 Sara Missaglia1,2†
Sara Missaglia1,2† Eleonora Martegani1,2†
Eleonora Martegani1,2† Corrado Angelini3
Corrado Angelini3 Rita Horvath4
Rita Horvath4 Veronika Karcagi5
Veronika Karcagi5 Endre Pal6,7
Endre Pal6,7 Daniela Tavian1,2*
Daniela Tavian1,2*- 1Laboratory of Cellular Biochemistry and Molecular Biology, CRIBENS, Catholic University of the Sacred Heart, Milan, Italy
- 2Department of Psychology, Catholic University of Sacred Heart, Milan, Italy
- 3Campus Pietro d’Abano, Department of Neurosciences, University of Padua, Padua, Italy
- 4Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 5Istenhegyi Genetic Diagnostic Centre, Molecular Genetic Laboratory, Budapest, Hungary
- 6Department of Neurology, Medical School, University of Pécs, Pécs, Hungary
- 7Department of Pathology, Neuropathology Unit, Medical School, University of Pécs, Pécs, Hungary
Neutral Lipid Storage Disease with Myopathy (NLSDM) is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues. It typically manifests with progressive skeletal myopathy, with an onset of around 35 years. In addition, some patients develop cardiomyopathy and liver dysfunction. Herein, we report the molecular characterization of a 27-year-old Hungarian patient and his family in whom two severe PNPLA2 mutations led to complete loss of ATGL production in the patient’s tissues. DNA sequencing revealed a nonsense (c.24G>A) and a frameshift mutation (c.798dupC) in the PNPLA2 gene. RNA analysis showed nonsense-mediated decay of the c.798dupC transcript, while c.24G>A was normally expressed in the patient. However, Western blot analysis did not detect ATGL protein production. From a clinical perspective, our patient exhibited pes planus, proximal muscle weakness of the lower limbs and elevated CK levels from the age of six. MRI and biopsy revealed lipid accumulation, and leukocytes showed Jordans’ anomaly. The muscle weakness progressed, and cardiomyopathy and hepatic steatosis were also observed recently. The case highlights two severe PNPLA2 mutations leading to complete ATGL deficiency and an unusual early-onset myopathy in childhood.
1 Introduction
Adipose Triglyceride Lipase (ATGL) is a key enzyme involved in lipid metabolism in human cells. Indeed, ATGL catalyzes the first step of triacylglycerols (TAGs) breakdown stored into lipid droplets (LDs), the main depots of neutral lipids in cells. TAGs are hydrolyzed into diacylglycerols (DAGs) and free fatty acids (FFAs) that are necessary during energy requirement when lipolysis pathway is activated. ATGL is highly expressed in white and brown adipocytes, but is also expressed in other tissues including heart, skeletal muscle, liver, pancreas, lung, retina, and immune cells (Schweiger et al., 2009; Schreiber et al., 2019). ATGL is encoded by patatin-like phospholipase domain containing 2 (PNPLA2) gene and is composed of 504 amino acids with a patatin domain (amino acids from 10 to 178) containing the active site residues (S47 and D166) at the N-terminal region. A hydrophobic LD binding site is localized in the C-terminal part (amino acids from 315 to 360) (Missaglia et al., 2019).
Mutations in PNPLA2 gene cause the development of a rare autosomal recessive muscle disorder known as Neutral Lipid Storage Disease with Myopathy (NLSDM, OMIM #610717) (Fischer et al., 2007). Indeed, changes in normal expression or function of ATGL lead to an abnormal accumulation of TAGs in multiple tissues and organs, especially in skeletal muscle and heart (Pasanisi et al., 2016; Missaglia et al., 2017).
Manifestation of NLSDM usually begins with proximal and distal muscle weakness of upper and lower limbs and elevated levels of serum creatine kinase (CK) (Laforêt et al., 2013; Kaneko et al., 2014). Then, all patients develop progressive skeletal muscle myopathy and almost half of them also present cardiomyopathy. Indeed, ATGL plays an important role in myocardial cytosolic lipolysis, and defects in FFAs metabolism can compromise proper heart functioning (Wang et al., 2024; Fonseka et al., 2025). In addition, symptoms can include hepatomegaly, diabetes mellitus, chronic pancreatitis, and sensorineural hearing loss (Kaneko et al., 2014). Moreover, histological analyses of patients’ blood reveal Jordans’ anomaly in leukocytes [3].
More than 70 PNPLA2 mutations have been described until now in almost 130 patients worldwide (Missaglia et al., 2022; Wang et al., 2024). NLSDM is a highly variable disease ranging from mild symptoms to more severe ones. Clinical phenotypes may depend on the type of mutations in PNPLA2 gene causing total or partial loss of enzyme activity. However, since the same mutation can lead to different clinical manifestation, a genotype-phenotype correlation is not clear yet (Pennisi et al., 2017; Zhang et al., 2019).
In this study, we reported the clinical and molecular characterization of the first Hungarian patient and his family members, in which two different mutations in PNPLA2 gene were identified.
2 Methods
2.1 Clinical evaluation
Muscle histopathology was assessed following the standard methodology outlined by Dubowitz. Haematoxylin and eosin (H&E) and Oil Red O (ORO) staining were performed on frozen cross-sections of muscle biopsies.
Peripheral blood samples treated with EDTA were collected and centrifuged at 3,300 × g for 10 min. The resulting buffy coats were smeared onto microscope slides, air-dried thoroughly, and fixed. Staining was performed using the May-Grünwald–Giemsa (MGG) and Oil Red O, following established protocols.
2.2 Molecular analysis
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Histopaque, following the manufacturer’s instructions. Genomic DNA was obtained using a Puregene DNA Isolation kit (Qiagen, Venlo, Netherlands), while total RNA was extracted with Trizol solution and reverse-transcribed as reported previously (Tavian et al., 2021). PNPLA2 exons were amplified and PCR products purified by NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Germany), as previously reported (Tavian et al., 2021).
ATGL protein expression was assessed using PBMCs fraction after protein extraction (RIPA buffer–ThermoFisher Scientific) and quantification (BCA protein assay–ThermoFisher Scientific), following manufacturer’s instructions. Anti-ATGL Polyclonal Antibody was purchased by Invitrogen (catalog #PA5-17436).
3 Case description
The patient is a Hungarian male of 27 years old, born to non-consanguineous parents (Supplementary Figure S1). Clinical manifestations began at age six when, during an orthopedic visit, the patient manifested with some foot weakness and pes planus. He had elevated CK levels (1350 IU/L), muscle biopsy revealed lipid accumulation, and he was positive for Jordans’ anomaly test (Figures 1A,D).
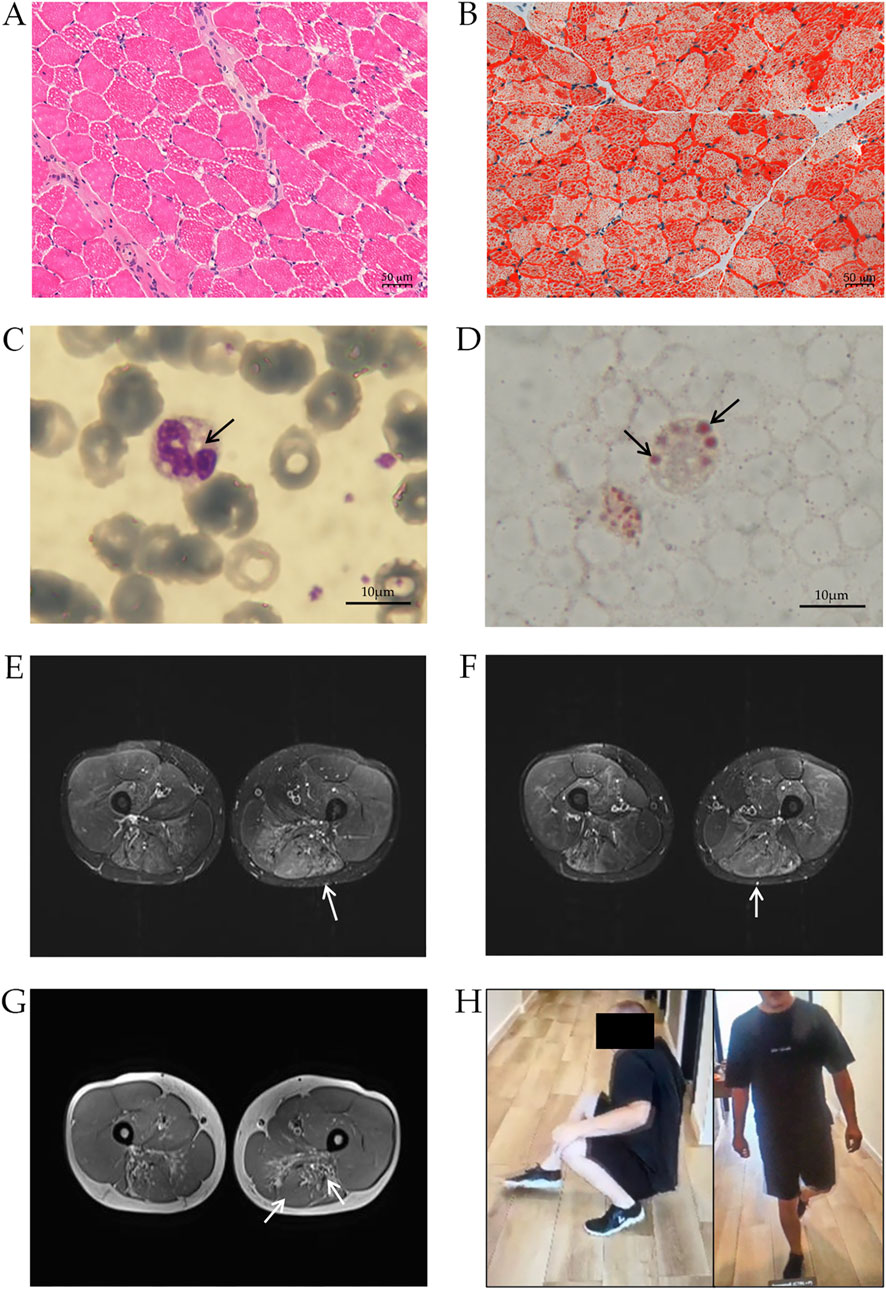
Figure 1. Clinical characterization of NLSDM patient. Muscle histopathology of patient’s biopsy, stained with haematoxylin-eosin (A) and Oil Red-O (B) Peripheral blood smear stained with May-Grünwald-Giemsa (C) and Oil Red-O (D) shows the presence of LDs (arrows) in leukocytes. (E) T2-weighted STIR. Pronounced oedema can be noted in the affected semitendinosus muscles (arrow). Currently, no significant atrophy can be detected in the quadriceps femoris muscles, however, diffuse, mild, symmetrical oedema is also noted in these muscles, predominantly in the distal portions. Subtle oedema can be noted in the adductors (arrow) and semitendinosus (arrow) (F). (G). MRI scan of the soft tissue of the thigh. Moderate atrophy can be identified in the flexor muscle groups symmetrically bilaterally, mainly in the distal portions of the biceps femoris muscles (arrow) and the semimembranosus muscles (arrow). (H) Patient’s photographs during Gowers’ maneuver (left) and during stepping gait (right).
At age 22, the patient suffered from a distal and proximal weakness of the lower limbs, with minimal weakness to upper limb and shoulder girdle. Magnetic Resonance Imaging (MRI) revealed diffuse oedema of thigh muscles (Figures 1E,F) and atrophy to thigh flexors muscles (biceps femoris, semimembranosus muscles) (Figure 1G), indicating a progression of the disease. Diffuse abnormalities were also observed in the legs, particularly in the anterior muscle compartment.
Patient’s upper limb musculature was normotrophic with moderate scapular winging (scapula alata). Manual muscle testing (MRC scale) showed muscle weakness in shoulder muscles 4+/5, arm rotator muscles 4+/5, without significant weakness in other muscle groups of the upper limbs. Hip muscles strength was retained. In the lower limbs, thigh muscles were slightly hypotrophic, while calf and lower leg muscles were markedly hypotrophic. Foot dorsiflexion was 2/5, and plantarflexion was 4-/5 (MRC scale). He has difficulty standing on tiptoe and could not stand on his heel. He could stand up easily from squatting, however he had difficulty during Gowers’ maneuver. There was areflexia in all limbs, and his gait was stepping (Figure 1H). Coordination and stance were normal, speech was intact, and he was alert and oriented. A cardiac MRI revealed hypertrophic cardiomyopathy (HCM) with inferoseptal hypertrophy of 15 mm. Recently, transient elastography has revealed hepatic steatosis.
The father is a 59-years-old overweight man, with lower physical fitness compared to his peers. He reported having less physical strength and exertional capacity at a young age, for example, he was unable to carry as much weight or cycle as long as other schoolmates. Currently, he does not experience difficulties in daily life, however his level of physical activity is quite low.
The mother, 52 years old, recently suffered from weakness of her upper limbs and had difficulty swallowing. The sister of the patient is a healthy woman of 30 years without clinical implications.
4 Molecular and genetic characterization
Diagnosis of NLSDM derived from genetic analysis of peripheral blood (Baldo et al., 2016). PNPLA2 exon sequencing revealed that the patient is a compound heterozygote carrying the nonsense mutation c.24G>A (p.W8*) in exon 2 and the frameshift mutation c.798dupC in exon 7 (Figure 2A). Sequencing analyses also revealed that the patient inherited from his mother the c.24G>A mutation, while the father carried the c.798dupC allele. The sister of the patient was a healthy control without mutations in PNPLA2 gene.
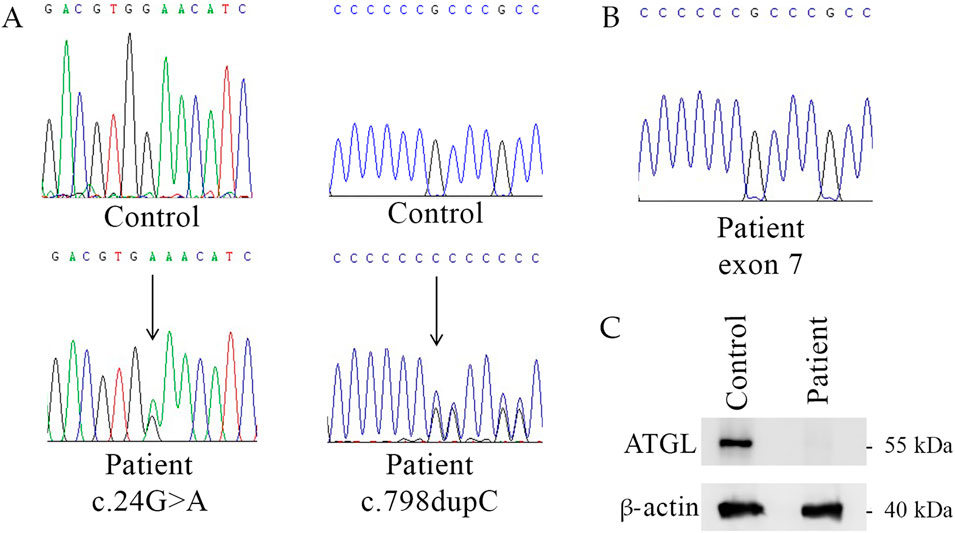
Figure 2. Genetic and molecular characterization of NLSDM patient. (A) Patient’s genomic DNA electropherograms of PNPLA2 exon 2 and exon 7. (B) cDNA electropherogram of exon 7 of NLSDM patient does not show the (C). 798dupC allele. (C) Western blot analyses for the expression of ATGL in the patient and his sister (healthy control).
To determine the pathogenetic impact of DNA mutations on ATGL production we analyzed the RNA extracted from patient’s blood sample. Sequencing of PNPLA2 exons from the patient revealed the c.24G>A mutation in exon 2, while c.798dupC allele was not detected (Figure 2B). This result suggests the absence of the mRNA carrying the frameshift mutation.
Western blot analyses revealed the absence of ATGL in patient’s sample, compared to his sister, the healthy control, as shown in Figure 2C.
5 Discussion
We presented the first NLSDM Hungarian family with a male patient characterized by an early onset at age 6. The average age of NLSDM onset is usually around the third/fourth decade (Zhang et al., 2019; Fu et al., 2023). However, a small minority of patients have been described manifesting symptoms during infancy or childhood (0–10 years) (Zhang et al., 2019; Fu et al., 2023). Early signs of NLSDM mainly appear as exercise intolerance, asymptomatic hyperCKemia, and rarely mild hypotonia and myopathy. In our patient pes planus and some mild foot muscle weakness in addition to elevated CK serum levels were observed originally. Current pathological conditions include persistent and progressive muscle weakness, more pronounced in distal lower extremities, and muscle atrophy in proximal lower limbs characterized by symmetrical progression. Hypertrophic cardiomyopathy and hepatic steatosis were recently observed. Cardiomyopathy was reported in half of 130 NLSDM cases (47%) and is mainly diagnosed after the third decade of age, indicating a late involvement of the heart. Liver involvement was found in less than 15% of the total of the cases and usually is presented as hepatomegaly, while rarely (5%) patients manifested fatty liver (Missaglia et al., 2019; Shi et al., 2021; Yamada et al., 2023).
Clinical symptoms of NLSDM are associated with abnormal accumulation of lipids in patients’ tissues due to the reduced or absent activity of ATGL lipase. The severity of manifestations is usually linked to the impairment in ATGL activity and, in general, mutations that lead to dramatic decrease in lipase production may undergo more serious clinical manifestation (Tavian et al., 2021). However, the relationship between genotype and phenotypic manifestation, including the age of onset, remains unclear.
Here we report a patient carrying a novel combination of mutated PNPLA2 alleles: the c.24G>A (p.W8*) variation, inherited from the mother, and the c.798dupC mutation of paternal origin, both showing the total loss of ATGL protein. The mutant transcript of c.24G>A allele should encode an eight-amino acid-long peptide (p.W8*), however it is likely that it has no functional relevance. The same mutation was previously identified in two Italian patients (Tavian et al., 2012). These two siblings were compound heterozygotes for the c.24G>A mutation and the c.516C>A (p.N172K) missense mutation. Previous functional studies showed that ATGL (N172K) preserved a partial lipase activity in patients’ tissues (Tavian et al., 2012). In these subjects clinical manifestations began late, at ages 25 and 40, with leg myalgia and cramps, respectively.
The frameshift mutation c.798dupC was previously identified by Jousserand and colleagues in a male patient who was homozygote for this PNPLA2 variation. He suffered from distal skeletal myopathy and severe cardiomyopathy, and his diagnosis was confirmed at age 54 (Jousserand et al., 2016). Nevertheless, the patient reported difficulties performing physical activities as early as his school years.
The c.798dupC mutation is predicted to produce an ATGL truncated protein of 266 wild type amino acids and further 39 residues (p.A267Rfs*40). Nevertheless, our molecular analyses highlighted the absence of the c.798dupC mutated transcript and of the ATGL truncated protein. The c.798dupC mutation causes a shift in the reading frame that alters the amino acid sequence starting from codon 267 and leads to a premature termination codon (PTC) at the end of exon 7. Given that the canonical PNPLA2 transcript comprises 10 exons, this PTC lies well upstream of several exon–exon junctions (exons 8–10) and thus fulfils the structural prerequisites for nonsense-mediated mRNA decay (NMD) activation (Lejeune, 2022; Miller and Pearce, 2014; Carrard and Lejeune, 2023). In the case of PNPLA2, the PTC generated by c.798dupC is expected to leave downstream intact EJCs, which can recruit the NMD machinery during the pioneer round of translation, leading to rapid degradation of the mutant transcript. This mechanism is particularly stringent when the PTC occurs in middle exons, such as exon 7 in PNPLA2. As described by Lejeune (Lejeune, 2022), these exons are typically flanked by multiple splice junctions that ensure EJC-dependent NMD activation, thereby preventing any stable accumulation of the mutant mRNA. This may explain the absence of detectable ATGL protein in the patient, since the transcript is likely degraded before any significant translation occurs.
Moreover, NMD efficiency varies significantly depending on cellular context, developmental stage, and type of tissue (Carrard and Lejeune, 2023). This variability is influenced by differential expression of core NMD factors, including UPF1, UPF3X, SMG6, and auxiliary modulators such as RNPS1 and PNRC2. The interaction of these factors defines the functional capacity of NMD in each cell type. This may be particularly relevant in NLSDM, where skeletal muscle is the principal tissue affected by LD accumulation due to defective ATGL activity.
Until now, more than 70 different PNPLA2 mutations have been reported, and 24 of them are frameshift mutations, predicted to produce a mutated ATGL protein. In these cases, the mechanism of NMD should be considered, since it may play an important role in degrading aberrant transcripts and dramatically reduce ATGL production (Samukawa et al., 2020).
The mother of our patient is heterozygote for the PNPLA2 gene, carrying c.24G>A allele. She recently manifested light signs of muscle weakness, underlining the pathogenic load of this variant. Exercise intolerance in youth was also manifested by the father of our patient, who has a wild-type allele and the c.798dupC mutation. However, we cannot exclude that symptoms manifested by parents could be associated with ageing rather than to the storage disease.
In summary, we reported a novel combination of two pathogenetic PNPLA2 variants in a male patient, resulting in a complete loss of ATGL production in the patient’s tissues and an early onset of the disease with progressive muscle weakness. NLSDM symptoms are heterogeneous, and the same genetic mutations may not always lead to identical clinical manifestations. This variability could be related to other factors, such as modifier genes, living environments, and patients’ lifestyles. The involvement of mRNA decay in the pathogenicity of NLSDM should also be taken into consideration since it could impair the production of ATGL lipase and worsen patients’ clinical conditions.
Data availability statement
The data that support the findings of this study are available from Daniela Tavian, Catholic University of the Sacred Heart, Milan (Italy).
Ethics statement
The study was conducted according to the guidelines of Declaration of Helsinki and approved by Comitato Etico Territoriale Lombardia 4 (protocol code n CET 73-25). The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SM: Writing – review and editing, Methodology. EM: Formal Analysis, Writing – original draft. CA: Investigation, Writing – review and editing. RH: Formal Analysis, Writing – review and editing. VK: Formal Analysis, Writing – review and editing. EP: Investigation, Writing – review and editing. DT: Writing – review and editing, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by European Union funding - Next Generation EU, Mission 4 Component 2 CUP J53D23019120001/Finanziato dall’Unione europea- Next Generation EU, Missione 4 Componente 2 CUP J53D23019120001. The authors acknowledge the Biobank of the Laboratory of Human Genetics–IRCCS Istituto Gaslini (former Galliera Genetic Biobank), member of the Network Telethon of Genetic Biobanks (Project No. GTB18001).
Acknowledgments
The authors sincerely thank the patient and his family members for their great kindness, availability, and cooperation. The authors acknowledge Viktoriia Fylymonenko for generous scientific assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1642442/full#supplementary-material
References
Baldo, C., Viotti, V., Maioli, E., Mogni, M., Castagnetta, M., Cavani, S., et al. (2016). Galliera genetic bank: a DNA and cell line biobank from patients affected by genetic diseases. Open J. Bioresour. 3, 1–5. doi:10.5334/ojb.15
Carrard, J., and Lejeune, F. (2023). Nonsense-mediated mRNA decay, a simplified view of a complex mechanism. BMB Rep. 56, 625–632. doi:10.5483/BMBRep.2023-0190
Fischer, J., Lefèvre, C., Morava, E., Mussini, J. M., Lafor̂t, P., Negre-Salvayre, A., et al. (2007). The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 39, 28–30. doi:10.1038/ng1951
Fonseka, O., Raja, R., Ross, C., Gare, S. R., Zhang, J., Hille, S. S., et al. (2025). XBP1s-EDEM2 prevents the onset and development of HFpEF by ameliorating cardiac lipotoxicity. Circulation 151, 1583–1605. doi:10.1161/CIRCULATIONAHA.124.072194
Fu, X., Yang, X., Wang, X., Jia, B., Ma, W., Xiong, H., et al. (2023). HyperCKemia: an early sign of childhood-onset neutral lipid storage disease with myopathy. Neuromuscul. Disord. 33, 81–89. doi:10.1016/j.nmd.2023.07.007
Jousserand, G., Streichenberger, N., and Petiot, P. (2016). Myopathie à surcharge lipidique secondaire à une nouvelle mutation du gène PNPLA2. Medecine/Sciences 32, 10–11. doi:10.1051/medsci/201632s203
Kaneko, K., Kuroda, H., Izumi, R., Tateyama, M., Kato, M., Sugimura, K., et al. (2014). A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: a case report and literature review. Neuromuscul. Disord. 24, 634–641. doi:10.1016/j.nmd.2014.04.001
Laforêt, P., Stojkovic, T., Bassez, G., Carlier, P. G., Clément, K., Wahbi, K., et al. (2013). Neutral lipid storage disease with myopathy: a whole-body nuclear MRI and metabolic study. Mol. Genet. Metab. 108, 125–131. doi:10.1016/j.ymgme.2012.12.004
Lejeune, F. (2022). Nonsense-mediated mRNA decay, a finely regulated mechanism. Biomedicines 10, 141. doi:10.3390/biomedicines10010141
Miller, J. N., and Pearce, D. A. (2014). Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. - Rev. Mutat. Res. 762, 52–64. doi:10.1016/j.mrrev.2014.05.001
Missaglia, S., Coleman, R. A., Mordente, A., and Tavian, D. (2019). Neutral lipid storage diseases as cellular model to study lipid droplet function. Cells 8, 187. doi:10.3390/cells8020187
Missaglia, S., Maggi, L., Mora, M., Gibertini, S., Blasevich, F., Agostoni, P., et al. (2017). Late onset of neutral lipid storage disease due to novel PNPLA2 mutations causing total loss of lipase activity in a patient with myopathy and slight cardiac involvement. Neuromuscul. Disord. 27, 481–486. doi:10.1016/j.nmd.2017.01.011
Missaglia, S., Tavian, D., and Angelini, C. (2022). Neutral lipid storage disease with myopathy: a 10-year follow-up case report. Eur. J. Transl. Myol. 32, 10645. doi:10.4081/ejtm.2022.10645
Pasanisi, M. B., Missaglia, S., Cassandrini, D., Salerno, F., Farina, S., Andreini, D., et al. (2016). Severe cardiomyopathy in a young patient with complete deficiency of adipose triglyceride lipase due to a novel mutation in PNPLA2 gene. Int. J. Cardiol. 207, 165–167. doi:10.1016/j.ijcard.2016.01.137
Pennisi, E. M., Arca, M., Bertini, E., Bruno, C., Cassandrini, D., D’amico, A., et al. (2017). Neutral lipid storage diseases: clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. Rare Dis. 12, 90–10. doi:10.1186/s13023-017-0646-9
Samukawa, M., Nakamura, N., Hirano, M., Morikawa, M., Sakata, H., Nishino, I., et al. (2020). Neutral lipid storage disease associated with the PNPLA2 gene: case report and literature review. Eur. Neurol. 83, 317–322. doi:10.1159/000508346
Schreiber, R., Xie, H., and Schweiger, M. (2019). Of mice and men: the physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta - Mol. Cell. Biol. Lipids 1864, 880–899. doi:10.1016/j.bbalip.2018.10.008
Schweiger, M., Lass, A., Zimmermann, R., Eichmann, T. O., and Zechner, R. (2009). Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. - Endocrinol. Metab. 297, E289–E296. doi:10.1152/ajpendo.00099.2009
Shi, J., Qu, Q., Liu, H., Zhang, Y., Cui, W., Chen, P., et al. (2021). Case report: PNPLA2 gene complex heterozygous mutation leading to neutral lipid storage disease with myopathy. Front. Integr. Neurosci. 14, 554724–554725. doi:10.3389/fnint.2020.554724
Tavian, D., Maggi, L., Mora, M., Morandi, L., Bragato, C., and Missaglia, S. (2021). A novel PNPLA2 mutation causing total loss of RNA and protein expression in two NLSDM siblings with early onset but slowly progressive severe myopathy. Genes. Dis. 8, 73–78. doi:10.1016/j.gendis.2019.07.006
Tavian, D., Missaglia, S., Redaelli, C., Pennisi, E. M., Invernici, G., Wessalowski, R., et al. (2012). Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: a strikingly low amount of lipase activity May preserve cardiac function. Hum. Mol. Genet. 21, 5318–5328. doi:10.1093/hmg/dds388
Wang, S., Wu, S., and Peng, D. (2024). Dilated cardiomyopathy caused by mutation of the PNPLA2 gene: a case report and literature review. Front. Genet. 15, 1415156. doi:10.3389/fgene.2024.1415156
Yamada, K., Yaguchi, H., Abe, M., Ishikawa, K., Tanaka, D., Oshima, Y., et al. (2023). Neutral lipid storage disease with myopathy with a novel homozygous PNPLA2 variant. Clin. Neurol. Neurosurg. 228, 107670. doi:10.1016/j.clineuro.2023.107670
Keywords: case report, early onset myopathy, NLSDM, ATGL, nonsense-mediated RNA decay, no protein production
Citation: Missaglia S, Martegani E, Angelini C, Horvath R, Karcagi V, Pal E and Tavian D (2025) Case Report: Pathogenic PNPLA2 variants and nonsense-mediated mRNA decay result in an early-onset neutral lipid storage disease with myopathy. Front. Genet. 16:1642442. doi: 10.3389/fgene.2025.1642442
Received: 06 June 2025; Accepted: 21 July 2025;
Published: 21 August 2025.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Antonia Ribes, Hospital clinic, BarcelonaAlessandra Ruggieri, IRCCS Carlo Besta Neurological Institute Foundation, Italy
Copyright © 2025 Missaglia, Martegani, Angelini, Horvath, Karcagi, Pal and Tavian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Tavian, ZGFuaWVsYS50YXZpYW5AdW5pY2F0dC5pdA==
†These authors have contributed equally to this work and share first authorship