 Qisheng Hu
Qisheng Hu Feng Zhu1†
Feng Zhu1† Yiqing Jiang
Yiqing Jiang- 1Department of Neurology, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Taizhou, China
- 2Department of Radiation Oncology, University of Miami Miller School of Medicine, Miami, FL, United States
Childhood-onset ataxia, intention tremor and hypotonia syndrome (ATITHS) is a rare neurological disorder that encompasses features of hereditary ataxia, hypotonia. To date, only one report has associated the pathogenic variant in the POU4F1 gene with ATITHS. We report the case of a 28-year-old male who presented with lifelong gait instability and hypokinesia. The brain magnetic resonance imaging of this patient revealed significant cerebellar atrophy. Genetic analysis identified a novel heterozygous nonsense variant in Pou structural domain class 4 transcription factor 1 (POU4F1), which is predicted to result in loss of normal protein function. Segregation analysis within the family confirmed the presence of this variant in multiple symptomatic relatives. We confirmed diagnosis of ATITHS for this patient. This report provides additional evidence linking this mutation to specific neurologic disorders. We emphasize the importance of genetic testing to determine genetic etiology in patients presenting with ATITHS.
Introduction
Childhood-onset ataxia, intention tremors and hypotonia syndrome (ATITHS) is a rare neurologic disorder characterized by features of hereditary ataxia and hypotonia. It is proposed that the heterozygous loss-of-function (LOF) variant in POU4F1 is the causative agent of this novel ataxia syndrome (Webb et al., 2021).
POU4F1, also known as BRN3A, encodes a class IV POU domain–containing transcription factor (Herr et al., 1988). Its associated phenotype is primarily expressed in the nervous system and plays a key role in neuronal development. The gene produces two forms: a long form that contains an additional 84 amino acids at the N-terminus and a short form lacks this region. The C-terminal POU domain, common to both forms, activates a number of other neuron-expressed genes that stimulate neuronal growth (Latchman, 1998).
To our knowledge, only one report has associated the pathogenic variant in the POU4F1 gene with ATITHS. It reported four independent patients with ataxia, intention tremor, and hypotonia, identified 4 de novo, heterozygous, LOF variants in POU4F1(Webb et al., 2021).
In this study, we applied the Whole Exome Sequencing (WES) and Validated Sanger Sequencing Analysis to identify pathogenic mutations in a Chinese patient with walking instability. We report a 28-year-old man with a history of lower limb weakness and gait instability since childhood. Genetic analysis revealed a novel heterozygous nonsense variant in the POU4F1. Analysis of the family segregation prompted its reclassification as pathogenic. This is the first reported case of a nonsense variant of POU4F1 associated with this ataxia syndrome. It is also the first report of ATITHS involving a multigenerational pedigree.
Case presentation
A 28-year-old Han Chinese male was admitted to Taizhou Hospital with a lifelong history of gait instability. Dizziness with intermittent nausea has been reported over the past year. The patient had been hypokinetic and developmentally delayed since childhood, with poor performance in physical education, He did not begin walking until the age of six and was unable to run long distances and jump high during childhood. The patient’s father informed us that the patient had a tremor as a child, which disappeared in adulthood. In adulthood, he continues to experience unsteady gait and difficulty with weight-bearing ambulation. On examination, he showed fair physical development, weight 65kg, height 163cm, along with concurrent cognitive impairment and poor learning ability.
In the physical examination, the patient exhibited a wide-based gait and positive linear tandem walk test (Supplementary Video S1). Examination may reveal a positive heel-to-shin test, suggesting possible cerebellar ataxia. Tendon reflexes were normal, and Babinski sign was negative. Mild bilateral calf muscles atrophy was observed, with increased flaccidity during movement. We noticed hypotonia of the lower limbs. However, muscle strength was preserved in all four limbs. Notably, the patient demonstrated abnormal ocular motility, characterized by impaired up and down vision and abduction in both eyes (Supplementary Video S1). Intelligence testing revealed cognitive impairment, with an MMSE score of 17 and a MoCA score of 12. Further neuropsychological testing demonstrated deficits in processing speed, working memory, situational memory, visuospatial abilities, and verbal comprehension. Magnetic resonance imaging (MRI) showed dilatation of the fourth ventricle and cerebellar atrophy as shown in Figure 1A. The patient’s electroencephalogram (EEG) was within the normal range.
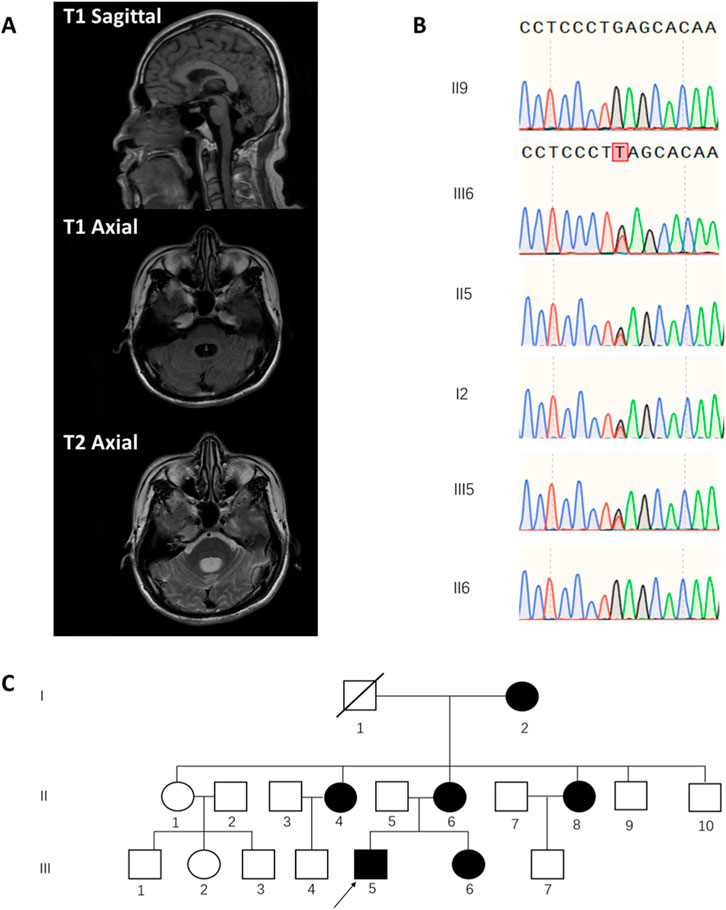
Figure 1. Clinical Imaging, Genetic Analysis, and Pedigree of a Patient with ATITHS Carrying a POU4F1 Nonsense Variant. (A) T1-and T2-weighted MRI showing dilatation of the fourth ventricle and cerebellar atrophy. (B) Sanger sequencing chromatograms showing the heterozygous nonsense variant, POU4F1. NM_006237.4:c.55G>T in the proband (III5), his sister (III6), mother (II6), and grandmother (I2), but absent in his healthy father (II5) and maternal uncle (II9). (C) Family pedigree chart showing segregation of the POU4F1 variant. (arrow indicates the proband).
A family survey revealed that the patient’s mother, sister, and maternal grandmother exhibited similar symptoms, including unsteady gait, bilateral knee inversion, and difficulty in weight-bearing ambulation. His sister and mother presented with noticeable ophthalmologic abnormalities, while sister underwent esotropia surgery 2 years ago. Additionally, the patient’s two maternal aunts were reported to have similar symptoms (Figure 1C for the family pedigree chart). Medical examinations and genetic sequencing could not be performed on all relatives owing to difficulties in obtaining informed consent from some family members.
To determine the genetic basis of this patient’s condition, we performed whole exome sequencing (WES) on DNA extracted from peripheral blood. We identified a heterozygous nonsense mutation, the POU4F1, NM_006237.4:c.55G>T(p.Glu19*) variant, The WES results were then validated by direct Sanger sequencing analysis. This variant leads to loss of normal protein function through nonsense-mediated mRNA decay or premature termination of amino acid translation. The pathogenicity of other LOF variants in POU4F1 has been previously reported and is supported by databases such as ClinVar and HGMD, fulfilling the ACMG PVS1 criterion. Additionally, this variant has not been previously reported in the literature or listed in the large population frequency database such as gnomAD, meeting the ACMG PM2 criterion. The absence of this variant in the peripheral blood DNA of the proband’s asymptomatic father supports its maternal inheritance and strengthens the evidence for segregation with disease within the maternal lineage.
We then performed Sanger sequencing analysis on peripheral blood DNA from the proband’s mother and asymptomatic maternal uncle, as well as hair follicle DNA from the sister and grandmother. The same variant was detected in the symptomatic mother, sister, and grandmother, but was absent in the healthy uncle in the same family (Figure 1B) which is consistent with ACMG evidence PP1. Considering the above results and in accordance with the ACMG guidelines, we classify the POU4F1, NM_006237.4:c.55G>T (p.Glu19*) variant classified as pathogenic, supported by evidence PVS1, PM2 and PP1.
Thus, the combination of clinical presentation and genetic analysis supports the diagnosis of ATITHS in this patient, associated with the POU4F1 c.55G>T (p.Glu19*) mutation. Although he is currently able to complete basic daily activities with intermittent muscle strength and balance rehabilitation, his weight-bearing remains reduced compared to healthy individuals. Given the likely pathogenic nature of this variant and its familial segregation, we recommend genetic counseling and prenatal diagnosis for future pregnancies in this family to prevent potential transmission of the mutant allele.
Discussion
ATITHS is a rare neurodevelopmental disorder inherited in an autosomal dominant manner, characterized by childhood-onset ataxia, intention tremor, and hypotonia, often resulting in delayed ambulation. Additional clinical features may include global developmental delay, mildly impaired intellectual development, speech delays or learning disabilities, and abnormal eye movements. The disease is rarely reported, and its full phenotypic spectrum is still being defined. Pathogenicity of the POU4F1 gene is believed to underlie the condition, consistent with its critical role in neuronal development.
Webb et al. first reported a pathogenic variant in POU4F1 associated with ATITHS in four unrelated patients (Webb et al., 2021). All four patients in their report presented with motor symptoms like ataxia. In the oldest patient, obligatory paroxysmal head lifting diminished over time. A similar pattern was observed in our study: the patient had increased motor abilities in adulthood compared to early childhood. Interestingly, the patient in our study had persistent ocular motility symptoms into adulthood. Furthermore, similar eye movement symptoms were reported in his mother and sister. Although oculomotor palsy was not explicitly described in the earlier report, three of the patients had strabismus and underwent esotropia surgery, suggesting that partial oculomotor palsy may have been present but underrecognized. These findings indicate that ophthalmologic symptoms represent a key clinical feature in patients with ATITHS, especially in older patients and may serve as an important diagnostic clue of the disease. A comparison between the present case and previously reported cases is summarized in Table 1.
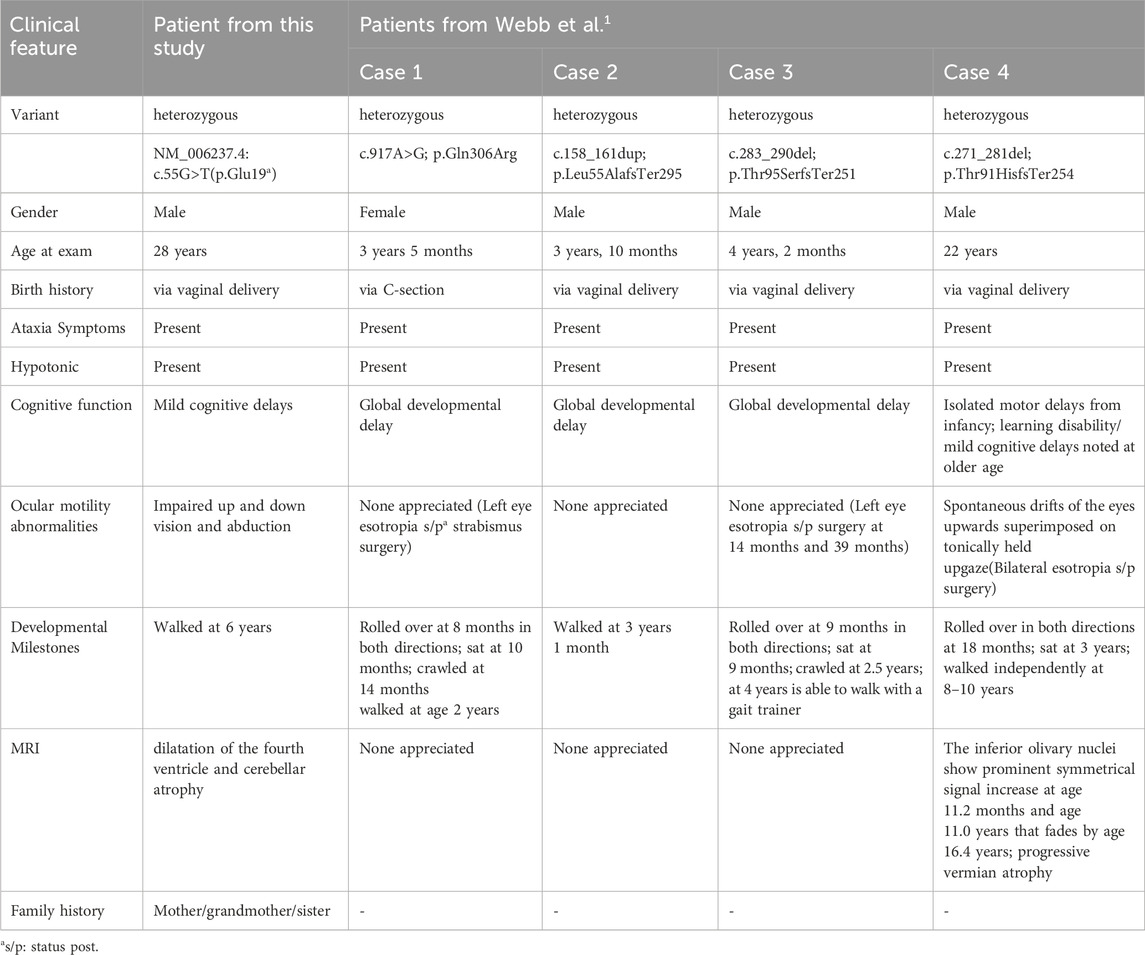
Table 1. Comparison of Clinical Characteristics Between the Present Case and Previously Reported ATITHS Patients with POU4F1 Variants by Webb et al.
Eye movement disorders are commonly attributed to peripheral neuropathy, while certain intracranial lesions can also cause impaired ocular motility (Musazadeh et al., 2004; Williams and Hoyt, 1989; Schreuders et al., 2012). Several brain regions are involved in the control of normal eye movements. The neural pathways responsible for smooth pursuit eye movements are believed to project to the cerebellum and then to the premotor regions of the brainstem after integration through the pontine nuclei. Both the cerebellar pons and the parvocellular pons receive inputs from the pontine nuclei, with the ventral parvocellular pons playing a particularly important role in smooth pursuit eye movements (Nagao et al., 1997; Rambold et al., 2002). In addition, other cerebellar structures, such as the oculomotor vermis (OMV) and the fastigial nucleus oculomotor (FOR), are also involved in maintaining smooth ocular tracking (Heinen and Keller, 1996). A range of eye movement symptoms observed in patients with ATITHS may be related to cerebellar atrophy involving these regions.
In this case, the patient did not exhibit significant palatal tremor at the time of examination at age 28. Neuropathologically, in most cases of palatal tremor, the lesion originates in the central pericentral fasciculus, which connects the red nucleus and the deep cerebellar nucleus to the inferior olive (Hainline et al., 2017). As reported in a previous study, serial MRI scans highlighted the disease progression, showing marked symmetric T2 hyperintensity in the inferior olive nucleus at younger ages, which tends to fade over time, while cerebellar atrophy progressively became more pronounced (Webb et al., 2021). Since our patient was first examined at age 28, the dominant MRI findings were cerebellar atrophy. Due to the lack of earlier imaging data, the longitudinal relationship between MRI changes and clinical features remains uncertain.
POU4F1 plays a critical role in the development of the sensory peripheral nervous system. In animal model, affected Pou4f1−/− neurons undergo apoptosis and are unable to properly innervate their peripheral target organs. This leads to loss of other functions in sensory axon growth (Eng et al., 2001; Xiang et al., 1997), which may further result in uncoordinated limb movement and impaired sucking in mice after birth (Xiang et al., 1996; Agarwala and Ragsdale, 2002). Its role in the central nervous system, particularly in regions such as the parietal nuclei, reinnervating nuclei, and the retina, has also been extensively investigated (Fedtsova et al., 2008; Badea et al., 2012; Quina et al., 2009).
Pou4f1 is expressed in the red nucleus (RN) during both embryonic development and adulthood, and it defines the RN signature. Deletion of Pou4f1 in mice leads to disorganization of the rubrospinal tracts (Martinez-Lopez et al., 2015), a system that plays an important role in the development of basic motor functions.
In conclusion, our study describes a male patient with adolescent peripheral neuropathy carrying the heterozygous nonsense mutation POU4F1 NM_006237.4:c.55G>T(p.Glu19*) chr13:79177407. As the first reported case of ATITHS with a complete family lineage, our study has described more neurological features of this particular neurological syndrome. Our findings further support a causal relationship between this novel mutation and ATITHS, contributing to its classification to pathogenic. We emphasize the importance of genetic testing to determine genetic etiology in patients presenting with ATITHS, which can significantly improve diagnostic accuracy, guide clinical management and inform genetic counseling for affected families.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Taizhou Hospital of Zhejiang Province. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
QH: Writing – original draft, Writing – review and editing, Data curation, Investigation. FZ: Writing – review and editing, Data curation, Investigation. WW: Conceptualization, Investigation, Writing – original draft. YX: Writing – original draft. HR: Investigation, Writing – original draft. YZ: Writing – original draft. YJ: Writing – review and editing. SK: Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Scientific Research Foundation of Taizhou Enze Medical Center (Group)-General Project (Grant No. 25EZD05). We declare that research support played no role in study design, in collection, analysis and interpretation of data, as well as in the writing and the decision to submit the report for publication.
Acknowledgments
We would like to express our gratitude to the family, who made this study possible. We acknowledge that the Guangzhou Kingmed Diagnostics Group Co., Ltd. (Address: No. 10, LuoXuan 3rd Road, Guangzhou International Bio Island) providing WES analysis for the participants in this study on 10/27/2024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1702803/full#supplementary-material
References
Agarwala, S., and Ragsdale, C. W. (2002). A role for midbrain arcs in nucleogenesis. Development 129 (24), 5779–5788. doi:10.1242/dev.00179
Badea, T. C., Williams, J., Smallwood, P., Shi, M., Motajo, O., and Nathans, J. (2012). Combinatorial expression of Brn3 transcription factors in somatosensory neurons: genetic and morphologic analysis. J. Neurosci. 32 (3), 995–1007. doi:10.1523/JNEUROSCI.4755-11.2012
Eng, S. R., Gratwick, K., Rhee, J. M., Fedtsova, N., Gan, L., and Turner, E. E. (2001). Defects in sensory axon growth precede neuronal death in Brn3a-deficient mice. J. Neurosci. 21 (2), 541–549. doi:10.1523/JNEUROSCI.21-02-00541.2001
Fedtsova, N., Quina, L. A., Wang, S., and Turner, E. E. (2008). Regulation of the development of tectal neurons and their projections by transcription factors Brn3a and Pax7. Dev. Biol. 316 (1), 6–20. doi:10.1016/j.ydbio.2007.12.040
Hainline, C., Neophytides, A., Borja, M. J., and Galetta, S. L. (2017). Progressive ataxia and palatal tremor. Neurol. Clin. Pract. 7, e37–e38. doi:10.1212/CPJ.0000000000000298
Heinen, S. J., and Keller, E. L. (1996). The function of the cerebellar uvula in monkey during optokinetic and pursuit eye movements: single-unit responses and lesion effects. Exp. Brain Res. 110 (1), 1–14. doi:10.1007/BF00241368
Herr, W., Sturm, R. A., Clerc, R. G., Corcoran, L. M., Baltimore, D., Sharp, P. A., et al. (1988). The POU domain: a large conserved region in the Mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes Dev. 2 (12A), 1513–1516. doi:10.1101/gad.2.12a.1513
Latchman, D. S. (1998). The Brn-3a transcription factor. Int. J. Biochem. Cell Biol. 30 (11), 1153–1157. doi:10.1016/s1357-2725(98)00090-9
Martinez-Lopez, J. E., Moreno-Bravo, J. A., Madrigal, M. P., Martinez, S., and Puelles, E. (2015). Red nucleus and rubrospinal tract disorganization in the absence of Pou4f1. Front. Neuroanat. 9, 8. doi:10.3389/fnana.2015.00008
Musazadeh, M., Hartmann, K., and Simon, F. (2004). Late onset esotropia as first symptom of a cerebellar tumor. Strabismus 12 (2), 119–123. doi:10.1080/09273970490517557
Nagao, S., Kitamura, T., Nakamura, N., Hiramatsu, T., and Yamada, J. (1997). Differences of the primate flocculus and ventral paraflocculus in the mossy and climbing fiber input organization. J. Comp. Neurol. 382 (4), 480–498. doi:10.1002/(sici)1096-9861(19970616)382:4<480::aid-cne5>3.0.co;2-z
Quina, L. A., Wang, S., Ng, L., and Turner, E. E. (2009). Brn3a and Nurr1 mediate a gene regulatory pathway for habenula development. J. Neurosci. 29, 14309–14322. doi:10.1523/jneurosci.2430-09.2009
Rambold, H., Churchland, A., Selig, Y., Jasmin, L., and Lisberger, S. G. (2002). Partial ablations of the flocculus and ventral paraflocculus in monkeys cause linked deficits in smooth pursuit eye movements and adaptive modification of the VOR. J. Neurophysiol. 87 (2), 912–924. doi:10.1152/jn.00768.2000
Schreuders, J., Thoe Schwartzenberg, G. W., Bos, E., and Versteegh, F. G. (2012). Acute-onset esotropia: should we look inside? J. Pediatr. Ophthalmol. Strabismus 4, e70–e72. doi:10.3928/01913913-20121127-05
Webb, B. D., Evans, A., Naidich, T. P., M Bird, L., Parikh, S., Fernandez Garcia, M., et al. (2021). Haploinsufficiency of POU4F1 causes an ataxia syndrome with hypotonia and intention tremor. Hum. Mutat. 42 (6), 685–693. doi:10.1002/humu.24201
Williams, A. S., and Hoyt, C. S. (1989). Acute comitant esotropia in children with brain tumors. Arch. Ophthalmol. 107 (3), 376–378. doi:10.1001/archopht.1989.01070010386029
Xiang, M., Gan, L., Zhou, L., Klein, W. H., and Nathans, J. (1996). Targeted deletion of the mouse POU domain gene Brn-3a causes selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proc. Natl. Acad. Sci. U. S. A. 93 (21), 11950–11955. doi:10.1073/pnas.93.21.11950
Keywords: ataxia, hypotonia, Pou4f1, case report, hypokinesia
Citation: Hu Q, Zhu F, Wang W, Xu Y, Ren H, Zheng Y, Jiang Y and Ke S (2025) Ataxia, intentional tremor and hypotonia syndrome caused by a novel POU4F1 gene mutation: a case report. Front. Genet. 16:1702803. doi: 10.3389/fgene.2025.1702803
Received: 10 September 2025; Accepted: 03 October 2025;
Published: 14 October 2025.
Edited by:
Salvatore Gallone, University of Turin, ItalyReviewed by:
Nazli Ayse Basak, Koç University, TürkiyeNazli Durmaz Celik, Eskişehir Osmangazi University, Türkiye
Copyright © 2025 Hu, Zhu, Wang, Xu, Ren, Zheng, Jiang and Ke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiqing Jiang, amlhbmd5cUBlbnplbWVkLmNvbQ==; Shaofa Ke, a2VzZkBlbnplbWVkLmNvbQ==
†These authors have contributed equally to this work and share first authorship