 Xiang Ding1,2†
Xiang Ding1,2† Runlang Su
Runlang Su- 1School of Mechanical and Electrical Information, Yiwu Industrial and Commercial College, Jinhua, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Fujian Port & Waterway Investigation and Design Institute Limited, Fuzhou, China
Embryonic development in honeybees is a critical stage that shapes the formation of organs and structures in adult bees. Although there has been substantial progress in transcriptome studies on honeybee embryogenesis, time-series transcriptomic and alternative splicing data in the embryonic development of the Asian honeybee (Apis cerana) remain limited. In this study, we conducted an in-depth analysis of RNA-seq data from public databases to examine the transcriptomic profiles at three key developmental stages of A. cerana embryos (24 h, 48 h, and 72 h), uncovering the dynamic changes in gene expression and alternative splicing across these stages. Results showed that the number of differentially expressed genes and alternative splicing events peaked at 24 and 48 h and then gradually decreased. Time-series transcriptomic analysis further identified key physiological and biochemical processes at these stages, reflecting a progression from foundational metabolism and cellular structure construction in the early stages to cell differentiation and organogenesis in the middle stage, and finally to functional structure refinement and behavioral trait formation in the later stage. Notably, our study highlighted the central role of alternative splicing and gene expression in driving key physiological and morphological changes during embryogenesis. We identified multiple key genes, including DMRT family genes, the Maelstrom (Mael) gene, and highly GO-enriched genes such as Dll, CaMKII, and Cnn. These genes not only play essential roles in structural formation but also support neurodevelopment and the emergence of complex behavioral patterns in adult bees. Gene expression and splicing patterns at different developmental stages provide new insights, revealing the early foundational mechanisms underlying limb development, behavior and memory, sensory organ development, and neural plasticity in honeybees.
1 Introduction
The Asian honeybee (Apis cerana) is one of the primary bee species widely cultivated in East Asia, with a rich beekeeping tradition in China, Japan, and India, contributing significantly to the beekeeping industry. Unlike Western honeybees, A. cerana has an extremely high sensitivity to scents, allowing it to make more effective use of dispersed nectar sources, making it well-suited to gather from a variety of nectar plants, whereas Western honeybees tend to prefer large-scale, single nectar sources. Additionally, A. cerana demonstrates outstanding disease resistance and adaptability, showing resilience to cold temperatures, lower feed requirements, and a longer foraging period (Alamancos et al., 2015). These characteristics give A. cerana a unique advantage in diverse ecological environments, contributing to sustainable agricultural and ecological development.
The development of honeybees consists of four life cycle stages: embryo, larva, pupa, and adult (Cantalapiedra et al., 2021). Embryonic development, the earliest and most critical stage, is precisely regulated by environmental factors and intracellular signals (Chen et al., 2016). During this stage, the rudimentary organs of the adult bee are gradualy formed, making the bee embryo an ideal model for genetic modification (Chen et al., 2011).
With the decreasing costs of long-read sequencing and advancements in sequencing technology, high-quality genome assembly for non-model organisms has become achievable. The first high-quality genome of the Italian honeybee was published in 2019 (Chen et al., 2019), followed by the successful assembly of the first high-quality A. cerana genome in 2020 (Alamancos et al., 2015). Improved reference genome assembly has provided multiple advantages for RNA-seq data analysis, including higher efficiency in read alignment, increased accuracy in identifying differentially expressed genes, and enhanced precision in detecting alternatively spliced transcripts. These benefits result from the high-quality genome’s reduction of the effects from single nucleotide polymorphisms (SNPs) and annotation inconsistencies, thereby improving the overall accuracy and reliability of transcriptomic studies (Cridge et al., 2017).
Proteomics and transcriptomics are key research areas in studying honeybee embryonic development. Previous studies have found distinct phosphorylation patterns in worker and drone bees during their respective developmental stages, with worker bee embryos showing significantly enhanced phosphorylation signals during germ layer formation and organogenesis (Chen et al., 2011). Given the 3-day duration of the honeybee embryonic development period, research has primarily focused on sampling at 24, 48, and 72 h to capture critical developmental stages (Chen et al., 2016; Chen et al., 2011; Fang et al., 2015; Garcia-Blanco, 2005). These studies lay an important foundation for understanding the molecular regulatory mechanisms during honeybee embryonic development.
Alternative splicing (AS) is a crucial focus in transcriptomics research, greatly enhancing protein diversity by enabling plants and animals to produce different protein isoforms to adapt to environmental changes. AS expands the coding capacity of genes, adding unparalleled complexity to transcriptomes and proteomes. This complexity varies across developmental stages, tissues, and environmental perturbations, significantly impacting gene expression regulation (Haas et al., 2013). Studies have shown that AS is prevalent across various species, correlating with traits like drought resistance (Heuer et al., 1995), agronomic traits (Hu et al., 2018), and heat resistance (Kim et al., 2015) in plants. Likewise, in animals such as brine shrimp, birds, zebrafish, and sea cucumbers, AS is closely linked to sex regulation (Li et al., 2009; Li and Kaufman, 1996; Liao et al., 2014; Lin et al., 2022). However, the spatiotemporal dynamics of AS during honeybee embryogenesis remain largely unexplored.
Through RNA-Seq analysis, time-series transcriptome analysis, and alternative splicing analysis, we identified stage-specific gene expression patterns during honeybee embryonic development. Our findings reveal several non-coding genes that may play key roles in embryonic differentiation and identify three core genes driving complex developmental processes. These core genes are pivotal in specific alternative splicing events, contributing to the regulation of limb development, photoreception, and memory function. Overall, these data highlight the diversity and complexity of AS in honeybee embryogenesis.
In summary, our research deepens the understanding of honeybee embryonic development and its molecular mechanisms, offering a comprehensive view of the physiological and biochemical processes involved. Our findings provide valuable candidate genes for future developmental research and hold significant potential for applications.
2 Materials and methods
2.1 Source of sequence data
The analyses in this study are based on publicly available sequence data from the NCBI GenBank database under accession number SRP131292 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP131292) (Chen et al., 2016). The datasets used and analyzed were obtained from this publication and can be requested from the corresponding author of the original study upon reasonable request.
2.2 Mapping and quality control of RNA-seq data
Before alignment, the raw data were quality-checked and cleaned using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Specifically, we used the default parameters of FastQC to assess quality scores (Q values), GC content, and the proportion of N bases in the reads. The filtered reads were then aligned to the reference genome (GCA_029169275.1) using Hisat2 (v2.2.1) with the following parameters: -max-intronlen 10,000 (maximum intron length set to 10,000 bp), --dta (optimization for downstream transcript assembly), and--phred33 (Phred+33 quality score encoding) (Wang et al., 2021a). The resulting SAM files were converted to BAM format, sorted, and indexed with Samtools (v1.6) (Ma et al., 2022). Quantification of aligned reads was performed using FeatureCounts (Patro et al., 2017). Finally, differential expression analysis was conducted using Trinity (v2.15.1) with the DESeq2 algorithm (Rogers et al., 2021).
2.3 GO and KEGG analysis
Using emapper. py (v2.1.10) with the eggNOG database (Roth et al., 2023), GO and KEGG annotations were performed on the protein sequence of the longest csd sequence in the reference genome of A. cerana. The annotated file was then converted into org. db format to facilitate GO and KEGG enrichment analysis using the clusterProfiler (v4.0) package in R (Setton and Sharma, 2018).
2.4 Temporal clustering analysis of genes
Time-series transcriptome analysis was conducted on significant genes in the expression matrix using the ClusterGVis package in R (https://github.com/junjunlabClusterGVis). By clustering similar genes, gene modules in different clusters were analyzed to understand gene expression patterns across various embryonic stages. The ClusterGVis package integrates fuzzy C-means (Mfuzz) and K-means clustering algorithms, allowing for efficient clustering analysis of gene expression matrices and providing insights into gene expression trends at each time point.
2.5 Alternative splicing analysis workflow
For alternative splicing analysis, filtered sequencing reads were aligned to the reference genome (GCA_029169275.1) using the Salmon software, which provides a fast and accurate tool for quantifying transcript expression. Salmon aligns the reads pseudo-selectively, allowing for quantification without the need to generate large alignment files. This approach is efficient in handling complex transcriptomes, especially when exploring alternative splicing events across multiple samples (Slabaugh et al., 2019). Next, alternative splicing events were identified using the SUPPA (Simulation of Uncertainty and Prediction of Alternative splicing) tool (Song et al., 2020; Tao et al., 2021). SUPPA leverages transcript-level quantification data from Salmon and annotation files (e.g., GTF format) to generate alternative splicing events, such as exon skipping, alternative 5′ and 3′ splice sites, and intron retention. By applying SUPPA’s splicing event generation functionality (suppa.py), the annotated files were processed to identify splicing variations across conditions, providing insights into how alternative splicing patterns may vary between developmental stages or treatment groups (Song et al., 2020; Tao et al., 2021).
3 Results
3.1 Overall RNA-Seq results and quality control
The Illumina RNA sequencing generated an average of 86.89 million (M) raw reads per sample. After filtering, an average of 86.59 million clean reads per sample was retained for subsequent transcriptome analysis. The average quality scores for clean reads showed Q20 and Q30 ratios of 100%, indicating high quality of the obtained clean reads. Additionally, the average GC content was 38.24% (Supplementary Table S1). We used HISAT2 software to perform alignment analysis with the reference genome. The sliding window density analysis showed that over 80% of the transcripts were uniquely aligned to the reference genome, with an average alignment rate of 83.33% across nine samples. Only 15.66% of the reads (7.97 million) failed to align to the genome (Supplementary Table S2).
3.2 Hierarchical clustering and principal component analysis of samples
To assess the consistency of sample collection and explore the transcriptomic relationships among embryos at 24, 48, and 72 h, we conducted hierarchical clustering and principal component analysis (PCA) on the global gene expression data of these samples. These two analytical methods summarize the similarities and differences among the samples. Prior to these analyses, we normalized the gene expression levels using the regularized log transformation method implemented in the limma package. The results showed that the 48-hour and 72-hour samples were more similar to each other, while both time points exhibited significant differences compared to the 24-hour samples, consistent with previous studies (Figure 1a). In the PCA plot (Figure 1b), embryo samples collected at the same time point clustered together, further validating the results of the hierarchical clustering analysis. Notably, the 24-hour samples were distinctly separated from the 48-hour and 72-hour samples along the first principal component (PC1), indicating that PC1 primarily reflects the gene expression differences between the 24-hour stage and subsequent developmental stages. Meanwhile, the 48-hour and 72-hour samples were differentiated along the second principal component (PC2), suggesting additional transcriptomic differences between these two time points. Moreover, we found that the Apd−2 and HEX70b gene had high loadings on both PC1 and PC2 (Supplementary Figure S1), indicating its significant role in distinguishing samples from different developmental stages. Both analyses demonstrate that gene expression at 24 h of embryonic development was significantly different from the subsequent two stages, suggesting that the most substantial changes and regulation in gene expression occur between 24 and 48 h.
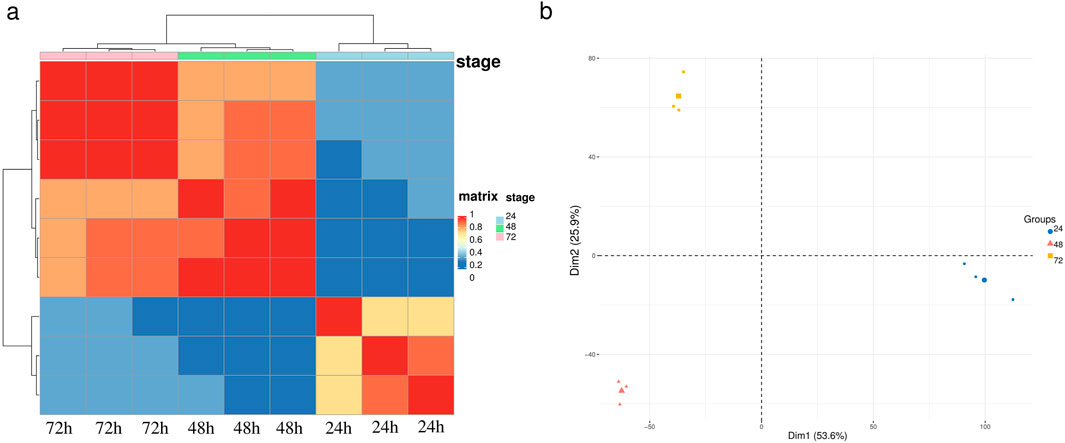
Figure 1. Sample clustering for embryos of A. cerana. (a) Heat map and hierarchical clustering of the embryo samples using the whole transcriptome data. (b) Principal component analysis (PCA) of the embryo samples using the whole transcriptome data.
3.3 Analysis of differentially expressed genes (DEGs)
The volcano plots (Figure 2b) visually illustrate the overall trends in gene expression levels across stages. Each point represents a gene, with the horizontal axis showing the logarithmic fold change in gene expression (log2 Fold Change) and the vertical axis showing the negative logarithm of the significance level (−log10 p-value). Significantly upregulated genes were distributed in the upper right of the plot, while significantly downregulated genes appear in the upper left. It is evident that the number of differentially expressed genes is highest during the 24-hour to 48-hour stage; the volcano plot displayed a more dispersed pattern, indicating the most dramatic changes in gene expression occur at this stage. This was consistent with the previous hierarchical clustering and principal component analysis results, further confirming that the period from 24 to 48 h was a critical turning point in embryonic development, during which significant reprogramming of gene expression occurs.
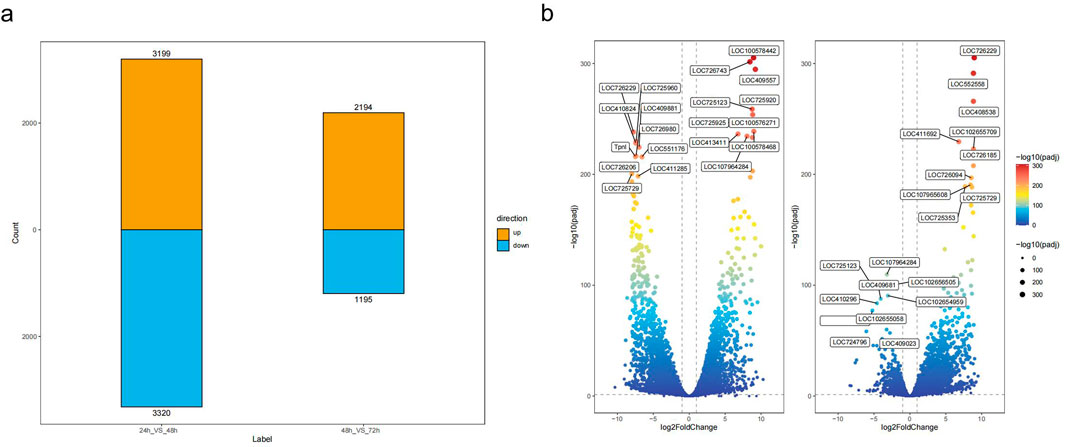
Figure 2. Differential Gene Expression Analysis. (a) Statistical summary of differentially expressed genes across the developmental stages, showing the count of upregulated (orange) and downregulated (blue) genes in the comparisons between 24 h vs. 48 h and 48 h vs. 72 h. (b) Volcano plots illustrating the differential expression of genes across three embryonic developmental stages. The left plot shows the comparison between 24 h vs. 48 h, and the right plot shows the comparison between 48 h vs. 72 h. Genes with |log2FoldChange| > 1 and -log10 (padj) > 0.05 are displayed, with labels indicating the most significantly differentially expressed genes. Gene color represents the degree of significance based on the -log10 (padj) values, and the size of the points reflects the fold change.
In contrast, from 48 h to 72 h, the number of significantly differentially expressed genes in the volcano plot decreases markedly, and the plot becomes more concentrated. This suggested that the degree of gene expression changes was reduced during this stage, and the embryonic development process enters a relatively stable period.
The genes showing significant differential expression in these volcano plots may be closely related to important physiological and biochemical processes, morphological formation, and functional differentiation at each embryonic stage. Upregulated genes may be involved in cell proliferation, differentiation, and the acquisition of specific functions, while downregulated genes may be associated with the regulation of the cell cycle and adjustments in metabolic activities. Further functional annotation and pathway enrichment analyses of these differentially expressed genes will help reveal their specific roles and regulatory mechanisms in embryonic development, providing important insights into the molecular basis of embryogenesis.
3.4 Temporal transcriptome analysis of embryonic development using k-means clustering
To further explore the dynamic changes in gene expression during embryonic development, we employed a k-means clustering algorithm to partition 9,492 genes into 10 clusters based on the similarity of their expression patterns. Detailed analysis of these clusters revealed significant enrichment of genes at specific developmental stages (Figure 3).
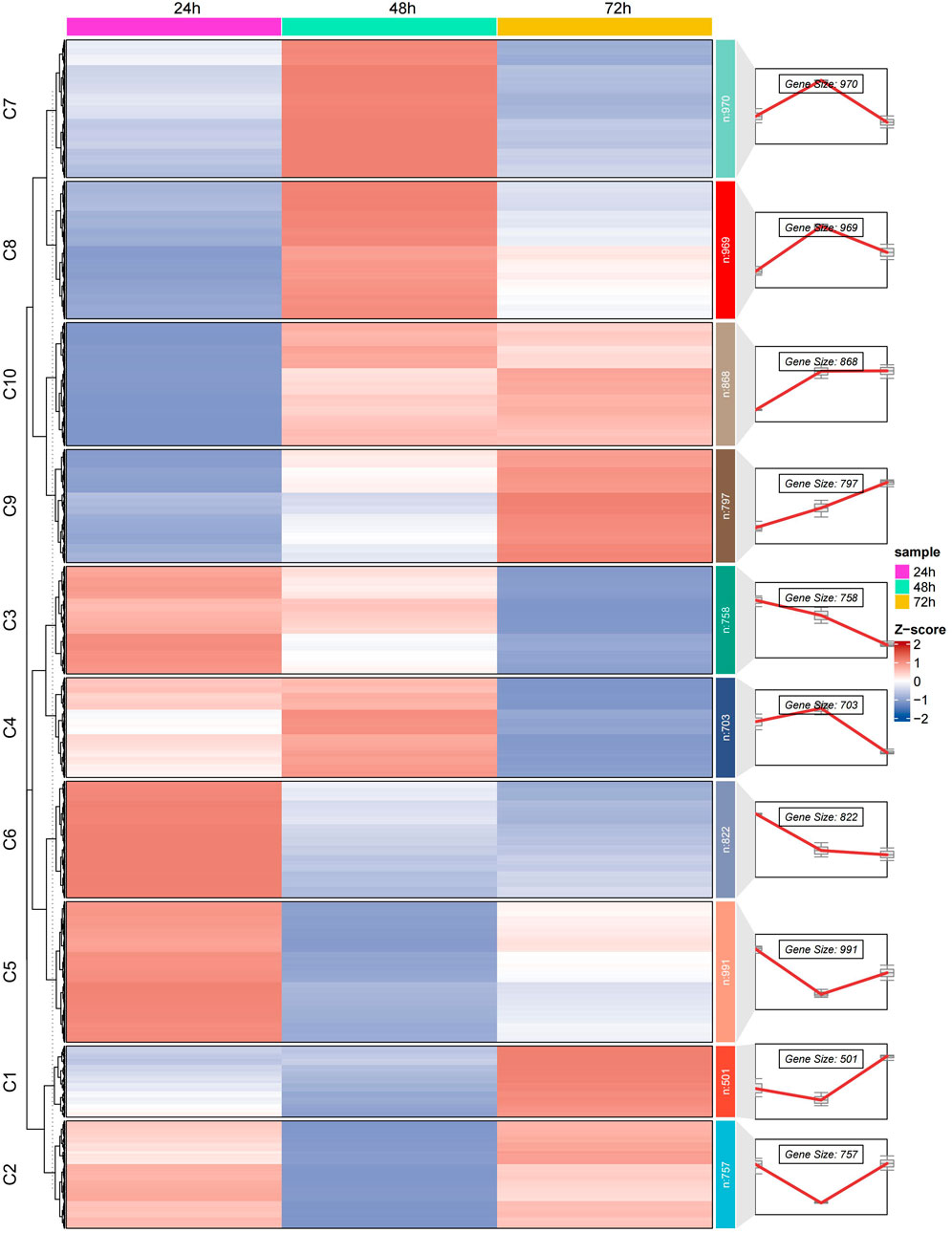
Figure 3. Illustrates the dynamic changes in gene expression during the embryonic development of the Asian honeybee (A. cerana). Using k-means clustering analysis, we divided the gene expression patterns into 10 clusters. The heatmap on the left side of the figure shows the gene expression levels at various developmental stages, while theline plots on the right depict the trends in gene expression at three key time points during embryonic development (24 h, 48 h, and 72 h). The clusters are labeled from C1 to C10.
At the 24-hour embryonic stage, genes in clusters C3, C5, and C6 exhibited significant enrichment. The associated Gene Ontology (GO) terms mainly include fatty acid derivative metabolic processes, biosynthetic processes of organic acids and carboxylic acids, key complexes related to transcription and translation, mRNA metabolism and splicing, and the metabolic processing of rRNA and ncRNA (Supplementary Tables S3, S4, S5). These results indicated that at the 24-hour stage, gene expression primarily involves fundamental metabolic processes and the establishment of gene expression machinery, emphasizing the construction of basic cellular components and functions required for early development.
Overall, these genes play crucial roles in multiple key processes and functions of cell biology and developmental biology throughout various stages of embryonic development. As development progresses into the 48-hour stage, genes in clusters C7 and C8 showed high expression levels. The related GO terms were concentrated on the positive regulation of biological and cellular processes, post-embryonic development, larval or pupal development, appendage morphogenesis, and RNA synthesis, modification, and processing (Supplementary Tables S6, S7). This suggested that at the 48-hour stage, genes were involved in developmental programs that initiated morphological changes and cell differentiation, promoting the formation of specific structures while continuing RNA processing activities. Further, at the 72-hour stage, genes in clusters C9 and C10 become predominant. The associated GO terms mainly pertain to protein synthesis processes, the structure and function of ribosomes, appendage development and morphogenesis (including imaginal disc-derived appendage development, appendage morphogenesis, and leg disc development), and specific molecular binding and enzymatic activities (such as signal receptor binding, heme binding, tetrapyrrole binding, and nucleotide monophosphate kinase activity) (Supplementary Tables S8, S9).
These resulted reflect that at the 72-hour stage, gene expression focuses on protein synthesis and the role of ribosomes as the core machinery for protein production, promoting further development of appendages and demonstrating diverse functions in molecular binding and catalysis, indicating an advanced stage of tissue formation and functional specialization.
3.5 Alternative splicing and its role in gene regulation during early embryonic development
Alternative splicing analysis validated the expression levels of different gene transcripts at three time points during embryonic development. The results showed that the numbers of alternatively spliced genes and splicing events at these three time points significantly overlapped with the distribution of differentially expressed genes. During the critical developmental stage from 24 to 48 h, the counts of all seven types of splicing events reached their highest levels, with Alternative First Exon (AF) events being the most abundant, totaling 255 (Figures 4a,b).
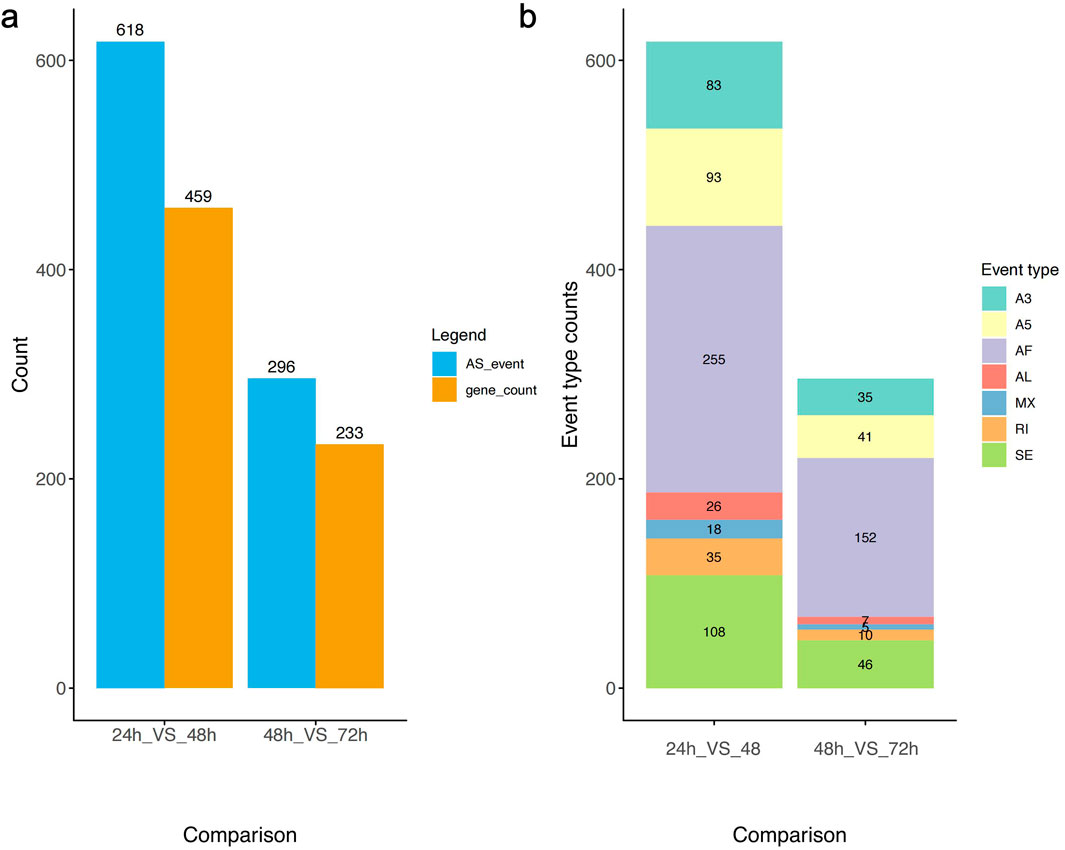
Figure 4. Differential isoforms expression analysis. (a) Variable splicing events and the corresponding gene count in three time points of A. cerana embryonic development. (b) Graph showing the number of seven types of alternative splicing events across three time points of A. cerana embryonic development.
The high frequency of AF events may be related to the complexity and diversity of gene expression regulation in the early stages of embryonic development. Between 24 and 48 h a pivotal developmental period embryonic cells undergo rapid proliferation and differentiation, necessitating precise and flexible mechanisms of gene expression regulation. AF events, by selecting different initial exons, can produce protein isoforms with varying N-terminal structures, thereby influencing protein localization, function, and interactions with other molecules. This mechanism allowed the same gene to perform diverse functions in different cell types or developmental stages.
3.6 GO enrichment in alternative splicing events
The results of GO enrichment analysis (Figure 5) indicate that significantly differentially spliced transcripts in embryos are primarily involved in biological processes such as morphogenesis, growth, cytoskeleton organization, and the formation of specific organs and appendage structures. Specifically, these functions encompass biological processes such as cytoskeleton organization, morphogenesis, neuromuscular junction formation, axon development, cell polarity, protein polymerization, cell junction organization, organ development, and cell structure formation (Supplementary Table S10). These processes include core functions such as cell differentiation, signal transduction, structural assembly, and cell-cell junctions, revealing the role of complex regulatory networks in maintaining and modulating the morphology and function of cells and tissues during development.
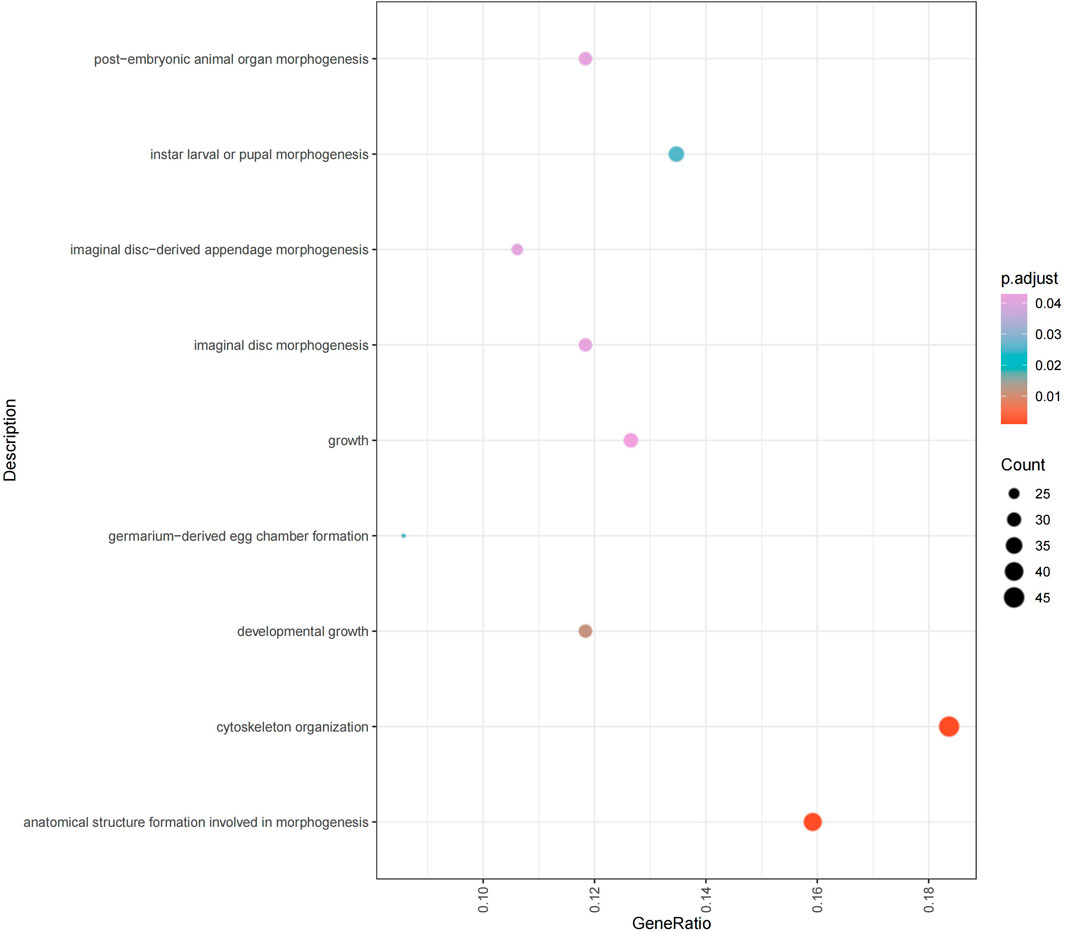
Figure 5. Embryonic development alternative splicing GO enrichment chart (p.adjust<0.05).
These findings suggest that alternative splicing events may play an essential role in regulating the formation and maintenance of specific cellular structures at critical stages of embryonic development. For instance, the dynamic regulation of cytoskeleton and cell junctions is crucial for maintaining cell morphology, differentiation, and migration, while regulation of protein polymerization and axon development may be pivotal in the formation of the nervous system. Additionally, these differentially spliced transcripts may have significant roles in the morphogenesis and physiological functions of specific organs and appendages (e.g., muscle and nervous system), highlighting the highly refined gene expression regulatory mechanisms during embryonic development. These insights provide important clues for further exploration of how alternative splicing mediates functional diversity of genes and precise regulation in specific developmental processes.
3.7 Alternative splicing dynamics of key genes Dll, CaMKII, and LOC408563 during embryonic development
To investigate the key genes involved in alternative splicing events during embryonic development, we selected three genes with the highest connectivity in GO enrichment analysis: Dll, CaMKII, and LOC408563. Our results showed that the Dll gene exhibited exon-skipping events at both the 24-h and 48-h time points, with each time point featuring unique exon skipping events. Additionally, at 72 h, an additional exon skipping event was observed compared to the 48-h time point (Figure 6).
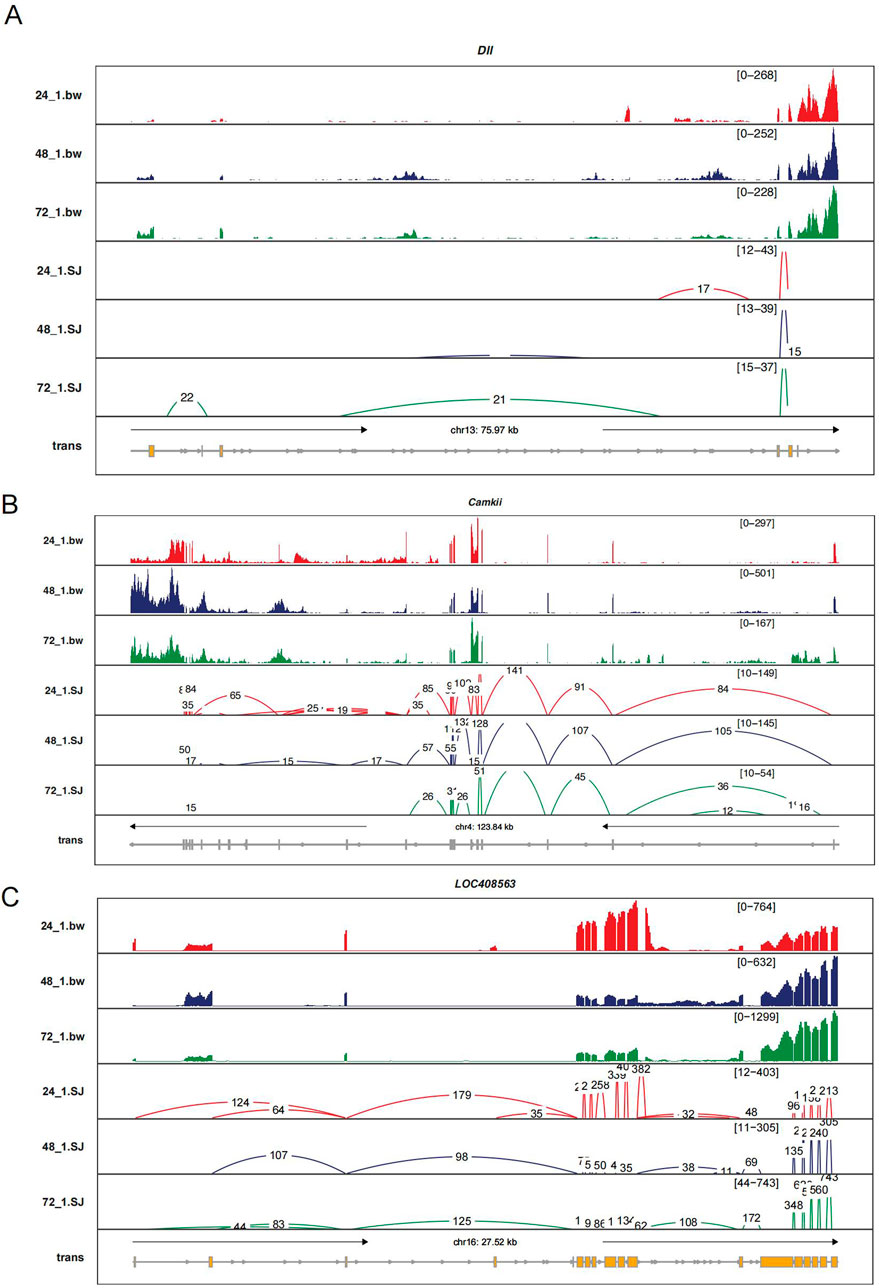
Figure 6. Sashimi Plot Illustrating Levels of Intron Retention and Exon Usage for Three Genes (Dll, CaMKII, LOC408563). (a) Gene Dll, (b) CaMKII, and (c) LOC408563. In the legend, yellow represents exons, with red, blue, and green lines corresponding to time points 24 h, 48 h, and 72 h, respectively. The connected line segments represent the gene segments that are linked together. (The data represented here only includes contigs with a count greater than 10, as we filtered out contigs with lower counts to focus on more reliable and significant splicing events.).
In the CaMKII gene, compared to 48 h, the 24-hour time point had two additional exon skipping events and one intron retention event. At 72 h, CaMKII showed one unique intron retention event but had three fewer exon skipping events and one fewer intron retention event. The LOC408563 gene exhibited one more exon skipping event and one more intron retention event at 24 h compared to 48 h. Conversely, at 48 h compared to 24 h, it had one unique intron retention event. Compared to 72 h, the 48-hour time point had two more exon skipping and one more intron retention event, while at 72 h, a unique intron retention event was present (Figure 6).
The changes in these splicing events at different developmental stages may suggest distinct roles for these genes in regulating embryonic development. Time-specific splicing patterns likely relate to the precise regulatory requirements of gene expression, with such dynamic splicing mechanisms enabling the production of different protein isoforms to meet the varying functional needs of cells during development. Thus, these alternative splicing events could represent an important aspect of the regulatory complexity in embryonic development.
To further validate the reliability of our public database-based analysis results, we performed independent verification of the identified differentially spliced genes using quantitative real-time PCR (qRT-PCR). Specifically, we selected three representative genes, Dll (XM_006560304, XM_006560302.3), CaMKII (XM_00650514.3), and LOC408563 (XM_006563138.3), and validated the transcripts corresponding to the relatively unique splicing events at the 24-hour, 48-hour, and 72-hour time points. The results demonstrated that the alternative splicing patterns of these three genes were highly consistent with those predicted by our bioinformatics analysis, confirming the accuracy and reliability of our analytical pipeline (Figure 7; Supplementary Table S11).
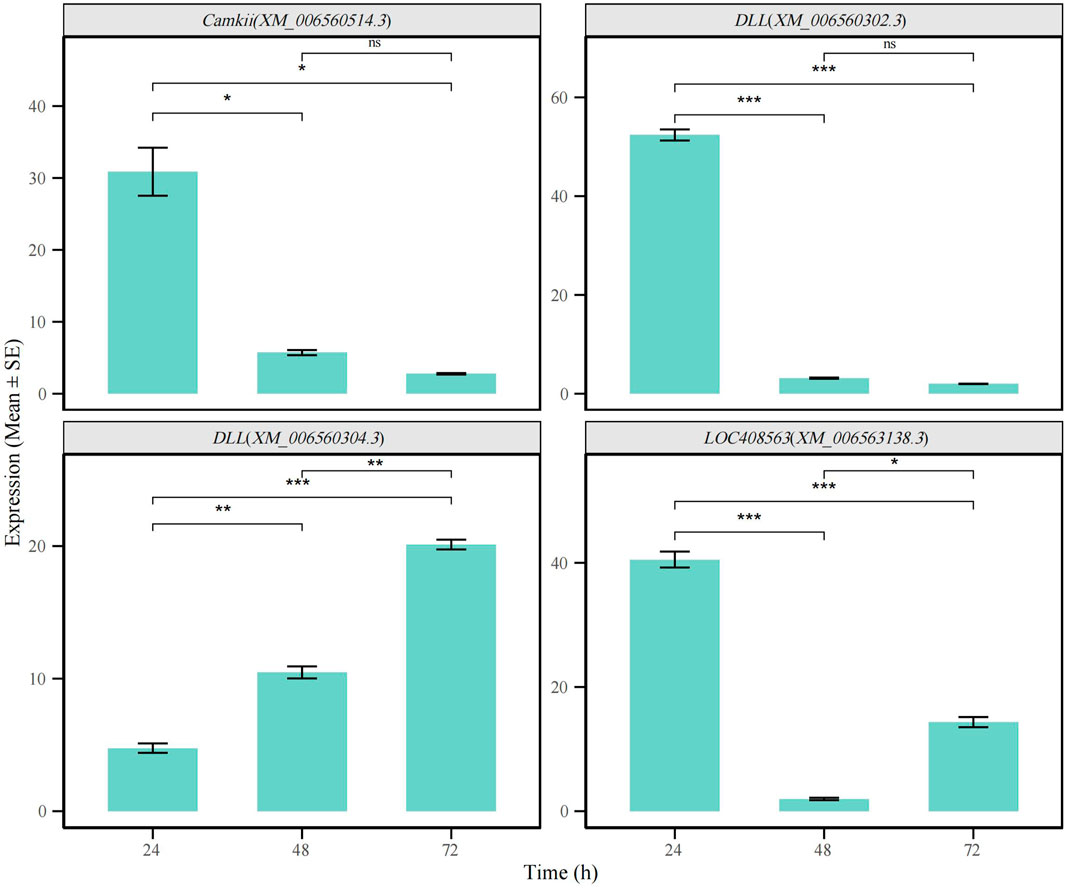
Figure 7. qRT-PCR validation of alternative splicing events in CaMKII, Dll, and LOC408563 genes across different time points (t-test,p < 0.05). (*p < 0.05, **p < 0.01, ***p < 0.001).
4 Discussion
To investigate the gene expression levels and alternative splicing changes in Asian domestic honeybee embryos against a high-quality genome background, we conducted RNA-seq analysis on embryos at three developmental time points: 24 h, 48 h, and 72 h. Compared with previous studies (Chen et al., 2016), the high-quality genome significantly enhanced alignment efficiency and the number and accuracy of identified differentially expressed genes (Figure 2a; Supplementary Table S2). Additionally, principal component analysis (PCA) and clustering heatmap analysis further validated the reliability and reproducibility of our data, with conclusions consistent with earlier research, including the expression of photoreception genes (Figures 1a,b).
Through volcano plot analysis, we identified genes with significant differential expression between different time points. Notably, LOC726743 exhibited significantly high expression during the 24 h–48 h period. This gene belongs to the DMRT transcription factor family, known in Drosophila with the dsx gene as a representative that regulates sex-specific behaviors (Trincado et al., 2018). This finding suggests that differential expression of DMRT family genes may have been activated early in honeybee embryonic development, participating in sex differentiation and the formation of worker bee-specific behavioral patterns.
Furthermore, we discovered that the gene LOC409557 (encoding the Maelstrom protein) showed significantly high expression during the 24 h–48 h period. The Maelstrom protein plays a crucial role in spermatogenesis in silkworms, and its high expression in honeybees may imply specific functions in worker bee development, particularly in regulating genome stability and cell differentiation (Viet et al., 2022). We also identified unknown function genes with significantly high expression at different time points, such as LOC100578442, LOC726229, and LOC552558.
To gain deeper insights into the dynamic changes in gene expression during embryonic development, we performed time-series transcriptome analysis. The results showed that honeybee embryonic development gradually transitions from basic metabolism and gene expression activities to advanced tissue formation and functional specialization (Figure 3). At the 24 h stage, non-coding RNA metabolic regulation plays a critical role in preparing for subsequent cell differentiation. By the 48 h stage, gene expression shifts toward processes of cell specialization and morphological changes, initiating the formation of appendages and differentiated cell structures. At the 72 h stage, gene expression focuses on protein synthesis and ribosomal activity, supporting further structural and functional diversity.
We also found that the distribution of alternative splicing events highly overlaps with differentially expressed genes, both peaking in number during the 24 h–48 h period (Figures 4a,b), indicating more active gene alternative splicing in early embryonic development. Among these, alternative first exon (AF) events occur frequently at this stage, possibly because early development requires highly flexible gene expression regulatory mechanisms to meet the demands of rapid cell proliferation and differentiation (Wallberg et al., 2019).
Through GO enrichment analysis of alternative splicing events, we found they play significant roles in morphogenesis, organ development, cytoskeleton remodeling, and growth regulation (Figure 5). This suggests that alternative splicing supports the structural and functional needs of embryos during rapid development by regulating the expression of genes related to cell structure and development.
Further analysis revealed three high-frequency genes highly associated with multiple GO terms: Dll, CaMKII, and LOC408563 (Figures 6a–c). The Dll gene plays an essential role in arthropod development, regulating the formation of mouthparts, limbs, spider spinnerets, and other appendage structures (Wang X. et al., 2023; Wang Y. et al., 2023). Based on these findings, we speculate that in honeybees, the Dll gene not only participates in limb development but may also play a key role in the formation of mouthparts and stingers. The CaMKII gene is also crucial in nervous system development, especially in learning and memory processes. The alternative splicing of the CaMKII gene may provide a molecular basis for honeybee nervous system development, contributing to the complex behavioral patterns and environmental adaptability of workers (Wang et al., 2020; Wu et al., 2021). For the LOC408563 gene, homology analysis indicates it corresponds to the Cnn gene. In Drosophila, the Cnn gene primarily regulates photoreception, cytoskeletal stability, maintenance of cell polarity and morphology, and plays a pivotal role in cell division and nervous system development (Zalcman et al., 2018; Zhang et al., 2024). Besides, based on previous studies, sex differentiation in both Apis cerana and Apis mellifera is regulated by the Csd gene, which controls the expression of the sex-specific gene Fem through splicing, and further affects the sex-specific splicing of the downstream gene Dsx. Therefore, sex differentiation in Apis cerana and Apis mellifera follows similar pathways and genes (Su et al., 2025; Wang X. et al., 2021). This aligns with the conclusions from the GO enrichment analysis in the original article (Chen et al., 2016), suggesting that gene expression and alternative splicing in the early embryonic stage may collectively influence photoreception and neural development. The alternative splicing of the Cnn gene not only ensures the normal progression of the cell cycle but also supports the development and functional specialization of specific tissues, particularly in nervous system development, potentially laying the foundation for the subsequent behavioral complexity of honeybees.
In conclusion, the alternative splicing of high-frequency genes plays a pivotal role in the formation of key structures in early honeybee embryos and the precise regulation of the nervous system and other physiological features. Through gene alternative splicing, honeybee embryos can adapt to different developmental needs, gradually establishing a complex behavioral foundation and physiological characteristics. These results provide important evidence for further exploring the molecular mechanisms of honeybee behavior and functional differentiation, and also reveal the significance of alternative splicing in the embryonic development process.
5 Conclusion
Our research utilized high-quality genome data and further mined transcriptome resources from public databases, enhancing the analytical precision of gene expression and alternative splicing in Asian domestic honeybee (A. cerana) embryos. Compared with the original research that only used draft genomes for alignment and focused solely on gene expression, we further explored the dynamic changes of alternative splicing events and time-series transcriptomes. The identified differentially expressed genes and alternative splicing events, especially key genes such as the DMRT family, Mael gene, Dll, CaMKII, and Cnn, may play important roles in honeybee embryonic development, sex differentiation, and the formation of complex behaviors.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, SRP131292.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
XD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. ZL: Investigation, Software, Writing – review and editing, Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft. LO: Investigation, Software, Writing – review and editing. ZW: Formal Analysis, Methodology, Supervision, Visualization, Writing – original draft. QX: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review and editing. RS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research is supported by the Yiwu Industrial and Commercial College Program: Response of Phytoplankton Community to Climate Events in the Northeastern South China Sea under Project XJKJ2502YB. This work was also supported by Jinhua Public Welfare Technology Application Research Project in 2024: Key Technology Application Research on Multiple Access for QoS Guarantee inWireless Communication Networks under Project 2024-4-260.
Conflict of interest
Authors ZL, LO, ZW, QX, and RS were employed by Fujian Port & Waterway Investigation and Design Institute Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1665548/full#supplementary-material
References
Alamancos, G. P., Pages, A., Trincado, J. L., Bellora, N., and Eyras, E. (2015). Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21, 1521–1531. doi:10.1261/rna.051557.115
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi:10.1093/molbev/msab293
Chen, G., Rogers, A. K., League, G. P., and Nam, S.-C. (2011). Genetic interaction of centrosomin and bazooka in apical domain regulation in drosophila photoreceptor. PLoS One 6, e16127. doi:10.1371/journal.pone.0016127
Chen, B., Piel, W. H., and Monteiro, A. (2016). Distal-less homeobox genes of insects and spiders: genomic organization, function, regulation and evolution. Insect Sci. 23, 335–352. doi:10.1111/1744-7917.12327
Chen, K., Chen, S., Xu, J., Yu, Y., Liu, Z., Tan, A., et al. (2019). Maelstrom regulates spermatogenesis of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 109, 43–51. doi:10.1016/j.ibmb.2019.03.012
Cridge, A. G., Lovegrove, M. R., Skelly, J. G., Taylor, S. E., Petersen, G. E. L., Cameron, R. C., et al. (2017). The honeybee as a model insect for developmental genetics. genesis 55, e23019. doi:10.1002/dvg.23019
Fang, Y., Feng, M., Han, B., Qi, Y., Hu, H., Fan, P., et al. (2015). Proteome analysis unravels mechanism underling the embryogenesis of the honeybee drone and its divergence with the worker (apis mellifera lingustica). J. Proteome Res. 14, 4059–4071. doi:10.1021/acs.jproteome.5b00625
Garcia-Blanco, M. A. (2005). Methods for the study of alternative splicing. Methods 37, 289–291. doi:10.1016/j.ymeth.2005.10.001
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi:10.1038/nprot.2013.084
Heuer, J. G., Li, K., and Kaufman, T. C. (1995). The drosophila homeotic target gene centrosomin (Cnn) encodes a novel centrosomal protein with leucine zippers and maps to a genomic region required for midgut morphogenesis. Development 121, 3861–3876. doi:10.1242/dev.121.11.3861
Hu, X., Ke, L., Wang, Z., and Zeng, Z. (2018). Dynamic transcriptome landscape of Asian domestic honeybee (apis cerana) embryonic development revealed by high-quality RNA sequencing. BMC Dev. Biol. 18, 11. doi:10.1186/s12861-018-0169-1
Kim, D., Langmead, B., and Salzberg, S. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi:10.1038/nmeth.3317
Li, K., and Kaufman, T. C. (1996). The homeotic target gene centrosomin encodes an essential centrosomal component. Cell 85, 585–596. doi:10.1016/S0092-8674(00)81258-1
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Lin, X., Liu, F., Meng, K., Liu, H., Zhao, Y., Chen, Y., et al. (2022). Comprehensive transcriptome analysis reveals sex-specific alternative splicing events in zebrafish gonads. Life 12, 1441. doi:10.3390/life12091441
Ma, B., Ma, C., Li, J., and Fang, Y. (2022). Revealing phosphorylation regulatory networks during embryogenesis of honey bee worker and drone (apis mellifera). Front. Cell Dev. Biol. 10, 1006964. doi:10.3389/fcell.2022.1006964
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi:10.1038/nmeth.4197
Rogers, T. F., Palmer, D. H., and Wright, A. E. (2021). Sex-specific selection drives the evolution of alternative splicing in birds. Mol. Biol. Evol. 38, 519–530. doi:10.1093/molbev/msaa242
Roth, J. F., Braunschweig, U., Wu, M., Li, J. D., Lin, Z.-Y., Larsen, B., et al. (2023). Systematic analysis of alternative exon-dependent interactome remodeling reveals multitasking functions of gene regulatory factors. Mol. Cell 83, 4222–4238.e10. doi:10.1016/j.molcel.2023.10.034
Setton, E. V. W., and Sharma, P. P. (2018). Cooption of an appendage-patterning gene cassette in the head segmentation of arachnids. Proc. Natl. Acad. Sci. 115, E3491–E3500. doi:10.1073/pnas.1720193115
Slabaugh, E., Desai, J. S., Sartor, R. C., Lawas, L. M. F., Jagadish, S. V. K., and Doherty, C. J. (2019). Analysis of differential gene expression and alternative splicing is significantly influenced by choice of reference genome. RNA 25, 669–684. doi:10.1261/rna.070227.118
Song, L., Pan, Z., Chen, L., Dai, Y., Wan, J., Ye, H., et al. (2020). Analysis of whole transcriptome RNA-Seq data reveals many alternative splicing events in soybean roots under drought stress conditions. Genes 11, 1520. doi:10.3390/genes11121520
Su, R., Chen, Y., Zhu, R., Ding, G., Dong, K., Feng, M., et al. (2025). Transcriptomic analysis reveals patterns of expression of stage-specific genes in early apis cerana Embryos. Genes (Basel) 16 (2), 187. doi:10.3390/genes16020187
Tao, W., Lee, J., Chen, X., Díaz-Alonso, J., Zhou, J., Pleasure, S., et al. (2021). Synaptic memory requires CaMKII. eLife 10, e60360. doi:10.7554/eLife.60360
Trincado, J. L., Entizne, J. C., Hysenaj, G., Singh, B., Skalic, M., Elliott, D. J., et al. (2018). SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19, 40. doi:10.1186/s13059-018-1417-1
Viet, D. N., Christiaens, O., De Vos, S., Smagghe, G., and Bossier, P. (2022). The sex-specific splicing of doublesex in Brine Shrimp Artemia franciscana. Genes 13, 1997. doi:10.3390/genes13111997
Wallberg, A., Bunikis, I., Pettersson, O. V., Mosbech, M. B., Childers, A. K., Evans, J. D., et al. (2019). A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics 20, 275. doi:10.1186/s12864-019-5642-0
Wang, Z. L., Zhu, Y. Q., Yan, Q., Yan, W. Y., Zheng, H. J., and Zeng, Z. J. (2020). A chromosome-scale assembly of the Asian honeybee Apis cerana Genome. Front. Genet. 11, 279. doi:10.3389/fgene.2020.00279
Wang, M., Chen, D., Zheng, H., Zhao, L., Xue, X., Yu, F., et al. (2021a). Sex-specific development in haplodiploid honeybee is controlled by the female-embryo-specific activation of thousands of intronic LncRNAs. Front. Cell Dev. Biol. 9, doi:10.3389/fcell.2021.690167
Wang, X., Lin, Y., Liang, L., Geng, H., Zhang, M., Nie, H., et al. (2021b). Transcriptional profiles of diploid mutant apis Mellifera embryos after knockout of csd by CRISPR/Cas9. Insects 12 (8), 704. doi:10.3390/insects12080704
Wang, X., Wang, Y., Wang, R., Gong, L., Wang, L., and Xu, J. (2023a). Genome-wide analysis of alternative splicing events responding to high temperatures in Populus tomentosa carr. Forests 14, 1878. doi:10.3390/f14091878
Wang, Y., Yang, Y., and Chen, M. (2023b). Identification of sex-specific splicing via comparative transcriptome analysis of gonads from sea cucumber Apostichopus japonicus. Comp. Biochem. Physiology Part D Genomics Proteomics 45, 101031. doi:10.1016/j.cbd.2022.101031
Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141. doi:10.1016/j.xinn.2021.100141
Zalcman, G., Federman, N., and Romano, A. (2018). CaMKII isoforms in learning and memory: localization and function. Front. Mol. Neurosci. 11, 445. doi:10.3389/fnmol.2018.00445
Keywords: Apis cerana, embryonic development, alternative splicing, time-seriestranscriptome, differentially expressed genes (DEGs)
Citation: Ding X, Liu Z, Ou L, Wang Z, Xu Q and Su R (2025) Time-series transcriptomics and alternative splicing analysis of embryonic development of the asian honeybee (Apis cerana). Front. Genet. 16:1665548. doi: 10.3389/fgene.2025.1665548
Received: 14 July 2025; Accepted: 28 August 2025;
Published: 02 October 2025.
Edited by:
Zemin Wang, Gansu Agricultural University, ChinaReviewed by:
Weiwei Liu, Chinese Academy of Sciences (CAS), ChinaXiaofei Wang, China Agricultural University, China
Copyright © 2025 Ding, Liu, Ou, Wang, Xu and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Runlang Su, c3VydW5sYW5nQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work