 Volker Spindler
Volker Spindler Jens Waschke
Jens Waschke- 1Department of Biomedicine, University of Basel, Basel, Switzerland
- 2Faculty of Medicine, Ludwig-Maximilians-Universität (LMU) Munich, Munich, Germany
Pemphigus is a severe autoimmune-blistering disease of the skin and mucous membranes caused by autoantibodies reducing desmosomal adhesion between epithelial cells. Autoantibodies against the desmosomal cadherins desmogleins (Dsgs) 1 and 3 as well as desmocollin 3 were shown to be pathogenic, whereas the role of other antibodies is unclear. Dsg3 interactions can be directly reduced by specific autoantibodies. Autoantibodies also alter the activity of signaling pathways, some of which regulate cell cohesion under baseline conditions and alter the turnover of desmosomal components. These pathways include Ca2+, p38MAPK, PKC, Src, EGFR/Erk, and several others. In this review, we delineate the mechanisms relevant for pemphigus pathogenesis based on the histology and the ultrastructure of patients’ lesions. We then dissect the mechanisms which can explain the ultrastructural hallmarks detectable in pemphigus patient skin. Finally, we reevaluate the concept that the spectrum of mechanisms, which induce desmosome dysfunction upon binding of pemphigus autoantibodies, finally defines the clinical phenotype.
Introduction
Pemphigus is a severe autoimmune-blistering skin disease caused by autoantibodies primarily targeting the desmosomal adhesion molecules desmogleins (Dsgs) 1 and 3 (1), which are required for the firm intercellular adhesion of keratinocytes. Autoantibodies targeting Dsg3 are found during the mucosal-dominant phase in pemphigus vulgaris (mPV) which is frequently followed by a mucocutaneous phase (mcPV) with additional epidermal blistering and characterized by the presence of both anti-Dsg3 and anti-Dsg1 antibodies. By contrast, in pemphigus foliaceus (PF), flaccid blisters are found in the skin only, and their formation is associated with the occurrence of anti-Dsg1 autoantibodies. Autoantibodies against other desmosomal cadherins such as desmocollin (Dsc) 3 are rarely detectable in PV or PF but can be pathogenic (2–5). The pathogenicity of autoantibodies against a range of non-desmosomal autoantibodies that are often present in patients’ sera in addition to anti-Dsg antibodies is unclear (6). We here focus on the mechanisms causing skin blistering in response to autoantibodies against Dsg1 and Dsg3. In addition to suppressing autoimmunity, which is the current basis for disease management (1), we believe that novel targeted therapeutic strategies are necessary to stabilize desmosomes in situations when autoantibodies are present. It is essential to better understand the regulation of desmosomal adhesion, and we aim to provide perspectives for future research by reevaluating the mechanisms leading to desmosome dysfunction, loss of keratinocyte cohesion, and blistering.
Histology and Ultrastructure of Patient’s Lesions Highlight Relevant Mechanisms
Cultured keratinocytes or mouse models can only partially reproduce the situation in patients. We believe that for the identification of relevant mechanisms in pemphigus pathogenesis, the careful evaluation of patients’ lesions is the gold standard. On the histological level, skin blistering in PV occurs by suprabasal splitting (Figure 1), whereas PF is characterized by superficial lesions restricted to granular or upper spinous layers of the epidermis (1). Although exceptions occur, PF splitting is usually found in the upper half of the epidermis, whereas PV affects the lower half. At least in some parts of typical PV but not PF lesions, the histological hallmark of a “tomb-stone appearance” of keratinocytes in the blister bottom can be detected (7, 8).
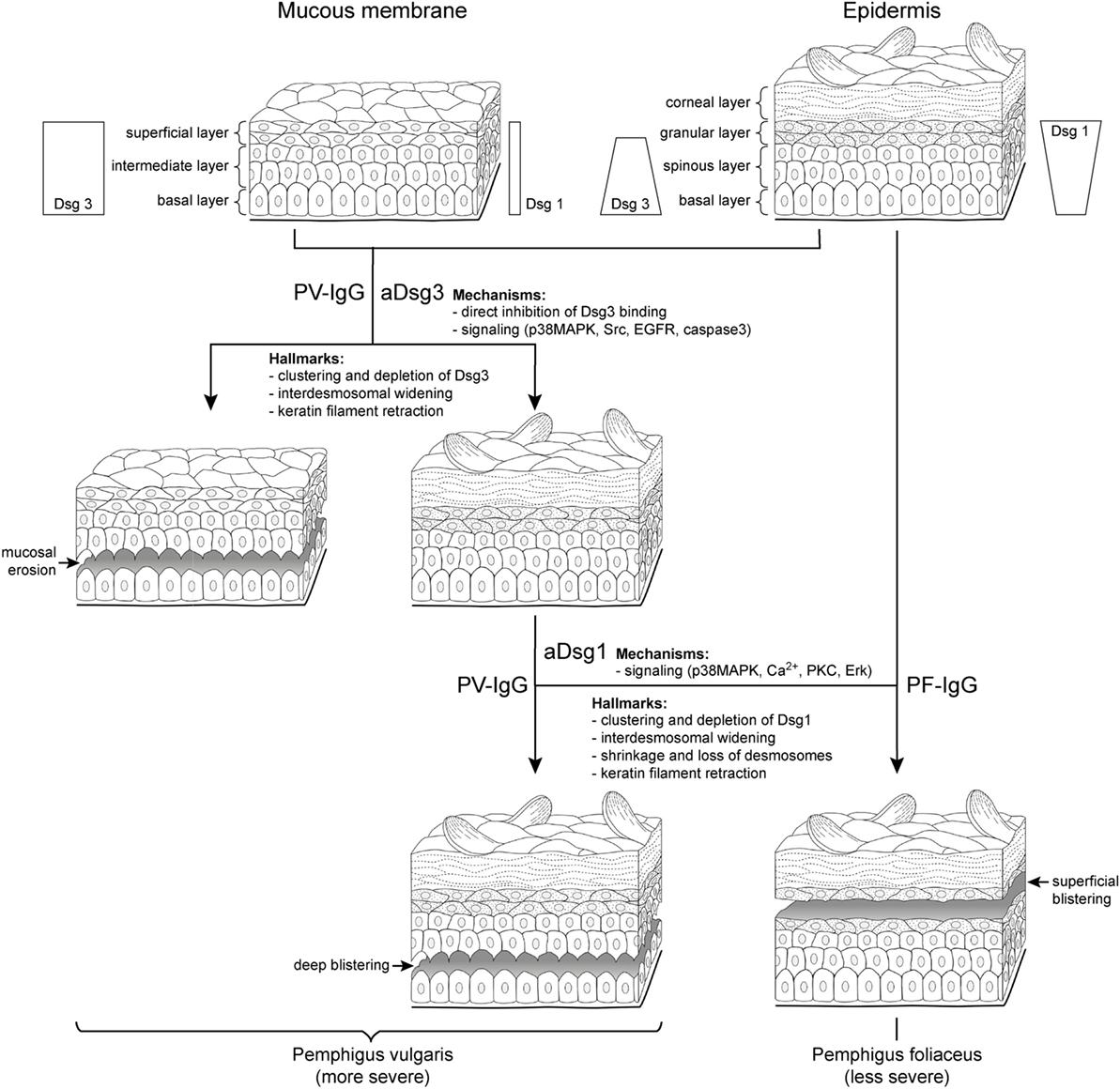
Figure 1. Clinical phenotypes correlate with autoantibody profiles and mechanisms causing desmosome dysfunction. It is well established that clinical phenotypes of pemphigus largely correlate with autoantibody profiles but the underlying mechanisms are not clear. In pemphigus vulgaris (PV) (left), mucosal erosions often precede epidermal blistering. During the mucosal-dominant phase, autoantibodies (PV-IgG) primarily against desmoglein (aDsg3) are pathogenic, which is the most abundant Dsg isoform in the mucosal epithelium. The occurrence of antibodies targeting Dsg1 (aDsg1), which is strongly expressed in the superficial epidermis compared to Dsg3, usually correlates with skin blistering, affecting the deep epidermis right above the basal layer. By contrast, in pemphigus foliaceus (PF), autoantibodies (PF-IgG) primarily against Dsg1 (aDsg1) cause superficial epidermal blistering. The different phenotypes are characterized by structural hallmarks in patients’ lesions, and the mechanisms causing desmosome dysfunction in response to autoantibody binding appear to be different for aDsg1 and aDsg3. Please note that the distribution of Dsg1 and Dsg3 in the epidermis is indicated for all layers except the for corneal layer.
By immunostaining, the clustering of Dsg3 together with autoantibodies has been detected in the mucosal and skin lesions in mcPV but also in unaffected epidermis in the mucosal-dominant type (mPV) in the absence of ultrastructural alterations of desmosomes (9, 10). By contrast, Dsg1 clustering was found in mcPV skin and PF epidermis only, i.e., when antibodies against Dsg1 were present (9, 10) (Figure 1). Since Dsg3 clustering was shown to be the structural correlate of Dsg3 depletion (11), these data indicate that the depletion of Dsg3 alone may be a primary mechanism in mucosal eroding but alone is not sufficient to cause epidermal blistering. Nevertheless, Dsg3 depletion may drastically worsen desmosome destabilization in the epidermis which is suggested by the fact that glucocorticoids rapidly improve the clinical phenotype (1) at least in part via Stat3-induced Dsg3 transcription increase (12).
On the ultrastructural level, smaller desmosomes were found only in conditions when patients presented with antibodies against Dsg1 such as in mcPV and PF but not in mPV (9, 10, 13, 14), suggesting that Dsg1 targeting is critical and may interfere with desmosome assembly or cause dismantling of existing desmosomes (Figure 1). Besides a reduced size, a general loss of desmosomes is present under all conditions where blistering occurred. Electron microscopy revealed the formation of double-membrane structures in PV and PF containing desmosomes with reduced size and altered morphology which may be the correlate for the depletion of extradesmosomal Dsg molecules and the uptake of entire desmosomes (13). Similarly, interdesmosomal widening, which is the first ultrastructural sign to be detected in pemphigus lesions, may be caused by the endocytosis of extradesmosomal Dsg1 rather than of Dsg3 (13, 15). This alone appears not to be sufficient for blister formation since it was detected also in the unaffected deep epidermis and the mucosa of PF patients but not in mPV with intact Dsg1 distribution.
Split desmosomes both with and without attached keratin filaments were detected by electron microscopy and SIM on the keratinocyte surface facing blisters in PF and mcPV (13, 14). Desmosome splitting can be induced by mechanical stress (14) and may be the ultrastructural correlate for the direct inhibition of Dsg binding. Since split desmosomes in this study were of reduced size, altered desmosome structure appears to be required, suggesting an additional role of impaired desmosome assembly or the depletion of desmosomal Dsg. The final hallmark described early for both PV and PF by electron microscopy is keratin retraction (16, 17) (Figure 1). Recently, keratin filament retraction was observed only when desmosomes were completely absent (13). This can be interpreted in the way that keratin filaments are not the cause but rather the consequence of desmosomal loss or the changes are temporally tightly correlated.
Apoptosis is not a major mechanism because cells displaying signs of apoptotic cell death are absent or sparse in PV and PF skin lesions and therefore cannot explain acantholysis of a significant epidermal area (13, 18, 19).
Autoantibody-Triggered Mechanisms Impairing Desmosome Turnover
As outlined earlier, split desmosomes, reduced desmosome numbers and size, and keratin retraction are ultrastructural hallmarks in pemphigus skin. Reduced desmosome size or numbers cannot be explained by the direct interference of pemphigus autoantibodies with Dsg binding but rather are a consequence of the altered turnover of desmosomal proteins. These changes are likely steered by intracellular-signaling pathways, which are modulated in response to autoantibody binding and represent potential pharmacologic targets. In principal, reduced desmosome size and numbers can result either from interference with desmosome assembly or from the enhanced disassembly of desmosomes. Available data suggest that in pemphigus, both mechanisms contribute to impaired desmosome turnover, shifting the balance toward an overall reduction of desmosomal components (20).
Desmosome assembly is tightly interwoven with adherens junction formation and appears to proceed in distinct steps (21) (Figure 2, left panel). Desmosomal cadherins are initially transported to the cell membrane in a microtubule- and kinesin-dependent process (22), which, in case of Dsg2, is enhanced by its palmitoylation (23). The precise mechanisms are unclear but once membrane-localized, desmosomal cadherins appear to cluster in an intermediate junction with E-cadherin, β-catenin, and plakoglobin and probably segregate to form desmosomes clusters later on (24, 25). Plakophilins (Pkps) are essential as they are required to assemble keratin-anchored DP pools in the cortical regions of the cell (26, 27). Pkp3 was shown to participate in transferring DP clusters to the membrane and to stabilize desmosomal cadherins in a Rap1-dependent manner (28). In addition, cortical actin and actin-binding proteins such as adducins and RhoA signaling are necessary for full desmosome assembly (29–31). Desmosomal molecules localize to lipid rafts and the raft-associated proteins Flotillin-1 and -2 (32, 33). In line with this, interference with lipid raft composition prevents both desmosomal assembly and disassembly, suggesting these lipid-enriched membrane domains to be hot spots for desmosome turnover. Compared to the assembly, the disassembly of desmosomes under physiologic conditions is poorly understood, which may be related to the relative chemical inaccessibility and the stability of maturated desmosomes (34).
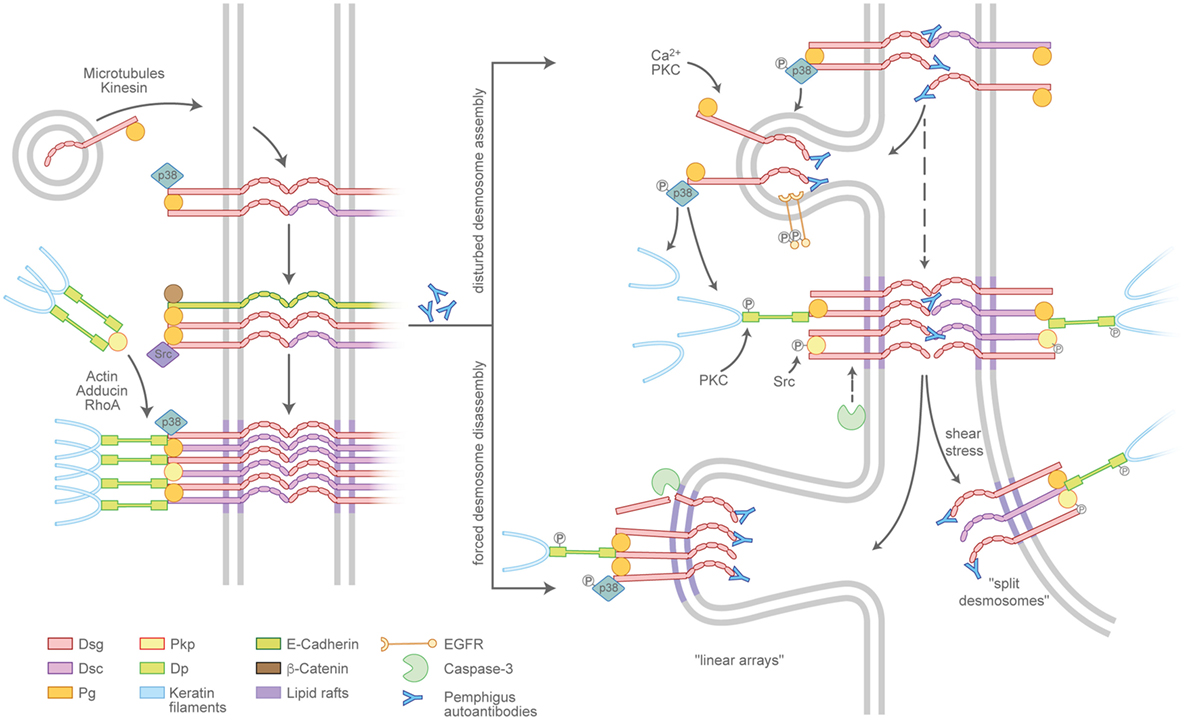
Figure 2. Pemphigus autoantibodies interfere with desmosome turnover. Left panel: a simplified view of the assembly pathway of desmosomes. Desmosomal cadherins and the plaque components desmoplakin (DP), plakophilins (Pkps), as well as the keratins are transported to the plasma membrane by distinct routes and may involve an intermediate complex containing adherens junction proteins. Right panel: Pemphigus autoantibodies interfere with the assembly of nascent desmosomes and promote the disassembly of existing desmosomes. The modulation of desmosome turnover is tied to changes of intracellular-signaling cascades which, for instance, lead to the phosphorylation or cleavage of desmosomal components. Linear arrays represent zones of desmosome disassembly. Split desmosomes may occur if the already-weakened desmosomes are exposed to shear stress.
Pemphigus autoantibodies interfere with desmosome turnover by enhancing the internalization of desmosomal components (Figure 2, right panel). However, a clear separation of mechanisms specifically affecting either the assembly or the disassembly pathway in most cases is not possible. It is conceivable that autoantibodies reach Dsgs located in extradesmosomal membrane pools more easily than those densely packed in a mature desmosome. In line with this scenario, the non-desmosomal pool of Dsg3 is the first to be depleted (35, 36). Consequently, the reduced availability of supply molecules leads to the destabilization of desmosomes and in the longer run may favor the depletion of desmosome-localized, now more easily accessible molecules. Thus, the reduced desmosome assembly route in pemphigus is at least in part a result of internalized extradesmosomal molecules, summarized as “desmoglein nonassembly depletion hypothesis” (9, 37). However, the situation apparently is more complex than a simple disbalance in supply and demand. Importantly, especially the extradesmosomal molecules are considered to serve as signaling scaffolds (38, 39). As an example, it was shown that a complex of Dsg3 and Pg binds p38MAPK and suppresses its activity (40, 41). Upon the loss of Dsg3 interaction, e.g., by steric hindrance through pemphigus autoantibodies, this suppressive function is abolished and p38MAPK is activated. Indeed, p38MAPK signaling was shown to promote Dsg3 internalization (42) as well as keratin retraction (41, 43), suggesting that p38MAPK is a central signaling molecule in desmosome turnover. Vice versa, keratin filaments were shown to influence both desmosome stability and Dsg3-binding characteristics in a signaling-dependent manner. Under physiologic conditions, the keratin-dependent suppression of p38MAPK signaling increases Dsg3-binding strength, whereas the suppression of PKC signaling through the adapter protein Rack-1 prevents DP phosphorylation, stabilizes Dsg3 in the desmosome (44, 45), and, together with Pkp1 (46), promotes a hyperadhesive state. These functions appear to be disturbed by autoantibody-induced keratin retraction (Vielmuth et al., Frontiers Immunol, this issue). Furthermore, Pkp3 is phosphorylated in response to autoantibody binding in an Src-dependent manner (47), which may be connected to Src being present in the intermediate E-cadherin/Dsg3 complex (25). Together, these data suggest a feed-forward loop from extradesmosomal complexes to desmosomes that, at least in part through interference with keratin-dependent signaling, destabilizes desmosome composition and function. As the precise mechanisms are unclear, it is possible that keratins affect both the assembly and the disassembly pathways of desmosomes.
The occurrence of desmosome disassembly can be concluded from observations that Dsg3 together with PV-IgG is excluded from desmosomes and internalized (48). This is supported by observations that IgG autoantibodies can access Dsgs in native desmosomes (49). Clusters of desmosomal molecules localizing in arrays perpendicular to the cell–cell border may represent sites of internalization of single desmosomal components or the entire complexes (50). These may correspond to the “double-membrane structures” that are visible in patients’ skin and are thought to be regions of internalization for partially dismantled desmosomes (13). Although all desmosomal proteins eventually are internalized, the degradation pathways of specific components are not uniform. Dsg3 as well as Pg colocalize with markers for endosomes and lysosomes, whereas DP and presumably other plaque proteins use different, yet unknown, routes (35, 42). Lipid rafts are thought to be important for desmosome disassembly, as the impairment of raft composition prevents the depletion at least of Dsg3 and reduces loss of cell cohesion (32) (Schlögl et al., this issue). In addition, the cleavage of Dsg3 by caspase-3 independent from apoptosis was observed and may promote internalization (51–53), the latter of which was shown to require EGFR signaling (54).
Split desmosomes are rarely detectable in keratinocyte cultures incubated with pemphigus antibodies in the absence of mechanical stress (55) but are enhanced when cultures are subjected to mechanical strain as well as in human skin models and in patients’ skin (14, 56). Furthermore, split desmosomes appear typically severely altered with reduced plaque sizes and aberrant keratin insertion. This may indicate that desmosome splitting occurs secondary to changes in desmosome composition. Collectively, interference with desmosome turnover can largely explain the ultrastructural hallmarks of pemphigus patients’ lesions.
Desmosome Dysfunction Defines the Clinical Phenotype of Pemphigus
All mechanisms described earlier finally lead to desmosome dysfunction, which impair keratinocyte cohesion. However, the functional interplay of the different mechanisms is not well understood at present. It is also unclear why epidermal splitting affects the deep epidermis in PV, whereas PF blisters are restricted to superficial epidermal layers (Figure 1). Besides homophilic adhesion, Dsgs were shown to undergo heterophilic interactions both with other Dsg isoforms and Dscs, respectively (5, 45, 57, 58). Moreover, the genetic deletion of Dsc3 causes a PV phenotype with suprabasal blistering in mice (59). On the other hand, the forced overexpression of Dsg2, a Dsg isoform not present in the adult human epidermis except hair follicles, which is upregulated in PV lesions, can compensate for the loss of Dsg1 in a PF mouse model (60, 61). Therefore, it is likely that all desmosomal cadherins (i.e., Dsg1–4, Dsc1–3) contribute to keratinocyte cohesion and epidermal integrity in a layer-specific manner.
In the mucosa, Dsg3 is the predominant desmosomal cadherin, and in the superficial epidermis Dsg1 is strongly expressed, whereas other isoforms of desmosomal cadherins are largely absent except of Dsc1 (Figure 1) (8). The cadherin distribution pattern can well explain the clinical phenotype of mPV and PF because the loss of function of the target molecules cannot be outbalanced by relevant amounts of other desmosomal cadherins (1). However, the Dsg compensation theory falls short in mcPV, because if Dsg1 and Dsg3 would specifically compensate for each other, the whole epidermis or at least the lower epidermis should disintegrate completely when considering that autoantibodies entering the epidermis from the dermis may be concentrated most in the first few layers of cells. Rather, epidermal splitting typically occurs right at the suprabasal level, i.e., between the basal keratinocytes and the first layer of the spinous layer cells where in intact epidermis as well as in PV lesions, Dsg1 and Dsg3 are expressed (8). This favors the hypothesis that different mechanisms contribute to desmosome dysfunction in a layer-specific manner.
The direct inhibition of Dsg3 binding by PV-IgG has been described under cell-free conditions as well as on the surface of living keratinocytes; however, it was ineffective to induce the complete loss of cell cohesion (62–64). By contrast, the interference of anti-Dsg1 autoantibodies with Dsg1 binding was detectable neither in cell-free AFM experiments (63, 65, 66) nor in studies with intact keratinocytes (Vielmuth 2018, Frontiers Immun, this issue). This suggests that the mechanisms underlying the loss of keratinocyte cohesion in pemphigus are autoantibody-specific. In this respect, it was reported recently that autoantibody profiles in pemphigus correlate with signaling patterns which makes it intriguing to speculate that specific signaling patterns may be defining the clinical phenotype (65). It has been reported that p38MAPK and Src can be activated by PV-IgG containing antibodies against Dsg3 as well as by AK23 which is Dsg3-specific (Figure 1). In addition, EGFR and caspase-3 activation in response to AK23 and PV-IgG was shown in mouse models (53, 54, 67). By contrast, the increase of intracellular Ca2+ as well as Erk activation was induced only by PV-IgG and PF-IgG containing Dsg1 autoantibodies, which was paralleled by p38MAPK activation (65). In this line of thoughts, the direct inhibition of Dsg3 binding together with the signaling of p38MAPK, Src, EGFR, and caspase-3 would be sufficient to cause mucosal erosions (Figure 1), whereas additional mechanisms such as Ca2+, which has been shown to be associated with PKC activation in keratinocytes treated with pemphigus antibodies (68, 69), as well as Erk would be required for epidermal blistering. If so, it could be postulated that one function of Dsg and Dsc isoforms expressed in the epidermis but not in the mucosa may be to further strengthen keratinocyte cohesion by regulating signaling pathway which control desmosome turnover and function.
For most of the pathways described earlier, it was shown that modulation is sufficient to largely reduce skin blistering in mice (6). However, mice differ from humans with respect to epidermal Dsg expression patterns. Moreover, the genetic deletion of Dsg3 or the inactivation of Dsg3 via incubation with AK23 is sufficient to cause skin blisters in mice (41, 70). This is different to the situation in mPV where the skin is usually not affected and indicates that Dsg3 is more critical for epidermal integrity in mice compared to humans. Therefore, to prevent misinterpretations, studies including ultrastructural analyses of human skin are required in addition to mouse models. Indeed, studies in human skin ex vivo indicate that the significance of p38MAPK for blister formation may be different in PV and PF. When using PV-IgG, the inhibition of p38MAPK was sufficient to abrogate blistering (11, 56) but not when PF-IgG was applied (71). On the other hand, it was shown that p38MAPK in response to PV-IgG and PF-IgG is involved in the reduction of desmosome length (56, 71). However, since the shortening of desmosomes and interdesmosomal widening were detectable also in the absence of blistering following treatment with AK23, this indicates that p38MAPK-mediated reduction of desmosome length and interdesmosomal widening may not be sufficient to cause skin blistering. Rather, the complete loss of desmosomes appears to be required (56) for which other mechanisms such as Ca2+, PKC, or Erk may be necessary to induce superficial epidermal blistering in PF (Figure 1). However, when bolstered by the direct inhibition of Dsg3 binding and Src activation, the panel of mechanisms causing desmosome dysfunction may be sufficient to cause suprabasal splitting as seen in mcPV. Taken together, it is likely that the clinical phenotype in pemphigus is caused by a complex set of mechanisms causing desmosome dysfunction.
Author Contributions
VS and JW conceived the perspective and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The study was supported by the Deutsche Forschungsgemeinschaft (DFG SP1300/1-3 to VS and FOR2497 to JW).
References
1. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers (2017) 3:17026. doi:10.1038/nrdp.2017.26
2. Mindorf S, Dettmann IM, Krüger S, Fuhrmann T, Rentzsch K, Karl I, et al. Routine detection of serum antidesmocollin autoantibodies is only useful in patients with atypical pemphigus. Exp Dermatol (2017) 26(12):1267–70. doi:10.1111/exd.13409
3. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol (2010) 177:2724–30. doi:10.2353/ajpath.2010.100483
4. Rafei D, Müller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, et al. IgG Autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol (2011) 178:718–23. doi:10.1016/j.ajpath.2010.10.016
5. Spindler V, Heupel W-M, Efthymiadis A, Schmidt E, Eming R, Rankl C, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J Biol Chem (2009) 284:30556–64. doi:10.1074/jbc.M109.024810
6. Spindler V, Eming R, Schmidt E, Amagai M, Grando S, Jonkman MF, et al. Mechanisms causing loss of keratinocyte cohesion in pemphigus. J Invest Dermatol (2017) 138(1):32–7. doi:10.1016/j.jid.2017.06.022
7. Ohata C, Ishii N, Furumura M. Locations of acantholysis in pemphigus vulgaris and pemphigus foliaceus. J Cutan Pathol (2014) 41:880–9. doi:10.1111/cup.12384
8. Waschke J. The desmosome and pemphigus. Histochem Cell Biol (2008) 130:21–54. doi:10.1007/s00418-008-0420-0
9. Oktarina DAM, van der Wier G, Diercks GFH, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol (2011) 165:552–62. doi:10.1111/j.1365-2133.2011.10463.x
10. van der Wier G, Pas HH, Kramer D, Diercks GFH, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J Invest Dermatol (2014) 134:2287–90. doi:10.1038/jid.2014.140
11. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One (2012) 7:e50696. doi:10.1371/journal.pone.0050696
12. Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight (2017) 2:92253. doi:10.1172/jci.insight.92253
13. Sokol E, Kramer D, Diercks GF, Kuipers J, Jonkman MF, Pas HH, et al. Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol (2015) 135:1763–70. doi:10.1038/jid.2015.109
14. Stahley SN, Warren MF, Feldman RJ, Swerlick RA, Mattheyses AL, Kowalczyk AP. Super-resolution microscopy reveals altered desmosomal protein organization in tissue from patients with pemphigus vulgaris. J Invest Dermatol (2016) 136:59–66. doi:10.1038/JID.2015.353
15. van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of acantholysis in pemphigus foliaceus re-examined from the current perspective. Br J Dermatol (2012) 167:1265–71. doi:10.1111/j.1365-2133.2012.11173.x
16. Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in pemphigus vulgaris. J Invest Dermatol (1961) 36:373–82. doi:10.1038/jid.1961.58
17. Wilgram GF, Caulfield JB, Madgic EB. An electron microscopic study of acantholysis and dyskeratosis in pemphigus foliaceus: with a special note on peculiar intracytoplasmic bodies. J Invest Dermatol (1964) 43:287–99. doi:10.1038/jid.1964.159
18. Janse IC, van der Wier G, Jonkman MF, Pas HH, Diercks GFH. No evidence of apoptotic cells in pemphigus acantholysis. J Invest Dermatol (2014) 134:2039–41. doi:10.1038/jid.2014.60
19. Schmidt E, Waschke J. Apoptosis in pemphigus. Autoimmun Rev (2009) 8:533–7. doi:10.1016/j.autrev.2009.01.011
20. Kitajima Y. 150(th) anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun Adhes (2014) 21:269–80. doi:10.3109/15419061.2014.943397
21. Nekrasova O, Green KJ. Desmosome assembly and dynamics. Trends Cell Biol (2013) 23:537–46. doi:10.1016/j.tcb.2013.06.004
22. Nekrasova OE, Amargo EV, Smith WO, Chen J, Kreitzer GE, Green KJ. Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J Cell Biol (2011) 195:1185–203. doi:10.1083/jcb.201106057
23. Roberts BJ, Svoboda RA, Overmiller AM, Lewis JD, Kowalczyk AP, Mahoney MG, et al. Palmitoylation of desmoglein 2 is a regulator of assembly dynamics and protein turnover. J Biol Chem (2016) 291:24857–65. doi:10.1074/jbc.M116.739458
24. Hatzfeld M, Keil R, Magin TM. Desmosomes and intermediate filaments: their consequences for tissue mechanics. Cold Spring Harb Perspect Biol (2017) 9:a029157. doi:10.1101/cshperspect.a029157
25. Rotzer V, Hartlieb E, Vielmuth F, Gliem M, Spindler V, Waschke J. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci (2015) 72:4885–97. doi:10.1007/s00018-015-1977-0
26. Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, et al. Plakophilin 2: a critical scaffold for PKCα that regulates intercellular junction assembly. J Cell Biol (2008) 181:605–13. doi:10.1083/jcb.200712133
27. Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, Huen AC, et al. Desmoplakin assembly dynamics in four dimensions. J Cell Biol (2005) 171:1045–59. doi:10.1083/jcb.200510038
28. Todorovic V, Koetsier JL, Godsel LM, Green KJ. Plakophilin 3 mediates Rap1-dependent desmosome assembly and adherens junction maturation. Mol Biol Cell (2014) 25:3749–64. doi:10.1091/mbc.E14-05-0968
29. Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, et al. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol Biol Cell (2010) 21:2844–59. doi:10.1091/mbc.E10-02-0131
30. Pasdar M, Li Z. Disorganization of microfilaments and intermediate filaments interferes with the assembly and stability of desmosomes in MDCK epithelial cells. Cell Motil Cytoskeleton (1993) 26:163–80. doi:10.1002/cm.970260207
31. Rötzer V, Breit A, Waschke J, Spindler V. Adducin is required for desmosomal cohesion in keratinocytes. J Biol Chem (2014) 289:14925–40. doi:10.1074/jbc.M113.527127
32. Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome assembly and disassembly are membrane raft-dependent. PLoS One (2014) 9:e87809. doi:10.1371/journal.pone.0087809
33. Vollner F, Ali J, Kurrle N, Exner Y, Eming R, Hertl M, et al. Loss of flotillin expression results in weakened desmosomal adhesion and pemphigus vulgaris-like localisation of desmoglein-3 in human keratinocytes. Sci Rep (2016) 6:28820. doi:10.1038/srep28820
34. Windoffer R, Borchert-Stuhltrager M, Leube RE. Desmosomes: interconnected calcium-dependent structures of remarkable stability with significant integral membrane protein turnover. J Cell Sci (2002) 115:1717–32.
35. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem (2006) 281:7623–34. doi:10.1074/jbc.M512447200
36. Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem (2007) 282:17866–76. doi:10.1074/jbc.M607963200
37. Kitajima Y. New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci (2013) 29:1–13. doi:10.1016/j.kjms.2012.08.001
38. Müller EJ, Williamson L, Kolly C, Suter MM. Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol (2008) 128:501–16. doi:10.1038/sj.jid.5701248
39. Waschke J, Spindler V. Desmosomes and extradesmosomal adhesive signaling contacts in pemphigus. Med Res Rev (2014) 34:1127–45. doi:10.1002/med.21310
40. Spindler V, Dehner C, Hubner S, Waschke J. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J Invest Dermatol (2014) 134:1655–64. doi:10.1038/jid.2014.21
41. Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest (2013) 123:800–11. doi:10.1172/JCI60139
42. Jolly PS, Berkowitz P, Bektas M, Lee H-E, Chua M, Diaz LA, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem (2010) 285:8936–41. doi:10.1074/jbc.M109.087999
43. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem (2005) 280:23778–84. doi:10.1074/jbc.M501365200
44. Dehner C, Rotzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am J Pathol (2014) 184:2528–36. doi:10.1016/j.ajpath.2014.05.016
45. Vielmuth F, Wanuske MT, Radeva MY, Hiermaier M, Kugelmann D, Walter E, et al. Keratins regulate the adhesive properties of desmosomal cadherins through signaling. J Invest Dermatol (2017) 138(1):121–31. doi:10.1016/j.jid.2017.08.033
46. Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J Invest Dermatol (2014) 134:1033–43. doi:10.1038/jid.2013.401
47. Cirillo N, AlShwaimi E, McCullough M, Prime SS. Pemphigus vulgaris autoimmune globulin induces Src-dependent tyrosine-phosphorylation of plakophilin 3 and its detachment from desmoglein 3. Autoimmunity (2014) 47:134–40. doi:10.3109/08916934.2013.866100
48. Aoyama Y, Nagai M, Kitajima Y. Binding of pemphigus vulgaris IgG to antigens in desmosome core domains excludes immune complexes rather than directly splitting desmosomes. Br J Dermatol (2010) 162:1049–55. doi:10.1111/j.1365-2133.2010.09672.x
49. Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. IgG Binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol (2004) 122:1145–53. doi:10.1111/j.0022-202X.2004.22426.x
50. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol (2011) 131:706–18. doi:10.1038/jid.2010.389
51. Cirillo N, Campisi G, Gombos F, Perillo L, Femiano F, Lanza A. Cleavage of desmoglein 3 can explain its depletion from keratinocytes in pemphigus vulgaris. Exp Dermatol (2008) 17:858–63. doi:10.1111/j.1600-0625.2008.00719.x
52. Hariton WVJ, Galichet A, Vanden Berghe T, Overmiller AM, Mahoney MG, Declercq W, et al. Feasibility study for clinical application of caspase-3 inhibitors in pemphigus vulgaris. Exp Dermatol (2017) 26(12):1274–7. doi:10.1111/exd.13458
53. Luyet C, Schulze K, Sayar BS, Howald D, Muller EJ, Galichet A. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS One (2015) 10:e0119809. doi:10.1371/journal.pone.0119809
54. Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem (2013) 288:9447–56. doi:10.1074/jbc.M112.438010
55. Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol (2009) 129:908–18. doi:10.1038/jid.2008.339
56. Egu DT, Walter E, Spindler V, Waschke J. Inhibition of p38MAPK signalling prevents epidermal blistering and alterations of desmosome structure induced by pemphigus autoantibodies in human epidermis. Br J Dermatol (2017) 177(6):1612–8. doi:10.1111/bjd.15721
57. Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, et al. Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci U S A (2016) 113:7160–5. doi:10.1073/pnas.1606272113
58. Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D. Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem (2011) 286:2143–54. doi:10.1074/jbc.M110.192245
59. Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci (2008) 121:2844–9. doi:10.1242/jcs.031518
60. Brennan D, Hu Y, Medhat W, Dowling A, Mahoney MG. Superficial dsg2 expression limits epidermal blister formation mediated by pemphigus foliaceus antibodies and exfoliative toxins. Dermatol Res Pract (2010) 2010:410278. doi:10.1155/2010/410278
61. Hartlieb E, Kempf B, Partilla M, Vigh B, Spindler V, Waschke J. Desmoglein 2 is less important than desmoglein 3 for keratinocyte cohesion. PLoS One (2013) 8:e53739. doi:10.1371/journal.pone.0053739
62. Heupel W-M, Müller T, Efthymiadis A, Schmidt E, Drenckhahn D, Waschke J. Peptides targeting the desmoglein 3 adhesive interface prevent autoantibody-induced acantholysis in pemphigus. J Biol Chem (2009) 284:8589–95. doi:10.1074/jbc.M808813200
63. Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol (2008) 181:1825–34. doi:10.4049/jimmunol.181.3.1825
64. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. J Invest Dermatol (2015) 135:3068–77. doi:10.1038/jid.2015.324
65. Walter E, Vielmuth F, Rotkopf L, Sardy M, Horvath ON, Goebeler M, et al. Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Sci Rep (2017) 7:3579. doi:10.1038/s41598-017-03697-7
66. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest (2005) 115:3157–65. doi:10.1172/JCI23475
67. Sayar BS, Ruegg S, Schmidt E, Sibilia M, Siffert M, Suter MM, et al. EGFR inhibitors erlotinib and lapatinib ameliorate epidermal blistering in pemphigus vulgaris in a non-linear, V-shaped relationship. Exp Dermatol (2014) 23:33–8. doi:10.1111/exd.12290
68. Osada K, Seishima M, Kitajima Y. Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol (1997) 108:482–7. doi:10.1111/1523-1747.ep12289726
69. Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y. Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol (1995) 104:33–7. doi:10.1111/1523-1747.ep12613469
70. Rotzer V, Hartlieb E, Winkler J, Walter E, Schlipp A, Sardy M, et al. Desmoglein 3-dependent signaling regulates keratinocyte migration and wound healing. J Invest Dermatol (2016) 136:301–10. doi:10.1038/JID.2015.380
71. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci (2017) 85:197–207. doi:10.1016/j.jdermsci.2016.12.010
Keywords: pemphigus, autoantibodies, cell adhesion, desmosomes, keratinocytes
Citation: Spindler V and Waschke J (2018) Pemphigus—A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front. Immunol. 9:136. doi: 10.3389/fimmu.2018.00136
Received: 29 November 2017; Accepted: 16 January 2018;
Published: 01 February 2018
Edited by:
Ralf J. Ludwig, University of Lübeck, GermanyReviewed by:
Robert Gniadecki, University of Alberta, CanadaHendri H. Pas, University Medical Center Groningen, Netherlands
Copyright: © 2018 Spindler and Waschke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Volker Spindler, dm9sa2VyLnNwaW5kbGVyQHVuaWJhcy5jaA==;
Jens Waschke, amVucy53YXNjaGtlQG1lZC51bmktbXVlbmNoZW4uZGU=