



- Department of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA, USA
Recent data with CD8+ T cells show that the initial phase of T cell receptor (TCR) binding to MHC–peptide (MHCp) is quickly followed by a second, stronger, binding phase representing the binding of CD8 to the MHCp. This second phase requires signaling by a Src-family kinase such as Lck. These data point out two aspects of the initial stage of TCR signaling that have not yet been clearly resolved. Firstly, how and by which Src-family kinase, is the initial phosphorylation of CD3ζ accomplished, given that the Lck associated with the co-receptors (CD4 or CD8) is not yet available. Secondly, what is the mechanism by which the co-receptor is brought close to the bound TCR before the co-receptor binds to MHCp?
Introduction
Recognition of antigen as MHC–peptide (MHCp) complexes by the T cell receptor (TCR) is the first stage of T cell activation, and therefore critical to all adaptive immune responses to pathogens and cancers, as well as to autoimmunity. The binding of TCR to MHCp initiates a protein kinase signaling cascade that branches out as it proceeds, to encompass various signaling pathways leading to changes in cell shape, adhesion, and motility, as well as to changes in gene transcription.
There are several models for how TCR interaction with MHCp can lead to initial triggering of the signaling cascade. These have been comprehensively reviewed (Germain, 2001; Krogsgaard and Davis, 2005; van der Merwe and Dushek, 2011), so will only be briefly mentioned here. They fall into several groups, aggregation, conformation/architectural changes, and separation models. Evidence from studies of T cells activated by APC or by soluble MHCp indicate that some kind of crosslinking or aggregation of two or more TCRs is required (Boniface et al., 1998; Cochran et al., 2000; Cebecauer et al., 2005). TCRs are grouped into microclusters during antigen recognition (Yokosuka et al., 2005; Varma et al., 2006), and there is also some evidence for pre-formed aggregations of TCRs existing on the cell surface (Schamel et al., 2005; Lillemeier et al., 2006, 2010; Kumar et al., 2011). Clustering of the TCRs by antigen recognition aided by weak interactions with endogenous MHCp and the recruitment of co-receptor-bound Lck to the site of recognition would then start the signaling cascade (Krogsgaard and Davis, 2005; Gascoigne, 2008). Conformational changes have been proposed as a mechanism for signal transduction, but apart from movement in the CDRs during binding to MHCp, conformation change upon antigen binding has only been found as a movement of part of the Cα domain, in a region predicted to interact with CD3ε (Kjer-Nielsen et al., 2003; Beddoe et al., 2009). Conformation changes in intracellular domains of CD3 seem to be involved in allowing signaling adaptor proteins to interact with CD3 (Gil et al., 2002) or for accessibility for immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation (Xu et al., 2008). A conformational/architectural model has received strong recent support whereby mechanical pulling by the TCR–MHCp interaction puts torque on the CD3 subunits, leading to signal transduction (Kim et al., 2009; Li et al., 2010). In the kinetic separation model, the interaction between short molecules like TCR and MHC at the immunological synapse requires that larger molecules such as cell adhesion molecules and the phosphatase CD45 are pushed out to the periphery. Removal of negative regulators of signaling could thus aid phosphorylation steps at the center of the synapse (Davis and van der Merwe, 2006). These different classes of model are not necessarily mutually exclusive, and indeed elements of each class seem to operate in T cell activation (van der Merwe and Dushek, 2011).
The very first stage of the TCR signaling cascade after TCR–MHCp binding is the phosphorylation of the CD3ζ subunits of the TCR. The TCR–CD3 complex consists of the TCRα and β chains that recognize MHCp, and the CD3γε, δε, and ζζ dimers that form the signal transduction modules of the TCR. All of these subunits are cell surface transmembrane proteins and all have at least one ITAM in their cytoplasmic domain. CD3γ, δ, and ε each have one ITAM, while CD3ζ has three. At the start of the signaling cascade, the CD3ζ ITAMs are phosphorylated, and are then bound by another kinase called ZAP70. Each ITAM has dual tyrosines so that when they are both phosphorylated, they form a high affinity binding site for the dual SH2 domains of ZAP70. After ZAP70 binds to CD3, the co-receptors CD4 or CD8, which are associated with the Src-family kinase Lck, become associated with the TCR–CD3 complex and bind to MHC. This stabilizes the TCR–MHCp interaction, and Lck continues the phosphorylation of CD3 elements, ZAP70 and the many other downstream targets.
A recent biophysical study on TCR and CD8 binding to MHCp demonstrated that the initial binding of TCR to MHCp induces, in a Src-family kinase-dependent manner, the binding of CD8 to the MHCp (Jiang et al., 2011). This illustrates two related and important problems in understanding T cell activation that have never been fully resolved. Firstly, how is the first CD3 phosphorylation step accomplished, and by which Src-family kinase(s)? Secondly, if the co-receptor is brought to the activated TCR before the co-receptor binds to MHC, but after TCR signaling has started, how is this initial recruitment to the TCR accomplished?
Rather little is known about these earliest steps in T cell activation. Lck is a major target for drug discovery, as small molecule inhibitors could be used as immunosuppressives, for example in autoimmunity, rheumatoid arthritis, or asthma. Current inhibitors are not very selective, also inhibiting other Src-family kinases or other kinases (Martin and Machacek, 2010). It is therefore important to understand exactly how Src-family kinases work in T cell activation.
TCR Binding to MHCp Augments CD8 Binding to MHC
It was shown in the early 1990s that stimulation of the TCR enhances the binding of CD8 to MHC class I (MHCI), and that this increase in binding requires tyrosine kinase activity (O’Rourke et al., 1990; O’Rourke and Mescher, 1992). Recently, a cell-based adhesion frequency assay was developed that enables investigation of molecular interactions at the cell surface with very fast time resolution (Huang et al., 2007, 2010; Jiang et al., 2011). This method is based on touching a T cell to a red blood cell coated with appropriate MHCp ligands, then retracting it, and measuring whether or not the red blood cell membrane is distorted. This process is repeated multiple times with the interaction allowed to continue for certain fixed periods. For each of these interaction times, an adhesion frequency is calculated. This method showed clearly that TCR and CD8 interactions with MHCp are biphasic (Jiang et al., 2011). The first phase – the initial TCR interaction with ligand – starts within the first 0.25 s after the cell surfaces make contact (Figure 1). After about 1 s of contact, a second, stronger, binding phase occurs. Initiation of the second phase requires a Src-family tyrosine kinase, as it does not occur when the inhibitor PP2 is present. The first phase is co-receptor-independent but the second phase is co-receptor dependent, as the second phase is blocked by anti-CD8 antibodies and does not occur when the CD8 binding site of MHCI is non-functional. Thus, the first phase is the TCR interaction. This is followed by a second phase of TCR-induced CD8 binding (Jiang et al., 2011). There is also a CD8–MHCI interaction that occurs without stimulation of the TCR. This is weaker than the TCR–MHCp interaction, but like that, it has single-stage kinetics and reaches a plateau at about 1 s (Huang et al., 2007). This CD8–MHCI interaction is sufficient to concentrate CD8 at the interface between a T cell and an antigen-presenting cell (Yachi et al., 2005, 2006; Gascoigne et al., 2010).
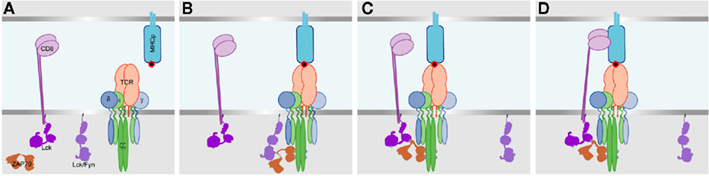
Figure 1. A model for initiation of TCR– CD3 phosphorylation, and the recruitment of the co-receptor. (A) Before TCR interaction with MHCp. Lck and/or Fyn are associated with the plasma membrane, and Lck also exists associated with CD8. Upon TCR binding to MHCp (B) the Fyn and/or non-CD8 bound Lck becomes associated with the TCR–CD3 complex, and phosphorylates CD3ζ ITAMs and then ZAP70 which binds to the phosphorylated CD3ζ ITAMs. (C) The SH2 domain of CD8 bound Lck then interacts with the phospho-ZAP70, pulling CD8 into proximity with the MHCp-interacting TCR. (D) The extracellular immunoglobulin domains then bind to the MHCp, stabilizing the TCR–MHCp interaction.
This two-step activation (Jiang et al., 2011) does not fit with the classical model of TCR triggering, where the co-receptor binding to MHCp is required for Lck to phosphorylate CD3 ITAMs. It is compatible with aggregation models such as the pseudodimer or co-receptor pre-concentration models, where the co-receptor’s extracellular domains bind to non-cognate MHCp but the co-receptor-bound Lck interacts with the antigen binding TCR/CD3 within the cell (Krogsgaard et al., 2007; Gascoigne, 2008). It is also consistent with those models requiring a conformation/architectural change due to antigen binding, providing that the initial phosphorylation of CD3 recruits the co-receptor through the interaction of Lck with phospho-ITAMs.
Initial Phosphorylation of TCR–CD3 in T Cell Activation
Several lines of evidence indicate that the initial phosphorylation of CD3ζ ITAMs and ZAP70 is caused by a Src-family kinase (Lck and/or Fyn) that is not bound to CD4 or CD8, yet the mechanism of this process that initiates the TCR signaling cascade is poorly defined (Acuto et al., 2008). It is clear that co-receptors are not absolutely required for TCR signaling as some T cells are independent of co-receptor, and the requirement for CD8 has been shown to be inversely related to the affinity of the TCR. TCRs with affinity above a certain threshold do not need co-receptor (Cho et al., 2001; Holler and Kranz, 2003). Another piece of evidence is that thymocyte development requires the presence of Lck, but co-receptors are not absolutely required for thymocyte development (Schilham et al., 1993; Van Laethem et al., 2007). In addition, the fact that CD8 does not bind MHC to stabilize TCR binding until after signaling has started argues this point strongly (Jiang et al., 2011). Lck can be co-precipitated with TCR/CD3 in mild detergents (Schraven et al., 1993; Stefanova et al., 2003), but although Lck can be co-capped with co-receptor (after crosslinking by suitable antibody), it does not co-cap with TCR (Ehrlich et al., 2002). Fyn can also be co-precipitated with the TCR in mild detergents (Samelson et al., 1990; Timson Gauen et al., 1992; Schraven et al., 1993; Fusaki et al., 1996). Thus there is evidence that both Lck and Fyn can associate in some way with TCR, but at very low stoichiometry. Also, because these experiments used detergents that do not disrupt lipid rafts, it is possible that the interactions were artifacts due to the detergent lysis method causing interactions between normally separate lipid domains.
Fyn or Lck individually can both perform the constitutive phosphorylation of TCR/CD3 required for maintenance of cell viability in vivo, but Lck-deficient cells have stronger defects in most aspects of signaling than Fyn-deficient cells. The signaling differences between T cells from Fyn and Lck knockout mice are summarized in Table 1. Because of the block in positive selection in Lck−/−mice, data on Lck-deficiency in mature T cells have come from a very elegant inducible transgenic system bred onto the Lck KO background. This transgene is turned on to allow normal T cell development, then off so that Lck expression is gradually lost. Transgenic Lck expression in mature T cells is, however, only about 10–20% of the normal amount (Seddon et al., 2000; Lovatt et al., 2006). For the most part, the defects in Fyn-deficient T cells were rather modest, but recent studies have shown that Fyn may in fact be a negative regulator of TCR signaling. Stimulation with antigen (or with crosslinking anti-CD3 plus anti-CD8) gave stronger and faster responses in the absence of Fyn (Filby et al., 2007). This is in contrast to earlier studies that showed that cells lacking Fyn gave lower responses than normal to anti-CD3 stimulation (as opposed to antigen or anti-TCR plus anti-co-receptor stimulation; Reviewed in Palacios and Weiss, 2004; Salmond et al., 2009b).
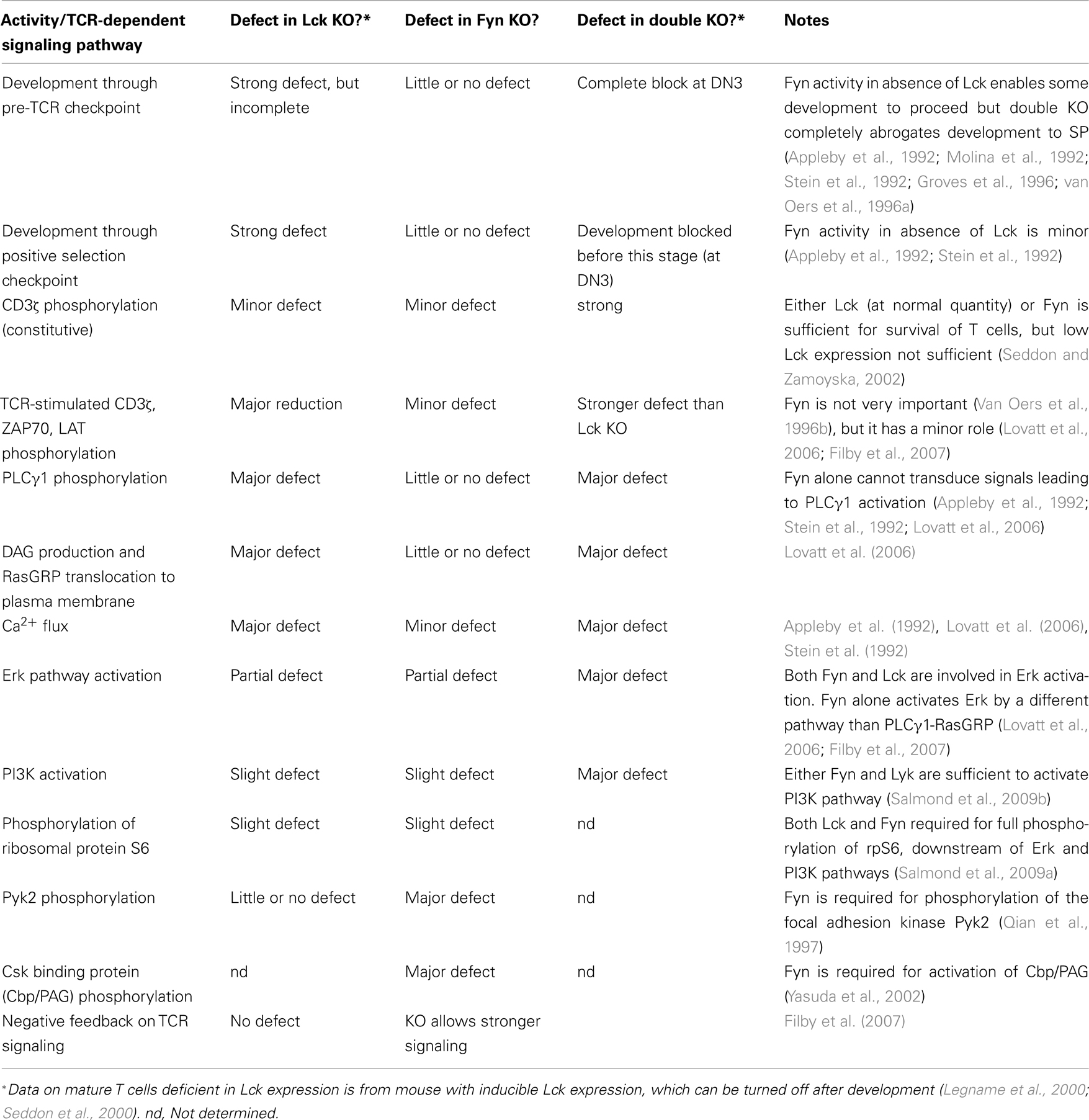
Table 1. Defects in TCR signaling and development in the absence of Lck and/or Fyn.
Most interestingly, in Lck knockout mature T cells, Fyn directs relatively strong Erk activation, but reduced LAT phosphorylation, while PLCγ1 phosphorylation and Ca2+ signaling do not occur (Lovatt et al., 2006; Filby et al., 2007). Fyn may therefore act through a different pathway than Lck to activate Erk. A direct interaction of Grb2–SOS with CD3ζ has been suggested (Chau and Madrenas, 1999; Methi et al., 2007), which could perhaps provide such a pathway. However, T cell activation is very strongly dependent on the RasGRP pathway to activate Ras and then Erk, and it is unclear if SOS has any crucial role (Dower et al., 2000; Priatel et al., 2002). It may also be important to consider that these studies in Lck or Fyn-deficient cell lines or knockout mice have not distinguished free Lck from that bound to the co-receptor.
Resting T cells and thymocytes contain substantial quantities of both activated Lck and Fyn, and stimulation through TCR or co-receptor does not increase the proportion of the activated kinases (Nika et al., 2010). Both Lck and Fyn are targeted to the inner leaflet of the plasma membrane in lipid rafts through palmitoylation and myristoylation of their amino termini. This fatty acylation is required for Fyn to interact with and phosphorylate CD3ζ ITAMs (van’t Hof and Resh, 1999). Lipid-raft targeting by fatty acylation is also the case for Lyn, a Src-family kinase that is important in initiation of BCR signaling. A recent experiment showed that Lyn interacts with the BCR after BCR crosslinking by binding to antigen, when Ag-induced BCR microclusters interact with lipid rafts (Sohn et al., 2008). A similar mechanism is likely to apply for the initiation of TCR signaling by Lck and/or Fyn.
Yet another complexity comes from single-molecule studies of the movement of cell membrane-associated molecules. Stimulation by very small clusters of anti-CD3 antibodies on a solid surface showed that a raft-targeted construct (the amino terminal of Lck with the fatty acylation sites) or full-length Lck both concentrated quickly at the stimulation site and were retained there (Ike et al., 2003). Diffusion was slower closer to the site of stimulation, and faster away from these sites. Directed movement of Lck was not seen, indicating that Lck was not brought to the signaling region by actin, but concentration at the stimulation site was blocked by drugs that either poisoned the cytoskeleton or dispersed lipid rafts. Clustering of the actin filaments at the site of activation (Wulfing and Davis, 1998) would slow diffusion away from the activation site, either because the actin mesh impedes free diffusion of the membrane-bound protein, or because of a “picket fence” of transmembrane proteins associated with the cytoskeleton. These phenomena would lead to “corralling” of Lck (and rafts) at the activation site (Ike et al., 2003). Others have described a similar phenomenon of microdomains caused by proteins rather than lipids, where it was noted that CD45 was excluded from microdomains that contained Lck (Douglass and Vale, 2005). This study also demonstrated that various T cell signaling proteins were able to move quickly from one microdomain to another. A protein such as annexin A6, which binds negatively charged phospholipids, lipid rafts, and F-actin, could organize and bridge different kinds of microdomains and/or rafts around signaling receptors (Cornely et al., 2011).
How is CD8 Recruited to the Activated TCR Before CD8 Interacts with MHC?
The data on biphasic TCR and CD8 interactions with MHCp show that in a very quick sequence, TCR binds MHCp, causing Src-family kinase activity, then CD8 binds to the MHC and stabilizes the interaction between the three molecules (Jiang et al., 2011). If CD8 does not bind to MHCp until the second stage, then how is the co-receptor initially recruited to the TCR? It has generally been assumed that this is accomplished by CD8 binding to MHC, but this does not fit with the new data. It has been suggested that CD8 is brought into the signaling complex passively, through an interaction of the Lck molecule attached to CD8 with one of the phospho-tyrosines produced by the initial signal on CD3ζ, or to phospho-ZAP70 (Jiang et al., 2011).
There is in fact some strong but rather old evidence supporting such a mechanism. A study using a chimera between CD4 and Lck demonstrated that the SH2 domain of Lck was involved in CD4 co-receptor activity: a mutation stopping the Lck SH2 from binding phosphotyrosine partially reduced co-receptor activity (Xu and Littman, 1993). This suggested that a protein–protein interaction mediated by Lck’s SH2 domain is important for co-receptor function, and indeed it has since been shown that after TCR stimulation, the Lck SH2 domain binds to phospho-ZAP70 bound to phospho-CD3ζ, and that this interaction causes the co-receptor to come into proximity with TCR (Thome et al., 1995, 1996). As mentioned above, much of the Lck in resting T cells is activated kinase, and this activity is not affected by TCR stimulation (Nika et al., 2010), but kinase activity is not required for the co-receptor function of CD4 (Collins and Burakoff, 1993; Xu and Littman, 1993). The CD4 co-receptor is bound to both active and inactive Lck (Nika et al., 2010), indicating that there is no simple explanation. The protein–protein interaction functions of Lck during TCR activation deserve more detailed analysis.
CD8 exists as both an αα homodimer and as the αβ heterodimer that is the main co-receptor for T cell activation. Both forms are equivalent in their ability to bind to classical MHCI, but CD8αα binds much more strongly than CD8αβ to the non-classical MHCI molecule H2-TL (Cheroutre and Lambolez, 2008). CD8αβ is a much stronger co-receptor than CD8αα, probably because of preferential localization of CD8αβ to lipid rafts where it is more likely to interact with Lck. We tested the ability of the two forms expressed on the same cell to be recruited to the site of antigen recognition, and found that there was very little difference between them (Rybakin et al., 2011). If H2-TL was present on the antigen-presenting cell, then the CD8αα form was preferentially recruited, leading us to the conclusion that the ability of the CD8 isoforms to be recruited to the immunological synapse was not directly related to their co-receptor ability, but simply to the strength of their interaction with MHCI.
Different Phases in the TCR Interaction with Co-Receptor
In previous studies from this lab, we used Foerster resonance energy transfer (FRET) microscopy in live cells to investigate the interaction of both CD4 and CD8 with TCR, using CD3ζ–CFP as FRET donor and CD4–YFP or CD8β–YFP as FRET acceptors (Zal et al., 2002; Gascoigne and Zal, 2004; Yachi et al., 2005, 2006; Gascoigne et al., 2009). We showed that the co-receptors interact closely with the TCR only when TCR recognizes antigen, even though both CD4 and CD8 can be recruited to the immunological synapse without any requirement for antigen recognition. This was most striking for CD8, which binds to non-antigenic MHCI molecules and can be concentrated in the immunological synapse in proportion to the number of MHCI proteins available (Yachi et al., 2005; Gascoigne et al., 2010). We found that the strength of the FRET signal between the TCR–CD3 complex (CD3ζ–CFP → CD8β–YFP FRET) corresponded closely to strength of T cell activation by particular peptides, and that differences in FRET kinetics during recognition of different MHCp ligands correlated closely to the ability of a particular ligand to induce positive or negative selection in thymocytes (Yachi et al., 2006; Mallaun et al., 2008). However, the kinetics of the close apposition of TCR and CD8 were slower than expected from other measures of T cell activation, as they developed over several minutes, peaking at about 10 min (Yachi et al., 2005, 2006). Early signaling events such as LAT phosphorylation or calcium flux occur within a few seconds, although other events like cytoskeletal rearrangement for example, are on a longer timescale of a few minutes (Huse et al., 2007). In more recent higher-resolution experiments using a supported lipid bilayer system, we have been able to image TCR–CD8 FRET in small regions within the immunological synapse (J. Casas and Nicholas R. J. Gascoigne, unpublished). These were too localized to show a signal above the background when FRET signals were averaged over the whole synapse, explaining why they were not noticed before. This leads us to believe that the strong FRET signals measured previously in synapses with antigen-presenting cells that were on the timescale of minutes (Yachi et al., 2005, 2006), represent the result of stabilization of TCR–MHCp binding by the co-receptor – the second phase of interaction identified in the cell–cell adhesion experiments (Jiang et al., 2011), rather than the first.
The TCR α-chain connecting peptide motif (α-CPM) is a highly conserved membrane-proximal extracellular sequence motif in the TCR α-chain. It is important for the ability of CD8 to associate with TCR to enhance T cell responses (Naeher et al., 2002; Mallaun et al., 2008), and for T cell activation and positive selection (Werlen et al., 2000). Mutation of the α-CPM reduced the stable FRET response between TCR and CD8 (Mallaun et al., 2008). The α-CPM is believed to work by interacting with membrane-proximal regions of CD8β in a “co-receptor zipper” mechanism, enabling Lck to completely phosphorylate the CD3 ITAMs (Palmer and Naeher, 2009). CD8β is reported to interact primarily with CD3δ (Doucey et al., 2003). The molecular basis of the interaction between the α-CPM and CD8β is not clear, as it is unlikely that CD8 could approach closely enough to the α-CPM to interact with it directly, due to the presence of the CD3 subunits. However, the transmembrane region of CD3εδ interacts with that of TCRα (Call et al., 2002), and structural models place CD3εδ and TCRα apposed to each other (Sun et al., 2004; Kuhns and Davis, 2007). It has been suggested that movement within the TCR’s α-CPM could apply torque to CD3εδ in the architectural/conformational change model of TCR triggering described earlier (Kim et al., 2010). It will be interesting to find out how the α-CPM mutation affects the two-stage TCR binding (Jiang et al., 2011). We predict that the α-CPM will be involved in stabilization of the TCR–CD8 interaction after the initial TCR–MHCp binding, so that mutation of the α-CPM will inhibit the second phase of TCR–MHCp binding without affecting the first phase.
Future Directions
The molecular details of the apparent adaptor function of Lck need to be more clearly worked out, and indeed, a similar function may exist in other Src-family kinases. Lck and Fyn molecules have overlapping but distinct roles at the beginning of TCR signal transduction, but the mechanisms for their initial interaction with TCR to start the signaling cascade are still somewhat obscure. Improvements in our ability to image TCR–CD8 FRET will allow us to investigate separately the early and later phases of the TCR–CD8 interactions. The early phase likely corresponds to the passive recruitment of CD8 to TCR by the Lck interacting with phospho-CD3ζ and/or phospho-ZAP70, whereas the later and stronger phase of FRET is likely the result of stabilization of TCR–MHCp binding by the co-receptor. In this regard, it is worth noting that the two-stage binding model that shows co-receptor stabilization of the TCR–MHCp interaction was performed in a MHCI-restricted system. CD8 has a clear ability to stabilize the TCR–MHCp interaction, whereas CD4 does not. Indeed, modeling studies suggest that the major role of co-receptors, particularly CD4, is simply to ferry Lck toward the site of TCR activation (Artyomov et al., 2010). We are currently analyzing the different functions of CD8 and Lck using mutational analyses and FRET microscopy. The future application of super-resolution microscopy techniques such as PALM and STORM (stochastic optical reconstruction microscopy) will enable a nanometer-scale understanding of the molecular interactions in the early phases of T cell activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grants GM065230 and AI074074 to Nicholas R. J. Gascoigne. This is manuscript 21444 from The Scripps Research Institute.
References
Acuto, O., Di Bartolo, V., and Micheli, F. (2008). Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat. Rev. Immunol. 8, 699–712.
Appleby, M. W., Gross, J. A., Cooke, M. W., Levin, S. D., Qian, X., and Perlmutter, R. (1992). Defective T cell receptor signaling in mice lacking the thymic isoform of p59-fyn. Cell 70, 751–763.
Artyomov, M. N., Lis, M., Devadas, S., Davis, M. M., and Chakraborty, A. K. (2010). CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. U.S.A. 107, 16916–16921.
Beddoe, T., Chen, Z., Clements, C. S., Ely, L. K., Bushell, S. R., Vivian, J. P., Kjer-Nielsen, L., Pang, S. S., Dunstone, M. A., Liu, Y. C., Macdonald, W. A., Perugini, M. A., Wilce, M. C., Burrows, S. R., Purcell, A. W., Tiganis, T., Bottomley, S. P., McCluskey, J., and Rossjohn, J. (2009). Antigen ligation triggers a conformational change within the constant domain of the αβ T cell receptor. Immunity 30, 777–788.
Boniface, J. J., Rabinowitz, J. D., Wulfing, C., Hampl, J., Reich, Z., Altman, J. D., Kantor, R. M., Beeson, C., McConnell, H. M., and Davis, M. M. (1998). Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity 9, 459–466.
Call, M. E., Pyrdol, J., Wiedmann, M., and Wucherpfennig, K. W. (2002). The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111, 967–979.
Cebecauer, M., Guillaume, P., Mark, S., Michielin, O., Boucheron, N., Bezard, M., Meyer, B. H., Segura, J. M., Vogel, H., and Luescher, I. F. (2005). CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex-peptide dimers. J. Biol. Chem. 280, 23820–23828.
Chau, L. A., and Madrenas, J. (1999). Phospho-LAT-independent activation of the ras-mitogen-activated protein kinase pathway: a differential recruitment model of TCR partial agonist signaling. J. Immunol. 163, 1853–1858.
Cheroutre, H., and Lambolez, F. (2008). Doubting the TCR coreceptor function of CD8alphaalpha. Immunity 28, 149–159.
Cho, B. K., Lian, K. C., Lee, P., Brunmark, A., McKinley, C., Chen, J., Kranz, D. M., and Eisen, H. N. (2001). Differences in antigen recognition and cytolytic activity of CD8+ and CD8− T cells that express the same antigen-specific receptor. Proc. Natl. Acad. Sci. U.S.A. 98, 1723–1727.
Cochran, J. R., Cameron, T. O., and Stern, L. J. (2000). The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity 12, 241–250.
Collins, T. L., and Burakoff, S. J. (1993). Tyrosine kinase activity of CD4-associated p56lck may not be required for CD4-dependent T-cell activation. Proc. Natl. Acad. Sci. U.S.A. 90, 11885–11889.
Cornely, R., Rentero, C., Enrich, C., Grewal, T., and Gaus, K. (2011). Annexin A6 is an organizer of membrane microdomains to regulate receptor localization and signalling. IUBMB Life 63, 1009–1017.
Davis, S. J., and van der Merwe, P. A. (2006). The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 7, 803–809.
Doucey, M. A., Goffin, L., Naeher, D., Michielin, O., Baumgartner, P., Guillaume, P., Palmer, E., and Luescher, I. F. (2003). CD3δ establishes a functional link between the T cell receptor and CD8. J. Biol. Chem. 278, 3257–3264.
Douglass, A. D., and Vale, R. D. (2005). Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 121, 937–950.
Dower, N. A., Stang, S. L., Bottorff, D. A., Ebinu, J. O., Dickie, P., Ostergaard, H. L., and Stone, J. C. (2000). RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 1, 317–321.
Ehrlich, L. I., Ebert, P. J., Krummel, M. F., Weiss, A., and Davis, M. M. (2002). Dynamics of p56lck translocation to the T cell immunological synapse following agonist and antagonist stimulation. Immunity 17, 809–822.
Filby, A., Seddon, B., Kleczkowska, J., Salmond, R., Tomlinson, P., Smida, M., Lindquist, J. A., Schraven, B., and Zamoyska, R. (2007). Fyn regulates the duration of TCR engagement needed for commitment to effector function. J. Immunol. 179, 4635–4644.
Fusaki, N., Matsuda, S., Nishizumi, H., Umemori, H., and Yamamoto, T. (1996). Physical and functional interactions of protein tyrosine kinases, p59fyn and ZAP-70, in T cell signaling. J. Immunol. 156, 1369–1377.
Gascoigne, N. R. J. (2008). Do T cells need endogenous peptides for activation? Nat. Rev. Immunol. 8, 895–900.
Gascoigne, N. R. J., Ampudia, J., Clamme, J. P., Fu, G., Lotz, C., Mallaun, M., Niederberger, N., Palmer, E., Rybakin, V., Yachi, P. P., and Zal, T. (2009). Visualizing intermolecular interactions in T cells. Curr. Top. Microbiol. Immunol. 334, 31–46.
Gascoigne, N. R. J., and Zal, T. (2004). Molecular interactions at the T cell-antigen-presenting cell interface. Curr. Opin. Immunol. 16, 114–119.
Gascoigne, N. R. J., Zal, T., Yachi, P. P., and Hoerter, J. A. H. (2010). Co-receptors and recognition of self at the immunological synapse. Curr. Top. Microbiol. Immunol. 340, 171–189.
Germain, R. N. (2001). The T cell receptor for antigen: signaling and ligand discrimination. J. Biol. Chem. 276, 35223–35226.
Gil, D., Schamel, W. W. A., Montoya, M., Sanchez-Madrid, F., and Alarcon, B. (2002). Recruitment of Nck by CD3ε reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 109, 901–912.
Groves, T., Smiley, P., Cooke, M. P., Forbush, K., Perlmutter, R. M., and Guidos, C. J. (1996). Fyn can partially substitute for Lck in T lymphocyte development. Immunity 5, 417–428.
Holler, P. D., and Kranz, D. M. (2003). Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 18, 255–264.
Huang, J., Edwards, L. J., Evavold, B. D., and Zhu, C. (2007). Kinetics of MHC-CD8 interaction at the T cell membrane. J. Immunol. 179, 7653–7662.
Huang, J., Zarnitsyna, V. I., Liu, B., Edwards, L. J., Jiang, N., Evavold, B. D., and Zhu, C. (2010). The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932–936.
Huse, M., Klein, L. O., Girvin, A. T., Faraj, J. M., Li, Q. J., Kuhns, M. S., and Davis, M. M. (2007). Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity 27, 76–88.
Ike, H., Kosugi, A., Kato, A., Iino, R., Hirano, H., Fujiwara, T., Ritchie, K., and Kusumi, A. (2003). Mechanism of Lck recruitment to the T-cell receptor cluster as studied by single-molecule-fluorescence video imaging. Chemphyschem 4, 620–626.
Jiang, N., Huang, J., Edwards, L. J., Liu, B., Zhang, Y., Beal, C. D., Evavold, B. D., and Zhu, C. (2011). Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity 34, 13–23.
Kim, S. T., Takeuchi, K., Sun, Z. Y., Touma, M., Castro, C. E., Fahmy, A., Lang, M. J., Wagner, G., and Reinherz, E. L. (2009). The αβ T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037.
Kim, S. T., Touma, M., Takeuchi, K., Sun, Z. Y., Dave, V. P., Kappes, D. J., Wagner, G., and Reinherz, E. L. (2010). Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of αβ T cell receptors. J. Immunol. 185, 2951–2959.
Kjer-Nielsen, L., Clements, C. S., Purcell, A. W., Brooks, A. G., Whisstock, J. C., Burrows, S. R., McCluskey, J., and Rossjohn, J. (2003). A structural basis for the selection of dominant αβ T cell receptors in antiviral immunity. Immunity 18, 53–64.
Krogsgaard, M., Juang, J., and Davis, M. M. (2007). A role for “self” in T-cell activation. Semin. Immunol. 19, 236–244.
Kuhns, M. S., and Davis, M. M. (2007). Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity 26, 357–369.
Kumar, R., Ferez, M., Swamy, M., Arechaga, I., Rejas, M. T., Valpuesta, J. M., Schamel, W. W., Alarcon, B., and van Santen, H. M. (2011). Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity 35, 375–387.
Legname, G., Seddon, B., Lovatt, M., Tomlinson, P., Sarner, N., Tolaini, M., Williams, K., Norton, T., Kioussis, D., and Zamoyska, R. (2000). Inducible expression of a p56Lck transgene reveals a central role for Lck in the differentiation of CD4 SP thymocytes. Immunity 12, 537–546.
Li, Y. C., Chen, B. M., Wu, P. C., Cheng, T. L., Kao, L. S., Tao, M. H., Lieber, A., and Roffler, S. R. (2010). Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol. 184, 5959–5963.
Lillemeier, B. F., Mortelmaier, M. A., Forstner, M. B., Huppa, J. B., Groves, J. T., and Davis, M. M. (2010). TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 11, 90–96.
Lillemeier, B. F., Pfeiffer, J. R., Surviladze, Z., Wilson, B. S., and Davis, M. M. (2006). Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 103, 18992–18997.
Lovatt, M., Filby, A., Parravicini, V., Werlen, G., Palmer, E., and Zamoyska, R. (2006). Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol. Cell. Biol. 26, 8655–8665.
Mallaun, M., Naeher, D., Daniels, M. A., Yachi, P. P., Hausmann, B., Luescher, I. F., Gascoigne, N. R. J., and Palmer, E. (2008). The T cell receptor’s α-chain connecting peptide motif promotes close approximation of the CD8 coreceptor allowing efficient signal initiation. J. Immunol. 180, 8211–8221.
Martin, M. W., and Machacek, M. R. (2010). Update on lymphocyte specific kinase inhibitors: a patent survey. Expert Opin. Ther. Pat. 20, 1573–1593.
Methi, T., Ngai, J., Vang, T., Torgersen, K. M., and Tasken, K. (2007). Hypophosphorylated TCR/CD3ζ signals through a Grb2-SOS1-Ras pathway in Lck knockdown cells. Eur. J. Immunol. 37, 2539–2548.
Molina, T. J., Kishihara, K., Siderovski, D. P., van Ewijk, W., Timms, A. N., Wakeham, A., Paige, C. J., Hartman, K. U., Veillette, A., Davidson, D., and Mak, T. W. (1992). Profound block in thymocyte development in mice lacking p56lck. Nature 357, 161–164.
Naeher, D., Luescher, I. F., and Palmer, E. (2002). A role for the α-chain connecting peptide motif in mediating TCR-CD8 cooperation. J. Immunol. 169, 2964–2970.
Nika, K., Soldani, C., Salek, M., Paster, W., Gray, A., Etzensperger, R., Fugger, L., Polzella, P., Cerundolo, V., Dushek, O., Höfer, T., Viola, A., and Acuto, O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777.
O’Rourke, A. M., and Mescher, M. F. (1992). Cytotoxic T-lymphocyte activation involves a cascade of signalling and adhesion events. Nature 358, 253–255.
O’Rourke, A. M., Rogers, J., and Mescher, M. F. (1990). Activated CD8 binding to class I protein mediated by the T cell receptor results in signalling. Nature 346, 187–189.
Palacios, E. H., and Weiss, A. (2004). Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000.
Palmer, E., and Naeher, D. (2009). Affinity threshold for thymic selection through a T-cell receptor-co-receptor zipper. Nat. Rev. Immunol. 9, 207–213.
Priatel, J. J., Teh, S. J., Dower, N. A., Stone, J. C., and Teh, H. S. (2002). RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity 17, 617–627.
Qian, D., Lev, S., van Oers, N. S., Dikic, I., Schlessinger, J., and Weiss, A. (1997). Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J. Exp. Med. 185, 1253–1259.
Rybakin, V., Clamme, J.-P., Ampudia, J., Yachi, P. P., and Gascoigne, N. R. J. (2011). CD8αα and CD8αβ isotypes are equally recruited to the immunological synapse through their ability to bind MHC class I. EMBO Rep. doi: 10.1038/embor.2011.209. [Epub ahead of print].
Salmond, R. J., Emery, J., Okkenhaug, K., and Zamoyska, R. (2009a). MAPK, phosphatidylinositol 3-kinase, and mammalian target of rapamycin pathways converge at the level of ribosomal protein S6 phosphorylation to control metabolic signaling in CD8 T cells. J. Immunol. 183, 7388–7397.
Salmond, R. J., Filby, A., Qureshi, I., Caserta, S., and Zamoyska, R. (2009b). T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev. 228, 9–22.
Samelson, L. E., Phillips, A. F., Luong, E. T., and Klausner, R. D. (1990). Association of the fyn protein-tyrosine kinase with the T-cell antigen receptor. Proc. Natl. Acad. Sci. U.S.A. 87, 4358–4362.
Schamel, W. W., Arechaga, I., Risueno, R. M., van Santen, H. M., Cabezas, P., Risco, C., Valpuesta, J. M., and Alarcon, B. (2005). Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J. Exp. Med. 202, 493–503.
Schilham, M. W., Fung-Leung, W. P., Rahemtulla, A., Kuendig, T., Zhang, L., Potter, J., Miller, R. G., Hengartner, H., and Mak, T. W. (1993). Alloreactive cytotoxic T cells can develop and function in mice lacking both CD4 and CD8. Eur. J. Immunol. 23, 1299–1304.
Schraven, B., Wild, M., Kirchgessner, H., Siebert, B., Wallich, R., Henning, S., Samstag, Y., and Meuer, S. C. (1993). Alterations of CD2 association with T cell receptor signaling molecules in “CD2 unresponsive” human T lymphocytes. Eur. J. Immunol. 23, 119–123.
Seddon, B., Legname, G., Tomlinson, P., and Zamoyska, R. (2000). Long-term survival but impaired homeostatic proliferation of Naive T cells in the absence of p56lck. Science 290, 127–131.
Seddon, B., and Zamoyska, R. (2002). TCR signals mediated by Src family kinases are essential for the survival of naive T cells. J. Immunol. 169, 2997–3005.
Sohn, H. W., Tolar, P., and Pierce, S. K. (2008). Membrane heterogeneities in the formation of B cell receptor-Lyn kinase microclusters and the immune synapse. J. Cell Biol. 182, 367–379.
Stefanova, I., Hemmer, B., Vergelli, M., Martin, R., Biddison, W. E., and Germain, R. N. (2003). TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4, 248–254.
Stein, P. L., Lee, H.-M., Rich, S., and Soriano, P. (1992). pp59-fyn Mutant mice display differential signaling in thymocytes and peripheral T cells. Cell 70, 741–750.
Sun, Z. Y., Kim, S. T., Kim, I. C., Fahmy, A., Reinherz, E. L., and Wagner, G. (2004). Solution structure of the CD3εδ ectodomain and comparison with CD3εγ as a basis for modeling T cell receptor topology and signaling. Proc. Natl. Acad. Sci. U.S.A. 101, 16867–16872.
Thome, M., Duplay, P., Guttinger, M., and Acuto, O. (1995). Syk and ZAP-70 mediate recruitment of p56lck/CD4 to the activated T cell receptor/CD3/ζ complex. J. Exp. Med. 181, 1997–2006.
Thome, M., Germain, V., DiSanto, J. P., and Acuto, O. (1996). The p56lck SH2 domain mediates recruitment of CD8/p56lck to the activated T cell receptor/CD3/ζ complex. Eur. J. Immunol. 26, 2093–2100.
Timson Gauen, L. K., Kong, A. N., Samelson, L. E., and Shaw, A. S. (1992). p59fyn Tyrosine kinase associates with multiple T-cell receptor subunits through its unique amino-terminal domain. Mol. Cell. Biol. 12, 5438–5446.
van der Merwe, P. A., and Dushek, O. (2011). Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55.
Van Laethem, F., Sarafova, S. D., Park, J. H., Tai, X., Pobezinsky, L., Guinter, T. I., Adoro, S., Adams, A., Sharrow, S. O., Feigenbaum, L., and Singer, A. (2007). Deletion of CD4 and CD8 coreceptors permits generation of αβT cells that recognize antigens independently of the MHC. Immunity 27, 735–750.
van Oers, N. S., Lowin-Kropf, B., Finlay, D., Connolly, K., and Weiss, A. (1996a). αβ T cell development is abolished in mice lacking both Lck and Fyn protein tyrosine kinases. Immunity 5, 429–436.
Van Oers, N. S. C., Killeen, N., and Weiss, A. (1996b). Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J. Exp. Med. 183, 1053–1062.
van’t Hof, W., and Resh, M. D. (1999). Dual fatty acylation of p59(Fyn) is required for association with the T cell receptor ζ chain through phosphotyrosine-Src homology domain-2 interactions. J. Cell Biol. 145, 377–389.
Varma, R., Campi, G., Yokosuka, T., Saito, T., and Dustin, M. L. (2006). T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25, 117–127.
Werlen, G., Hausmann, B., and Palmer, E. (2000). A motif in the αβ T-cell receptor controls positive selection by modulating ERK activity. Nature 406, 422–426.
Wulfing, C., and Davis, M. M. (1998). A receptor/cytoskeletal movement triggered by costimulation during T cell activation. Science 282, 2266–2269.
Xu, C., Gagnon, E., Call, M. E., Schnell, J. R., Schwieters, C. D., Carman, C. V., Chou, J. J., and Wucherpfennig, K. W. (2008). Regulation of T cell receptor activation by dynamic membrane binding of the CD3ε cytoplasmic tyrosine-based motif. Cell 135, 702–713.
Xu, H., and Littman, D. R. (1993). A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell 74, 633–643.
Yachi, P. P., Ampudia, J., Gascoigne, N. R. J., and Zal, T. (2005). Nonstimulatory peptides contribute to antigen-induced CD8-T cell receptor interaction at the immunological synapse. Nat. Immunol. 6, 785–792.
Yachi, P. P., Ampudia, J., Zal, T., and Gascoigne, N. R. J. (2006). Altered peptide ligands induce delayed and reduced CD8-TCR interaction – a role for CD8 in distinguishing antigen quality. Immunity 25, 203–211.
Yasuda, K., Nagafuku, M., Shima, T., Okada, M., Yagi, T., Yamada, T., Minaki, Y., Kato, A., Tani-Ichi, S., Hamaoka, T., and Kosugi, A. (2002). Cutting edge: Fyn is essential for tyrosine phosphorylation of Csk-binding protein/phosphoprotein associated with glycolipid-enriched microdomains in lipid rafts in resting T cells. J. Immunol. 169, 2813–2817.
Yokosuka, T., Sakata-Sogawa, K., Kobayashi, W., Hiroshima, M., Hashimoto-Tane, A., Tokunaga, M., Dustin, M. L., and Saito, T. (2005). Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6, 1253–1262.
Keywords: Src-family kinases, T cell receptor, CD8, Lck, Fyn
Citation: Gascoigne NRJ, Casas J, Brzostek J and Rybakin V (2011) Initiation of TCR phosphorylation and signal transduction. Front. Immun. 2:72. doi: 10.3389/fimmu.2011.00072
Received: 02 September 2011;
Accepted: 21 November 2011;
Published online: 07 December 2011.
Edited by:
Bernard Malissen, Centre d’Immunologie de Marseille Luminy, FranceCopyright: © 2011 Gascoigne, Casas, Brzostek and Rybakin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nicholas R. J. Gascoigne, Department of Immunology and Microbial Science, IMM1, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. e-mail:Z2FzY29pZ25lQHNjcmlwcHMuZWR1