 Joey Riepsaame1
Joey Riepsaame1 Adri van Oudenaren1
Adri van Oudenaren1 Berlinda J. H. den Broeder1
Berlinda J. H. den Broeder1 Wilfred F. J. van IJcken2Joris Pothof3†
Wilfred F. J. van IJcken2Joris Pothof3† Pieter J. M. Leenen1*†
Pieter J. M. Leenen1*†- 1Department of Immunology, Erasmus University Medical Center, Rotterdam, Netherlands
- 2Erasmus Center for Biomics, Erasmus University Medical Center, Rotterdam, Netherlands
- 3Department of Genetics, Erasmus University Medical Center, Rotterdam, Netherlands
Dendritic cell (DC) maturation is a tightly regulated process that requires coordinated and timed developmental cues. Here we investigate whether microRNAs are involved in this process. We identify microRNAs in mouse GM-CSF-generated, monocyte-related DC (GM-DC) that are differentially expressed during both spontaneous and LPS-induced maturation and characterize M-CSF receptor (M-CSFR), encoded by the Csf1r gene, as a key target for microRNA-mediated regulation in the final step toward mature DC. MicroRNA-22, -34a, and -155 are up-regulated in mature MHCIIhi CD86hi DC and mediate Csf1r mRNA and protein down-regulation. Experimental inhibition of Csf1r-targeting microRNAs in vitro results not only in sustained high level M-CSFR protein expression but also in impaired DC maturation upon stimulation by LPS. Accordingly, over-expression of Csf1r in GM-DC inhibits terminal differentiation. Taken together, these results show that developmentally regulated microRNAs control Csf1r expression, supplementing previously identified mechanisms that regulate its transcription and protein surface expression. Furthermore, our data indicate a novel function for Csf1r in mouse monocyte-derived DC, showing that down-regulation of M-CSFR expression is essential for final DC maturation.
Introduction
Dendritic cells (DC) constitute a heterogeneous population of leukocytes that interconnect the innate and the adaptive immune response, in particular through their capacity to activate naïve T lymphocytes (1). DC depend on several growth factors for their proliferation, survival, and differentiation, most importantly Flt3L, GM-CSF, and M-CSF (2). Flt3L drives the development of various DC populations, in particular plasmacytoid and conventional DC (cDC), in peripheral tissues and lymphoid organs in the steady-state (3), whereas GM-CSF is important in generating inflammatory, monocyte-derived TNF/iNOS-producing DC (TipDC) (1, 4). Inactivation of M-CSF or its receptor in vivo results in a significant decrease in DC numbers (5, 6) and shift in DC subset composition (7), including a complete absence of epidermal Langerhans cells (8) and monocyte-derived DC in the intestinal lamina propria (9). Interestingly, M-CSF has been shown to induce also plasmacytoid and cDC development, in addition to development of macrophages, from BM cells of normal and Flt3L-knock out mice (10, 11). These observations underline a critical role of M-CSF signaling in the development of several DC populations.
Dendritic cells can initiate various types of T-cell responses, depending in part on the developmental status of the DC interacting with the T cells. Immature DC (iDC) characterized in mice as CD11c+MHC class IIlowCD86low cells, are specialized in taking up and processing antigens but are poor immune stimulators and may induce tolerance. In contrast, mature DC (mDC), characterized as CD11c+MHC class IIhiCD86hi cells, induce cell-mediated and/or humoral immune responses (12, 13). Thus, tight regulation of DC maturation is required to maintain a proper immune balance.
MicroRNAs are an important class of regulators involved in differentiation and cell fate decisions (14, 15). They represent an extensive family of short (∼22 nt) single-stranded non-coding RNAs that regulate gene expression at a post-transcriptional level by binding to the 3′untranslated region (3′UTR) of mRNAs, thereby causing translational inhibition of the target mRNA primarily as a result of mRNA degradation (16). In recent years, microRNAs have emerged as important regulators of immune function, which has been demonstrated in particular by in vivo gain- or loss-of-function microRNA studies (17, 18). Thus far, however, most studies linking microRNAs with the immune system have focused on T and B lymphocytes, while only a limited number of studies have focused on their role in DC development and function (19, 20). Studies using human cells have shown that the microRNA expression profiles alter during DC development (20–24). Manipulating microRNA expression affects DC function in both human and mouse (21, 25). Here, we approached the question whether microRNAs are involved in regulating mouse monocyte-derived DC maturation focusing on the final stages where CD11c+MHC class IIlowCD86low iDC develop into CD11c+MHC class IIhiCD86hi mDC. We determined the microRNA expression profiles of different mouse GM-DC maturation stages during GM-CSF-stimulated development in vitro. A set of microRNAs is described, which expression is prominently up-regulated during both the spontaneous and lipopolysaccharide (LPS)-induced transition of iDC to mDC. Csf1r, the gene encoding the growth factor receptor M-CSFR (c-Fms, M-CSFR, CD115), is identified as a predominant common target regulated by the induced miR-22, miR-34a, and miR-155. Moreover, we show that down-regulation of M-CSFR expression is a prerequisite for final DC maturation.
Materials and Methods
Animals
Female C57BL/6J mice were obtained from Harlan (Horst, Netherlands) and were kept under specific pathogen-free conditions at the animal facility of the Erasmus MC, Rotterdam, Netherlands. Housing, care, and experimental handling were performed in accordance with Dutch legal regulations. Ethical approval was obtained after protocol review by the independent animal experiment committee DEC Consult, and registered under permit numbers EUR1408 (128-08-05), EUR1738 (128-09-02), EMC2135 (128-10-10), and EMC2759 (128-12-07).
DC Maturation In vitro
Monocyte-derived DC were generated by GM-CSF stimulation of bone marrow (BM) precursors as described previously (26). These cells are indicated as GM-DC. Briefly, BM cells isolated from 8 to 13-week-old C57BL/6 mice were cultured in RPMI-1640 medium (Lonza, Belgium) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, and 20 ng/ml rmGM-CSF (Biosource International, Camarillo, CA, USA). Cells were kept in a humidified incubator at 37°C with 5% CO2. At day 0, BM leukocytes were seeded at 3 × 105 per ml in either 100 mm dishes (BD Biosciences), 12-wells plates (Nunc) or 96-wells round-bottom plates (Nunc). At day 3, fresh culture medium was added to the plates and at day 6, half of the medium was replaced. To induce enforced GM-DC maturation, 100 ng/ml LPS (Escherichia coli strain 055:B5, Sigma) was added on day 6. Alternatively, plasmacytoid DC (pDC) and cDC were generated by Flt3L stimulation of BM precursors essentially as described by Naik et al. with minor modifications (27). To this end, BM cells isolated from 8 to 13-week-old female C57BL/6 mice and erythrocytes were lysed by treatment for 2 min with 0.155 M NH4Cl. Then, BM cells were extensively washed and cultured in RPMI-1640 (Lonza, Belgium) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, and 200 ng/ml Flt3L (Peprotech, Rocky Hill, NJ, USA), and seeded at 4 × 106 per 2 ml in six-wells plates (Nunc). At day 7, cells were washed to remove free Flt3L, and stimulated for 24 h with 10 μg/ml CpG (ODN 2395, InvivoGen, San Diego, CA, USA).
To assess the role of TNF-α converting enzyme (TACE) in M-CSFR down-regulation, TACE inhibitors TMI-1 and TMI-2 (28, 29) were used in a final concentration of 10 and 15 μM, respectively (kindly provided by Dr. B. Scholte, Erasmus, MC, Netherlands). The functional inhibition during overnight cultures was tested by determining the TACE-mediated decrease of M-CSFR expression on BM monocytes. To that end, freshly isolated BM cells (106 per ml) were cultured at 37°C for 24 h in a 12-well plate in RPMI-1640 with 10% FCS and antibiotics as described before, but without additional growth factors. Inhibitors were added from the start of the culture. As TACE inducer E. coli LPS (O55 B5, Sigma) was used in a final concentration of 100 ng/ml. Cells were harvested after 24 h. Similarly, TACE activity was inhibited in GM-DC cultures by adding inhibitors during the last 24 h of a 7-day culture in combination, either or not in the presence of LPS. Expression of M-CSFR/CD115 was determined as described below.
Flow Cytometry and Cell Sorting
For cell labeling, incubations were performed in staining buffer (PBS pH 7.8, 1% BSA, 0.01% sodium azide) on ice for 30 min. Reagents used were fluorescent conjugates of CD11b (M1/70), CD11c (HL3), CD86 (GL1), CD115 (anti-M-CSFR, clone AFS98), mMGL/CD301 (ER-MP23), MHC class II I-A/I-E (M5/114.15.2), SiglecH (eBio440c), and rat-anti-mouse IgG-Alexa488 and streptavidin-Alexa633. These antibodies were obtained from BD Biosciences, eBioscience, Molecular Probes or prepared as purified Ig from hybridomas created in our lab. Cells were analyzed by flow cytometry using a FACSCalibur or FACSCanto II (Becton Dickinson) and FlowJo Analysis Software (Tree Star, Ashland, OR, USA). Sorting of cells was performed using a FACSAria Cell Sorter (Becton Dickinson).
MicroRNA Microarray Hybridization and Analysis
Total RNA was extracted using acid-phenol:chloroform (Ambion) extraction and enriched for microRNAs using a mirVana microRNA isolation kit (Ambion) according to the manufacturer’s protocols. RNA was labeled using a ULS™ aRNA labeling kit (Kreatech Diagnostics, Amsterdam). 1.5 μg of total RNA was incubated with Cy3-ULS for 30 min at 85°C and purified to remove unbound Cy3-ULS. Labeled RNA was hybridized on miRCURY LNA microRNA arrays (probe set 8.0; Exiqon, Vedbaek, Denmark) at 60°C for 16 h using a Tecan 4800 hybridization station. Slides were washed and immediately scanned using a Tecan LS Reloaded microarray laser scanner. Microarray data extraction, normalization, and data analysis were carried out as described (30). Heatmaps were generated using the TM4 microarray software suite (31). Significance Analysis of Microarrays (SAM) analysis was carried out on sorted DC populations obtained from three biological replicates, implementing a false discovery rate (FDR) ≤ 10% and a minimum 1.5-fold change in expression. All data are MIAME compliant. Raw data have been deposited in ArrayExpress and are accessible under numbers A-MEXP-2085 and E-MEXP-3311.
Csf1r-3′UTR Luciferase Reporter Assay
The full-length Csf1r-3′UTR was cloned into the XhoI/NotI site downstream the coding sequence of Renilla luciferase in the psiCHECK-2 luciferase reporter vector (Promega). Cloning primers Csf1r-3′UTR: FW 5′-GGATTCCTCGAGTCCTGCCGCT-CTCTACGT-3′ and RE 5′-GGATTCGCGGCCGCCTGGCTGTGTTAATGCTGTT-AGTT-3′. Mutant Csf1r-3′UTR constructs were generated by introducing three basepair mismatches into each seed region of the corresponding miR-22, -34a, and -155 binding sites (Csf1r-3′UTR mut all) or the miR-22 site alone (Csf1r-3′UTR 22mut) [outsourced to Genscript (Piscataway, USA)]. HEK293T cells, described by Stewart et al. (32), were plated in a 48-well plate at a density of 6 × 104 cells per well and then co-transfected the next day with 10 ng psiCHECK-2 vector containing the full 3′UTR of Csf1r mRNA, together with miR-22, -34a, -155, and control over-expression oligonucleotides (Ambion) at 50 nM final concentration using Lullaby transfection reagent (Boca Scientific). Luciferase activity was measured 48 h later using the Dual Glow luciferase kit (Promega) in a TopCount NXT microplate luminescence counter (Packard Instrument Company, Connecticut, USA). Transfections were performed in duplicate and repeated three times in independent experiments. Statistical analysis was by unpaired two-tailed Student’s t test; p-values of less than 0.05 were considered significant.
Transient MicroRNA Inhibition in GM-DC
3 × 105 BM cells were cultured in 12-well plates to generate GM-DC. These cells were transfected on day 4 of culture with 50 nM anti-miR microRNA inhibitors (Ambion), mixed with siGLO Cy3-labeled non-targeting anti-miR oligonucleotides (Dharmacon) at a 5:1 ratio. Control samples were treated with a control non-targeting inhibitor (Dharmacon). Transient transfection was accomplished using DharmaFECT1 reagent (Dharmacon) according to the manufacturer’s protocol.
Transient Csf1r Over-Expression in GM-DC
The complete ORF of mouse Csf1r was purchased as a full-length cDNA clone (Open Biosystems, IMAGE accession no. 30436119). GM-DC (8 × 106 cells) were co-electroporated at day 6 of culture with 8 μg of the Csf1r cDNA clone and 2 μg pEGFP-C1 control vector (Clontech) using an Amaxa nucleofector apparatus (Lonza; program Y-01) and Amaxa mouse macrophage nucleofector kit (Lonza) according to the manufacturer’s instructions. For control experiments, cells were co-electroporated with 8 μg pCMV-SPORT6 vector and 2 μg pEGFP-C1 vector.
Quantitative Real-Time PCR of microRNAs and Csf1r
The quantification of mature miR-155, -34a, and -22 expression levels was carried out with 1 μg of total RNA using the miScript PCR System (Qiagen) according to the manufacturer’s instructions. ΔCt values for each microRNA were normalized to tubulin reference gene. miR-155 FW primer: 5′-TTAATGCTAATTGTGATAGGGG-3′. miR-34a FW primer: 5′-TGGCAGTGTCTTAGCTGGTTGT-3′. miR-22 FW primer: 5′-AAGCTGCCAGTTGAAGAACTGT-3′. Tubulin primers: FW 5′-CAGACCAACCACT-GCTACAT-3′ and RE 5′-AGGGAATGAAGTTGGCCAGT-3′.
Ex vivo Analysis of M-CSFR Expression
FITC painting of mice was performed as described previously (33). Briefly, mice were painted on the shaved back with 250 μl of 1% FITC (Sigma) in 1:1 acetone:dibutylphthalate (Sigma) and draining axillary and brachial lymph nodes (LN) were collected 24 h afterward. Mesenteric LN were taken as a control. Cells were isolated by mechanical disruption of the LN, without enzymatic treatment, and stained with CD301/mMGL (ER-MP23), CD11b, CD86, anti-MHC class II I-A/I-E, and CD115 antibodies and analyzed by flow cytometry. To assess expression of M-CSFR expression in myeloid cells in the skin, ears from 8 to 13-week-old C57BL/6 mice were collected and frozen in Tissue-Tek OCT embedding medium (Sakura Finetek, Zoeterwoude, Netherlands) and cut into 6 μm-thick sections. Cryosections were fixed and stained as described earlier (33). Optimally titrated goat-anti-rat IgG-Alexa546 and streptavidin-Alexa633 were used to detect unlabeled antibodies and biotinylated antibodies, respectively. Images were acquired using a Leica TCS SP5 confocal microscope.
Statistical Analysis
Statistical analysis between experimental and control groups was carried out using unpaired two-tailed Student’s t test (unless stated otherwise) with the Graphpad Prism 5 software package. P-values of less than 0.05 were considered significant. Error bars represent mean ± SEM from at least three experiments.
Results
MicroRNA Expression Profiles Change during DC Development
To investigate which microRNAs are differentially expressed during monocyte-derived DC development in vitro, we performed microRNA profiling of distinct DC maturation stages isolated from 7 days GM-CSF-stimulated BM cultures, which were either or not additionally stimulated for 16 h with LPS (i.e., forced vs. spontaneous maturation). Different populations of GM-DC were sorted based on differential expression of maturation markers CD11c, MHC class II, and CD86 (Figure 1A). We then performed profiling of 328 different microRNAs using locked nucleic acid-based microRNA arrays. In total, 14 microRNAs were found to be differentially expressed in iDC to mDC development (Significance Analysis of Microarrays criteria: FDR ≤ 10%; fold change ≥ 1.5 or ≤−1.5, Figure 1B). The changes during spontaneous or LPS-induced maturation appeared to be very similar. Of all microRNAs that were screened, miR-155 showed the most abundant increase, reaching 11- and 17-fold up-regulation in the transition of iDC to mDC or iDC to LPS-mDC, respectively. These findings support data from Ceppi et al. (34) who found high levels of miR-155 in human monocyte-derived DC upon LPS stimulation. Conversely, miR-200b and -215 levels were down-regulated approximately threefold in both mDC and LPS-mDC compared to iDC. These results demonstrate that microRNA levels change during DC development in vitro and that iDC and mDC are characterized by distinct microRNA expression profiles.
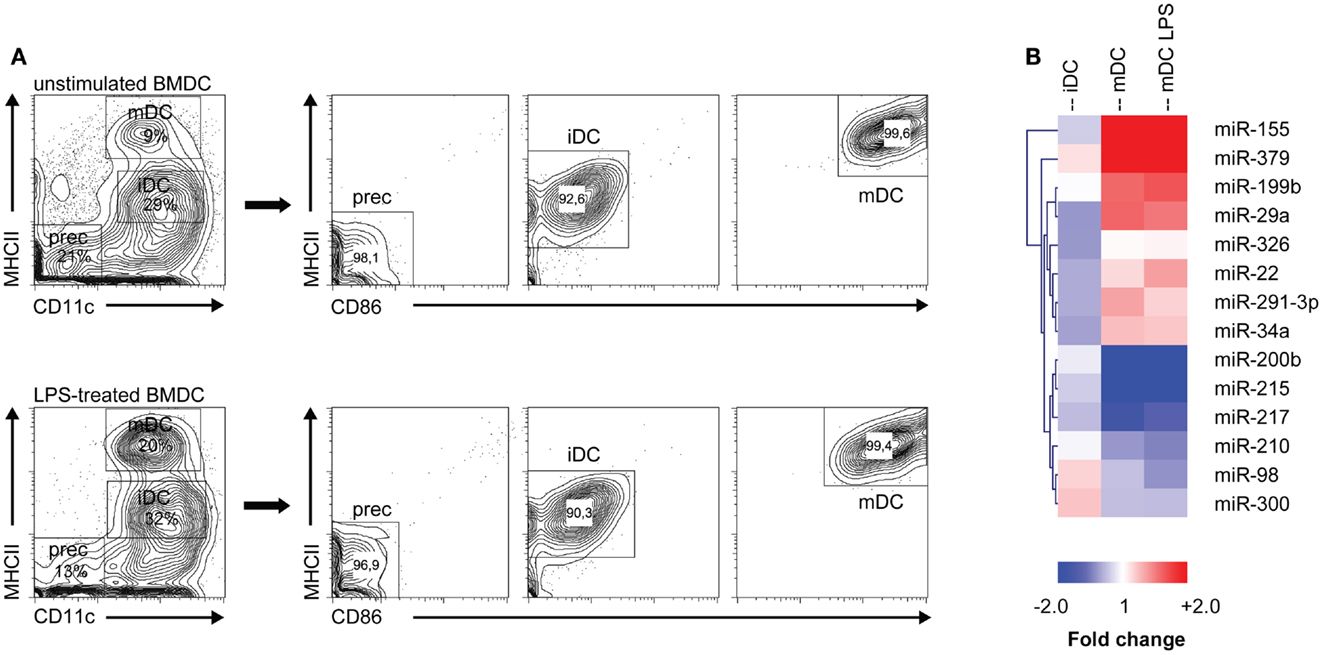
Figure 1. MicroRNA expression is altered during DC development. (A) Different DC subsets from either unstimulated or LPS-stimulated DC cultures were flow cytometrically sorted according to their differential expression of CD11c, MHC class II, and CD86. Prec, precursor cells; iDC, immature DC; mDC, mature DC. (B) Heat map of differentially expressed microRNAs (criteria: false discovery rate ≤ 10%; fold change ≥ 1.5 or ≤−1.5). Each column represents all microRNAs differentially expressed between the DC populations depicted in the heading (iDC, mDC, mDC LPS) and the precursor population. MicroRNA cluster analysis was performed using hierarchical clustering and Euclidean distance measurement.
M-CSFR is a Target of miR-22, -34a, and -155, Which are Up-Regulated during Final DC Maturation
To investigate which developmental genes are regulated by microRNAs in the final maturation step from iDC to mDC we compared the list of differentially expressed microRNAs with one compiled for genes known to be involved in the development of myeloid cells (DC/macrophage/neutrophil) as listed in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database (35) (KEGG entry: mmu04640; Hematopoietic cell lineage – Mus musculus). Using the Targetscan algorithm (36), we identified that 10 out of these 30 genes have predicted microRNA target sites in their 3′UTR conserved across mammals. Subsequently, we compared these 10 genes to our microRNA profiling data (Figure 1B), and found that only Csf1r, the gene encoding M-CSFR, and Kitl (SCF, stem cell factor) were potentially regulated by microRNAs differentially expressed in GM-DC. From these, M-CSFR was the most likely target for microRNA regulation in iDC to mDC transition as three out of four conserved predicted binding sites in the 3′UTR of Csf1r mRNA were targeted by differentially expressed microRNAs, i.e., miR-22, -34a, and -155 (Figure 2A). Other microRNA target prediction algorithms (incl. PicTar, EIMMo) confirmed the miR-22, -34a, and -155 binding sites in both human and mouse Csf1r-3′UTR (not shown). Interestingly, expression of all three microRNAs was up-regulated upon iDC to (LPS-) mDC transition in our array, which we could confirm by quantitative RT-PCR (Figure 2B). In accordance, we found a strong down-regulation of Csf1r mRNA levels in mDC compared to iDC (Figure 2C).
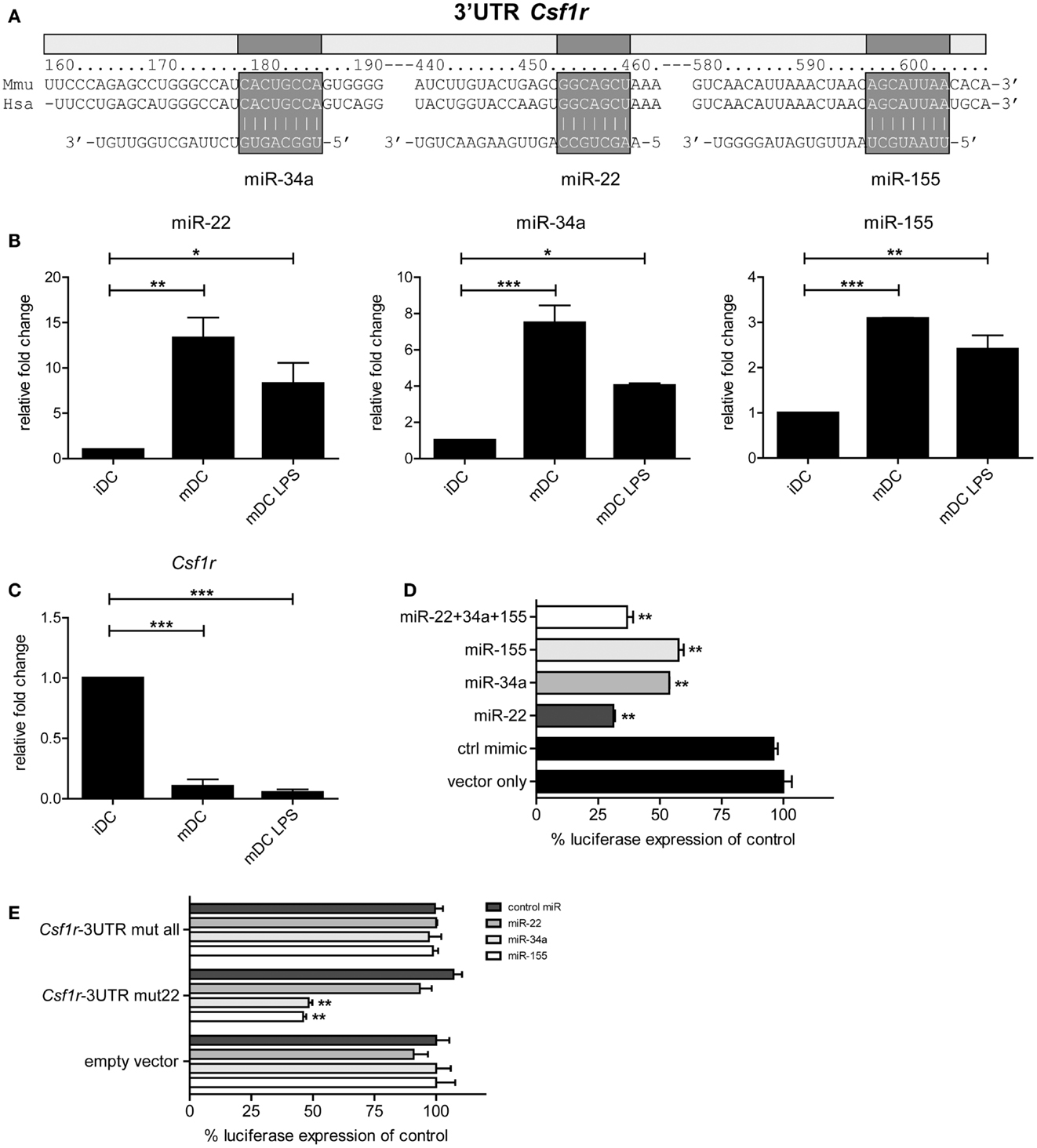
Figure 2. Csf1r is a verified target of miR-22, -34a, and -155. (A) Schematic representation of the Csf1r-3′UTR showing the predicted conserved microRNA binding sites for miR-22, -34a, and -155. Expression of (B) miR-22, -34a, and -155 and (C) Csf1r mRNA in different sorted DC populations that were used for microRNA profiling, assessed using qPCR. Relative expression changes were calculated using the 2-ΔΔCt method and expressed as fold change over tubulin controls. Data are expressed as means ± SEM from three experiments; *p < 0.05, **p < 0.01, ***p < 0.001 compared with iDC control. (D,E) The full-length Csf1r-3′UTR was cloned downstream a Renilla luciferase (RLuc) gene (psiCHECK-2 vector). HEK293T cells were transiently transfected with the psiCHECK-2 reporter vector containing (D) the wild-type mouse Csf1r-3′UTR or (E) a mutant Csf1r-3′UTR harboring three nucleotide point mutations in each seed region of the corresponding miR-22, -34a, and miR-155 binding sites (Csf1r-3′UTR mut all), or in the miR-22 site only (Csf1r-3′UTR mut22). The original psiCHECK-2 vector (containing no 3′UTR insert) was used as a control (empty vector). Cells were co-transfected with the hairpin precursors of miR-22, miR-34a, miR-155, or control oligonucleotide, all at a final concentration of 50 nM. A ubiquitously expressed firefly luciferase gene, also encoded by the vector, was used to normalize transfection efficiency. Luciferase activity was measured 48 h after transfection. Data are expressed as means ± SEM from three experiments.
To test whether miR-22, -34a, and -155 actually can regulate Csf1r expression through direct 3′UTR interactions, we cloned the complete 3′UTR of Csf1r into the psiCHECK-2 reporter vector downstream the coding sequence of Renilla luciferase. This vector, which also encoded firefly luciferase as an internal control, was transfected into HEK293T cells. When miR-155- or miR-34a precursor microRNAs were co-transfected we observed that Renilla luciferase expression was repressed approximately 50% compared to controls (Figure 2D). Co-transfection of miR-22 precursor inhibited Renilla luciferase expression even almost 75%. The combination of all three microRNAs did not further down-regulate luciferase expression. These findings confirm and extend the work of O’Connell et al. (37) and Lu et al. (24) who have shown that Csf1r is a validated target of miR-155. To verify whether the repressive effects were microRNA-specific, we repeated these experiments with Csf1r-3′UTR reporter constructs harboring point mutations in all three (miR-22, -34a, and -155) microRNA binding sites or in just one (miR-22) microRNA binding site. Indeed, no repressive effects of microRNAs could be observed when mutants were tested containing alterations in all binding sites, indicating the specificity of microRNA targeting (Figure 2E). Mutation of the miR-22 binding site alone (but not the miR-34a and -155 binding sites) completely impaired miR-22-mediated Csf1r-3′UTR repression, whereas the repressive effects of miR-34a and -155 remained unaffected, with similar levels of repression as in our initial experiments with the wild-type mouse Csf1r-3′UTR (Figure 2E).
Taken together, these data demonstrate that miR-22, -34a, and -155 are up-regulated in the final step of iDC to mDC maturation and that all three microRNAs can directly regulate Csf1r expression by targeting its mRNA via 3′UTR interactions.
M-CSFR Protein Expression is Down-Regulated in the Final Phase of DC Maturation
Based on the differential expression of Csf1r-regulating microRNAs and their Csf1r target mRNA, we predicted that M-CSFR protein expression decreased during the transition of iDC to mDC. Therefore, we followed M-CSFR expression in GM-CSF-stimulated BM cultures over time by flow cytometry. To induce final maturation, part of the cultures was treated with LPS on day 6. Indeed, M-CSFR expression was rapidly down-regulated during both spontaneous and LPS-induced transition of iDC to mDC, accompanied by the acquisition of high level MHC class II and CD86 expression (Figure 3A).
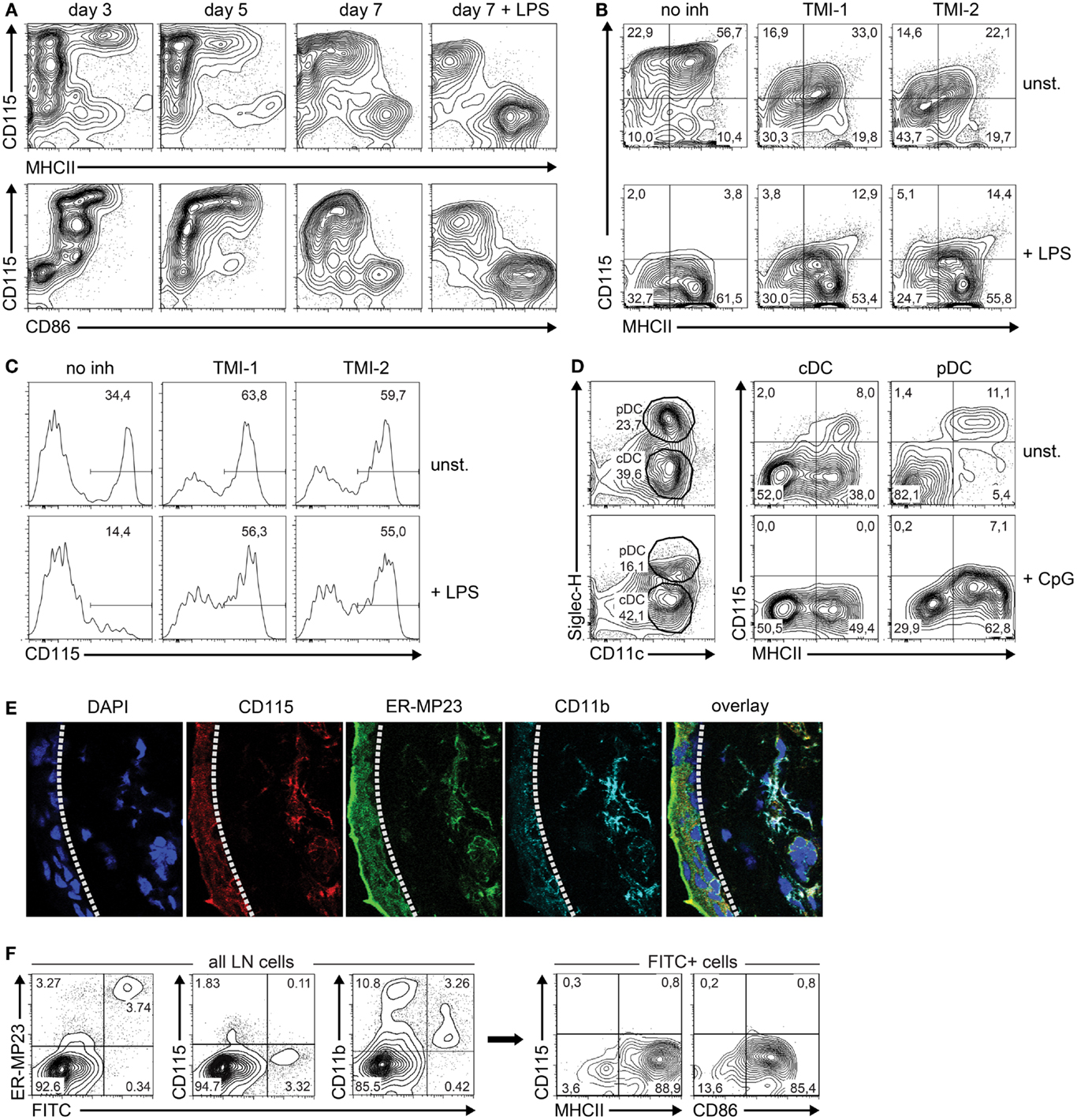
Figure 3. M-CSFR protein expression is regulated during DC maturation. (A) Flow cytometry contour plots show cell surface M-CSFR (CD115) protein expression over time plotted against the DC maturation markers MHC class II and CD86 in GM-CSF-stimulated DC cultures. LPS (100 ng/ml) was added on day 6 as a positive control to induce DC maturation. (B) Effect of TACE inhibition by TMI-1 or -2 on M-CSFR/CD115 expression during the final 24 h of GM-DC culture, in presence or absence of LPS-stimulated maturation. (C) Effect of TACE inhibition during overnight culture of fresh BM on spontaneous (top panel) and LPS-induced (lower panel) down-regulation of M-CSFR/CD115 expression on BM monocytes, identified by gating of CD11b+Ly-6ChiLy-6Gneg cells (not shown). (D) Detection of M-CSFR/CD115 expression on mature and immature in vitro-derived FL-cDC and pDC. Mouse BM cells were cultured for 8 days with Flt3L and surface marker expression was analyzed using flow cytometry. CpG was added on day 7 to induce DC maturation. Viable DC were gated using side scatter and negative DAPI staining as criteria (not shown). Next, differential SiglecH and CD11c expression was used to distinguish between pDC (CD11c+SiglecH+) and cDC (CD11c+SiglecH–). CD115 expression was plotted against the DC maturation marker MHC class II for both cDC and pDC. Results are shown of a representative experiment of three with similar outcome. (E) Macrophages and M-CSFR-dependent immature DC in sections of mouse ear skin dermis were identified by CD115 (red), mMGL/CD301 (green), CD11b (cyan), and DAPI (blue), and detected by confocal microscopy. The dashed line indicates the border of the epidermis (left) and dermis (right). (F) Analysis of skin-derived FITC+ cells migrated to skin-draining LN. Mice were skin-painted on shaved back with a 1% FITC solution. After 24 h, skin-draining LN were isolated, processed into single-cell suspensions by mechanical means only and analyzed using flow cytometry. FITC+ cells emigrated from the skin were found to represent primarily CD11c+MHCIIhiCD86hi cells lacking CD115 expression. Results of (E,F) are shown of a representative experiment of three with similar outcome.
Although our results suggest that microRNAs are responsible for the observed down-regulation of M-CSFR expression, we cannot exclude other mechanisms that might also be involved in this process. In particular, LPS is known to stimulate activation of the transmembrane protease TNF-α-converting enzyme (TACE), which cleaves M-CSFR from the surface (38). To study a putative contribution of TACE-mediated M-CSFR cleavage to decreasing expression during final maturation, GM-DC were incubated with and without TACE inhibitors TMI-1 and -2 (28, 29) before enforcing DC maturation with LPS during overnight culture. We found that M-CSFR levels decreased significantly in LPS-stimulated DC compared to unstimulated cells, despite inhibition of TACE (Figure 3B). TMI-1 and -2 treatment of GM-DC by itself already caused a decrease in M-CSFR expression, which was not caused by decreased cell viability (not shown). The efficacy of the TACE inhibitors under these conditions is indicated by the inhibition of spontaneous and LPS-induced decrease of membrane-bound M-CSFR on BM monocytes [compare left panels in Figure 3C to approximately 80% M-CSFR-expressing BM monocytes upon isolation (39)] (Figure 3C). Collectively, these data suggest that TACE-mediated shedding does not play a major role in reducing M-CSFR expression on maturing GM-DC.
To investigate whether down-regulation of surface M-CSFR expression is restricted to maturing GM-DC, we cultured freshly isolated BM in the presence of Flt3L to generate cDC (FL-cDC) and pDC and assessed surface M-CSFR levels by flow cytometry. We observed that only a minor subset of CD11c+SiglecH− FL-cDC and CD11c+SiglecH+ pDC expressed M-CSFR (Figure 3D). Although enforced maturation with CpG in these cultures led to a further reduction in M-CSFR levels, we conclude that M-CSFR expression by iDC and down-regulation during their maturation is not a universal characteristic of Flt3L-generated cDC and pDC in vitro.
Next, we asked whether M-CSFR expression is regulated in DC in vivo as well. To that end, we focused on DC present in the skin, and assessed their M-CSFR expression in situ and after stimulated migration to skin-draining LN. The skin harbors various cells of myeloid origin, such as Langerhans cells in the epidermis, and macrophages/iDC in the dermis (1, 33, 40). Both epidermal and dermal populations of cells are able to incorporate skin-applied antigens and migrate subsequently to the draining LN, where they appear as phenotypically mDC (33, 41). Although flow cytometric analysis is the preferred method to quantify protein expression in mixed cell populations, we observed that the sample preparation of mouse skin tissue into single-cell suspensions using enzymatic digestion did not allow the use of flow cytometry due to enzymatic cleavage of surface-bound M-CSFR (not shown). Therefore, we analyzed M-CSFR expression by skin mononuclear phagocytes in situ by less sensitive confocal microscopy of tissue sections. We labeled mouse ear skin cryosections with CD115 – an antibody directed against M-CSFR – together with mMGL (ER-MP23) and CD11b antibodies to identify all dermal macrophages and iDC of the partially M-CSFR-dependent CD103neg subset (7) and analyzed expression of these markers (Figure 3E). Cells that stain positively for the C-type lectin mMGL (CD301) are dermal macrophages and iDC that migrate to draining LN upon antigen uptake (33, 42). The images demonstrate that CD115 co-localizes with CD11b in the skin epidermis as well as with CD11b and mMGL in the dermis of the mouse ear. Similar observations were made in sections taken from mouse back skin (not shown). This indicates that both epidermal Langerhans cells and dermal mononuclear phagocytes uniformly express M-CSFR in situ, in agreement with previous findings by Hume et al. at the mRNA level (43).
To assess whether these CD115-positive mononuclear phagocytes in the skin down-regulate expression upon maturation during migration to draining LN, we skin-painted FITC onto shaved back skin of mice and analyzed the phenotype of the FITC+ cells that had emigrated from the skin to skin-draining LN 24 h later (Figure 3F). The majority of LN-immigrating, FITC-transporting cells from the skin express mMGL (ER-MP23) at a high level, reminiscent of their dermal origin. These findings are in agreement with our previous results (33) and with those of Irimura and colleagues (42). The skin-derived cells, expressing CD115 in situ, have lost CD115 expression upon LN arrival. Furthermore, FITC-positive LN cells have retained CD11b at an intermediate or low level, related to a DC nature, rather than the high level CD11b+ myelomonocytic cells, which are FITC-negative and partially M-CSFR+ (not shown). In agreement with their DC identity, gated FITC-positive LN cells show high levels of MHC class II and CD86 expression.
Together, our data demonstrate that M-CSFR protein is expressed by DC at the precursor and iDC stage, but down-regulated in the final stage of DC maturation. We show this during in vitro maturation of DC derived from BM precursors stimulated with GM-CSF, and our in vivo findings on maturation of connective tissue DC that migrate to skin-draining LN support this notion. In agreement with this, Cheong et al. have shown that monocytes down-regulate M-CSFR expression upon maturation to inflammatory LN DC in vivo (44).
Inhibition of miR-22, -34a, and -155 Dysregulates M-CSFR Expression and Blocks DC Maturation
At this point, it is reasonable to hypothesize that up-regulation of miR-22, -34a, and -155 is causally involved with Csf1r down-regulation. To substantiate this, we investigated whether M-CSFR protein down-regulation in maturing DC in vitro could be prevented by inhibiting miR-22, -34a, and -155 using microRNA inhibitor oligonucleotides. First, we used siGLO Cy3-labeled non-targeting anti-miR oligonucleotides to evaluate transfection efficiency, generally varying between 30 and 40% (Figure 4A). Then we inhibited microRNA-22, -34a, or -155 on day 4 of GM-DC culture and observed that M-CSFR down-regulation upon LPS-induced DC maturation at day 7 was significantly prevented (Figure 4B), demonstrating that miR-22, -34a, and -155 regulate M-CSFR expression in DC in the final stage of maturation. Interestingly, in these experiments we observed that not only down-regulation of M-CSFR was reduced on cells treated miR-22, -34a, or -155 inhibitors, compared to the cells transfected with control inhibitor, but that also the frequency of cells with a mDC phenotype (CD11c+MHCIIhiCD86hi) was significantly lower (p < 0.0001) (Figure 4C). This suggests that these microRNAs are directly involved with the final step in DC maturation. To investigate whether microRNA-mediated M-CSFR down-regulation was functionally involved in this step, we enforced Csf1r expression in developing GM-DC using an expression vector containing the complete ORF of mouse Csf1r. Cells were co-transfected with an EGFP expression vector at a 1:4 ratio (EGFP: Csfr1) to enable selection for successfully transfected cells (Figure 4D). In these experiments we found that enforced Csf1r expression significantly (p = 0.0013) impaired up-regulation of DC maturation markers MHC class II and CD86 upon stimulation with LPS (Figures 4E,F).
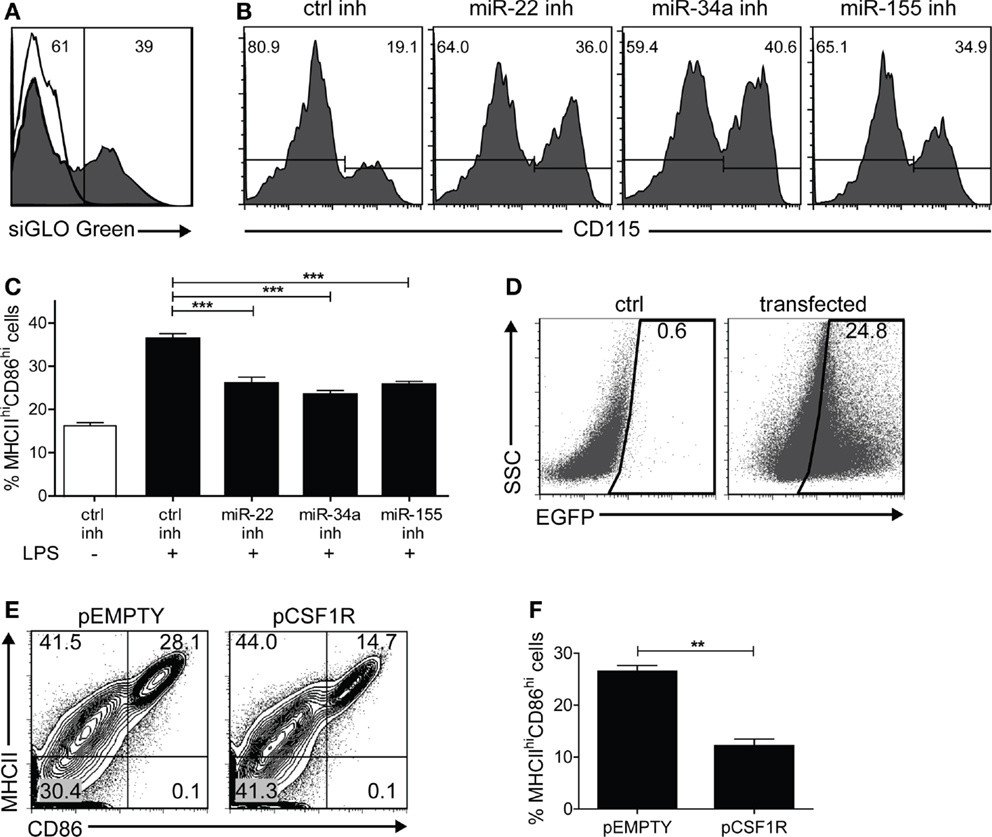
Figure 4. In vitro inhibition of miR-155/-34a/-22 prevents LPS-induced M-CSFR down-regulation and DC maturation. BM cultures stimulated with GM-CSF were transfected with different microRNA oligonucleotide inhibitors on day 4, together with siGLO Cy3-labeled non-targeting anti-miR oligonucleotides at a ratio of 5:1, and exposed to LPS on day 6. Cell surface marker expression was analyzed on day 7. (A) Only successfully transfected cells (siGLO+) were selected for analysis. The open histogram represents non-transfected cells. (B) Inhibition of miR-22, -34a, and -155 prevented LPS-induced CD115 down-regulation compared to control inhibitor alone. Results are shown of a representative experiment of five with similar results. (C) Frequency of mDC (MHCIIhiCD86hi) in cultures stimulated or not with LPS and transfected with control inhibitor or miR-22, -34a, or -155 inhibitor. Data are expressed as mean ± SEM from five experiments; ***p < 0.0001. (D) BM cultures stimulated with GM-CSF were nucleofected on day 6 of culture with an expression vector encoding Csf1r (pCSF1R) or the empty backbone vector (pEMPTY) together with an EGFP expression vector in a 4:1 ratio to assess transfection efficiency. LPS was added on day 8 to enforce DC maturation. In this experiment, transfection led to detectable EGFP expression in 24% of the cells. 7AAD staining confirmed that both vectors used did not cause significant differences in cell viability as a result of transfection (data not shown). (E,F) Cell surface marker expression of Csf1r-transfected EGFP+ cells was analyzed on day 9 using flow cytometry. Contour plots of transfected cells (E) and quantitation of viable MHCIIhiCD86hiEGFP+ mDC (F) after overnight LPS exposure. Data are expressed as mean ± SEM from three experiments; **p = 0.0013.
In summary, these results show that in vitro inhibition of miR-22, -34a, and -155 reduces LPS-induced M-CSFR down-regulation in GM-DC. Moreover, our findings suggest that down-regulation of M-CSFR in DC is required for full DC maturation.
Discussion
In this study we provide evidence that microRNAs play an important role in mouse GM-DC development by down-regulating M-CSFR expression in the final maturation step in which DC acquire the mature MHC class IIhi CD86hi phenotype (Figure 5). We show that the transition from CD11c+ MHC class IImed CD86−/lo iDC to CD11c+ MHC class IIhi CD86hi mDC coincides with an up-regulation of some microRNAs and down-regulation of others. Noteworthy in this respect is that spontaneously in vitro matured mDC and LPS-induced mDC show remarkably similar microRNA expression profiles. This suggests that spontaneous, GM-CSF-induced and LPS-induced maturation proceed along similar routes, at least on a microRNA level.
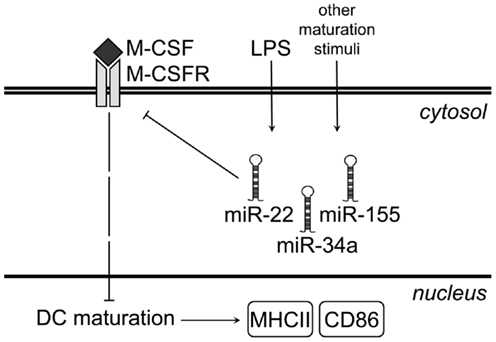
Figure 5. Proposed model for microRNA-mediated regulation of M-CSFR upon LPS-induced GM-DC maturation. Under steady-state conditions, immature GM-DC express high levels of M-CSFR. As a result of M-CSFR signaling, expression of maturation markers such as MHC class II and CD86 remains low. LPS stimulation of iDC or other maturation-inducing stimuli up-regulate miR-22, -34a, and -155, which bind to Csf1r mRNA and prevent translation of M-CSFR protein. This results in down-regulation of M-CSFR protein at the cell surface and allows up-regulation of both MHC class II and CD86.
Based on our profiling results, we have identified Csf1r as a primary target of microRNA-mediated regulation. The role of the Csf1r gene in myeloid cell development has been studied at several levels, but its function in DC development has remained underexposed (45). MacDonald et al. have demonstrated in the MacGreen mouse model, in which the Csf1r promoter directs the expression of EGFP, that the Csf1r gene is generally expressed by DC in vivo (6). Moreover, the significant reduction of DC in mice lacking Csf1r indicates that this gene is required for optimal expansion of DC (5–7). Although the MacGreen mouse model shows transcriptional activity of the Csf1r promoter throughout DC differentiation in GM-CSF-driven BM cultures, it does not necessarily indicate presence of the M-CSFR protein product (6). Our data demonstrate that Csf1r expression is actively down-regulated in the final phase of DC maturation at both the mRNA and protein level via microRNAs.
MicroRNAs are involved in the regulation of M-CSFR expression both directly and indirectly during myeloid cell differentiation. MicroRNAs 17-5p, -20a, and -106 regulate M-CSFR indirectly through targeting of the AML1/Runx1 transcription factor (46). In early myeloid precursor cells these microRNAs suppress AML1/Runx1 protein expression, leading to limited Csf1r transcription and inhibition of monocytic differentiation and maturation. In our profiling experiments, we observed no major changes in miR-17-5p, -20a, or -106 expression during DC development, suggesting that this regulation does not play an important role in later stages. O’Connell and colleagues have previously validated a significant number of targets of miR-155, including Csf1r (37), a finding that was recently confirmed by Lu et al. (24). However, these studies did not address the question whether microRNA-mediated regulation of Csf1r influences myeloid cell development. Our data provide evidence that final DC differentiation requires microRNA-mediated regulation of Csf1r. Additionally, we show here that miR-22 and miR-34a also directly target Csf1r and are up-regulated upon final DC maturation.
Tight regulation of Csf1r expression is essential for proper myeloid cell development and occurs at several levels. At a transcriptional level, the Csf1r gene is transactivated by several myeloid transcription factors, including PU.1, Runx1, C/EBPα, and several Ets family members through binding to its TATA box-deficient promoter located ∼300 bp upstream of the transcriptional start site (47–49). A highly conserved intronic enhancer named FIRE is located in the first intron downstream of the Csf1r promoter and contains additional Sp1 and Egr-2 binding motifs (50, 51). At the protein level, membrane-bound M-CSFR dimerizes after binding of its ligand M-CSF to macrophages, resulting in a number of modifications to the cell surface receptor including tyrosine/serine phosphorylation, ubiquitination, and subsequent internalization of the M-CSF/M-CSFR complex before its intralysosomal degradation (52). Additionally, various pro-inflammatory stimuli cause enzymatic cleavage of the M-CSFR ectodomain by TNFα-converting enzyme (TACE)-mediated shedding in vitro (38, 53, 54). However, in vivo down-regulation of surface M-CSFR expression under inflammatory conditions can also be TACE-independent, since TACE was not responsible for absence of M-CSFR expression from monocytes generated during severe L. monocytogenes infection (39). Our current experiments indicated that TACE also did not play a major role in M-CSFR down-regulation in final GM-DC maturation in vitro. Other post-translational modifications to the M-CSFR protein, such as ubiquitination or sumoylation, probably also contribute to M-CSFR instability. For instance, binding of M-CSF to M-CSFR induces conformational changes to the receptor leading to M-CSFR ubiquitination and subsequently altered protein half-life (55, 56).
Here, we have shown that an additional level of Csf1r expression regulation occurs, on top of transcriptional and post-translational control, through the action of microRNAs miR-22, -34a, and -155. Interestingly, two studies have indicated that DC from Mir155−/− mice show impaired expression of maturation markers after LPS stimulation and have impaired T cell stimulatory capacity (57, 58). In contrast, Lu et al. reported no such differences in miR-155-deficient DC after LPS stimulation (24), but the levels of LPS used to induce DC maturation in the latter study were at least 10-fold higher than those used in our and other studies (57, 58). Therefore, it is possible that hyper-stimulation by LPS annuls the attenuated DC maturation phenotype caused by miR-155 deletion. Direct comparison of M-CSFR-regulating microRNA levels and M-CSFR mRNA and protein expression in LPS- vs. spontaneously matured DC (Figures 2B,C, and 3A) indicates that differences at microRNA level, although important, do not provide a full explanation for differences at mRNA and protein level, and therefore post-translational mechanisms described above probably play an important additional role. Together, this shows that post-transcriptional microRNA-mediated regulation of Csf1r mRNA expression complements its transcriptional regulation, in line with the notion that Csf1r is stringently regulated during myelopoiesis.
To address the role of M-CSFR during final GM-DC maturation, we inhibited its down-regulation via microRNA inhibitors or transiently over-expressed Csf1r in GM-DC. Both approaches resulted in a significant inhibition of LPS-induced final DC maturation. Interestingly, mice lacking the Csf1r gene show major alterations in different subsets of DC, where especially the CD11b+ CD103− subset is affected (6, 7). Therefore, both absence of Csf1r as well as enforced expression lead to aberrant DC development. This underscores that regulation of the levels of this receptor throughout DC differentiation is crucial. Our analysis of M-CSFR expression by skin and LN DC suggests that this regulation also occurs during in vivo maturation of DC. In agreement with these findings, Cheong and colleagues have shown that mouse blood monocytes, which are recruited to peripheral LN after i.v. injection of Gram-negative bacteria or LPS, also lose their M-CSFR expression upon in vivo differentiation to DC in the draining LN (44). It is tempting to speculate that especially expression of miR-155, stimulated by the inflammatory conditions (59), is involved with M-CSFR down-regulation in these infection-induced monocytes and monocyte-derived DC. The importance of post-transcriptional regulation of Csf1r is further supported by Sasmono et al. who have found that mouse neutrophilic granulocytes contain significant amounts of Csf1r mRNA, but do not express the corresponding protein product (60). Involvement of microRNAs in this regulation has not been shown, however.
How might M-CSFR-mediated inhibition of DC differentiation operate at the molecular level? Although speculative, a possible scenario is that M-CSFR triggering leads to sustained activity of the PI-3K/Akt pathway (52). This then inhibits LPS-induced activation of p38 MAPK, JNK and NF-κB which are important for the expression of mDC characteristics such as MHC class II and CD86 (61, 62). This view is supported by a study showing that GM-DC from mice lacking SHIP, a negative regulator of the PI-3K pathway, have an immature phenotype and mature poorly in response to LPS (63). Notably, this apparent block in DC maturation could be inverted by treating these cells with PI-3K inhibitors LY294002 or Wortmannin, indicating that PI-3K is a negative regulator of DC maturation. Therefore, M-CSFR-mediated activation of the PI-3K pathway might block final LPS-induced DC maturation. Additionally, M-CSFR-signaling elevates levels of c-Fos (64), whereas miR-155-mediated down-regulation of c-Fos was recently shown to be involved with DC maturation and function (58). It should be stressed, however, that M-CSFR signaling involves a variety of pathways, including those mediated by Ras, Jak/STAT, or β-catenin. Collectively, these downstream mechanisms are responsible for the final outcome of M-CSFR signaling in macrophages (52, 65). Which of these pathways is responsible for inhibition of DC maturation by M-CSFR activity remains to be demonstrated.
In summary, we have elucidated a previously unknown molecular mechanism regulating the final step in monocyte-derived DC maturation, which acts upstream of M-CSFR signaling and where the receptor itself is subject to microRNA-mediated control. Our work demonstrates that decreasing M-CSFR signaling contributes to enable final GM-DC maturation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to acknowledge Edwin de Haas and Benjamin Bartol for operating the cell sorter and Drs. G. van Cappellen and A. Houtsmuller for help with confocal microscopy. TACE inhibitors TMI-1 and -2 were kindly provided by Dr. B. Scholte (Erasmus MC, Department of Cell Biology).
References
1. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
2. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol (2012) 12:101–13. doi:10.1038/nri3149
3. Onai N, Obata-Onai A, Schmid MA, Manz MG. Flt3 in regulation of type I interferon-producing cell and dendritic cell development. Ann N Y Acad Sci (2007) 1106:253–61. doi:10.1196/annals.1392.015
4. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol (2007) 7:19–30. doi:10.1038/nri1996
5. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi:10.1182/blood.V99.1.111
6. MacDonald KP, Rowe V, Bofinger HM, Thomas R, Sasmono T, Hume DA, et al. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J Immunol (2005) 175:1399–405.
7. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206:3115–30. doi:10.1084/jem.20091756
8. Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai XM, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol (2006) 7:265–73. doi:10.1038/ni1307
9. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity (2009) 31:513–25. doi:10.1016/j.immuni.2009.08.010
10. Fancke B, Suter M, Hochrein H, O’Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood (2008) 111:150–9. doi:10.1182/blood-2007-05-089292
11. O’Keeffe M, Fancke B, Hochrein H. The generation of plasmacytoid and conventional dendritic cells with M-CSF. Methods Mol Biol (2010) 595:187–93. doi:10.1007/978-1-60761-421-0_12
13. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol (2007) 7:543–55. doi:10.1038/nri2103
14. Wienholds E, Plasterk RH. MicroRNA function in animal development. FEBS Lett (2005) 579:5911–22. doi:10.1016/j.febslet.2005.07.070
15. Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell (2010) 7:36–41. doi:10.1016/j.stem.2010.06.012
16. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136:215–33. doi:10.1016/j.cell.2009.01.002
17. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol (2008) 9:839–45. doi:10.1038/ni.f.209
18. Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell (2009) 136:26–36. doi:10.1016/j.cell.2008.12.027
19. Turner ML, Schnorfeil FM, Brocker T. MicroRNAs regulate dendritic cell differentiation and function. J Immunol (2011) 187:3911–7. doi:10.4049/jimmunol.1101137
20. Mildner A, Chapnik E, Manor O, Yona S, Kim KW, Aychek T, et al. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood (2013) 121:1016–27. doi:10.1182/blood-2012-07-445999
21. Hashimi ST, Fulcher JA, Chang MH, Gov L, Wang S, Lee B. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood (2009) 114:404–14. doi:10.1182/blood-2008-09-179150
22. Holmstrom K, Pedersen AW, Claesson MH, Zocca MB, Jensen SS. Identification of a microRNA signature in dendritic cell vaccines for cancer immunotherapy. Hum Immunol (2009) 71:67–73. doi:10.1016/j.humimm.2009.10.001
23. Jansen BJ, Sama IE, Eleveld-Trancikova D, Van Hout-Kuijer MA, Jansen JH, Huynen MA, et al. MicroRNA genes preferentially expressed in dendritic cells contain sites for conserved transcription factor binding motifs in their promoters. BMC Genomics (2011) 12:330. doi:10.1186/1471-2164-12-330
24. Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood (2011) 117:4293–303. doi:10.1182/blood-2010-12-322503
25. Kuipers H, Schnorfeil FM, Fehling HJ, Bartels H, Brocker T. Dicer-dependent microRNAs control maturation, function, and maintenance of Langerhans cells in vivo. J Immunol (2010) 185:400–9. doi:10.4049/jimmunol.0903912
26. Nikolic T, Bunk M, Drexhage HA, Leenen PJ. Bone marrow precursors of nonobese diabetic mice develop into defective macrophage-like dendritic cells in vitro. J Immunol (2004) 173:4342–51.
27. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol (2005) 174:6592–7.
28. Zhang Y, Xu J, Levin J, Hegen M, Li G, Robertshaw H, et al. Identification and characterization of 4-[[4-(2-butynyloxy)phenyl]sulfonyl]-N-hydroxy-2,2-dimethyl-(3S)thiomorpholinecar boxamide (TMI-1), a novel dual tumor necrosis factor-alpha-converting enzyme/matrix metalloprotease inhibitor for the treatment of rheumatoid arthritis. J Pharmacol Exp Ther (2004) 309:348–55.
29. Canault M, Leroyer AS, Peiretti F, Leseche G, Tedgui A, Bonardo B, et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol (2007) 171:1713–23. doi:10.2353/ajpath.2007.070021
30. Pothof J, Verkaik NS, Van Ijcken W, Wiemer EA, Ta VT, Van Der Horst GT, et al. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J (2009) 28:2090–9. doi:10.1038/emboj.2009.156
31. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques (2003) 34:374–8.
32. Stewart N, Bacchetti S. Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology (1991) 180:49–57. doi:10.1016/0042-6822(91)90008-Y
33. Dupasquier M, Stoitzner P, Wan H, Cerqueira D, Van Oudenaren A, Voerman JS, et al. The dermal microenvironment induces the expression of the alternative activation marker CD301/mMGL in mononuclear phagocytes, independent of IL-4/IL-13 signaling. J Leukoc Biol (2006) 80:838–49. doi:10.1189/jlb.1005564
34. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A (2009) 106:2735–40. doi:10.1073/pnas.0811073106
35. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res (2000) 28:27–30. doi:10.1093/nar/28.1.27
36. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell (2005) 120:15–20. doi:10.1016/j.cell.2004.12.035
37. O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med (2008) 205:585–94. doi:10.1084/jem.20072108
38. Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol (2001) 166:1583–9.
39. Drevets DA, Schawang JE, Mandava VK, Dillon MJ, Leenen PJ. Severe Listeria monocytogenes infection induces development of monocytes with distinct phenotypic and functional features. J Immunol (2010) 185:2432–41. doi:10.4049/jimmunol.1000486
40. Dupasquier M, Stoitzner P, Van Oudenaren A, Romani N, Leenen PJ. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. J Invest Dermatol (2004) 123:876–9. doi:10.1111/j.0022-202X.2004.23427.x
41. Kumamoto Y, Denda-Nagai K, Aida S, Higashi N, Irimura T. MGL2 Dermal dendritic cells are sufficient to initiate contact hypersensitivity in vivo. PLoS ONE (2009) 4:e5619. doi:10.1371/journal.pone.0005619
42. Chun KH, Imai Y, Higashi N, Irimura T. Migration of dermal cells expressing a macrophage C-type lectin during the sensitization phase of delayed-type hypersensitivity. J Leukoc Biol (2000) 68:471–8.
43. Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood (2003) 101: 1155–63. doi:10.1182/blood-2002-02-0569
44. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143:416–29. doi:10.1016/j.cell.2010.09.039
45. Merad M, Manz MG. Dendritic cell homeostasis. Blood (2009) 113:3418–27. doi:10.1182/blood-2008-12-180646
46. Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol (2007) 9:775–87. doi:10.1038/ncb1613
47. Roberts WM, Shapiro LH, Ashmun RA, Look AT. Transcription of the human colony-stimulating factor-1 receptor gene is regulated by separate tissue-specific promoters. Blood (1992) 79:586–93.
48. Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol (1994) 14:373–81.
49. Bonifer C, Hume DA. The transcriptional regulation of the colony-stimulating factor 1 receptor (csf1r) gene during hematopoiesis. Front Biosci (2008) 13:549–60. doi:10.2741/2700
50. Himes SR, Tagoh H, Goonetilleke N, Sasmono T, Oceandy D, Clark R, et al. A highly conserved c-fms gene intronic element controls macrophage-specific and regulated expression. J Leukoc Biol (2001) 70:812–20.
51. Krysinska H, Hoogenkamp M, Ingram R, Wilson N, Tagoh H, Laslo P, et al. A two-step, PU.1-dependent mechanism for developmentally regulated chromatin remodeling and transcription of the c-fms gene. Mol Cell Biol (2007) 27:878–87. doi:10.1128/MCB.01915-06
52. Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol (2004) 14:628–38. doi:10.1016/j.tcb.2004.09.016
53. Wilhelmsen K, van der Geer P. Phorbol 12-myristate 13-acetate-induced release of the colony-stimulating factor 1 receptor cytoplasmic domain into the cytosol involves two separate cleavage events. Mol Cell Biol (2004) 24:454–64. doi:10.1128/MCB.24.1.454-464.2004
54. Hiasa M, Abe M, Nakano A, Oda A, Amou H, Kido S, et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-alpha converting enzyme (TACE). Blood (2009) 114:4517–26. doi:10.1182/blood-2009-04-215020
55. Kallies A, Rosenbauer F, Scheller M, Knobeloch KP, Horak I. Accumulation of c-Cbl and rapid termination of colony-stimulating factor 1 receptor signaling in interferon consensus sequence binding protein-deficient bone marrow-derived macrophages. Blood (2002) 99:3213–9. doi:10.1182/blood.V99.9.3213
56. Xiong Y, Song D, Cai Y, Yu W, Yeung YG, Stanley ER. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. J Biol Chem (2011) 286:952–60. doi:10.1074/jbc.M110.166702
57. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316:608–11. doi:10.1126/science.1139253
58. Dunand-Sauthier I, Santiago-Raber ML, Capponi L, Vejnar CE, Schaad O, Irla M, et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood (2011) 117:4490–500. doi:10.1182/blood-2010-09-308064
59. O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol (2010) 10:111–22. doi:10.1038/nri2708
60. Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ, et al. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J Leukoc Biol (2007) 82:111–23. doi:10.1189/jlb.1206713
61. Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood (2000) 96:1039–46.
62. Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem (2002) 277:32124–32. doi:10.1074/jbc.M203298200
63. Antignano F, Ibaraki M, Kim C, Ruschmann J, Zhang A, Helgason CD, et al. SHIP is required for dendritic cell maturation. J Immunol (2010) 184:2805–13. doi:10.4049/jimmunol.0903170
64. Jack GD, Zhang L, Friedman AD. M-CSF elevates c-Fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood (2009) 114:2172–80. doi:10.1182/blood-2008-11-191536
Keywords: mouse, dendritic cells, microRNAs, M-CSR receptor, Csf1r
Citation: Riepsaame J, van Oudenaren A, den Broeder BJH, van IJcken WFJ, Pothof J and Leenen PJM (2013) MicroRNA-mediated down-regulation of M-CSF receptor contributes to maturation of mouse monocyte-derived dendritic cells. Front. Immunol. 4:353. doi: 10.3389/fimmu.2013.00353
Received: 22 July 2013; Accepted: 16 October 2013;
Published online: 30 October 2013.
Edited by:
Burkhard Ludewig, Kantonal Hospital St. Gallen, SwitzerlandCopyright: © 2013 Riepsaame, van Oudenaren, den Broeder, van IJcken, Pothof and Leenen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pieter J. M. Leenen, Department of Immunology, Erasmus University Medical Center, P.O. Box 2040, 3000 CA Rotterdam, Netherlands e-mail:cC5sZWVuZW5AZXJhc211c21jLm5s
†Joris Pothof and Pieter J. M. Leenen have contributed equally to this work.