 Raphael Carapito
Raphael Carapito Ismail Aouadi1,2,3
Ismail Aouadi1,2,3 Wassila Ilias
Wassila Ilias Seiamak Bahram
Seiamak Bahram- 1ImmunoRhumatologie Moléculaire, INSERM UMR_S1109, LabEx TRANSPLANTEX, Centre de Recherche d’Immunologie et d’Hématologie, Faculté de Médecine, Fédération de Médecine Translationnelle de Strasbourg (FMTS), Université de Strasbourg, Strasbourg, France
- 2Laboratoire International Associé (LIA) INSERM, Strasbourg (France) – Nagano (Japan), Strasbourg, France
- 3Fédération Hospitalo-Universitaire (FHU) OMICARE, Strasbourg, France
- 4Laboratoire Central d’Immunologie, Pôle de Biologie, Nouvel Hôpital Civil, Hôpitaux Universitaires de Strasbourg, Strasbourg, France
Natural killer group 2, member D (NKG2D) is an invariant activatory receptor present on subsets of natural killer and T lymphocytes. It stimulates the cytolytic effector response upon engagement of its various stress-induced ligands NKG2D ligands (NKG2DL). Malignant transformation and conditioning treatment prior to hematopoietic cell transplantation (HCT) are stress factors leading to the activation of the NKG2D/NKG2DL signaling in clinical settings. In the context of HCT, NKG2D-bearing cells can kill both tumor and healthy cells expressing NKG2DL. The NKG2D/NKG2DL engagement has therefore a key role in the regulation of one of the most salient issues in allogeneic HCT, i.e., maintaining a balance between graft-vs.-leukemia effect and graft-vs.-host disease. The present review summarizes the current state of our knowledge pertaining to the role of the NKG2D and NKG2DL in HCT.
Introduction
Hematopoietic cell transplantation (HCT) is a widely used curative treatment for a variety of both malignant and non-malignant hematological diseases (1). HCT involves the intravenous infusion of stem cells collected from bone marrow, peripheral blood, or umbilical cord blood, in order to reestablish hematopoietic function in patients whose bone marrow or immune system is damaged, defective or therapeutically ablated. Post-HCT reconstitution of the immune system is a crucial step for remission and involves, among other immunological cells, cytotoxic lymphocytes. Because of their ability to selectively kill malignant or infected cells, these lymphocytes are key players in immune surveillance. They include natural killer (NK) and T cells, which share the expression of the activatory natural killer group 2, member D (NKG2D) receptor. While important for killing malignant cells [graft-vs.-leukemia (GVL) effect] and protecting the immunosuppressed patient undergoing HCT against opportunistic infections, these cells can also provoke adverse outcomes where graft-vs.-host disease (GVHD) is the most dramatic.
Natural killer group 2, member D is a C-type lectin-like type II transmembrane protein encoded by the “killer cell lectin-like receptor K1” (KLRK1) gene, embedded within the NK-gene complex on human chromosome 12. It functionally assembles into a hexameric structure where each NKG2D monomer is associated with a DNAX-activating protein 10 (DAP10) dimer in man, an adapter protein bearing a YXXM motif that recruits phosphoinositide 3-kinase. The NKG2D receptor is present on NK cells, CD8+ αβ, subsets of CD4+ αβ, as well as γδ T lymphocytes as well as NKT cells (2, 3). It is a genetically invariant (very few alleles have been reported) receptor that stimulates effector responses upon engagement of various stress-inducible ligands; hereafter called NKG2D ligands (NKG2DL). The function of NKG2D is therefore directly linked to the biology of its ligands, which expression is absent or low in most “normal” cells, but induced by cell stress, whether caused by infection, malignant transformation, or conditioning treatments prior to HCT (3–5).
In human, there are two families of NKG2DL: the “MHC class I chain-related genes” (MIC) and “UL16-binding proteins” (ULBPs) genes; the latter also known as retinoic acid early transcripts 1 (RAET1). The first family includes the two non-conventional MHC-I loci MICA and MICB encoded within the MHC; at the centromeric extremity of the MHC class I region (6). These genes encode highly glycosylated MHC class I-like proteins, which are expressed at the cell surface independently of β2-microglobulin and cytosolic peptides. The genomic and protein structure of MICA and MICB is similar to those of conventional MHC class I genes and molecules. The proteins are composed of three extracellular domains (α1, α2, and α3), a transmembrane region, and a cytoplasmic tail. Unlike other non-conventional MHC class I genes, a high degree of diversity has been documented for MICA and MICB genes: 106 alleles for MICA and 42 for MICB thus far (http://hla.alleles.org/data/index.html). MICA polymorphism has been associated with a number of diseases such as autoimmune disorders (7, 8) and cancer (9, 10), but also with allograft rejection and GVHD (11–16). The second NKG2DL family encompasses six functional genes: RAET1I/ULBP1, RAET1H/ULBP2, RAET1N/ULBP3, RAET1E/ULBP4, RAET1G/ULBP5, and RAET1L/ULBP6. These genes are also localized on chromosome 6, but on its opposite arm with respect to the MHC, between cytogenetics bands 6q24.2 and 25.3. In contrast to MIC proteins, all ULBP/RAET1s lack an α3 domain, and RAET1I/ULBP1, RAET1H/ULBP2, RAET1N/ULBP3, and RAET1L/ULBP6 are attached to the membrane via a glycosylphosphatidylinositol anchor. ULBP/RAET1 genes appear to be less polymorphic as MICA and MICB. This may, however, not reflect the reality as only about 300 individuals have been sequenced thus far. In mice, there are three families of NKG2DL: Rae-1/RAET1 (five known proteins), H60 (three known proteins), and MULT-1 (one known protein) but as evidenced early-on no orthologous MIC genes (17).
Several converging lines of evidences indicate that the NKG2D–NKG2DL interaction is a key event in the regulation of the immune response following HCT, especially with respect to GVHD and GVL effect. First, NKG2DL proteins are mainly expressed in cells of fibroblastic and epithelial origin (18), which is in accordance with the localized tissues expression of GVHD (skin, liver, and gut). Second, in line with a possible involvement in the GVL effect, NKG2DL are known to be upregulated in tumor cells (19–21). Third, the systemic inflammatory response to conditioning regimens can serve as a danger/stress signal, which is needed to induce NKG2DL expression (22). Fourth, the DNA damage pathway that is activated in response to ionizing radiation and chemotherapy is central in the upregulation of NKG2DL (23). Fifth, neutralization of the NKG2D receptors by antibodies prevents graft rejection in mice (24, 25). Finally, blockade of the NKG2D/NKG2DL interaction by antibodies directed against NKG2D, attenuate GVHD while allowing CD8+ T cells to regain their GVL activity (26).
Here, we review the current knowledge of the role of both NKG2D and NKG2DL in HCT at the levels of genetic polymorphism, protein expression regulation, and antibody production.
Genetic Polymorphism of NKG2D and NKG2DL
Both human NKG2DL gene families have been studied with respect to genotype–phenotype relationship in the context of HCT. While several recent publications have analyzed the influence of MIC genes in HCT, a single study focused on the role of the ULBP/RAET1 gene family. In this study, Antoun and coworkers used a cohort of 371 patient/donor pairs of HLA-matched related allografts to analyze a total of 18 single nucleotide polymorphisms (SNPs) in the four most polymorphic members of this gene family, i.e., RAET1N/ULBP3, RAET1E/ULBP4, RAET1G/ULBP5, and RAET1L/ULBP6 (27). The only gene that could be related to clinical outcomes was RAET1L/ULBP6. Thanks to SNP haplotype structure analysis of this gene, the authors could associate the presence of the RAET1L*02 allele in patients with an improved overall survival (55% in patients with RAET1L*02 vs. 39% in patients without RAET1L*02, P = 0.003) and relapse-free survival (44% in patients with RAET1L*02 vs. 25% in patients without RAET1L*02, P < 0.001). In addition, this allele was found to be associated with a reduced risk of relapse, with 11.5% of risk for RAET1L*02 homozygotes compared to 29.1% for RAET1L*01 heterozygotes and 38.2% for the RAET1L*02 negative patients (P = 0.001 between the homozygous groups). Of note, neither the identity, nor the matching of the donors’ alleles had an impact on clinical outcomes.
With regards to MIC genes, in the last decade several genetic studies have collectively concluded to a potential role of MICA as a novel transplantation antigen. The very first hint toward a significant role of MICA/B matching in HCT came from an MHC “beta block” matching study (28). In this work, a small cohort of 44 donor/recipient pairs of unrelated allogeneic HCT was used to determine the correlation of the so-called “beta block”—HLA-B, -C, MICA, and MICB—matching on patient survival. The “beta block” spans 300 kb, harbors the immunology-related genes HLA-B, HLA-C, MICA, and MICB genes, and represents one of the four “MHC blocks” including also the alpha (cluster around HLA-A), gamma (cluster around Bf and C4), and delta (cluster around HLA-DR and -DQ) blocks (29). Patients who were HLA-B and -C matched showed an increased survival when they were additionally matched for the MHC beta block (59 vs. 16%, P = 0.04) or MICA and MICB (66 vs. 25%, P = 0.05). In addition, as patients who were beta block matched were matched at both HLA-B and MICB but not HLA-C and MICA, the authors could show that in this subset of patients, MICA- and HLA-C-matched patients had a significantly improved survival of 69% (P = 0.05).
Other studies have specifically analyzed the influence of the MICA gene by three different approaches: (i) focused analysis of the genotype of a single polymorphism at amino acid position 129 in patients (12, 15), (ii) matching of donors and patients genotypes at the same position; 129 (14), and (iii) matching of donor and patients at the MICA allele level (13, 16, 30, 31).
The polymorphism at amino acid position 129 involving a substitution of a methionine for a valine is known to lower the binding affinity of MICA for NKG2D, probably through a conformational change since this position is not directly involved in NKG2D binding (32–34). Patients’ genotype at position 129 was firstly analyzed in a cohort of 211 matched related allogeneic HCT patients, and it was shown that those bearing the MICA-129 Val/Val genotype—i.e., the lower binding affinity variant—were at a higher risk of developing chronic GVHD (63 vs. 45% at 3 years; P = 0.03) (12). Moreover, the relapse incidence in patients with the MICA-129 Met/Met genotype was higher than in those with other genotypes (51 vs. 19%, P = 0.007). A second group analyzed 452 patients that underwent matched unrelated (67.9% of patients) and related (31.6% of patients) allogeneic HCT and evidenced that the patients with the MICA-129 Met genotype had an increased probability of overall survival (HR = 0.77, P = 0.0445) and that the mortality specifically due to acute GVHD was reduced (HR = 0.57, P = 0.0400) (15). The improved survival of MICA-129 Met carriers was also observed in the subset of patients that received an MICA-129-matched transplant (n = 404, HR = 0.73, P = 0.0226). However, this study unexpectedly also reported an increased risk of acute GVHD (HR = 1.92, P = 0.0371) in MICA-129 Met/Met homozygote carriers, i.e., the higher binding affinity variant. As this was particularly the case for anti-thymocyte globulin (ATG) treated patients who have a lowered CD8+ T repertoire, the authors argued that the residual T CD8+ cells may be activated faster in the presence of two MICA-129 Met variants. In the case of heterozygote carriers, the beneficial effect was indeed attributed to the subgroup of patients not treated with ATG, indicating that such a faster activation may not matter if a full CD8+ T cell repertoire is present. Finally, a third publication by Askar et al. reported that neither the patient’s nor the donor’s genotype at position 129 had an impact on survival or GVHD (30). The authors could only detect an association between donor MICA-129 non-Val/Val genotypes and slower platelet engraftment (HR = 1.4; 95% CI: 1.109–1.985; P = 0.02).
More recently, a group analyzed not only the MICA-129 genotype of the patient but also the one of the donor and by this mean the role of the matching of donor and recipients at this position with respect to various clinical outcomes after unrelated HCT (14). They analyzed 2,172 patients that underwent unrelated HCT with donors HLA-matched at various grades (10/10, 9/10, and 8/10). They showed that matching of MICA-129 genotypes between donors and patients was important to increase overall survival and disease free survival and to lower the risk for acute GVHD. In the 10/10 matched group, multivariate analysis revealed indeed a clear association of MICA-129 mismatches with overall survival (HR = 1.77, 95% CI: 1.22–2.57, P = 0.003), disease free survival (HR = 1.77, 95% CI: 1.26–2.50, P = 0.001), and acute grade III–IV aGvHD (prevalence of 16.3 vs. 11.0%).
Finally, the third type of analysis was to consider, as it is done for classical HLA genes, the matching of donors and recipients at the MICA allele level. By analyzing a cohort of 172 matched pairs of unrelated HCT, Parmar et al. showed a higher rate of grade II–IV acute GVHD in MICA-mismatched vs. -matched patients, taking only account of MICA mismatches in the GVHD direction (75 vs. 39%, P = 0.02) (16). Askar and coworkers also studied an equally small cohort of 177 10/10 matched donor–recipient pairs (31). They did find a link between MICA mismatches and grade II–IV acute GVHD but in univariate analysis only; this link was reenforced in patients with mismatches at both MICA and HLA-DPB1 who had a significantly greater risk to develop grade II to IV acute GVHD (HR = 2.51; 95% CI: 1.30–4.87; P < 0.01). More recently, we were able to definitely establish the role of MICA mismatches in HCT based on the analysis of a multicenter cohort of 922 10/10 HLA-matched HCT. MICA mismatches were significantly associated with an increased incidence of grade III–IV acute GVHD (HR = 1.83; 95% CI: 1.50–2.23; P < 0.001), chronic GVHD (HR = 1.50; 95% CI: 1.45–1.55; P < 0.001), and non-relapse mortality (HR = 1.35; 95% CI: 1.24–1.46; P < 0.001). The increased risk of GVHD was mirrored by a lower risk of relapse (HR = 0.50; 95% CI: 0.43–0.59; P < 0.001), indicating a possible GVL effect. A few weeks later in 2016, Askar and coworkers, however, published contradictory results using a cohort of 713 patients and their unrelated HLA-matched 10/10 (n = 552) or 9/10 (n = 161) donors (30). They showed an absence of MICA mismatch effect on all major clinical outcomes apart an unexpected significantly higher incidence of relapse in MICA-mismatched vs. -matched patients (HR = 1.7; 95% CI: 1.2–2.4; P = 0.003). In their data, there was, however, a clear trend for an association between MICA mismatches and a higher risk of acute GVHD grades II–IV (HR = 1.4; 95% CI: 1.1–1.9; P = 0.013). The various genetic studies on NKG2D/NKG2DL polymorphisms or donor/recipient matching in HCT and their main findings are summarized in Table 1.
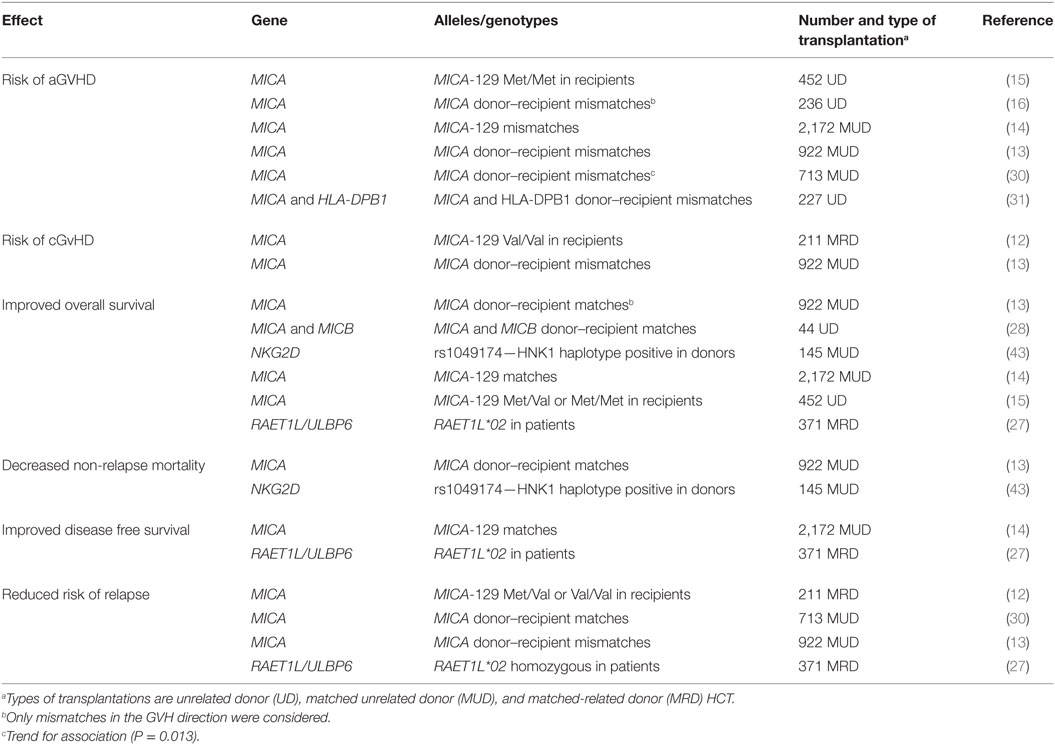
Table 1. Summary of genetic studies of natural killer group 2, member D (NKG2D)/NKG2D ligands polymorphisms or donor/recipient matching in hematopoietic cell transplantation (HCT).
Globally, these findings clearly support a role of MICA as a transplantation antigen in HCT. Concerning the pathophysiological explanation of the observed associations, there are two non-exclusive possible molecular mechanisms that could be involved. Integrating the fact that MICA expression is upregulated in intestinal tissues of GVHD patients and that NKG2D is induced on CD8+ and CD56+ cells after HCT (see next section); it is likely that a mismatch between donor/recipient MICA molecules directly affects the strength of recognition/signaling and hence cytotoxicity by NKG2D bearing T/NK cells and/or the intestine-enriched Vδ1-bearing γδ T cells (both receptors able to recognize MICA directly). While the direct link between MICA mismatches and increased expression needs to be experimentally demonstrated, two groups reported variable NKG2D-mediated NK-cell activation and T CD8+ co-stimulation depending on specific MICA alleles. Tonnerre et al. showed a higher NK-cell activation by allogeneic endothelial cells expressing the MICA A5.1 alleles (35), and Isernhagen et al. demonstrated an increased CD8+ T cell co-activation in response to the MICA-129 Met variant (15). Alternatively, MICA could act as a minor histocompatibility antigen, i.e., a source for polymorphic peptides presented by cognate and/or donor MHC class I molecules and hence participate in the GVHD pathophysiology through its contribution to alloreactivity. These two possibilities are schematized in Figure 1. Of note, NKG2D engagement by MICA could also play an indirect role in the enhancement of CD8+ T cytolysis, which is known to elicit GVHD (1). Although it has been shown that NKG2D may trigger cytotoxicity alone in expanded CD8+ T cells, cultured in the presence of IL-2 (36), several studies in human and in mouse reported that NKG2D rather acts as a co-stimulatory molecule in CD8+ T cells (26, 37–40). In T cells, NKG2D indeed associates with an adaptor protein, DAP10, where it provides a co-stimulatory signal by activating intracellular signaling pathways (37, 40, 41). By providing T cell co-stimulation, NKG2D ligand engagement may therefore enhance CD8+ T cell-mediated cytotoxicity of recipient cells, thereby triggering GVHD (42).
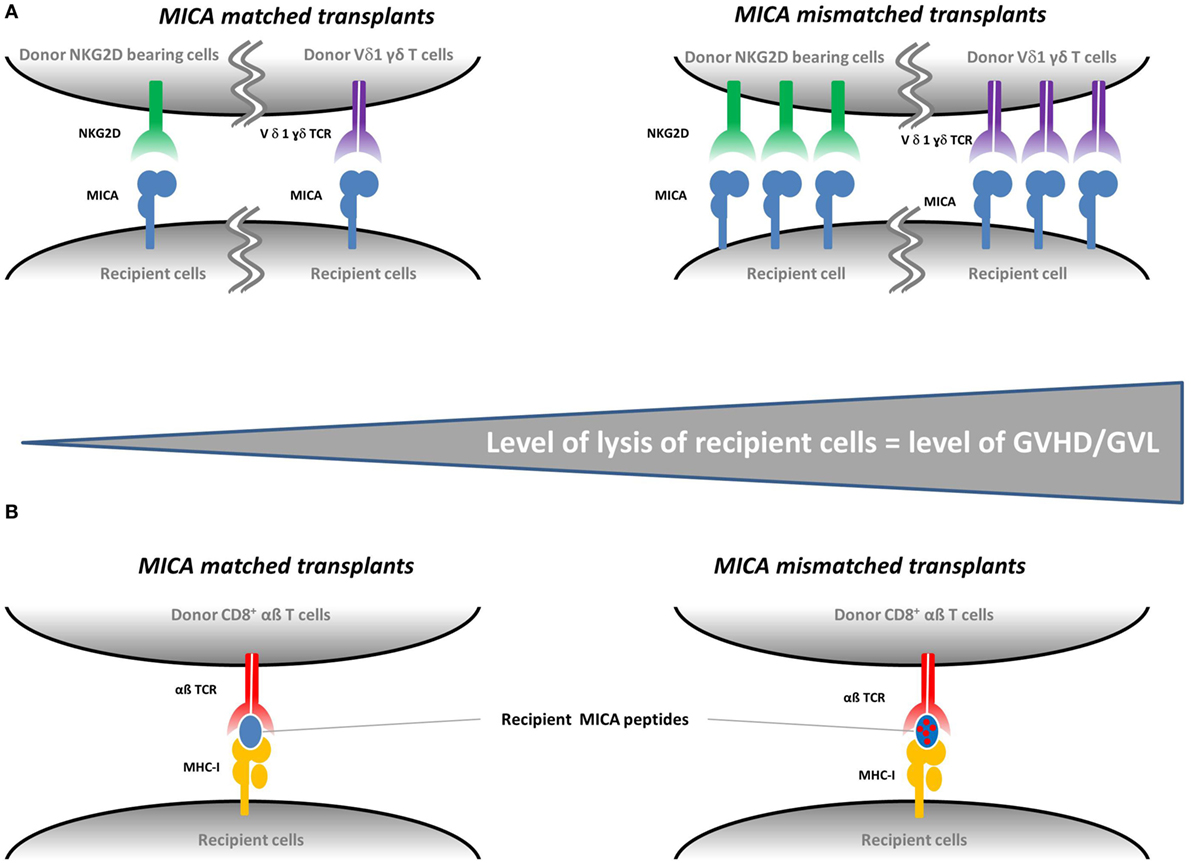
Figure 1. Proposed mechanisms for the role of MICA mismatches in graft-vs.-host disease (GVHD). There are two non-exclusive possible molecular mechanisms involved in the development of GVHD in the context of MICA-mismatched transplants. Panel (A) illustrates how an increased MICA expression—possibly induced by MICA mismatches—could lead to an increased cytolysis of recipient cells mediated by the natural killer group 2, member D (NKG2D) on donor natural killer cells and/or the TCR of donor Vδ1-bearing γδ T cells and/or donor T CD8+ cells as a co-stimulatory function. GVHD and graft-vs.-leukemia (GVL) can be triggered if the target cells of the recipients are healthy or malignant, respectively. Panel (B) illustrates how MICA could act as a minor histocompatibility antigen. If the donor is matched for MICA, the recipient MICA-derived peptides are recognized as «self» by donor cells. If the donor is mismatched for MICA, the presented recipient MICA-derived peptides are recognized as «non-self» and recipient cells are lysed by donor CD8+ T cells.
To conclude this section, a single study on NKG2D receptor polymorphism has to be mentioned. As mentioned above and in contrast to its ligands, NKG2D is not polymorphic. Only two SNPs with a minor allele frequency >0.01 have been reported in the coding sequence of the gene: the synonymous rs1049172 and the non-synonymous rs2255336. But the most studied SNP is the rs1049174 in the 3′-untranslated region of the gene, which enables to distinguish two haplotypes: HNK1 (high NK cytotoxicity) and LNK1 (low NK cytotoxicity). The HNK1 haplotype has been associated with improved overall survival (HR = 0.44; 95% CI: 0.23–0.85; P = 0.01) as well as transplant related mortality (HR = 0.42; 95% CI: 0.21–0.86; P = 0.02) after unrelated HLA-matched unrelated HCT (43).
Upregulation of NKG2D and NKG2DLs
Besides the role of donor/recipient compatibility detailed in the previous chapter, the stress-induced upregulation of NKG2DLs in patient’s tissues may per se contribute to the development of GVHD. In contrast to solid organ transplantation, where it is established that MIC antigens are expressed in transplanted organs and may cause early graft rejection (44, 45), only few studies analyzed NKG2DL protein expression in the context of HCT. By immunochemical staining, Dulphy et al. showed a strong expression of MICA on epithelial cells of intestinal biopsies from acute GVHD patients (46). Gannagé et al. confirmed this observation and also demonstrated that during GVHD, MICA/B expression was induced in skin and liver biopsies as well (5). In addition, they showed that in vitro, in immortalized human keratinocytes and colorectal carcinoma fibroblasts both MICA/B and ULBP1–3 were upregulated in response to TNF-α or γ-irradiation. Total body irradiation that is regularly used in HCT could therefore be a trigger for NKG2DL expression. But other types of pretransplant treatments such as chemotherapies could also play this role, e.g., the DNA-hypomethylating drug 2-deoxy-5-azacytidine (decitabine), which has been shown to upregulate MICB in cell lines (22). Finally, cancer cells by themselves have a propensity for NKG2DL upregulation. This was demonstrated by Sconocchia and coworkers who found an MICA/B overexpression on chronic myelogenous leukemia CD34 cells but not on normal CD34 cells and showed that this upregulation correlated with the ability of soluble MICA (sMICA)/B to bind to the NKG2D receptor (47).
The latter mentioned soluble form of NKG2DLs corresponds to a well-known immune escape mechanism in cancer cells. NKG2DLs and especially MICA have indeed the ability to be released from the cell surface through various mechanisms including alternative splicing, phosphatidylinositol-specific phospholipase C-mediated cleavage, proteolytic shedding, or exosome secretion [reviewed in Ref. (17)]. The soluble form of NKG2DL downregulates NKG2D and thereby impairs the cytolysis activity of NK and T effector cells (48). By doing so, tumor cells evade immune surveillance (48–50). Soluble MIC and ULBP proteins have been identified in the sera of patients with various tumor types including breast, lung, colon, and ovarian carcinoma, glioma, neuroblastoma, leukemia, and melanoma (48, 50–55). Following HCT, the serum levels of sMICA have been shown to increase and the presence of these molecules to confer susceptibility to chronic GVHD (sMICA > 80 pg/mL associated with chronic GVHD incidences of 82 vs. 46%, at 3 years; P = 0.001) (12, 56). Opposite to this increase of the GVH effect on recipient cells, high levels of sMICA were shown to decrease the GVL effect as shown by the higher rate of relapse (sMICA > 80 pg/mL associated with relapse rates of 37 vs. 17%, P = 0.05) (12). This trend toward a lowered cytolytic activity against cancer cells was confirmed in solid tumors (prostate cancer), where a higher level of sMICA has been associated with a decreased cytotoxicity of NK cells isolated from tumor biopsies (57).
The molecular mechanisms of expression regulation of NKG2DLs are still poorly understood. However, experimental data on two pathways are available: the DNA damage response and regulation by cellular and viral miRNA. Data from both mice and human indicate that NKG2DL are induced by DNA damaging agents such as 5-aza-2′-deoxycytidine (decitabine), a conditioning treatment used in myelodysplastic syndromes (22, 23, 58). The upregulation is thought to be due to promoter DNA demethylation in combination with the activation of the DNA damage pathway involving the protein kinases ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (23). For miRNA regulation, both viral (Cytomegalovirus) and cellular miRNA have been shown to modify the expression of various NKG2DLs and thereby to influence NK-cell responses (59–62).
For the ligand expressing cells to be efficiently targeted and killed, their receptor on effector cells must be expressed and active as well. Although the role of NKG2D in tumor immunity is not to be proven anymore as shown by the multiple studies demonstrating its involvement in NK cell-mediated killing of various cancer cells (63–65), its specific action in the context of HCT has only been scarcely analyzed. Only a handful of studies have indeed reported active NKG2D upregulation during HCT. Lu et al. firstly observed an increase in CD16+CD56− NK cells in peripheral blood of a cord blood transplanted patients (66). When cultured in vitro in presence of IL-2, these cells became CD16+CD56+NKG2D+ and were able to lyse the patient’s ULBP2-expressing leukemic cells. These results indicate that in vivo, after transplantation, mature NK cells derived from this NK-cell subset may contribute to the killing of leukemic cells expressing NKG2DL. Dulphy et al. also showed an expansion of CD56bright NK cells shortly after HLA matched related and unrelated HCT (46). NKG2D was expressed on these cells, which were also shown to be functional as they produced high amounts of IFN-γ and were competent for degranulation. In line with these two reports, Picardi et al. showed an upregulation of NKG2D on CD8+ and/or CD56+ cells between 30 and 90 days posttransplant in HLA-matched allogeneic and autologous HCT, coinciding with the engraftment period (67). The authors suggest that this upregulation, together with a concomitant upregulation of inhibitory NKG2 receptors, is the sign of an immune “reeducation” and a general stress response to pretransplant chemotherapy and posttransplant repopulation. The regulation of NKG2D expression is thought to be modulated by cytokines such as IL-15, which ultimately control the levels of NK cytotoxicity (68). Of note, one study reports that NKG2D expression by NK cells was not affected after fully haplo-mismatched-related donor HCT, challenging the importance of the interaction between NKG2D and its ligands for the NK lysis of leukemic cells at least in this transplant setting (69).
Natural killer cells are the first lymphocyte population to be reconstituted following HCT and are important in mediating the GVL effect (70). NK cells could be subtyped into three categories based on the following surface makers: CD56dim/CD16+ (hereafter termed CD56dim), CD56bright/CD16+/− (hereafter termed CD56bright) and CD56−/CD16+ (hereafter termed CD56neg) NK cells. The CD56dim cells represent up to 90% of circulating NK cells and are considered as the most cytotoxic subset. The CD56bright subtype comprises up to 10% of circulating NK cells but is the major NK subtype in tissues and secondary lymphoid organs. This subset is conventionally known as the cytokine-producing subset of NK cells but can also become cytotoxic upon appropriate activation. CD56neg NK cells are found in small numbers in healthy individuals and at elevated levels in individuals chronically infected with HIV-1 and HCV. All three subsets of NK cells express NKG2D (71, 72). After allogeneic HCT, there is an early expansion of the cytokine-producing CD56bright NK-cell subset, followed by an expansion of the dominant CD56dim subset, characterized by its higher cytotoxic activity (46). In a study focusing on the immune-regulatory role of NK cells, it was shown that NKG2D plays a major role in NK cell-mediated lysis of activated CD4+ T cells. Using anti-NKG2D antibodies, the authors evidenced equivalent NKG2D-mediated degranulation by CD56dim and CD56bright NK cells (73). In ovarian cancer patients, it was shown that CD56bright NK cells had increased expression of NKG2D (74). Finally, CD56neg NK cells were shown to be increased in cord blood recipients, suggesting that this subset may contribute to the killing of NKG2D expressing leukemic cells (66).
The function of NK cells is modulated by the balance between a number of activating (including NKG2D) and inhibitory receptors. NKG2 and killer immunoglobulin-like receptors (KIRs) are the two main receptor families involved in this regulation and both encompass activatory and inhibitory receptors. KIR receptors interact with HLA class I molecules and, in contrast to NKG2D, are very diverse with regard to gene polymorphism and content, expression level, and expression pattern (75). In the context of HCT, KIR–ligand mismatches in the GVH direction were shown to trigger donor-vs.-recipient NK-cell alloreactivity (76). Overall, together with other receptors such as immunoglobulin-like receptors or leukocyte-associated immunoglobulin-like receptor, KIR and NKG2 receptors regulate the alloreactivity of NK cells during HCT (77). Altogether, these data strongly suggest that the induction of the NKG2D/NKG2DL axis might be involved in tissue damage during GVHD or GVL reactions thanks to NKG2DLs and NKG2D expression by host or cancer cells, respectively. A summary of the impacts of NKG2DL and NKG2D upregulation during HCT is presented in Figure 2.
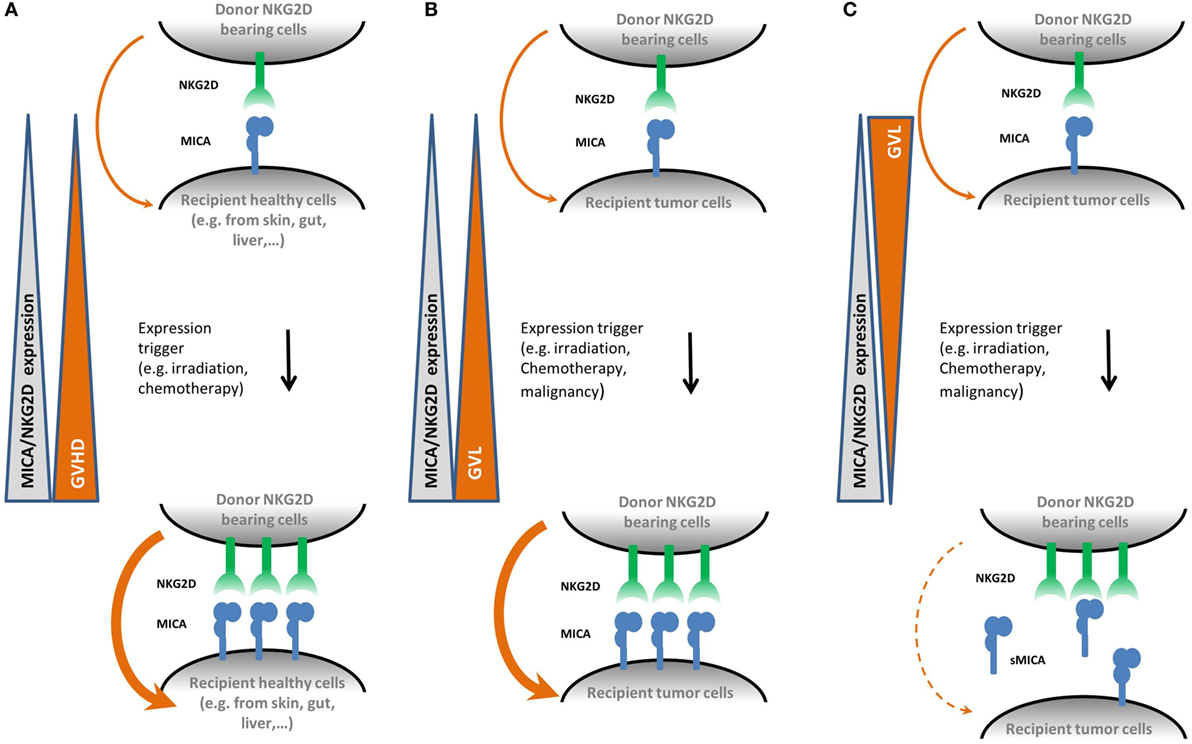
Figure 2. Proposed mechanisms for the role of natural killer group 2, member D (NKG2D) and MICA upregulation in graft-vs.-host disease (GVHD) and graft-vs.-leukemia (GVL) effect mediated by natural killer (NK) cells. Panel (A) shows that following hematopoietic cell transplantation (HCT) conditioning regimen, healthy recipient cells upregulate membrane-bound MICA expression. As NKG2D is known to be upregulated during HCT as well, it could therefore participate to an increased cytolysis activity alone or as a co-stimulatory signal on donor T CD8+ cells, thereby promoting the development of GVHD. Panel (B) shows the equivalent situation on recipient cancer cells. Here, upregulation of membrane bound MICA expression promotes the GVL effect. Panel (C) illustrates the effect of soluble MICA (sMICA) production by recipient cancer cells. sMICA leads to the downregulation of NKG2D and thereby to an impairment of the cytolysis activity of NK effector cells, i.e., lower the GVL effect.
Anti-MICA Antibodies
In the field of transplantation, studies on antibodies against NKG2DL have so far exclusively focused on MICA. The presence of anti-MICA antibodies is a well-known risk factor for both acute and chronic rejection after transplantation of kidney (78, 79), pancreas (45), or heart (80). In HCT, three groups have analyzed the role of anti-MICA antibodies. Boukouaci et al. showed that the presence of anti-MICA antibodies with concomitant low serum levels of sMICA (<80 pg/mL) was associated with a lower incidence of chronic GVHD (35% when MICA antibodies positive and sMICA < 80pg/mL vs. 81% when MICA antibodies negative or borderline and sMICA > 80 pg/mL, P = 0.0004) (12). The authors hypothesized that these antibodies have a neutralizing activity on sMICA and supported this theory by the observed trend for a protective effect of anti-MICA antibodies against relapse (3.26% when MICA antibodies negative vs. 6% when MICA antibodies positive, P = 0.05). In a second study focusing on a cohort of 70 pediatric patients that received cord blood transplantation, Ansari et al. showed that the presence of anti-MICA antibodies was associated with a reduced platelet recovery after transplantation (26.5% when MICA antibodies negative vs. 80.5% when MICA antibodies positive, P = 0.04) (81). Finally, Flaxa et al. could observe immunization against MICA before and after transplantation in a small series of eight granulocyte transfused patients that underwent HCT, but those antibodies did not significantly affect overall survival or the incidence of GVHD (82).
In summary, in contrast to solid organ transplantation very limited MICA immunization data are available for HCT. Thus, at this stage, any definitive conclusion on the role of anti-MICA or more generally anti-NKG2DL antibodies in HCT cannot be drawn without performing further experiments. Several studies, however, confirm that such antibodies could be involved in GVL. Jinushi et al. demonstrated that MICA antibodies can efficiently opsonize and participate in the complement-mediated lysis of cancer cells (83). In a follow-up study, the same group showed that in monoclonal gammopathy of undetermined significance patients, high levels of anti-MICA antibodies antagonize sMICA and stimulate dendritic cell cross-presentation of tumor antigens (49). This observation had previously been made Groh et al. in an in vitro assay: by loading dendritic cells with anti-MICA opsonized breast, melanoma, or ovarian tumor lines, tumor antigen cross-presentation was promoted and thereby primed antitumor effector CD4 and CD8 T cell responses (84).
In the field of organ transplantation, the production of anti-MICA antibodies has been associated with donor/recipient MICA mismatching (85). Another study showed that the genetic variant MICA A5.1 (a microsatellite marker in exon 5 encoding the transmembrane region of the protein) increased the surface expression of MICA and augmented the release of soluble and exosomal forms of MICA from endothelial cells (35). In addition, the authors demonstrate a significant association between donor/recipient MICA A5.1 mismatches and anti-MICA allo-immunization, particularly when donors carry the MICA A5.1 variant (P = 0.0104). In other fields of research such as cancer and infectious diseases, several MICA polymorphisms have been reported to affect MICA shedding including an SNP (rs2596542) in the promoter region (86–89), the microsatellite MICA A5.1 (35, 90, 91), and the MICA-129 Met/Val dimorphism in α2 domain of the MICA protein (88, 92, 93).
Conclusion
Despite some isolated conflicting results and sometimes scarce data, current research globally converges to the postulate that the NKG2D/NKG2DL axis is a new player in HCT. At the genetic level, most studies came to the conclusion that MICA mismatches between donor and recipients, either at amino acid position 129 or at the allelic level are deleterious and that whenever possible, an MICA-matched donor should be chosen for transplantation. Upregulation of both NKG2DLs and NKG2D during HCT have been demonstrated by various groups and associated with GVHD and GVL effects. Finally, recent data suggest a possible protecting role of anti-MICA antibodies via an antagonizing effect on sMICA that ultimately leads to promotion of GVL. Additional studies are, however, needed to definitely confirm these findings and to better understand the pathophysiological role of NKG2D and NKG2DLs in GVHD and GVL effect.
Author Contributions
RC and SB wrote the manuscript and prepared the figures; WI and IA contributed to work reported in the manuscript.
Conflict of Interest Statement
SB is the scientific founder and a (minority) shareholder in BIOMICA SAS.
Funding
This work is published under the framework of the LABEX TRANSPLANEX (ANR-11-LABX-0070_TRANSPLANTEX) that benefits from funding from the French government; funds managed by the French National Research Agency (ANR) as part of the «Investments for the future» program and the INSERM UMR_S 1109. Our laboratory further supported by Genomax, the Strasbourg School of Medicine Next Generation Sequencing center, the Institut Universitaire de France (IUF), the Partenariat Hubert Curien GUNDISHAPUR by the ministères des Affaires étrangères et du développement international (MAEDI) and Enseignement supérieur et de la Recherche (MESR), the Institut de Recherche pour le développement (IRD), the AVIESAN’s ITMO-cancer, IDEX UNISTRA, the Association pour la Recherche contre le Cancer (ARC), the Alsace Cancer association, and the European regional development fund (European Union) INTERREG V program (project no. 3.2 TRIDIAG).
References
1. Forman JS, Negrin SR, Antin HJ, Appelbaum RF. Thomas’ Hematopoietic Cell Transplantation. 5th ed. New York: Wiley-Blackwell (2016).
2. Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev (2010) 235(1):267–85. doi:10.1111/j.0105-2896.2010.00893.x
3. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
4. Gonzalez S, Lopez-Soto A, Suarez-Alvarez B, Lopez-Vazquez A, Lopez-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol (2008) 29(8):397–403. doi:10.1016/j.it.2008.04.007
5. Gannagé M, Buzyn A, Bogiatzi SI, Lambert M, Soumelis V, Dal Cortivo L, et al. Induction of NKG2D ligands by gamma radiation and tumor necrosis factor-alpha may participate in the tissue damage during acute graft-versus-host disease. Transplantation (2008) 85(6):911–5. doi:10.1097/TP.0b013e31816691ef
6. Bahram S, Bresnahan M, Geraghty DE, Spies T. A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci U S A (1994) 91(14):6259–63. doi:10.1073/pnas.91.14.6259
7. Amroun H, Djoudi H, Busson M, Allat R, El Sherbini SM, Sloma I, et al. Early-onset ankylosing spondylitis is associated with a functional MICA polymorphism. Hum Immunol (2005) 66(10):1057–61. doi:10.1016/j.humimm.2005.09.004
8. Kirsten H, Petit-Teixeira E, Scholz M, Hasenclever D, Hantmann H, Heider D, et al. Association of MICA with rheumatoid arthritis independent of known HLA-DRB1 risk alleles in a family-based and a case control study. Arthritis Res Ther (2009) 11(3):R60. doi:10.1186/ar2683
9. Jumnainsong A, Romphruk AV, Jearanaikoon P, Klumkrathok K, Romphruk A, Luanrattanakorn S, et al. Association of polymorphic extracellular domains of MICA with cervical cancer in northeastern Thai population. Tissue Antigens (2007) 69(4):326–33. doi:10.1111/j.1399-0039.2006.00754.x
10. Douik H, Ben Chaaben A, Attia Romdhane N, Romdhane HB, Mamoghli T, Fortier C, et al. Association of MICA-129 polymorphism with nasopharyngeal cancer risk in a Tunisian population. Hum Immunol (2009) 70(1):45–8. doi:10.1016/j.humimm.2008.10.008
11. Sumitran-Holgersson S. Relevance of MICA and other non-HLA antibodies in clinical transplantation. Curr Opin Immunol (2008) 20(5):607–13. doi:10.1016/j.coi.2008.07.005
12. Boukouaci W, Busson M, Peffault de Latour R, Rocha V, Suberbielle C, Bengoufa D, et al. MICA-129 genotype, soluble MICA, and anti-MICA antibodies as biomarkers of chronic graft-versus-host disease. Blood (2009) 114(25):5216–24. doi:10.1182/blood-2009-04-217430
13. Carapito R, Jung N, Kwemou M, Untrau M, Michel S, Pichot A, et al. Matching for the nonconventional MHC-I MICA gene significantly reduces the incidence of acute and chronic GVHD. Blood (2016) 128(15):1979–86. doi:10.1182/blood-2016-05-719070
14. Fuerst D, Neuchel C, Niederwieser D, Bunjes D, Gramatzki M, Wagner E, et al. Matching for the MICA-129 polymorphism is beneficial in unrelated hematopoietic stem cell transplantation. Blood (2016) 128(26):3169–76. doi:10.1182/blood-2016-05-716357
15. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, von Bonin F, et al. The MICA-129 dimorphism affects NKG2D signaling and outcome of hematopoietic stem cell transplantation. EMBO Mol Med (2015) 7(11):1480–502. doi:10.15252/emmm.201505246
16. Parmar S, Del Lima M, Zou Y, Patah PA, Liu P, Cano P, et al. Donor-recipient mismatches in MHC class I chain-related gene A in unrelated donor transplantation lead to increased incidence of acute graft-versus-host disease. Blood (2009) 114(14):2884–7. doi:10.1182/blood-2009-05-223172
17. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev (2015) 267(1):88–116. doi:10.1111/imr.12328
18. Schrambach S, Ardizzone M, Leymarie V, Sibilia J, Bahram S. In vivo expression pattern of MICA and MICB and its relevance to auto-immunity and cancer. PLoS One (2007) 2(6):e518. doi:10.1371/journal.pone.0000518
19. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res (2002) 62(21):6178–86.
20. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res (2003) 63(24):8996–9006.
21. Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood (2003) 102(4):1389–96. doi:10.1182/blood-2003-01-0019
22. Tang KF, He CX, Zeng GL, Wu J, Song GB, Shi YS, et al. Induction of MHC class I-related chain B (MICB) by 5-aza-2’-deoxycytidine. Biochem Biophys Res Commun (2008) 370(4):578–83. doi:10.1016/j.bbrc.2008.03.131
23. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi:10.1038/nature03884
24. Ogasawara K, Benjamin J, Takaki R, Phillips JH, Lanier LL. Function of NKG2D in natural killer cell-mediated rejection of mouse bone marrow grafts. Nat Immunol (2005) 6(9):938–45. doi:10.1038/ni1236
25. Beilke JN, Benjamin J, Lanier LL. The requirement for NKG2D in NK cell-mediated rejection of parental bone marrow grafts is determined by MHC class I expressed by the graft recipient. Blood (2010) 116(24):5208–16. doi:10.1182/blood-2010-05-285031
26. Karimi MA, Bryson JL, Richman LP, Fesnak AD, Leichner TM, Satake A, et al. NKG2D expression by CD8+ T cells contributes to GVHD and GVT effects in a murine model of allogeneic HSCT. Blood (2015) 125(23):3655–63. doi:10.1182/blood-2015-02-629006
27. Antoun A, Vekaria D, Salama RA, Pratt G, Jobson S, Cook M, et al. The genotype of RAET1L (ULBP6), a ligand for human NKG2D (KLRK1), markedly influences the clinical outcome of allogeneic stem cell transplantation. Br J Haematol (2012) 159(5):589–98. doi:10.1111/bjh.12072
28. Kitcharoen K, Witt CS, Romphruk AV, Christiansen FT, Leelayuwat C. MICA, MICB, and MHC beta block matching in bone marrow transplantation: relevance to transplantation outcome. Hum Immunol (2006) 67(3):238–46. doi:10.1016/j.humimm.2006.02.012
29. Abraham LJ, Leelayuwat C, Grimsley G, Degli-Esposti MA, Mann A, Zhang WJ, et al. Sequence differences between HLA-B and TNF distinguish different MHC ancestral haplotypes. Tissue Antigens (1992) 39(3):117–21. doi:10.1111/j.1399-0039.1992.tb01920.x
30. Askar M, Sobecks R, Wang T, Haagenson M, Majhail N, Madbouly A, et al. MHC class I chain-related gene A (MICA) donor-recipient mismatches and MICA-129 polymorphism in unrelated donor hematopoietic cell transplantations has no impact on outcomes in acute lymphoblastic leukemia, acute myeloid leukemia, or myelodysplastic syndrome: a center for International Blood and Marrow Transplant Research Study. Biol Blood Marrow Transplant (2016) 23(3):436–44. doi:10.1016/j.bbmt.2016.11.021
31. Askar M, Sun Y, Rybicki L, Zhang A, Thomas D, Kalaycio M, et al. Synergistic effect of major histocompatibility complex class I-related chain a and human leukocyte antigen-DPB1 mismatches in association with acute graft-versus-host disease after unrelated donor hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20(11):1835–40. doi:10.1016/j.bbmt.2014.07.019
32. Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol (2001) 2(5):443–51. doi:10.1038/87757
33. Li P, Willie ST, Bauer S, Morris DL, Spies T, Strong RK. Crystal structure of the MHC class I homolog MIC-A, a gammadelta T cell ligand. Immunity (1999) 10(5):577–84. doi:10.1016/S1074-7613(00)80057-6
34. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics (2001) 53(4):279–87. doi:10.1007/s002510100325
35. Tonnerre P, Gerard N, Chatelais M, Poli C, Allard S, Cury S, et al. MICA variant promotes allosensitization after kidney transplantation. J Am Soc Nephrol (2013) 24(6):954–66. doi:10.1681/ASN.2012080814
36. Verneris MR, Karimi M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood (2004) 103(8):3065–72. doi:10.1182/blood-2003-06-2125
37. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol (2001) 2(3):255–60. doi:10.1038/85321
38. Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol (2005) 174(8):4480–4. doi:10.4049/jimmunol.174.8.4480
39. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity (2002) 17(1):19–29. doi:10.1016/S1074-7613(02)00333-3
40. Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, et al. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J Immunol (2005) 175(5):2825–33. doi:10.4049/jimmunol.175.5.2825
41. Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev (2009) 227(1):150–60. doi:10.1111/j.1600-065X.2008.00720.x
42. Suarez-Alvarez B, Lopez-Vazquez A, Baltar JM, Ortega F, Lopez-Larrea C. Potential role of NKG2D and its ligands in organ transplantation: new target for immunointervention. Am J Transplant (2009) 9(2):251–7. doi:10.1111/j.1600-6143.2008.02526.x
43. Espinoza JL, Takami A, Onizuka M, Sao H, Akiyama H, Miyamura K, et al. NKG2D gene polymorphism has a significant impact on transplant outcomes after HLA-fully-matched unrelated bone marrow transplantation for standard risk hematologic malignancies. Haematologica (2009) 94(10):1427–34. doi:10.3324/haematol.2009.008318
44. Zwirner NW, Marcos CY, Mirbaha F, Zou Y, Stastny P. Identification of MICA as a new polymorphic alloantigen recognized by antibodies in sera of organ transplant recipients. Hum Immunol (2000) 61(9):917–24. doi:10.1016/S0198-8859(00)00162-2
45. Hankey KG, Drachenberg CB, Papadimitriou JC, Klassen DK, Philosophe B, Bartlett ST, et al. MIC expression in renal and pancreatic allografts. Transplantation (2002) 73(2):304–6. doi:10.1097/00007890-200201270-00029
46. Dulphy N, Haas P, Busson M, Belhadj S, Peffault de Latour R, Robin M, et al. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol (2008) 181(3):2227–37. doi:10.4049/jimmunol.181.3.2227
47. Sconocchia G, Lau M, Provenzano M, Rezvani K, Wongsena W, Fujiwara H, et al. The antileukemia effect of HLA-matched NK and NK-T cells in chronic myelogenous leukemia involves NKG2D-target-cell interactions. Blood (2005) 106(10):3666–72. doi:10.1182/blood-2005-02-0479
48. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419(6908):734–8. doi:10.1038/nature01112
49. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A (2008) 105(4):1285–90. doi:10.1073/pnas.0711293105
50. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol (2003) 171(12):6891–9. doi:10.4049/jimmunol.171.12.6891
51. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol (2006) 176(8):5108–16. doi:10.4049/jimmunol.176.8.5108
52. Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, et al. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol (2005) 43(6):1013–20. doi:10.1016/j.jhep.2005.05.026
53. Marten A, von Lilienfeld-Toal M, Buchler MW, Schmidt J. Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int J Cancer (2006) 119(10):2359–65. doi:10.1002/ijc.22186
54. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169(8):4098–102. doi:10.4049/jimmunol.169.8.4098
55. Salih HR, Goehlsdorf D, Steinle A. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol (2006) 67(3):188–95. doi:10.1016/j.humimm.2006.02.008
56. Boukouaci W, Al-Daccak R, Dulphy N, Lauden L, Amokrane K, Fortier C, et al. Soluble MICA-NKG2D interaction upregulates IFN-gamma production by activated CD3-CD56+ NK cells: potential impact on chronic graft versus host disease. Hum Immunol (2013) 74(12):1536–41. doi:10.1016/j.humimm.2013.08.281
57. Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest (2004) 114(4):560–8. doi:10.1172/JCI22206
58. Bashir Q, William BM, Garcia-Manero G, de Lima M. Epigenetic therapy in allogeneic hematopoietic stem cell transplantation. Rev Bras Hematol Hemoter (2013) 35(2):126–33. doi:10.5581/1516-8484.20130034
59. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol (2008) 9(9):1065–73. doi:10.1038/ni.1642
60. Leong JW, Sullivan RP, Fehniger TA. Natural killer cell regulation by microRNAs in health and disease. J Biomed Biotechnol (2012) 2012:632329. doi:10.1155/2012/632329
61. Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J Immunol (2009) 182(1):39–43. doi:10.4049/jimmunol.182.1.39
62. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res (2012) 72(2):460–71. doi:10.1158/0008-5472.CAN-11-1977
63. Castriconi R, Dondero A, Negri F, Bellora F, Nozza P, Carnemolla B, et al. Both CD133+ and CD133- medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur J Immunol (2007) 37(11):3190–6. doi:10.1002/eji.200737546
64. Pende D, Cantoni C, Rivera P, Vitale M, Castriconi R, Marcenaro S, et al. Role of NKG2D in tumor cell lysis mediated by human NK cells: cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur J Immunol (2001) 31(4):1076–86. doi:10.1002/1521-4141(200104)31:4<1076::AID-IMMU1076>3.0.CO;2-Y
65. El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res (2007) 67(18):8444–9. doi:10.1158/0008-5472.CAN-06-4230
66. Lu X, Kondo Y, Takamatsu H, Ohata K, Yamazaki H, Takami A, et al. CD16+ CD56- NK cells in the peripheral blood of cord blood transplant recipients: a unique subset of NK cells possibly associated with graft-versus-leukemia effect. Eur J Haematol (2008) 81(1):18–25. doi:10.1111/j.1600-0609.2008.01073.x
67. Picardi A, Mengarelli A, Marino M, Gallo E, Benevolo M, Pescarmona E, et al. Up-regulation of activating and inhibitory NKG2 receptors in allogeneic and autologous hematopoietic stem cell grafts. J Exp Clin Cancer Res (2015) 34:98. doi:10.1186/s13046-015-0213-y
68. Boyiadzis M, Memon S, Carson J, Allen K, Szczepanski MJ, Vance BA, et al. Up-regulation of NK cell activating receptors following allogeneic hematopoietic stem cell transplantation under a lymphodepleting reduced intensity regimen is associated with elevated IL-15 levels. Biol Blood Marrow Transplant (2008) 14(3):290–300. doi:10.1016/j.bbmt.2007.12.490
69. Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al Jijakli A, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood (2005) 105(10):4135–42. doi:10.1182/blood-2004-10-4113
70. Ullah MA, Hill GR, Tey SK. Functional reconstitution of natural killer cells in allogeneic hematopoietic stem cell transplantation. Front Immunol (2016) 7:144. doi:10.3389/fimmu.2016.00144
71. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi:10.1016/S1471-4906(01)02060-9
72. Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol (2001) 31(10):3121–7. doi:10.1002/1521-4141(2001010)31:10<3121::AID-IMMU3121>3.0.CO;2-4
73. Nielsen N, Odum N, Urso B, Lanier LL, Spee P. Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS One (2012) 7(2):e31959. doi:10.1371/journal.pone.0031959
74. Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol (2009) 183(8):4921–30. doi:10.4049/jimmunol.0901226
75. Rajalingam R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol Biol (2012) 882:391–414. doi:10.1007/978-1-61779-842-9_23
76. Velardi A, Ruggeri L, Mancusi A, Aversa F, Christiansen FT. Natural killer cell allorecognition of missing self in allogeneic hematopoietic transplantation: a tool for immunotherapy of leukemia. Curr Opin Immunol (2009) 21(5):525–30. doi:10.1016/j.coi.2009.07.015
77. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
78. Mizutani K, Terasaki P, Bignon JD, Hourmant M, Cesbron-Gautier A, Shih RN, et al. Association of kidney transplant failure and antibodies against MICA. Hum Immunol (2006) 67(9):683–91. doi:10.1016/j.humimm.2006.06.002
79. Zou Y, Stastny P, Susal C, Dohler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357(13):1293–300. doi:10.1056/NEJMoa067160
80. Suarez-Alvarez B, Lopez-Vazquez A, Gonzalez MZ, Fdez-Morera JL, Diaz-Molina B, Blanco-Gelaz MA, et al. The relationship of anti-MICA antibodies and MICA expression with heart allograft rejection. Am J Transplant (2007) 7(7):1842–8. doi:10.1111/j.1600-6143.2007.01838.x
81. Ansari M, Uppugunduri CR, Ferrari-Lacraz S, Bittencourt H, Gumy-Pause F, Chalandon Y, et al. The clinical relevance of pre-formed anti-HLA and anti-MICA antibodies after cord blood transplantation in children. PLoS One (2013) 8(8):e72141. doi:10.1371/journal.pone.0072141
82. Flaxa J, Rosner A, Holig K, Bornhauser M, Wassmuth R. Methodological and clinical aspects of alloimmunization after granulocyte transfusion in patients undergoing allogeneic stem cell transplantation. Tissue Antigens (2015) 85(2):93–103. doi:10.1111/tan.12487
83. Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A (2006) 103(24):9190–5. doi:10.1073/pnas.0603503103
84. Groh V, Li YQ, Cioca D, Hunder NN, Wang W, Riddell SR, et al. Efficient cross-priming of tumor antigen-specific T cells by dendritic cells sensitized with diverse anti-MICA opsonized tumor cells. Proc Natl Acad Sci U S A (2005) 102(18):6461–6. doi:10.1073/pnas.0501953102
85. Cox ST, Stephens HA, Fernando R, Karasu A, Harber M, Howie AJ, et al. Major histocompatibility complex class I-related chain A allele mismatching, antibodies, and rejection in renal transplantation. Hum Immunol (2011) 72(10):827–34. doi:10.1016/j.humimm.2011.05.004
86. Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat Genet (2011) 43(5):455–8. doi:10.1038/ng.809
87. Kumar V, Yi Lo PH, Sawai H, Kato N, Takahashi A, Deng Z, et al. Soluble MICA and a MICA variation as possible prognostic biomarkers for HBV-induced hepatocellular carcinoma. PLoS One (2012) 7(9):e44743. doi:10.1371/journal.pone.0044743
88. Tong HV, Toan NL, Song LH, Bock CT, Kremsner PG, Velavan TP. Hepatitis B virus-induced hepatocellular carcinoma: functional roles of MICA variants. J Viral Hepat (2013) 20(10):687–98. doi:10.1111/jvh.12089
89. Lo PH, Urabe Y, Kumar V, Tanikawa C, Koike K, Kato N, et al. Identification of a functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS One (2013) 8(4):e61279. doi:10.1371/journal.pone.0061279
90. Lu M, Xia B, Ge L, Li Y, Zhao J, Chen F, et al. Role of major histocompatibility complex class I-related molecules A*A5.1 allele in ulcerative colitis in Chinese patients. Immunology (2009) 128(1 Suppl):e230–6. doi:10.1111/j.1365-2567.2008.02953.x
91. Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, et al. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res (2010) 70(2):481–9. doi:10.1158/0008-5472.CAN-09-1688
92. Zhao J, Jiang Y, Lei Y, Zou K, Wang C, Huang S, et al. Functional MICA-129 polymorphism and serum levels of soluble MICA are correlated with ulcerative colitis in Chinese patients. J Gastroenterol Hepatol (2011) 26(3):593–8. doi:10.1111/j.1440-1746.2010.06524.x
Keywords: natural killer group 2, member D, NKG2D ligands, hematopoietic cell transplantation, graft-vs.-host disease, MHC class I chain-related gene, MICA
Citation: Carapito R, Aouadi I, Ilias W and Bahram S (2017) Natural Killer Group 2, Member D/NKG2D Ligands in Hematopoietic Cell Transplantation. Front. Immunol. 8:368. doi: 10.3389/fimmu.2017.00368
Received: 08 February 2017; Accepted: 14 March 2017;
Published: 27 March 2017
Edited by:
Zhenhua Dai, Guangdong Provincial Academy of Chinese Medical Sciences, ChinaReviewed by:
Ralf Dressel, Universitätsmedizin Göttingen, GermanyAkifumi Takaori-Kondo, Kyoto University, Japan
Copyright: © 2017 Carapito, Aouadi, Ilias and Bahram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raphael Carapito, Y2FyYXBpdG9AdW5pc3RyYS5mcg==;
Seiamak Bahram, c2lhbWFrQHVuaXN0cmEuZnI=