransmitter synthesis and release, which could underlie many of the neurochemical disturbances observed in schizophrenia. Mutations in BCL9 may impair the Wnt signaling pathway, which is crucial for neuronal differentiation and the formation of neural networks. Mutations in CHD1L could interfere with chromatin remodeling during early neurodevelopment, impacting the expression of key genes necessary for neuronal differentiation. Moreover, as the nervous system matures, mutations in GJA5 and GJA8 could disrupt the connectivity and synchronization of neuronal signals, further impairing the function of neural circuits. The combined effects of these genetic alterations can ultimately lead to dysfunction in critical brain regions such as the cortex and hippocampus, which are involved in cognitive functions, perception, and memory. This results in the hallmark symptoms of schizophrenia, including cognitive impairments, hallucinations, delusions, and negative symptoms. Thus, we suppose that the integrated effects of these genetic variations form a complex network of pathogenic mechanisms that contribute to the broad clinical spectrum of schizophrenia.
Despite significant progress in current research, identifying consistent genetic markers remains challenging due to clinical heterogeneity and genetic diversity of schizophrenia. While CNVs, such as the 1q21.1 microdeletion, are implicated in schizophrenia, the precise mechanisms through which these genetic alterations contribute to the spectrum of symptoms are still unclear. Additionally, the interaction between genetic and environmental factors requires further exploration. Future research should focus on integrating multi-omics approaches, including genomics, transcriptomics, and proteomics, to better understand the molecular pathways involved in schizophrenia. Moreover, advanced animal models and iPSC technologies offer promising avenues for further investigation into the relationship between 1q21.1 and schizophrenia pathogenesis.
Author contributions
XG: Methodology, Investigation, Writing – review and editing, Writing – original draft. JG: Writing – review and editing, Investigation. XL: Writing – review and editing. TH: Writing – review and editing, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Key Research and Development Program of China (Grant Nos 2022YFC2703300 and 2022YFC2703302), and the Natural Science Foundation of Sichuan Province (Grant No. 2025ZNSFSC0701).
Acknowledgments
The authors would like to express sincere gratitude to the Editorial Office of Frontiers in Genetics for their valuable assistance with journal statistics and support related to this Research Topic.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1612654/full#supplementary-material
References
Rare chromosomal deletions and duplications (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455 (7210), 237–241. doi:10.1038/nature07239
Biological insights from 108 schizophrenia (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510), 421–427. doi:10.1038/nature13595
Adachi, S., Jigami, T., Yasui, T., Nakano, T., Ohwada, S., Omori, Y., et al. (2004). Role of a BCL9-related beta-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of wnt signaling. Cancer Res. 64 (23), 8496–8501. doi:10.1158/0008-5472.CAN-04-2254
Agarwal, S. M., Caravaggio, F., Costa-Dookhan, K. A., Castellani, L., Kowalchuk, C., Asgariroozbehani, R., et al. (2020). Brain insulin action in schizophrenia: something borrowed and something new. Neuropharmacology 163, 107633. doi:10.1016/j.neuropharm.2019.05.010
Ahel, D., Horejsí, Z., Wiechens, N., Polo, S. E., Garcia-Wilson, E., Ahel, I., et al. (2009). Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Sci. (New York, NY) 325 (5945), 1240–1243. doi:10.1126/science.1177321
Ahmad, R., Sportelli, V., Ziller, M., Spengler, D., and Hoffmann, A. (2018). Tracing early neurodevelopment in schizophrenia with induced pluripotent stem cells. Cells 7 (9), 140. doi:10.3390/cells7090140
Bai, D. (2014). Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 588 (8), 1238–1243. doi:10.1016/j.febslet.2014.02.064
Belforte, N., Agostinone, J., Alarcon-Martinez, L., Villafranca-Baughman, D., Dotigny, F., Cueva Vargas, J. L., et al. (2021). AMPK hyperactivation promotes dendrite retraction, synaptic loss, and neuronal dysfunction in glaucoma. Mol. Neurodegener. 16 (1), 43. doi:10.1186/s13024-021-00466-z
Benes, F. M. (2011). Regulation of cell cycle and DNA repair in post-mitotic GABA neurons in psychotic disorders. Neuropharmacology 60 (7-8), 1232–1242. doi:10.1016/j.neuropharm.2010.12.011
Bergen, S. E., Ploner, A., Howrigan, D., O'Donovan, M. C., Smoller, J. W., Sullivan, P. F., et al. (2019). Joint contributions of rare copy number variants and common SNPs to risk for schizophrenia. Am. J. psychiatry 176 (1), 29–35. doi:10.1176/appi.ajp.2018.17040467
Bernier, R., Steinman, K. J., Reilly, B., Wallace, A. S., Sherr, E. H., Pojman, N., et al. (2016). Clinical phenotype of the recurrent 1q21.1 copy-number variant. official J. Am. Coll. Med. Genet. 18 (4), 341–349. doi:10.1038/gim.2015.78
Birnbaum, R., and Weinberger, D. R. (2017). Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 18 (12), 727–740. doi:10.1038/nrn.2017.125
Bobela, W., Nazeeruddin, S., Knott, G., Aebischer, P., and Schneider, B. L. (2017). Modulating the catalytic activity of AMPK has neuroprotective effects against α-synuclein toxicity. Mol. Neurodegener. 12 (1), 80. doi:10.1186/s13024-017-0220-x
Boen, R., Kaufmann, T., van der Meer, D., Frei, O., Agartz, I., Ames, D., et al. (2024). Beyond the global brain differences: intraindividual variability differences in 1q21.1 distal and 15q11.2 BP1-BP2 deletion carriers. Biol. psychiatry 95 (2), 147–160. doi:10.1016/j.biopsych.2023.08.018
Brockschmidt, A., Chung, B., Weber, S., Fischer, D. C., Kolatsi-Joannou, M., Christ, L., et al. (2012). CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT). Nephrol. Dial. Transplant. 27 (6), 2355–2364. doi:10.1093/ndt/gfr649
Brodie, S. A., Rodriguez-Aulet, J. P., Giri, N., Dai, J., Steinberg, M., Waterfall, J. J., et al. (2019). 1q21.1 deletion and a rare functional polymorphism in siblings with thrombocytopenia-absent radius-like phenotypes. Cold Spring Harb. Mol. case Stud. 5 (6), a004564. doi:10.1101/mcs.a004564
Brunetti-Pierri, N., Berg, J. S., Scaglia, F., Belmont, J., Bacino, C. A., Sahoo, T., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40 (12), 1466–1471. doi:10.1038/ng.279
Bryll, A., Skrzypek, J., Krzyściak, W., Szelągowska, M., Śmierciak, N., Kozicz, T., et al. (2020). Oxidative-antioxidant imbalance and impaired glucose metabolism in schizophrenia. Biomolecules 10 (3), 384. doi:10.3390/biom10030384
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5 (6), e1000536. doi:10.1371/journal.pgen.1000536
Bulut-Karslioglu, A., Jin, H., Kim, Y. K., Cho, B., Guzman-Ayala, M., Williamson, A. J. K., et al. (2021). Chd1 protects genome integrity at promoters to sustain hypertranscription in embryonic stem cells. Nat. Commun. 12 (1), 4859. doi:10.1038/s41467-021-25088-3
Busè, M., Cuttaia, H. C., Palazzo, D., Mazara, M. V., Lauricella, S. A., Malacarne, M., et al. (2017). Expanding the phenotype of reciprocal 1q21.1 deletions and duplications: a case series. Italian J. Pediatr. 43 (1), 61. doi:10.1186/s13052-017-0380-x
Ceroni, F., Aguilera-Garcia, D., Chassaing, N., Bax, D. A., Blanco-Kelly, F., Ramos, P., et al. (2019). New GJA8 variants and phenotypes highlight its critical role in a broad spectrum of eye anomalies. Hum. Genet. 138 (8-9), 1027–1042. doi:10.1007/s00439-018-1875-2
Chapman, G., Alsaqati, M., Lunn, S., Singh, T., Linden, S. C., Linden, D. E. J., et al. (2022). Using induced pluripotent stem cells to investigate human neuronal phenotypes in 1q21.1 deletion and duplication syndrome. Mol. psychiatry 27 (2), 819–830. doi:10.1038/s41380-021-01182-2
Cheng, W., van der Meer, D., Parker, N., Hindley, G., O'Connell, K. S., Wang, Y., et al. (2022). Shared genetic architecture between schizophrenia and subcortical brain volumes implicates early neurodevelopmental processes and brain development in childhood. Mol. psychiatry 27 (12), 5167–5176. doi:10.1038/s41380-022-01751-z
Christiansen, J., Dyck, J. D., Elyas, B. G., Lilley, M., Bamforth, J. S., Hicks, M., et al. (2004). Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circulation Res. 94 (11), 1429–1435. doi:10.1161/01.RES.0000130528.72330.5c
Costain, G., Silversides, C. K., and Bassett, A. S. (2016). The importance of copy number variation in congenital heart disease. NPJ genomic Med. 1, 16031. doi:10.1038/npjgenmed.2016.31
Decrock, E., De Bock, M., Wang, N., Bultynck, G., Giaume, C., Naus, C. C., et al. (2015). Connexin and pannexin signaling pathways, an architectural blueprint for CNS physiology and pathology? Cell. Mol. life Sci. CMLS 72 (15), 2823–2851. doi:10.1007/s00018-015-1962-7
de la Roche, M., Worm, J., and Bienz, M. (2008). The function of BCL9 in wnt/Beta-Catenin signaling and colorectal cancer cells. BMC cancer 8, 199. doi:10.1186/1471-2407-8-199
Dienel, G. A. (2019). Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99 (1), 949–1045. doi:10.1152/physrev.00062.2017
Dong, S., Zou, T., Zhen, F., Wang, T., Zhou, Y., Wu, J., et al. (2024). Association of variants in GJA8 with familial acorea-microphthalmia-cataract syndrome. Eur. J. Hum. Genet. EJHG. 32 (4), 413–420. doi:10.1038/s41431-023-01503-9
Dou, D., Zhao, H., Li, Z., Xu, L., Xiong, X., Wu, X., et al. (2017). CHD1L promotes neuronal differentiation in human embryonic stem cells by upregulating PAX6. Stem cells Dev. 26 (22), 1626–1636. doi:10.1089/scd.2017.0110
Edwards, S. D., Schulze, K. V., Rosenfeld, J. A., Westerfield, L. E., Gerard, A., Yuan, B., et al. (2021). Clinical characterization of individuals with the distal 1q21.1 microdeletion. Am. J. Med. Genet. Part A 185 (5), 1388–1398. doi:10.1002/ajmg.a.62104
Eze, U. C., Bhaduri, A., Haeussler, M., Nowakowski, T. J., and Kriegstein, A. R. (2021). Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 24 (4), 584–594. doi:10.1038/s41593-020-00794-1
Freyberg, Z., Ferrando, S. J., and Javitch, J. A. (2010). Roles of the Akt/GSK-3 and wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. psychiatry 167 (4), 388–396. doi:10.1176/appi.ajp.2009.08121873
Gordon, A., Forsingdal, A., Klewe, I. V., Nielsen, J., Didriksen, M., Werge, T., et al. (2021). Transcriptomic networks implicate neuronal energetic abnormalities in three mouse models harboring autism and schizophrenia-associated mutations. Mol. psychiatry 26 (5), 1520–1534. doi:10.1038/s41380-019-0576-0
Gudmundsson, O. O., Walters, G. B., Ingason, A., Johansson, S., Zayats, T., Athanasiu, L., et al. (2019). Attention-deficit hyperactivity disorder shares copy number variant risk with schizophrenia and autism spectrum disorder. Transl. psychiatry 9 (1), 258. doi:10.1038/s41398-019-0599-y
Guo, R., and Haldeman-Englert, C. R. (1993). “1q21.1 recurrent deletion,” in GeneReviews(®). Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and A. Amemiya (Seattle (WA): University of Washington, Seattle).
Hardie, D. G. (2007). AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell. Biol. 8 (10), 774–785. doi:10.1038/nrm2249
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell. Biol. 13 (4), 251–262. doi:10.1038/nrm3311
Harvard, C., Strong, E., Mercier, E., Colnaghi, R., Alcantara, D., Chow, E., et al. (2011). Understanding the impact of 1q21.1 copy number variant. Orphanet J. rare Dis. 6, 54. doi:10.1186/1750-1172-6-54
He, C. W., Liao, C. P., and Pan, C. L. (2018). Wnt signalling in the development of axon, dendrites and synapses. Open Biol. 8 (10), 180116. doi:10.1098/rsob.180116
Henkel, N. D., Wu, X., O'Donovan, S. M., Devine, E. A., Jiron, J. M., Rowland, L. M., et al. (2022). Schizophrenia: a disorder of broken brain bioenergetics. Mol. psychiatry 27 (5), 2393–2404. doi:10.1038/s41380-022-01494-x
Hoseth, E. Z., Krull, F., Dieset, I., Mørch, R. H., Hope, S., Gardsjord, E. S., et al. (2018). Exploring the wnt signaling pathway in schizophrenia and bipolar disorder. Transl. psychiatry 8 (1), 55. doi:10.1038/s41398-018-0102-1
Inestrosa, N. C., Montecinos-Oliva, C., and Fuenzalida, M. (2012). Wnt signaling: role in alzheimer disease and schizophrenia. J. neuroimmune Pharmacol. official J. Soc. NeuroImmune Pharmacol. 7 (4), 788–807. doi:10.1007/s11481-012-9417-5
Iyegbe, C. O., and O'Reilly, P. F. (2022). Genetic origins of schizophrenia find common ground. Nature 604 (7906), 433–435. doi:10.1038/d41586-022-00773-5
Jensen, M., Hoerndli, F. J., Brockie, P. J., Wang, R., Johnson, E., Maxfield, D., et al. (2012). Wnt signaling regulates acetylcholine receptor translocation and synaptic plasticity in the adult nervous system. Cell. 149 (1), 173–187. doi:10.1016/j.cell.2011.12.038
Jiang, B. H., Chen, W. Y., Li, H. Y., Chien, Y., Chang, W. C., Hsieh, P. C., et al. (2015). CHD1L regulated PARP1-Driven pluripotency and chromatin remodeling during the early-stage cell reprogramming. Stem cells Dayt. Ohio 33 (10), 2961–2972. doi:10.1002/stem.2116
Kendall, K. M., Rees, E., Escott-Price, V., Einon, M., Thomas, R., Hewitt, J., et al. (2017). Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol. psychiatry 82 (2), 103–110. doi:10.1016/j.biopsych.2016.08.014
Kimura, H., Tanaka, S., Kushima, I., Koide, T., Banno, M., Kikuchi, T., et al. (2015). Association study of BCL9 gene polymorphism rs583583 with schizophrenia and negative symptoms in Japanese population. Sci. Rep. 5, 15705. doi:10.1038/srep15705
Kuwabara, T., Hsieh, J., Muotri, A., Yeo, G., Warashina, M., Lie, D. C., et al. (2009). Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 12 (9), 1097–1105. doi:10.1038/nn.2360
Lapato, A. S., and Tiwari-Woodruff, S. K. (2018). Connexins and pannexins: at the junction of neuro-glial homeostasis and disease. J. Neurosci. Res. 96 (1), 31–44. doi:10.1002/jnr.24088
Legge, S. E., Santoro, M. L., Periyasamy, S., Okewole, A., Arsalan, A., and Kowalec, K. (2021). Genetic architecture of schizophrenia: a review of major advancements. Psychol. Med. 51 (13), 2168–2177. doi:10.1017/S0033291720005334
Levinson, D. F., Duan, J., Oh, S., Wang, K., Sanders, A. R., Shi, J., et al. (2011). Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. psychiatry 168 (3), 302–316. doi:10.1176/appi.ajp.2010.10060876
Li, J., Zhou, G., Ji, W., Feng, G., Zhao, Q., Liu, J., et al. (2011). Common variants in the BCL9 gene conferring risk of schizophrenia. Archives general psychiatry 68 (3), 232–240. doi:10.1001/archgenpsychiatry.2011.1
Luo, X., Huang, L., Han, L., Luo, Z., Hu, F., Tieu, R., et al. (2014). Systematic prioritization and integrative analysis of copy number variations in schizophrenia reveal key schizophrenia susceptibility genes. Schizophr. Bull. 40 (6), 1285–1299. doi:10.1093/schbul/sbu045
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 148 (6), 1223–1241. doi:10.1016/j.cell.2012.02.039
Markkanen, E., Meyer, U., and Dianov, G. L. (2016). DNA damage and repair in schizophrenia and autism: implications for cancer comorbidity and beyond. Int. J. Mol. Sci. 17 (6), 856. doi:10.3390/ijms17060856
Marshall, C. R., Howrigan, D. P., Merico, D., Thiruvahindrapuram, B., Wu, W., Greer, D. S., et al. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49 (1), 27–35. doi:10.1038/ng.3725
Mefford, H. C., Sharp, A. J., Baker, C., Itsara, A., Jiang, Z., Buysse, K., et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359 (16), 1685–1699. doi:10.1056/NEJMoa0805384
Merkle, F. T., and Alvarez-Buylla, A. (2006). Neural stem cells in Mammalian development. Curr. Opin. Cell. Biol. 18 (6), 704–709. doi:10.1016/j.ceb.2006.09.008
Mitterauer, B. (2009). Loss of function of glial gap junctions May cause severe cognitive impairments in schizophrenia. Med. hypotheses 73 (3), 393–397. doi:10.1016/j.mehy.2009.04.003
Mitterauer, B. J. (2011). Possible role of glia in cognitive impairment in schizophrenia. CNS Neurosci. and Ther. 17 (5), 333–344. doi:10.1111/j.1755-5949.2009.00113.x
Mollon, J., Almasy, L., Jacquemont, S., and Glahn, D. C. (2023). The contribution of copy number variants to psychiatric symptoms and cognitive ability. Mol. psychiatry 28 (4), 1480–1493. doi:10.1038/s41380-023-01978-4
Muraleedharan, R., and Dasgupta, B. (2022). AMPK in the brain: its roles in glucose and neural metabolism. FEBS J. 289 (8), 2247–2262. doi:10.1111/febs.16151
Muraleedharan, R., Gawali, M. V., Tiwari, D., Sukumaran, A., Oatman, N., Anderson, J., et al. (2020). AMPK-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell. Rep. 32 (9), 108092. doi:10.1016/j.celrep.2020.108092
Nagy, S., Maurer, G. W., Hentze, J. L., Rose, M., Werge, T. M., and Rewitz, K. (2018). AMPK signaling linked to the schizophrenia-associated 1q21.1 deletion is required for neuronal and sleep maintenance. PLoS Genet. 14 (12), e1007623. doi:10.1371/journal.pgen.1007623
Ng, C. H., Guan, M. S., Koh, C., Ouyang, X., Yu, F., Tan, E. K., et al. (2012). AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in drosophila models of parkinson's disease. J. Neurosci. official J. Soc. Neurosci. 32 (41), 14311–14317. doi:10.1523/JNEUROSCI.0499-12.2012
Ni, X., Valente, J., Azevedo, M. H., Pato, M. T., Pato, C. N., and Kennedy, J. L. (2007). Connexin 50 gene on human chromosome 1q21 is associated with schizophrenia in matched case control and family-based studies. J. Med. Genet. 44 (8), 532–536. doi:10.1136/jmg.2006.047944
Nielsen, J., Fejgin, K., Sotty, F., Nielsen, V., Mørk, A., Christoffersen, C. T., et al. (2017). A mouse model of the schizophrenia-associated 1q21.1 microdeletion syndrome exhibits altered mesolimbic dopamine transmission. Transl. psychiatry 7 (11), 1261. doi:10.1038/s41398-017-0011-8
Notaras, M., Lodhi, A., Dündar, F., Collier, P., Sayles, N. M., Tilgner, H., et al. (2022). Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol. psychiatry 27 (3), 1416–1434. doi:10.1038/s41380-021-01316-6
Odemis, S., Tuzun, E., Gulec, H., Semiz, U. B., Dasdemir, S., Kucuk, M., et al. (2016). Association between polymorphisms of DNA repair genes and risk of schizophrenia. Genet. Test. Mol. biomarkers 20 (1), 11–17. doi:10.1089/gtmb.2015.0168
O'Donovan, M. C., Craddock, N., Norton, N., Williams, H., Peirce, T., Moskvina, V., et al. (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40 (9), 1053–1055. doi:10.1038/ng.201
Okerlund, N. D., and Cheyette, B. N. (2011). Synaptic wnt signaling-a contributor to major psychiatric disorders? J. Neurodev. Disord. 3 (2), 162–174. doi:10.1007/s11689-011-9083-6
Pahlevan, K. M., Giordano, A., Starvaggi Cucuzza, C., Venkata, S. B. T., Samudyata, S., Lemée, M. V., et al. (2024). A genetic-epigenetic interplay at 1q21.1 locus underlies CHD1L-mediated vulnerability to primary progressive multiple sclerosis. Nat. Commun. 15 (1), 6419. doi:10.1038/s41467-024-50794-z
Panaccione, I., Napoletano, F., Forte, A. M., Kotzalidis, G. D., Del Casale, A., Rapinesi, C., et al. (2013). Neurodevelopment in schizophrenia: the role of the wnt pathways. Curr. Neuropharmacol. 11 (5), 535–558. doi:10.2174/1570159X113119990037
Peterson, B. S., Kaur, T., Sawardekar, S., Colibazzi, T., Hao, X., Wexler, B. E., et al. (2023). Aberrant hippocampus and amygdala morphology associated with cognitive deficits in schizophrenia. Front. Cell. Neurosci. 17, 1126577. doi:10.3389/fncel.2023.1126577
Rash, J. E., Yasumura, T., Dudek, F. E., and Nagy, J. I. (2001). Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. official J. Soc. Neurosci. 21 (6), 1983–2000. doi:10.1523/JNEUROSCI.21-06-01983.2001
Rees, E., Kendall, K., Pardiñas, A. F., Legge, S. E., Pocklington, A., Escott-Price, V., et al. (2016). Analysis of intellectual disability copy number variants for association with schizophrenia. JAMA psychiatry 73 (9), 963–969. doi:10.1001/jamapsychiatry.2016.1831
Rees, E., Moskvina, V., Owen, M. J., O'Donovan, M. C., and Kirov, G. (2011). De novo rates and selection of schizophrenia-associated copy number variants. Biol. psychiatry 70 (12), 1109–1114. doi:10.1016/j.biopsych.2011.07.011
Robicsek, O., Ene, H. M., Karry, R., Ytzhaki, O., Asor, E., McPhie, D., et al. (2018). Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophr. Bull. 44 (2), 432–442. doi:10.1093/schbul/sbx077
Robicsek, O., Karry, R., Petit, I., Salman-Kesner, N., Müller, F. J., Klein, E., et al. (2013). Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. psychiatry 18 (10), 1067–1076. doi:10.1038/mp.2013.67
Ronnett, G. V., Ramamurthy, S., Kleman, A. M., Landree, L. E., and Aja, S. (2009). AMPK in the brain: its roles in energy balance and neuroprotection. J. Neurochem. 109 (1), 17–23. doi:10.1111/j.1471-4159.2009.05916.x
Rosenfeld, J. A., Traylor, R. N., Schaefer, G. B., McPherson, E. W., Ballif, B. C., Klopocki, E., et al. (2012). Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur. J. Hum. Genet. EJHG. 20 (7), 754–761. doi:10.1038/ejhg.2012.6
Sasabayashi, D., Yoshimura, R., Takahashi, T., Takayanagi, Y., Nishiyama, S., Higuchi, Y., et al. (2021). Reduced hippocampal subfield volume in schizophrenia and clinical high-risk state for psychosis. Front. Psychiatry 12, 642048. doi:10.3389/fpsyt.2021.642048
Shah, S. Z. A., Zhao, D., Hussain, T., and Yang, L. (2017). Role of the AMPK pathway in promoting autophagic flux via modulating mitochondrial dynamics in neurodegenerative diseases: insight into prion diseases. Ageing Res. Rev. 40, 51–63. doi:10.1016/j.arr.2017.09.004
Sherrington, R., Brynjolfsson, J., Petursson, H., Potter, M., Dudleston, K., Barraclough, B., et al. (1988). Localization of a susceptibility locus for schizophrenia on chromosome 5. Nature 336 (6195), 164–167. doi:10.1038/336164a0
Smeland, O. B., and Andreassen, O. A. (2022). Schizophrenia: genetic insights with clinical potential. Nat. Rev. Neurol. 18 (3), 129–130. doi:10.1038/s41582-021-00613-6
Sønderby, I. E., van der Meer, D., Moreau, C., Kaufmann, T., Walters, G. B., Ellegaard, M., et al. (2021). 1q21.1 distal copy number variants are associated with cerebral and cognitive alterations in humans. Transl. psychiatry 11 (1), 182. doi:10.1038/s41398-021-01213-0
Souza, R. P., Tiwari, A. K., Chowdhury, N. I., Ceddia, R. B., Lieberman, J. A., Meltzer, H. Y., et al. (2012). Association study between variants of AMP-Activated protein kinase catalytic and regulatory subunit genes with antipsychotic-induced weight gain. J. psychiatric Res. 46 (4), 462–468. doi:10.1016/j.jpsychires.2012.01.010
Stefansson, H., Rujescu, D., Cichon, S., Pietiläinen, O. P., Ingason, A., Steinberg, S., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455 (7210), 232–236. doi:10.1038/nature07229
Szecówka, K., Misiak, B., Łaczmańska, I., Frydecka, D., and Moustafa, A. A. (2023). Copy number variations and schizophrenia. Mol. Neurobiol. 60 (4), 1854–1864. doi:10.1007/s12035-022-03185-8
Upadhyai, P., Amiri, E. F., Guleria, V. S., Bielas, S. L., Girisha, K. M., and Shukla, A. (2020). Recurrent 1q21.1 deletion syndrome: report on variable expression, nonpenetrance and review of literature. Clin. Dysmorphol. 29 (3), 127–131. doi:10.1097/MCD.0000000000000327
van Erp, T. G., Hibar, D. P., Rasmussen, J. M., Glahn, D. C., Pearlson, G. D., Andreassen, O. A., et al. (2016). Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol. psychiatry 21 (4), 547–553. doi:10.1038/mp.2015.63
Wagh, V. V., Vyas, P., Agrawal, S., Pachpor, T. A., Paralikar, V., and Khare, S. P. (2021). Peripheral blood-based gene expression studies in schizophrenia: a systematic review. Front. Genet. 12, 736483. doi:10.3389/fgene.2021.736483
Wang, L., Chen, K., and Chen, Z. (2021). Structural basis of ALC1/CHD1L autoinhibition and the mechanism of activation by the nucleosome. Nat. Commun. 12 (1), 4057. doi:10.1038/s41467-021-24320-4
Wang, X., and Jia, J. (2023). Magnolol improves Alzheimer's disease-like pathologies and cognitive decline by promoting autophagy through activation of the AMPK/mTOR/ULK1 pathway. Biomed. and Pharmacother. 161, 114473. doi:10.1016/j.biopha.2023.114473
Weber, S., Landwehr, C., Renkert, M., Hoischen, A., Wühl, E., Denecke, J., et al. (2011). Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol. Dial. Transplant. 26 (1), 136–143. doi:10.1093/ndt/gfq400
Wegrzyn, D., Juckel, G., and Faissner, A. (2022). Structural and functional deviations of the hippocampus in schizophrenia and schizophrenia animal models. Int. J. Mol. Sci. 23 (10), 5482. doi:10.3390/ijms23105482
Weinberger, D. R. (2017). Future of days past: neurodevelopment and schizophrenia. Schizophr. Bull. 43 (6), 1164–1168. doi:10.1093/schbul/sbx118
Xiong, X., Lai, X., Li, A., Liu, Z., and Ma, N. (2021). Diversity roles of CHD1L in normal cell function and tumorigenesis. Biomark. Res. 9 (1), 16. doi:10.1186/s40364-021-00269-w
Xu, C., Aragam, N., Li, X., Villla, E. C., Wang, L., Briones, D., et al. (2013). BCL9 and C9orf5 are associated with negative symptoms in schizophrenia: meta-analysis of two genome-wide association studies. PloS one 8 (1), e51674. doi:10.1371/journal.pone.0051674
Yang, A. J. T., Mohammad, A., Tsiani, E., Necakov, A., and MacPherson, R. E. K. (2022). Chronic AMPK activation reduces the expression and alters distribution of synaptic proteins in neuronal SH-SY5Y cells. Cells 11 (15), 2354. doi:10.3390/cells11152354
Yang, Z., Li, M., Hu, X., Xiang, B., Deng, W., Wang, Q., et al. (2017). Rare damaging variants in DNA repair and cell cycle pathways are associated with hippocampal and cognitive dysfunction: a combined genetic imaging study in first-episode treatment-naive patients with schizophrenia. Transl. psychiatry 7 (2), e1028. doi:10.1038/tp.2016.291
Yoon, J., and Mao, Y. (2021). Dissecting molecular genetic mechanisms of 1q21.1 CNV in neuropsychiatric disorders. Int. J. Mol. Sci. 22 (11), 5811. doi:10.3390/ijms22115811
Zechner, D., Fujita, Y., Hülsken, J., Müller, T., Walther, I., Taketo, M. M., et al. (2003). beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 258 (2), 406–418. doi:10.1016/s0012-1606(03)00123-4
Keywords: schizophrenia, distal 1q21.1 microdeletion, PRKAB2, BCL9, CHD1L
Citation: Guo X, Guo J, Liu X and Hu T (2025) A review on the relationship between the distal 1q21.1 microdeletion and schizophrenia. Front. Genet. 16:1612654. doi: 10.3389/fgene.2025.1612654
Received: 16 April 2025; Accepted: 18 July 2025;
Published: 28 July 2025.
Edited by:
Itaru Kushima, Nagoya University, JapanReviewed by:
Hiroki Ishiguro, University of Yamanashi, JapanCopyright © 2025 Guo, Guo, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijing Liu, bHhqaW5nODgxOEAxNjMuY29t; Ting Hu, aHV0aW5nNDEyM0AxNjMuY29t