 Aurore Saudemont
Aurore Saudemont Laurent Jespers1
Laurent Jespers1 Timothy Clay
Timothy Clay- 1GlaxoSmithKline, Stevenage, United Kingdom
- 2GlaxoSmithKline, Collegeville, PA, United States
Ex vivo manipulations of autologous patient’s cells or gene-engineered cell therapeutics have allowed the development of cell and gene therapy approaches to treat otherwise incurable diseases. These modalities of personalized medicine have already shown great promises including product commercialization for some rare diseases. The transfer of a chimeric antigen receptor or T cell receptor genes into autologous T cells has led to very promising outcomes for some cancers, and particularly for hematological malignancies. In addition, gene-engineered cell therapeutics are also being explored to induce tolerance and regulate inflammation. Here, we review the latest gene-engineered cell therapeutic approaches being currently explored to induce an efficient immune response against cancer cells or viruses by engineering T cells, natural killer cells, gamma delta T cells, or cytokine-induced killer cells and to modulate inflammation using regulatory T cells.
Introduction
Cell and gene therapy is an emerging field with the high potential to offer a curative therapy. Gene therapy is defined as the use of genetic material such as DNA to manipulate a patient’s cells, and cell therapy is defined as the administration of live whole cells or of a specific cell population to a patient. In many diseases, cell and gene therapies are combined as gene engineering cell therapeutics in the development of promising therapies for the treatment of an acquired or inherited disease. The number of applications for gene engineering cell therapeutics is increasing at a very rapid pace, with these applications being at different development stages from preclinical to clinical.
Autologous gene engineering cell therapeutics have the potential to correct the underlying genetic cause of some monogenic disorders and potentiate immune responses against cancers to provide sustained clinical responses (1–5). In addition, one of the main advantages of autologous therapies is their full major histocompatibility complex (MHC) compatibility leading to a better engraftment and persistence of the cells and a low risk of graft versus host disease (GvHD). Gene transfer into autologous hematopoietic stem cells (HSC) has shown potential especially in treating primary immunodeficiencies such as X-linked severe combined immunodeficiency (X-SCID) or adenosine deaminase deficiency–SCID. The transfer of a chimeric antigen receptor (CAR) or T cell receptor (TCR) genes into autologous T cells allows redirecting the genetically engineered T cells towards specific antigens expressed on cancer cells or presented as peptides on MHC molecules, respectively. In particular, the transfer of autologous CD19-CAR T cells in patients with hematological malignancies has been very successful, achieving impressive remission rates (6). Notably, the Food and Drug Administration (FDA) recently approved the first CAR T cell therapy, Kymriah® (or tisagenlecleucel), for patients with B cell acute lymphoblastic leukemia (ALL). In addition, another CAR therapy was approved by the FDA, “Yescarta®” (axicabtagene ciloleucel), for the treatment of adult patients with certain types of non-Hodgkin lymphoma.
However, some of the trials testing gene engineering cell therapeutics have not been without setbacks such as the incidence of insertional mutagenesis observed in the first clinical trials for X-SCID, which has led to the design of new vectors allowing reducing their potential for insertional mutagenesis. This also highlighted the clear need for long-term follow-up for the patients receiving these live gene engineering cell therapeutics. In addition, several deaths linked to neurotoxicity in patients treated with CD19-CAR T cells have been reported and the cytokines produced after infusion of the product can lead to adverse effects such as cytokine release syndrome (CRS) that many patients experienced, highlighting the fact that we still need to gain a better understanding of the effects of gene engineering cell therapeutics in patients so as to make these therapies safer. Here, we review the latest gene engineered cell therapeutic approaches being currently explored preclinically but emphasizing those that have been clinically tested (Figure 1), to induce an efficient immune response against cancer cells or viruses by engineering T cells, natural killer (NK) cells, gamma delta T cells or cytokine-induced killer (CIK) cells and to modulate inflammation by using regulatory T cells (Tregs).
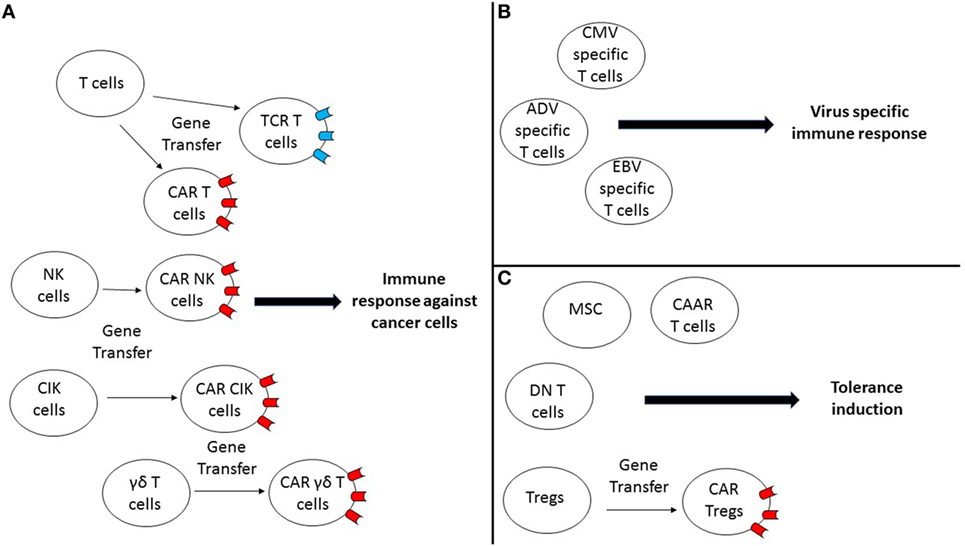
Figure 1. Gene-engineered cell therapeutic approaches are currently explored preclinically and clinically to induce potent immunity against cancer, infection, or to induce tolerance. (A) Different gene-engineered cell therapeutic approaches using either T cells, natural killer (NK) cells, cytokine-induced killer (CIK) cells, or γδ T cells are being explored to induce an efficient immune response against cancer cells. Notably, these different cell types can be reprogrammed by gene transfer of a T cell receptor (TCR) or a chimeric antigen receptor (CAR), so they can target efficiently specific antigens expressed by cancer cells. (B) Virus-specific T cells can be used as a cell therapy approach to restore virus-specific immunity in patients. (C) Different approaches are being explored to induce tolerance for different indications by either using mesenchymal stem cells (MSC), double negative (DN) T cells, CAR T cells, or regulatory T cells (Tregs)-based approaches or by explored manipulating Tregs (CAR-Tregs).
Cell and Gene Therapies for Oncology
Different cell types can be used to develop gene engineering cell therapeutics to induce an immune response against tumor cells. Here, we will review the use of TCR- or CAR-modified T cells (TCR-T or CAR-T), NK cells, CIK cells, and gamma delta T cells for oncology.
Autologous TCR-T and CAR-T
T cell engineering provides the possibility to generate antigen-specific T cells for many types of cancer. The identification of a graft versus leukemia (GvL) effect and subsequent recognition that T cells played a role provided an early indicator of the potential of T cells to mediate antitumor effects and led to the use of donor lymphocyte infusion (DLI). Subsequent studies of autologous tumor infiltrating lymphocytes (TIL) in melanoma patients for whom suitable TIL could be isolated and expanded successfully, allowing adoptive transfer, led to significant clinical responses (7, 8). The difficulty to isolate TIL from some patients’ tumors, especially with non-melanoma tumors, led to the concept of modifying autologous peripheral blood T cells to redirect them to recognize tumors offering an alternative approach to develop T cell therapies. Two principal strategies were developed, one using natural TCR that recognized human leukocyte antigen (HLA)-restricted tumor-associated antigen peptides (9–12) and the other seeking to use antibody recognition domains linked to TCR signaling molecules in CAR (13–15).
Both strategies have been continually refined over the last 15 years, with promising early data culminating in a number of medicines in late-stage clinical development and two approved medicines (Tables 1 and 2). The first clinical study of TCR-T was performed by Morgan et al. using a wild-type human TCR specific for a MART-1 epitope presented by HLA-A*0201, and the feasibility of this approach to modify autologous T cells ex vivo was demonstrated (12). There were some initial signs of clinical activity with 2/17 melanoma patients undergoing a partial response, and no related toxicities were observed. Subsequent studies included using TCR of mouse origin (16–20) and affinity-enhanced TCR (16, 21–23). A variety of different tumor antigen targets have also been evaluated in different tumor types (Table 1). The most advanced TCR-T clinical asset, NY-ESO-1-specific TCR-T cells, has shown particularly promising clinical responses in multiple myeloma (MM) and synovial sarcoma (19, 23, 24), with a 61% response rate and a 38% overall survival (OS) at 3 years (19, 24, 25) and a 91% response rate and a median OS of 3 years in MM in the context of autologous hematopoietic stem cell transplantation (HSCT) (23, 26). As shown in Table 1, improved clinical response rates have been observed in multiple TCR-T studies compared to the original study of Morgan et al. including complete responses, but there have also been some serious, sometimes fatal adverse events.
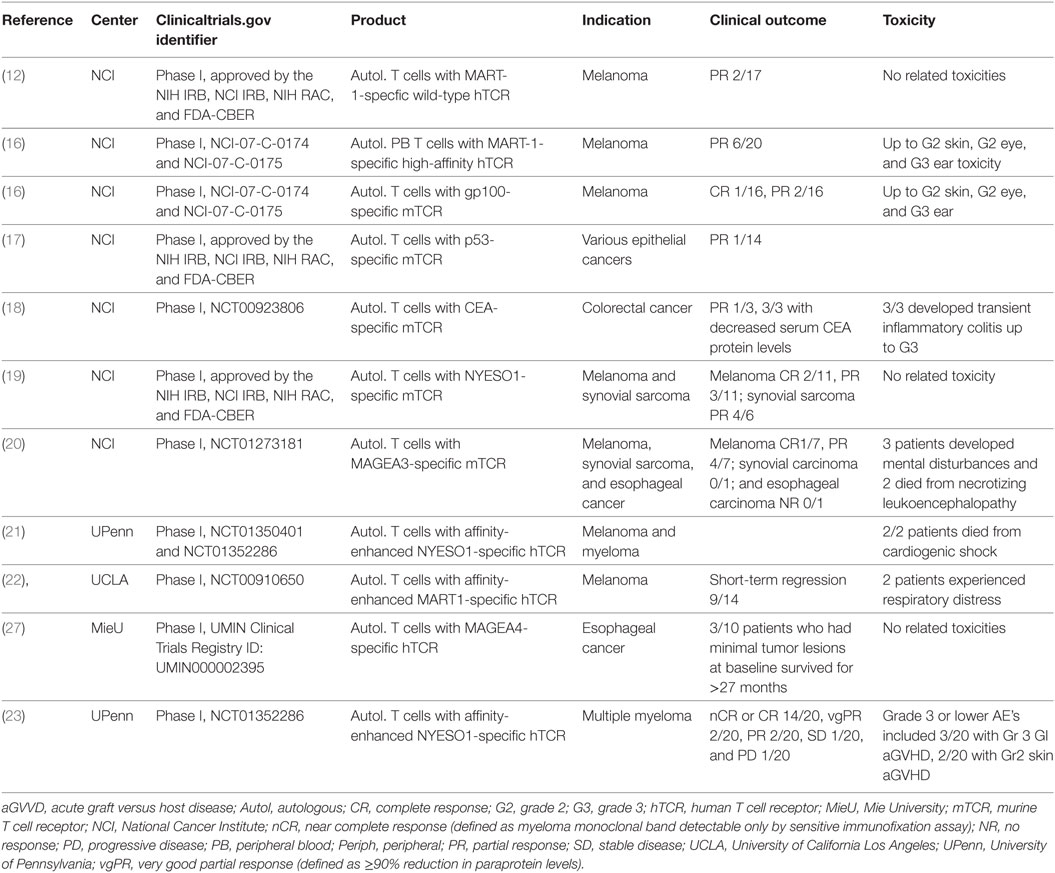
Table 1. Summary of key published clinical results of T cell receptor gene-modified T cell therapies.
Early CAR-T studies, using what is now termed a first-generation construct design with the antigen-binding domain scFV linked to CD3 zeta or FcR γ chain as a signaling domain, provided evidence of feasibility for CAR-T cell production but lacked clinical efficacy in HIV-infected patients or patients with solid tumors (Table 2) (28–30, 32, 33). Indeed, activation through the CD3zeta chain or FcR γ chain was insufficient to produce productive immunity (43, 44). Second-generation construct designs were developed by addition of a costimulatory domain (typically CD28 or 41BB) to CD3zeta to simultaneously provide both activation and co-stimulatory signals [several recent reviews have addressed CAR design in detail (45–49)]. The CAR-T field was truly energized when Carl June and colleagues at the University of Pennsylvania, using second generation CD19-specific CAR-T cells, achieved two complete responses in the treatment of three patients with refractory advanced chronic lymphocytic leukemia (CLL) using anti-CD19 CAR T cells (4, 35). Subsequently, CD19 CAR-T cell therapy has generated complete and durable remissions in patients with refractory and relapsed B cell malignancies (5, 6, 37, 50). As previously mentioned, the FDA has recently approved the CD19 CAR-T cell approach from Novartis, Kymriah®, to treat patients with B cell ALL and the CD19 CAR-T cell approach from Kite Pharma, YESCARTA™, for the treatment of diffuse large B-cell lymphoma.
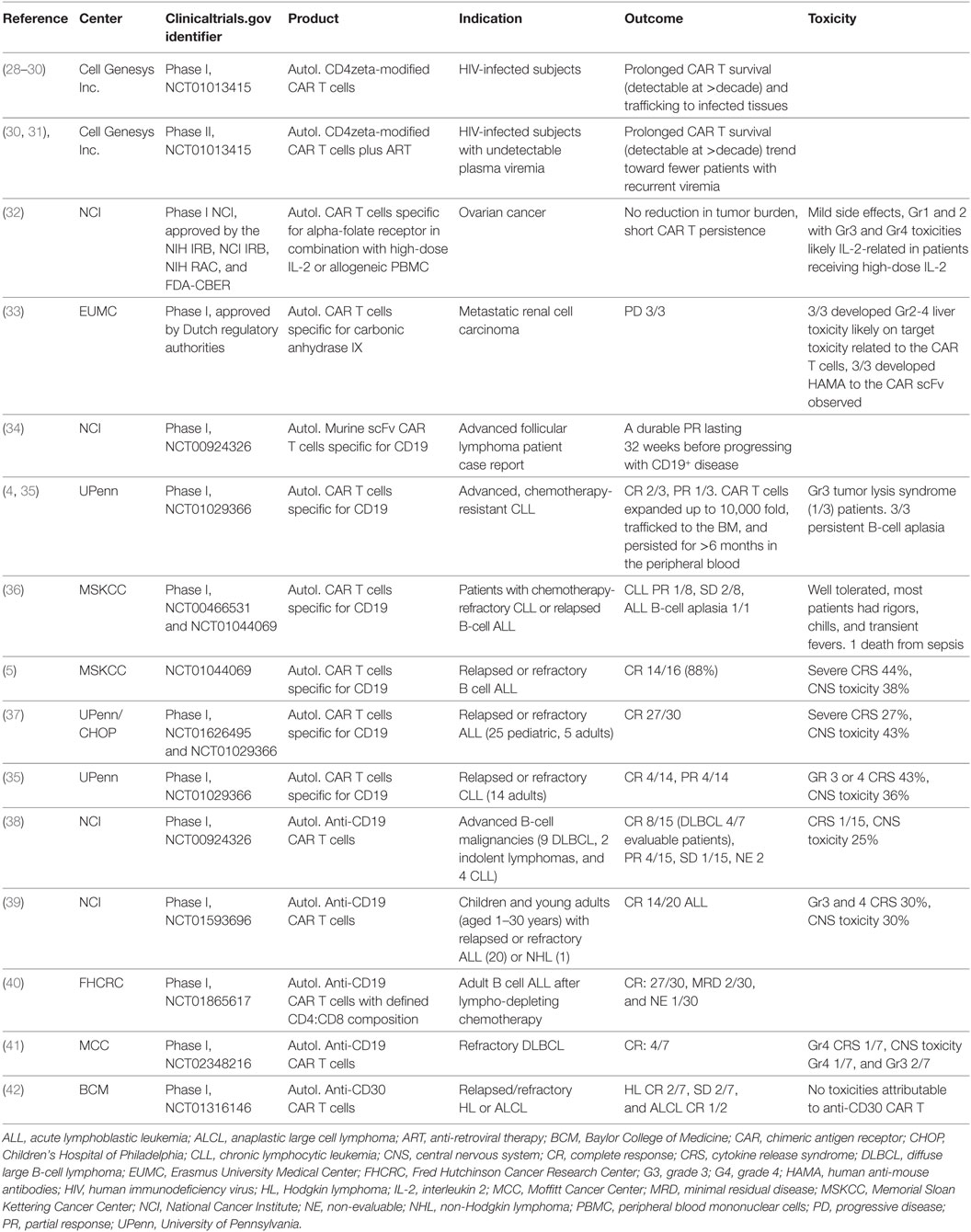
Table 2. Summary of some key published clinical results of CAR-gene-modified T cell therapies.
These autologous CAR-T and TCR-T cell therapies show great promise but there remain many aspects of these approaches that need to be further refined. The highly potent nature of these modified T cells produces dramatic tumor regressions; however, toxicities have also been observed. There have been a number of deaths on TCR-T and CAR-T trials. A detailed review of toxicity is beyond the scope of this review, but it is important to briefly underscore the risks. Some of these deaths have been due to apparent on target, off tumor toxicity, where expression of the target antigen on normal tissues occurs. For example, a patient died from rapid respiratory failure and multiorgan dysfunction in a CAR-T trial targeting ErbB2 in patients suffering from lung carcinoma, due to the recognition of ErbB2 on normal lung cells (51). Both Morgan et al. and Linette et al. reported instances where TCR-T targeting different MAGEA3 epitopes led to reactivity against proteins with amino acid sequence homolog to the target epitope in the brain (MAGEA12) and the heart (Titin), respectively (20, 21) (Table 1). In various CD19 CAR-T trials, patient deaths have been reported due to neurotoxicity caused by cerebral edemas [reviewed in Ref. (52)]. Here, the exact mechanism is less clear. Other severe side effects, such as CRS and tumor lysis syndrome, can also mean that patients often require aggressive support in an intensive care unit setting (53).
Another area for improvement is to induce better efficacy in more indications. Indeed, clinical response rates and the durability of response in some hematologic malignancies and all solid tumors are currently lower than those seen with CD19 CAR-T in ALL. The tumor microenvironment in solid tumors is a hostile environment for T cells, and low persistence of gene-engineered T cells with low efficacy has been observed. Since there is a dearth of truly tumor-specific targets because most tumor-associated antigens are also naturally expressed on some normal tissues, there is a need for new targets and ways to better control “on target, off-tumor” toxicity. Additional strategies will likely be needed to improve the effectiveness of CAR-T and TCR-T in the face of these obstacles [reviewed in Ref. (49, 54)]. In addition, the scale-up in manufacturing capability to supply thousands of patients has yet to be demonstrated and will be critical for the success of these autologous T cell approaches.
NK Cells
Natural killer cells are defined as CD56+CD3− cells and are innate immune lymphocytes able to exert cytotoxicity against tumor cells and virally infected cells without prior stimulation. Because of their antitumor activity, NK cells have been put forward as potential candidates for cancer immunotherapy. Cell therapy using autologous NK cells has proven to be safe and feasible when treating hematological malignancies such as CLL or different types of solid tumors but with however quite limited clinical responses (55–59).
The first evidence that allogeneic NK cells could be more cytolytic against tumor cells than autologous NK cells stems from the study by Ruggeri et al., who showed that allogeneic NK cells were very potent effectors of the GvL especially in the case of a killer-cell immunoglobulin-like receptors ligand incompatibility in the graft versus host direction in patients who received HLA-mismatched HSCT (60). In addition, the infusion of allogeneic NK cells together with interleukin (IL)-2 has shown some promising results in patients with different types of advanced cancers (61, 62) and several groups reported similar results using allogeneic NK cells mainly in the context of haploidentical HSCT (63–69) (Table 3). Although NK cells are short-lived, they may provide a potential off-the-shelf therapy with limited toxicity as they do not induce GvHD. A recent first-time-in-human study has also been reported exploring the use of umbilical cord blood (CB)-derived NK cells in MM (70).
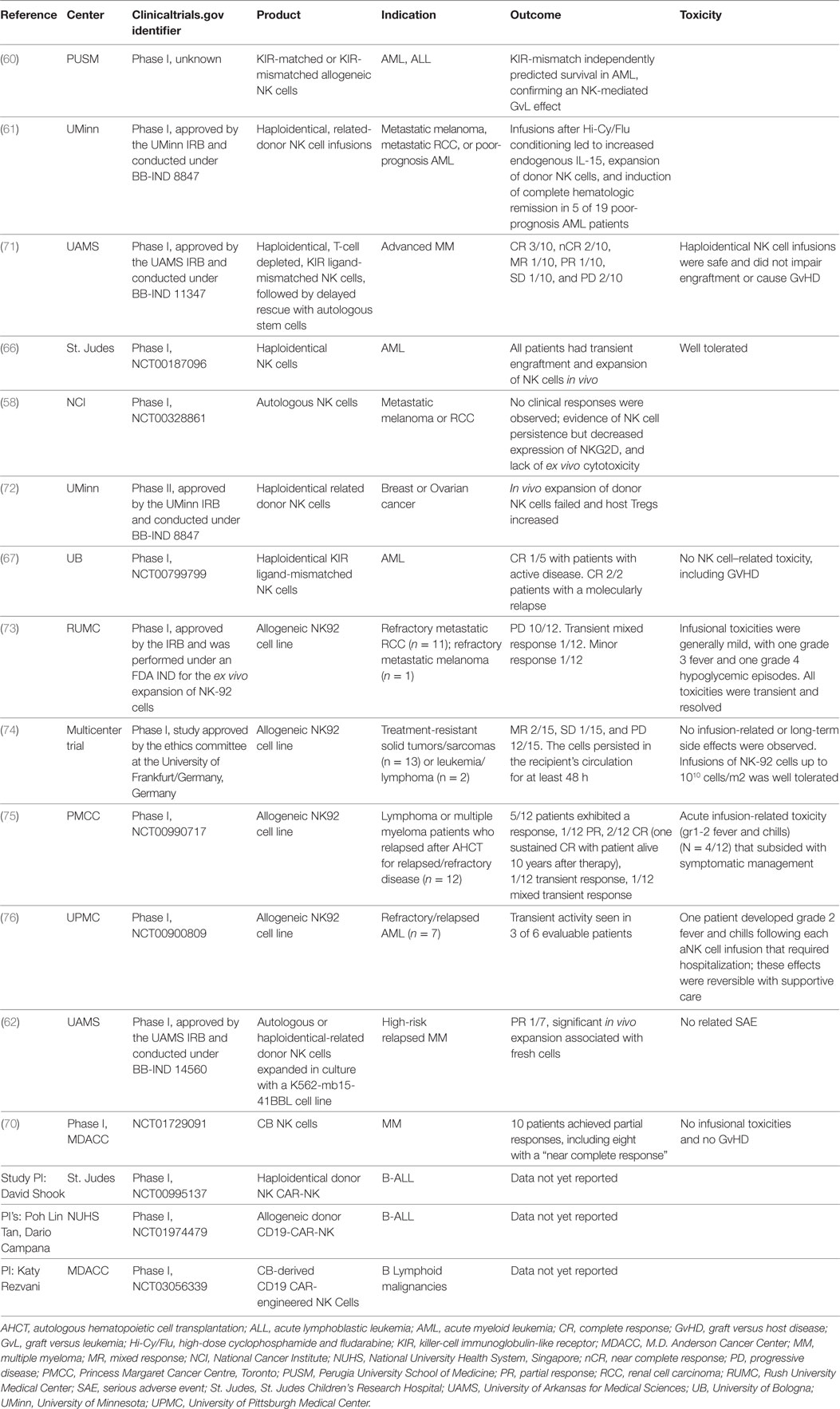
Table 3. Summary of some key published clinical results of NK cell therapies, including CAR-NK cells.
A number of gene engineering cell therapy using human NK cell lines have been established. In particular, NK92 has been investigated as a therapy, either unmodified or with a variety of different genetic modifications. The first human clinical trial report using NK92 cells by Arai et al. showed some initial signs of clinical activity (73). Subsequently, a multicenter trial enrolled 15 patients, showing as well signs of clinical activity (74). Recently, clinical trials results with NK92 cells were reported by Keating and colleagues at the Princess Margaret Cancer Centre, Toronto (NCT00990717) (75). In that study, 5 of 12 patients exhibited a clinical response, including 2 complete responses, 1 of which is sustained and ongoing 10 years posttherapy. Boyiadzis et al. also recently reported results of a clinical trial of NK92 cells in 7 refractory/relapsed AML patients with 3/6 showing transient clinical responses (76).
Since NK92 cells do not express CD16 and therefore cannot mediate ADCC, a NK92 variant that has been engineered to express CD16 and intracellularly retained IL-2 (haNK) to be combined with monoclonal antibody therapies and clinical development is underway (e.g., NCT03027128). No clinical results with haNK cells have yet been reported.
Natural killer cells could also be used for the generation of CAR NK cell products for immunotherapy to enhance their effector function by providing antigen specificity. This could be especially effective in the case of cancers resistant to NK cell killing. Moreover, CAR-NK cells could be used to bridge the patient’s immunity during HSCT or prior to administration of another therapy such as CAR-T cells. Preclinical studies using CAR-NK cells have shown promising results in vitro and in animal models against different types of solid tumors (77, 78) and hematological malignancies (79–81). Thus, first-time-in human testing is underway. Notably, Shook and colleagues at St. Judes Children’s Research Hospital, Memphis, USA, have completed enrollment in a trial of CD19 CAR-expressing NK cells for B-Lineage ALL (NCT00995137), and results are awaited. The CAR used was a second-generation design with a 4-1BB costimulatory domain linked to the CD3 zeta chain. Tan, Campana and colleagues at the National University Health System in Singapore are using the identical CAR construct in an actively recruiting trial of haploidentical CD19 CAR-expressing NK cells for B-Lineage ALL (NCT01974479). The number of early clinical trials of CAR-NK is increasing year-on-year. The Chinese company PersonGen BioTherapeutics has opened four studies to enrollment in 2016, using a third-generation CAR design with the relevant scFV attached to CD28 and 41BB costimulatory domains and the CD3 zeta chain in transduced allogeneic NK92 cells. The trials are focused on different targets and indications: CD7+ leukemia/lymphomas in adults (NCT02742727), CD33+ myeloid malignancies in children and adults (NCT02944162), refractory CD19+ malignancies in children and adults as bridge to HSCT transplant (NCT02892695), and MUC1+ solid tumors including malignant glioma of brain, colorectal carcinoma, gastric carcinoma, hepatocellular carcinoma (HCC), non-small-cell lung cancer (NSCLC), pancreatic carcinoma, and triple-negative basal-like breast carcinoma (NCT02839954). Early in 2017, Rezvani and colleagues at the MD Anderson Cancer Center, Houston, TX, USA and Bellicum Pharmaceuticals, Houston, TX, USA opened a trial of umbilical and CB-derived CAR-engineered NK cells for B lymphoid malignancies that is currently recruiting patients (NCT03056339).
Similarly, CAR-modified NK92 cells have been tested in preclinical studies and shown promising results (82–84). Consequently, several CAR-modified NK92 clinical studies are underway but currently no clinical results have been published. Overall, all the ongoing clinical trials will provide key data on safety and efficacy of gene engineering NK cell therapeutics. It will be interesting to evaluate whether NK cell therapies can be as effective as their T cell counterparts for certain indications and whether CAR-NK cells lead to similar adverse effects such as CRS and neurotoxicity.
CIK Cells
Cytokine-induced killer cells are a heterogeneous effector CD3+ CD8+ cell population that exhibit non-MHC-restricted cytotoxicity [reviewed in Ref. (85)]. There has been a long history of clinical studies testing CIK cells, with evidence of antitumor effects of CIK cells against hematologic malignancies and solid tumors, along with studies exploring their antivirus potential and anti-GVHD potential. The first human clinical trial of autologous peripheral blood-derived CIK transfected to express human IL-2 by electroporation demonstrated the safety in patients with metastatic renal cell carcinoma, colorectal carcinoma, and lymphoma (86) (Table 4). In one patient with follicular lymphoma grade I, a pre-existing bone marrow involvement as the only sign of disease resolved after CIK cell therapy and was scored as a complete clinical response.
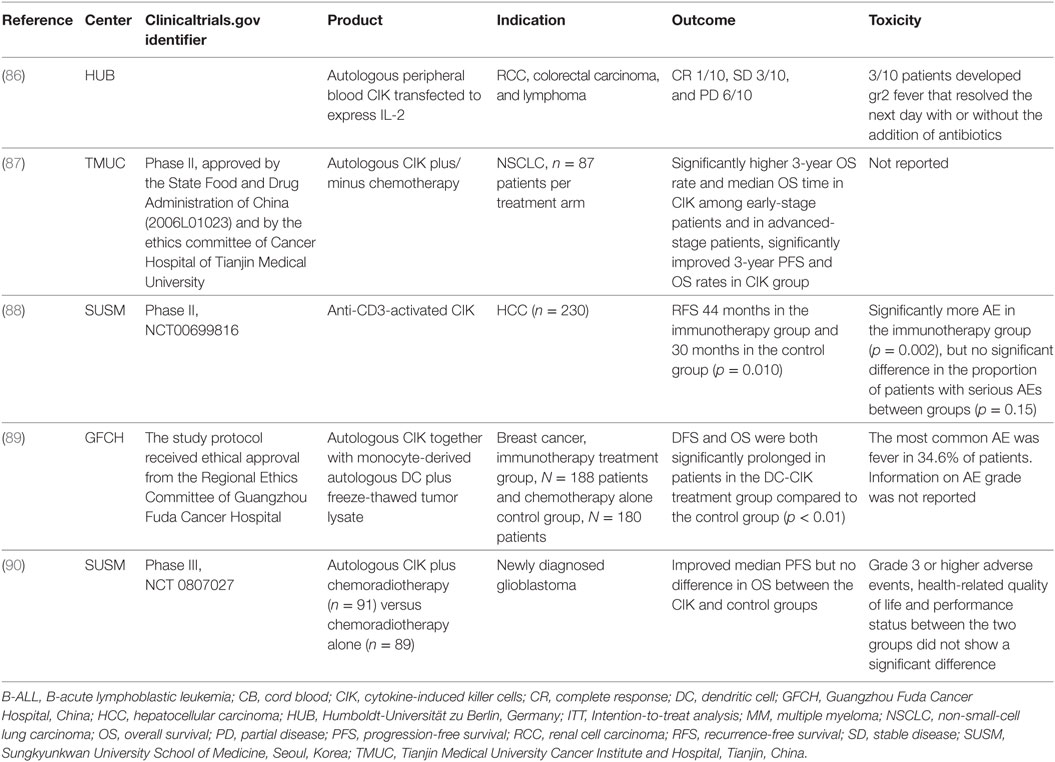
Table 4. Summary of some key published clinical results of CIK cell therapies.
Various CIK clinical trials have combined CIK cells with other therapies, including mAb therapies, chemotherapies, radiotherapy, and dendritic cell (DC) therapy. An in-depth review of all CIK clinical studies is beyond the scope of this article because more than 100 clinical trials of CIK have been reported, and readers are referred to Gao et al (85), for a review (91). Some examples of key clinical studies are listed in Table 4. In 2012, a randomized phase II clinical study showed that CIK therapy could enhance the efficacy of conventional chemotherapy in patients with NSCLC (87). The study was designed to evaluate the clinical efficacy of CIK cell immunotherapy following regular chemotherapy in patients with NSCLC after surgery. Among early-stage patients, the 3-year OS rate and median OS time in the immunotherapy arm were significantly higher than those in the no immunotherapy arm. Among the advanced-stage patients, the 3-year OS rates of immunotherapy arm were significantly higher than those of the no immunotherapy arm. Lee et al. demonstrated that adjuvant immunotherapy with anti-CD3-activated CIK cells could increase the recurrence-free survival (RFS) and OS of patients with HCC (88). Patients who received CIK immunotherapy after curative treatment for HCC had a 14-month median RFS benefit compared to the no immunotherapy control group. Another recent report described results of a study comparing chemotherapy followed by CIK immunotherapy including DCs versus chemotherapy alone in stage IV breast adenocarcinoma (89). Over a 10-year period, a total of 368 patients meeting the inclusion criteria were assigned to the study groups. CIK immunotherapy consisted of monocyte-derived autologous DC plus freeze-thawed tumor lysate, with CIK prepared from non-adherent cells from the DC culture process. OS rates were significantly prolonged in patients in the DC-CIK treatment group compared with the patients in the control chemotherapy alone group. Finally, a recent phase III trial has evaluated CIK immunotherapy with radiotherapy-temozolomide (TMZ) versus TMZ alone for the treatment of newly diagnosed glioblastomas (90). A total of 180 patients were randomly assigned to the CIK immunotherapy (n = 91) or control group (n = 89). The addition of CIK cell immunotherapy to standard chemoradiotherapy with TMZ resulted in a statistically significant improvement in PFS. However, the CIK immunotherapy group did not show evidence of a beneficial effect on OS rates. Other notable studies in clinical development but yet to report clinical results are seeking to combine CIK with mAb anti-PD1/PDL1 strategies (NCT02886897, NCT03190811, NCT03360630, NCT03282435, and NCT03146637).
Studies have begun to explore the use of CIK cells as a CAR cell carrier. Multiple preclinical studies have demonstrated promising antitumor activity against a variety of tumors and targets [for example, Ref. (92–96)]. Formula Pharmaceuticals recently announced the opening of the first clinical trial of CAR-modified allogeneic CIK cells using its non-viral gene delivery approach (http://www.formulapharma.com/news/2017_09_18_formula_press_release.asp).
The next few years will see clinical results from CAR-modified NK cell and CAR-modified CIK cell trials and will determine if these approaches are efficacious and can complement or perhaps replace CAR-T/TCR-T approaches.
Gamma Delta T Cells
Gamma-delta (γδ) T cells represent less than 5% of circulating T cells and unlike conventional αβ T cells, the TCR repertoire of γδ T cells is very restricted. They can be cytotoxic and secrete cytokines like conventional αβ T cells, but γδ T cells do not require MHC antigen presentation for antigen recognition. γδ T cells are also more widely distributed in tissues throughout the body, in addition to also being present in the typical T cell compartments, the lymph nodes, and spleen, where most αβ T-cells reside. Thus, γδ T cells could potentially offer differentiated activity from αβ T cells, and some investigators are beginning to explore their utility as CAR carriers. Indeed, one specific subset of γδ T cells identified by their expression of a Vg9Vd2 TCR can be expanded using bisphosphonates and are known to recognize in an MHC-independent manner phosphoantigens, and these tend to be enriched in tumors (97). The MHC-independent nature of the Vg9Vd2 antigen recognition also means that they are not alloreactive and therefore should not cause GvHD. Deniger et al. recently reported on a methodology to expand polyclonal γδ T cells in vitro and electroporation with Sleeping Beauty transposon and transposase to enforce expression of a CD19 CAR (98). The demonstrated killing of CD19+ tumor cell lines and adoptive transfer of CAR γδ T cells reduced growth of CD19+ leukemia xenografts in mice. Clinical translation of CAR γδ T cell approaches will be needed to determine if these cell carriers provide advantages over CAR αβ T cells. In that regard, Chen and colleagues at the Fuda Cancer Hospital, Guangzhou, China have recently opened four studies for enrollment to assess safety and efficiency of autologous peripheral blood mononuclear cell-derived γδ T cells against lung cancer (NCT03183232) and liver cancer (NCT03183219), breast cancer (NCT03183206), and pancreatic cancer (NCT03180437) respectively (Table 5).

Table 5. Summary of some key clinical studies evaluating gamma-delta T cell therapies.
Cell and Gene Therapies Using Genome Engineering
A further step in simplifying supply and broadening access to donor cells is to engineer partially matched allogeneic cells. This approach is attractive for situations where dramatic clinical efficacy can be mediated rapidly and where perhaps sustained persistence of the cell product is not a requirement. One such example was recently reported whereby unmatched allogeneic donor T cells were lentivirally transduced to express an anti-CD19 CAR, followed by gene editing with TALENs to remove expression of the endogenous TCR alpha chain to prevent GvHD together with removal of CD52 to render them insensitive to the lymphodepleting agent Alemtuzumab (99). These TCR-ablated T cells were then administered to two relapsed refractory B-acute lymphoblastic leukemia patients as temporary “bridge to transplant” designed to eradicate their leukemia before a subsequent planned allogeneic HSCT. Recently, it has also been shown that gene editing could be used to generate high numbers of redirected NY-ESO-1 T cells with a predominant stem and memory T cell phenotype that were tested preclinically in vitro and in a mouse model of MM without inducing GvHD (100).
The “holy grail” would be an entirely “universal donor,” long-lived cell product providing an “off-the-shelf” cell therapy acceptable to any patient irrespective of their HLA and TCR specificity. Technically, genome editing technology offers the possibility to engineer such universal donor cells in a controlled manner. Cells could be modified ex vivo using gene editing to knockout expression of HLA class I and potentially HLA class II molecules to prevent GvHD that could otherwise lead to graft rejection. It has been shown that elimination of HLA class I molecules using zinc finger nucleases was possible in primary T cells as well as in embryonic stem cells (101, 102). Furthermore, the removal of HLA-A molecules in HSC had no impact on their function as the cells could engraft in immuno-compromised mice (103). Depending upon the donor cell type, the removal of other molecules such as the TCR that could induce GvHD would be required. Additionally, other cell engineering may be necessary such as the introduction of a β-2 microglobulin (β-2M) gene fusion to a non-polymorphic HLA-E, F, or G gene in order to avoid NK cell-mediated lysis of the cell product that would otherwise occur if the engineered cells are completely devoid of class I molecules (102, 104).
Zhao, June and colleagues have reported on preclinical studies combining lentiviral delivery of a CAR with electroporation of Cas9 mRNA and gRNAs targeting endogenous TCR, β-2M, and PD1 simultaneously, to generate gene-disrupted allogeneic CAR T cells deficient of TCR, HLA class I molecule, and PD1 by multiplex genome editing (105, 106). A clinical trial is planned to be undertaken at the University of Pennsylvania, University of California San Francisco, and the MD Anderson Cancer Center, USA. Lu and colleagues at Sichuan University’s West China Hospital in Chengdu in China have reported the first treatment with CRISPR-modified autologous peripheral blood T cells in lung cancer patients that had been disrupted for PD1 using CRISPR/Cas9 (NCT02793856). These and other early studies will evaluate the safety of genome-edited T cells and pave the way for the more extensive multiplex genome editing required to create universal cell products.
Once clinically validated, the genome editing solutions could equally be applied to different cell types such as T cells, DCs, or HSC. However, it remains to be demonstrated that such “universal” cells could readily engraft and provide long-term protection without inducing toxicity. In addition, the less than 100% efficiency achieved for each edit will require purification of “fully edited” from “partially edited” cells, which is a significant obstacle that is yet to be resolved. There may also be unidentified molecules that may need to be edited for a cell to be truly “invisible” to the host immune system and persist for the life of the individual.
Cell Therapy to Control Infection
Infections are still one of the main complications post-HSCT contributing to mortality and morbidity of the procedure. Therefore, a lot of efforts have been dedicated in developing cell therapy approaches to control infection such as DLI as an alternative to more conventional therapies. Notably, it has been shown by several groups that the infusion of matched donor-derived virus-specific T cells specific for cytomegalovirus, Epstein–Barr virus, or adenovirus can restore virus-specific immunity and control infection or could be used prophylactically in transplanted patients (107–114) (Table 6). In addition, the use of multivirus-specific T cells has also been reported to be effective in either preventing or controlling viral infection (115–118) (Table 6). However, this approach is not applicable to seronegative donors, patients undergoing immunosuppressive regimens, or for urgent use because of the time required to generate the cell product. In these specific conditions, the use of partially matched allogeneic virus-specific T cells has shown great promise and efficacy in controlling infection (116, 119, 120) (Table 6), highlighting the possibility to develop banks of allogenic virus-specific T cells (113, 121) whereby the virus-specific T cells of one donor could be expanded and banked for immediate use, which could allow treating several patients with the cells from the same donor thus decreasing the cost of the therapy.
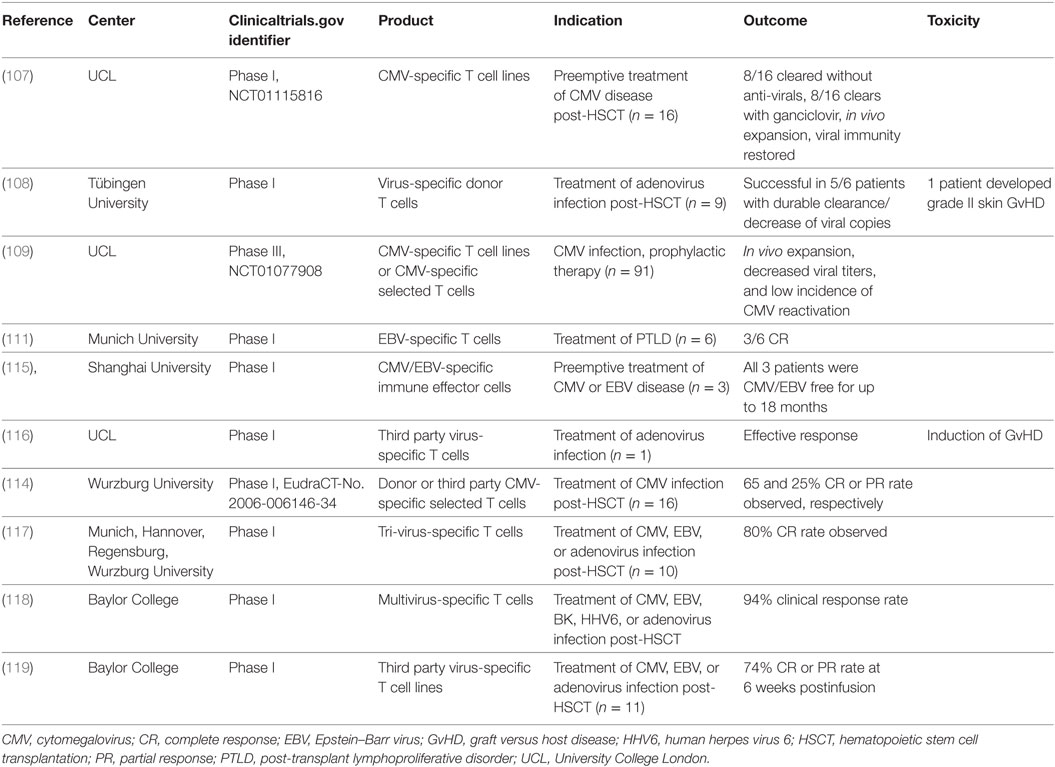
Table 6. Summary of some key published clinical studies evaluating virus-specific T cell therapies.
Cell and Gene Therapies to Induce Tolerance
Several cell types are capable to regulate immune responses and thus induce tolerance. NK cells have shown to have regulatory functions aside their cytotoxic activity in the context of HSCT, impacting on acute GvHD (60). In addition, different studies suggest that a higher number of NK cells and NK cell alloreactivity might reduce GvHD development (122–124). Moreover, double negative (DN) T cells have regulatory functions and play key roles in tolerance induction post-transplantation and in autoimmune diseases (125, 126). DN T cells have been reported to suppress immune responses, in particular T cell responses and can improve allograft survival in experimental mouse models of cardiac transplantation (127). Mesenchymal stem cells (MSC) are multipotent stem cells that also have anti-inflammatory and regenerative properties. Their regulatory functions have been evaluated using in vitro and in vivo disease models (128). In addition, the infusion of autologous and allogeneic MSCs has been tested in numerous phase I studies as immunosuppressive regimen in the context of organ transplant as well as treatment for GvHD and different autoimmune diseases (129, 130).
Regulatory T cells are key players in inducing tolerance and maintaining immune homeostasis. They constitute 5–10% of CD4 T cells in peripheral blood and are defined as CD4+ CD25+ CD127lowFoxP3+ (131). They have been shown to be able to regulate the functions of numerous immune cells such as CD4/CD8 T cells, DCs, NK cells, and B cells (132). Consequently, they have been evaluated when considering a cell therapy approach to induce tolerance especially for autoimmune diseases. Treg therapies have been shown to be safe and feasible with some promising results in clinical trials using autologous Tregs for GvHD (133–136) and autoimmunity such as diabetes (137, 138) and Crohn’s disease (139) as well as third-party allogeneic Tregs (136) (Table 7). All clinical studies have been done using polyclonal expanded Tregs; however, it has been shown that antigen-specific Tregs are more potent at suppressing undesired immune responses as compared to polyclonal Tregs. Some groups have studied the possibility to generate antigen-specific T cells by transferring a CAR into polyclonal expanded Tregs and showed their potency in vitro as well as in animal models (140–143). However, this approach is still to be tested clinically.
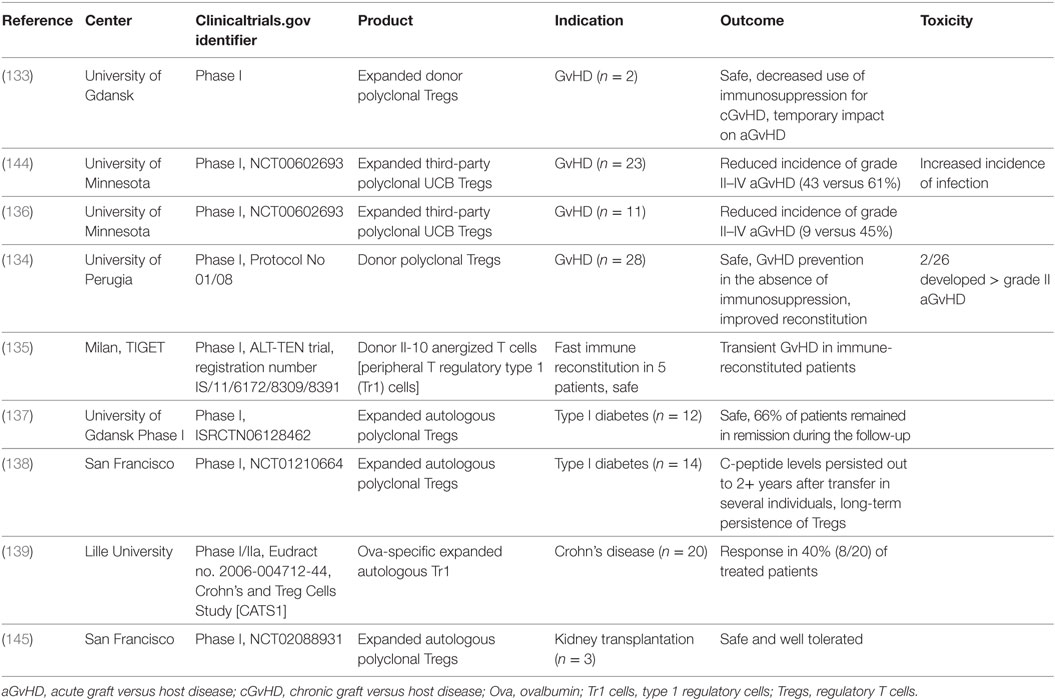
Table 7. Clinical studies evaluating Tregs therapies.
Another approach to induce tolerance could be the transfer of a chimeric autoantibody receptor (CAAR) into T cells to induce cytotoxicity against autoreactive T or B cells. Such an approach has demonstrated exciting results in vitro and in vivo in the case of CAAR T cells redirected against desmogelin-3 (DSG3) as a therapeutic approach for pemphigus vulgaris where B cells secrete autoantibodies against DSG3, which is an antigen expressed in the skin and mucosa (146). This study showed effective killing of autoreactive cells by DSG3-CAAR T cells in vitro and in vivo while sparing keratinocytes with no off-target effects. This approach showed promising results to potentially treat autoantibody mediated immune diseases.
Numerous cell types with regulatory functions have now been tested in preclinical studies showing promising results. Further evaluation in clinical studies is warranted but these data will be key to evaluate whether gene engineering cell therapeutics could be as successful in inducing tolerance in chronic diseases as these types of approaches have been for rare diseases and oncology.
Concluding Remarks
After many years of endeavor, the field of cell and gene therapy is starting to deliver remarkable clinical benefits in some patients, particularly in certain hematological malignancies. While this is a very exciting moment for the field, more work remains to be done to further develop and optimize therapies. More relevant disease models that can help predict not only efficacy but also adverse effects are much needed. In addition, making gene engineering cell therapeutics safer is key by adding, for example, safety or kill switch to the therapy or expression control elements. As this review shows, many cell types are being explored in early clinical trials and the results of these studies will provide important insights. A major factor in realizing the potential of cell and gene therapies in large numbers of patients and in more common diseases is the efficient and cost-effective manufacturing of gene engineering cell therapeutics. Advances in automation are a key component that needs to be addressed (147). The pace of clinical development is accelerating and this means that the next few years will provide many important learnings that will hopefully translate into new therapeutic options for patients.
Author Contributions
AS, LJ, and TC all contributed to write the manuscript.
Conflict of Interest Statement
All authors are employee of GlaxoSmithKline.
Funding
All the authors are supported by funding from GlaxoSmithKline.
References
1. Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med (2010) 363:355–64. doi:10.1056/NEJMoa1000164
2. Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med (2010) 363:147–55. doi:10.1056/NEJMoa0905955
3. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med (2011) 3:97ra80. doi:10.1126/scitranslmed.3002716
4. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3:95ra73. doi:10.1126/scitranslmed.3002842
5. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6:224ra225. doi:10.1126/scitranslmed.3008226
6. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368:1509–18. doi:10.1056/NEJMoa1215134
7. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst (1994) 86:1159–66. doi:10.1093/jnci/86.15.1159
8. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol (2016) 34:2389–97. doi:10.1200/JCO.2016.66.7220
9. Dembic Z, Haas W, Weiss S, Mccubrey J, Kiefer H, Von Boehmer H, et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature (1986) 320:232–8. doi:10.1038/320232a0
10. Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol (1999) 163:507–13.
12. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:126–9. doi:10.1126/science.1129003
13. Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, et al. Immuno-gene therapy of established prostate tumors using chimeric receptor-redirected human lymphocytes. Cancer Res (2003) 63:2470–6.
14. June CH, Levine BL. T cell engineering as therapy for cancer and HIV: our synthetic future. Philos Trans R Soc Lond B Biol Sci (2015) 370:20140374. doi:10.1098/rstb.2014.0374
15. Riviere I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther (2017) 25:1117–24. doi:10.1016/j.ymthe.2017.03.034
16. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi:10.1182/blood-2009-03-211714
17. Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res (2010) 16:5852–61. doi:10.1158/1078-0432.CCR-10-1280
18. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19:620–6. doi:10.1038/mt.2010.272
19. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi:10.1200/JCO.2010.32.2537
20. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi:10.1097/CJI.0b013e3182829903
21. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi:10.1182/blood-2013-03-490565
22. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res (2014) 20:2457–65. doi:10.1158/1078-0432.CCR-13-3017
23. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med (2015) 21:914–21. doi:10.1038/nm.3910
24. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res (2015) 21:1019–27. doi:10.1158/1078-0432.CCR-14-2708
25. Mackall C, D’angelo S, Grupp S, Glod J, Druta M, Chow W, et al. Open label non-randomized multi-cohort pilot study of genetically engineered NY-ESO-1 specific NY-ESO-1c259 SPEAR T-cellsTM in HLA-A*02+ patients with synovial sarcoma (NCT01343043). Ann Oncol (2016) 27:1075–1075. doi:10.1093/annonc/mdw378.29
26. Rapoport AP, Stadtmauer EA, Vogl DT, Weiss BM, Binder-Scholl GK, Brewer JE, et al. Adoptive transfer of gene-modified T-cells engineered to express high-affinity TCRs for cancer-testis antigens (CTAs) NY-ESO-1 or Lage-1, in MM patients post auto-SCT. Blood (2012) 120:472–472.
27. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res (2015) 21:2268–77. doi:10.1158/1078-0432.CCR-14-1559
28. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood (2000) 96:785–93.
29. Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood (2000) 96:467–74.
30. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med (2012) 4:132ra153. doi:10.1126/scitranslmed.3003761
31. Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther (2002) 5:788–97. doi:10.1006/mthe.2002.0611
32. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12:6106–15. doi:10.1158/1078-0432.CCR-06-1183
33. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24:e20–2. doi:10.1200/JCO.2006.05.9964
34. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116:4099–102. doi:10.1182/blood-2010-04-281931
35. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
36. Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118:4817–28. doi:10.1182/blood-2011-04-348540
37. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi:10.1056/NEJMoa1407222
38. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi:10.1200/JCO.2014.56.2025
39. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi:10.1016/S0140-6736(14)61403-3
40. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126:2123–38. doi:10.1172/JCI85309
41. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther (2017) 25:285–95. doi:10.1016/j.ymthe.2016.10.020
42. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest (2017) 127(9):3462–71. doi:10.1172/JCI94306
43. Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med (1995) 181:1653–9. doi:10.1084/jem.181.5.1653
44. Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood (2000) 96:1999–2001.
45. Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol (2013) 10:267–76. doi:10.1038/nrclinonc.2013.46
46. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3:388–98. doi:10.1158/2159-8290.CD-12-0548
47. June CH, Maus MV, Plesa G, Johnson LA, Zhao Y, Levine BL, et al. Engineered T cells for cancer therapy. Cancer Immunol Immunother (2014) 63:969–75. doi:10.1007/s00262-014-1568-1
48. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov (2015) 14:499–509. doi:10.1038/nrd4597
49. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res (2017) 27:38–58. doi:10.1038/cr.2016.154
50. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:177ra138. doi:10.1126/scitranslmed.3005930
51. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi:10.1038/mt.2010.24
52. Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med (2017) 9(9):1183–97. doi:10.15252/emmm.201607485
53. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127:3321–30. doi:10.1182/blood-2016-04-703751
54. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell (2017) 168:724–40. doi:10.1016/j.cell.2017.01.016
55. Burns LJ, Weisdorf DJ, Defor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi:10.1038/sj.bmt.1704086
56. Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, et al. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase i trial. Clin Cancer Res (2004) 10:3699–707. doi:10.1158/1078-0432.CCR-03-0683
57. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383–5. doi:10.7150/jca.2.383
58. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17:6287–97. doi:10.1158/1078-0432.CCR-11-1347
59. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med (2015) 13:277. doi:10.1186/s12967-015-0632-8
60. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi:10.1126/science.1068440
61. Miller JS, Soignier Y, Panoskaltsis-Mortari A, Mcnearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
62. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (2015) 38:24–36. doi:10.1097/CJI.0000000000000059
63. Koehl U, Sorensen J, Esser R, Zimmermann S, Gruttner HP, Tonn T, et al. IL-2 activated NK cell immunotherapy of three children after haploidentical stem cell transplantation. Blood Cells Mol Dis (2004) 33:261–6. doi:10.1016/j.bcmd.2004.08.013
64. Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kuhne T, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia (2004) 18:1835–8. doi:10.1038/sj.leu.2403524
65. Bachanova V, Burns LJ, Mckenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother (2010) 59:1739–44. doi:10.1007/s00262-010-0896-z
66. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28:955–9. doi:10.1200/JCO.2009.24.4590
67. Curti A, Ruggeri L, D’addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118:3273–9. doi:10.1182/blood-2011-01-329508
68. Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant (2013) 48:433–8. doi:10.1038/bmt.2012.162
69. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, Mckenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123:3855–63. doi:10.1182/blood-2013-10-532531
70. Shah N, Li L, Mccarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol (2017) 177:457–66. doi:10.1111/bjh.14570
71. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143:641–53. doi:10.1111/j.1365-2141.2008.07340.x
72. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13:98–107. doi:10.3109/14653249.2010.515582
73. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10:625–32. doi:10.1080/14653240802301872
74. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15:1563–70. doi:10.1016/j.jcyt.2013.06.017
75. Williams BA, Law AD, Routy B, Denhollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget (2017) 8:89256–68. doi:10.18632/oncotarget.19204
76. Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy (2017) 19:1225–32. doi:10.1016/j.jcyt.2017.07.008
77. Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U S A (2008) 105:17481–6. doi:10.1073/pnas.0804788105
78. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res (2009) 15:4857–66. doi:10.1158/1078-0432.CCR-08-2810
79. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood (2005) 106:376–83. doi:10.1182/blood-2004-12-4797
80. Muller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother (2008) 57:411–23. doi:10.1007/s00262-007-0383-3
81. Chu Y, Hochberg J, Yahr A, Ayello J, Van De Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell non-Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res (2015) 3:333–44. doi:10.1158/2326-6066.CIR-14-0114
82. Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology (2013) 2:e26527. doi:10.4161/onci.26527
83. Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology (2014) 3:e28147. doi:10.4161/onci.28147
84. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep (2015) 5:11483. doi:10.1038/srep11483
85. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front Immunol (2017) 8:774. doi:10.3389/fimmu.2017.00774
86. Schmidt-Wolf IG, Finke S, Trojaneck B, Denkena A, Lefterova P, Schwella N, et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br J Cancer (1999) 81:1009–16. doi:10.1038/sj.bjc.6690800
87. Li R, Wang C, Liu L, Du C, Cao S, Yu J, et al. Autologous cytokine-induced killer cell immunotherapy in lung cancer: a phase II clinical study. Cancer Immunol Immunother (2012) 61:2125–33. doi:10.1007/s00262-012-1260-2
88. Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology (2015) 148:1383–91.e6. doi:10.1053/j.gastro.2015.02.055
89. Lin M, Liang S, Jiang F, Xu J, Zhu W, Qian W, et al. 2003-2013, a valuable study: autologous tumor lysate-pulsed dendritic cell immunotherapy with cytokine-induced killer cells improves survival in stage IV breast cancer. Immunol Lett (2017) 183:37–43. doi:10.1016/j.imlet.2017.01.014
90. Kong DS, Nam DH, Kang SH, Lee JW, Chang JH, Kim JH, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget (2017) 8:7003–13. doi:10.18632/oncotarget.12273
91. Guo Y, Han W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin J Cancer (2015) 34:6. doi:10.1186/s40880-015-0002-1
92. Schlimper C, Hombach AA, Abken H, Schmidt-Wolf IG. Improved activation toward primary colorectal cancer cells by antigen-specific targeting autologous cytokine-induced killer cells. Clin Dev Immunol (2012) 2012:238924. doi:10.1155/2012/238924
93. Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol Ther (2013) 21:2268–77. doi:10.1038/mt.2013.192
94. Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol (2013) 161:389–401. doi:10.1111/bjh.12282
95. Ren X, Ma W, Lu H, Yuan L, An L, Wang X, et al. Modification of cytokine-induced killer cells with chimeric antigen receptors (CARs) enhances antitumor immunity to epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol Immunother (2015) 64:1517–29. doi:10.1007/s00262-015-1757-6
96. Merker M, Pfirrmann V, Oelsner S, Fulda S, Klingebiel T, Wels WS, et al. Generation and characterization of ErbB2-CAR-engineered cytokine-induced killer cells for the treatment of high-risk soft tissue sarcoma in children. Oncotarget (2017) 8:66137–53. doi:10.18632/oncotarget.19821
97. Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med (2003) 197:163–8. doi:10.1084/jem.20021500
98. Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther (2013) 21:638–47. doi:10.1038/mt.2012.267
99. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9:eaaj2013. doi:10.1126/scitranslmed.aaj2013
100. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited central memory and memory stem T cells to treat multiple myeloma without inducing GvHD. Blood (2017) 130(5):606–18. doi:10.1182/blood-2016-08-732636
101. Riolobos L, Hirata RK, Turtle CJ, Wang PR, Gornalusse GG, Zavajlevski M, et al. HLA engineering of human pluripotent stem cells. Mol Ther (2013) 21:1232–41. doi:10.1038/mt.2013.59
102. Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood (2013) 122:1341–9. doi:10.1182/blood-2013-03-478255
103. Torikai H, Mi T, Gragert L, Maiers M, Najjar A, Ang S, et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci Rep (2016) 6:21757. doi:10.1038/srep21757
104. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol (2017) 35:765–72. doi:10.1038/nbt.3860
105. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res (2017) 23:2255–66. doi:10.1158/1078-0432.CCR-16-1300
106. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget (2017) 8:17002–11. doi:10.18632/oncotarget.15218
107. Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet (2003) 362:1375–7. doi:10.1016/S0140-6736(03)14634-X
108. Feuchtinger T, Matthes-Martin S, Richard C, Lion T, Fuhrer M, Hamprecht K, et al. Safe adoptive transfer of virus-specific T-cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation. Br J Haematol (2006) 134:64–76. doi:10.1111/j.1365-2141.2006.06108.x
109. Mackinnon S, Thomson K, Verfuerth S, Peggs K, Lowdell M. Adoptive cellular therapy for cytomegalovirus infection following allogeneic stem cell transplantation using virus-specific T cells. Blood Cells Mol Dis (2008) 40:63–7. doi:10.1016/j.bcmd.2007.07.003
110. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood (2010) 115:925–35. doi:10.1182/blood-2009-08-239186
111. Moosmann A, Bigalke I, Tischer J, Schirrmann L, Kasten J, Tippmer S, et al. Effective and long-term control of EBV PTLD after transfer of peptide-selected T cells. Blood (2010) 115:2960–70. doi:10.1182/blood-2009-08-236356
112. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood (2012) 119:2644–56. doi:10.1182/blood-2011-08-371971
113. Bollard CM, Heslop HE. T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood (2016) 127:3331–40. doi:10.1182/blood-2016-01-628982
114. Neuenhahn M, Albrecht J, Odendahl M, Schlott F, Dossinger G, Schiemann M, et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia (2017) 31(10):2161–71. doi:10.1038/leu.2017.16
115. Dong L, Gao ZY, Chang LJ, Liang Y, Tan XY, Liu JH, et al. Adoptive transfer of cytomegalovirus/Epstein-Barr virus-specific immune effector cells for therapeutic and preventive/preemptive treatment of pediatric allogeneic cell transplant recipients. J Pediatr Hematol Oncol (2010) 32:e31–7. doi:10.1097/MPH.0b013e3181bf5e2d
116. Qasim W, Derniame S, Gilmour K, Chiesa R, Weber M, Adams S, et al. Third-party virus-specific T cells eradicate adenoviraemia but trigger bystander graft-versus-host disease. Br J Haematol (2011) 154:150–3. doi:10.1111/j.1365-2141.2011.08579.x
117. Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, Liu H, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther (2013) 21:2113–21. doi:10.1038/mt.2013.151
118. Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med (2014) 6:242ra283. doi:10.1126/scitranslmed.3008825
119. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood (2013) 121:5113–23. doi:10.1182/blood-2013-02-486324
120. Naik S, Nicholas SK, Martinez CA, Leen AM, Hanley PJ, Gottschalk SM, et al. Adoptive immunotherapy for primary immunodeficiency disorders with virus-specific T lymphocytes. J Allergy Clin Immunol (2016) 137:1498–505.e1. doi:10.1016/j.jaci.2015.12.1311
121. O’Reilly RJ, Prockop S, Hasan AN, Koehne G, Doubrovina E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant (2016) 51:1163–72. doi:10.1038/bmt.2016.17
122. Asai O, Longo DL, Tian ZG, Hornung RL, Taub DD, Ruscetti FW, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest (1998) 101:1835–42. doi:10.1172/JCI1268
123. Lundqvist A, Mccoy JP, Samsel L, Childs R. Reduction of GVHD and enhanced antitumor effects after adoptive infusion of alloreactive Ly49-mismatched NK cells from MHC-matched donors. Blood (2007) 109:3603–6. doi:10.1182/blood-2006-05-024315
124. Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood (2010) 115:4293–301. doi:10.1182/blood-2009-05-222190
125. Zhang ZX, Yang L, Young KJ, Dutemple B, Zhang L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med (2000) 6:782–9. doi:10.1038/72303
126. Ford MS, Young KJ, Zhang Z, Ohashi PS, Zhang L. The immune regulatory function of lymphoproliferative double negative T cells in vitro and in vivo. J Exp Med (2002) 196:261–7. doi:10.1084/jem.20020029
127. Zhang ZX, Lian D, Huang X, Wang S, Sun H, Liu W, et al. Adoptive transfer of DNT cells induces long-term cardiac allograft survival and augments recipient CD4(+)Foxp3(+) Treg cell accumulation. Transpl Immunol (2011) 24:119–26. doi:10.1016/j.trim.2010.11.003
128. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8:726–36. doi:10.1038/nri2395
129. Ghannam S, Bouffi C, Djouad F, Jorgensen C, Noel D. Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res Ther (2010) 1:2. doi:10.1186/scrt2
130. English K, Wood KJ. Mesenchymal stromal cells in transplantation rejection and tolerance. Cold Spring Harb Perspect Med (2013) 3:a015560. doi:10.1101/cshperspect.a015560
131. Wing K, Ekmark A, Karlsson H, Rudin A, Suri-Payer E. Characterization of human CD25+ CD4+ T cells in thymus, cord and adult blood. Immunology (2002) 106:190–9. doi:10.1046/j.1365-2567.2002.01412.x
132. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol (2012) 3:51. doi:10.3389/fimmu.2012.00051
133. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol (2009) 133:22–6. doi:10.1016/j.clim.2009.06.001
134. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi:10.1182/blood-2010-10-311894
135. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front Immunol (2014) 5:16. doi:10.3389/fimmu.2014.00016
136. Brunstein CG, Miller JS, Mckenna DH, Hippen KL, Defor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127:1044–51. doi:10.1182/blood-2015-06-653667
137. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets – results of one year follow-up. Clin Immunol (2014) 153:23–30. doi:10.1016/j.clim.2014.03.016
138. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi:10.1126/scitranslmed.aad4134
139. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology (2012) 143:.e1201–2. doi:10.1053/j.gastro.2012.07.116
140. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther (2014) 22:1018–28. doi:10.1038/mt.2014.41
141. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17:931–43. doi:10.1111/ajt.14185
142. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant (2017) 17:917–30. doi:10.1111/ajt.14175
143. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood (2017) 129:238–45. doi:10.1182/blood-2016-07-727834
144. Brunstein CG, Miller JS, Cao Q, Mckenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi:10.1182/blood-2010-07-293795
145. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant (2017) 17(11):2945–54. doi:10.1111/ajt.14415
146. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353:179–84. doi:10.1126/science.aaf6756
Keywords: cell therapy, gene therapy, autologous, allogeneic, ex vivo therapy
Citation: Saudemont A, Jespers L and Clay T (2018) Current Status of Gene Engineering Cell Therapeutics. Front. Immunol. 9:153. doi: 10.3389/fimmu.2018.00153
Received: 21 September 2017; Accepted: 17 January 2018;
Published: 05 February 2018
Edited by:
Ulrike Koehl, Hannover Medical School, GermanyReviewed by:
Reinhard Henschler, Swiss Red Cross, SwitzerlandEvelyn Ullrich, Goethe University Frankfurt, Germany
Copyright: © 2018 Saudemont, Jespers and Clay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aurore Saudemont, YXVyb3JlLnguc2F1ZGVtb250QGdzay5jb20=